Embed Size (px)
Citation preview

Original article
Efficacy and safety of pramipexole in idiopathic restless legs syndrome:
A polysomnographic dose-finding study—The PRELUDE study*
Markku Partinen a,e,*,1, Kari Hirvonen a,b,2, Leni Jama a,2, Anniina Alakuijala a,c,2,
Christer Hublin d,e,2, Ilkka Tamminen f,2, Juergen Koester g,2, Juergen Reess h,2
a Skogby Sleep Clinic, Rinnekoti Research Centre, Kumputie 3, FI-02980 Espoo, Finlandb Neurotest Tampere Oy, Hatanpaan Valtatie 1, FIN-33100 Tampere, Finland
c Department of Clinical Neurophysiology, University Hospital of Helsinki, P.O. Box 340, FI-00029 Helsinki, Finlandd Finnish Institute of Occupational Health, Brain at Work Research Center, Topeliuksenkatu 41 a A, FI-00250 Helsinki, Finland
e Department of Neurology, University of Helsinki, FI-00014 Helsinki, Finlandf Medical Division, Boehringer Ingelheim Finland Ky, Tammasaarenkatu 5, FI-00180 Helsinki, Finland
g Medical Division, Boehringer Ingelheim International GmbH, Binger Str. 173, 55216 Ingelheim, Germanyh Department of Clinical Research CNS, Boehringer Ingelheim Pharma GmbH and Co. KG, Birkendorferstr.65, 88397 Biberach, Germany
Received 22 November 2005; received in revised form 16 February 2006; accepted 6 March 2006
Abstract
Background and purpose: To evaluate the effects of pramipexole (0.125–0.75 mg/d) on polysomnographic (PSG) measures and patient and
clinician ratings of restless legs syndrome (RLS).
Patients and methods: Patients (nZ109) with moderate to severe RLS were randomized to placebo or fixed doses of pramipexole during a
3-week, double-blind, placebo-controlled, dose-finding study.
Results: In each pramipexole dose group, the periodic limb movements during time in bed index (PLMI) decreased significantly, compared
with placebo (adjusted mean difference in log-transformed data: 0.125 mg,K1.54; 0.25 mg,K1.93; 0.50 mg,K1.89; and 0.75 mg,K1.52;
P!0.0001). At all doses, International RLS Study Group Rating Scale (IRLS) scores were also significantly reduced, with the greatest
adjusted mean reduction in the 0.50 mg group (K17.01). At all but the lowest pramipexole dose, the percentage of responders (R50%
reduction of IRLS score) was substantially higher than for placebo (61.9–77.3, vs 33.3%). In the pramipexole groups, 50.0–77.3% of patients
rated their condition as ‘much better’ or ‘very much better’, compared with 38.1% of patients in the placebo group (PZ0.0139 for the
0.50 mg dose). Clinical global impressions (CGI) scale ratings of ‘much improved’ or ‘very much improved’ were given to 61.9–86.4% of
patients in the pramipexole groups, compared with 42.9% in the placebo group (P!0.05 for the 0.25, 0.50, and 0.75 mg groups).
Pramipexole was well tolerated and did not produce somnolence at any dose.
Conclusion: Pramipexole is effective and safe in the treatment of both objective and subjective facets of RLS.
q 2006 Elsevier B.V. All rights reserved.
Keywords: Restless legs syndrome; RLS; Periodic limb movements; Sleep; Polysomnography; Pramipexole; Placebo-controlled
Sleep Medicine 7 (2006) 407–417
www.elsevier.com/locate/sleep
1389-9457/$ - see front matter q 2006 Elsevier B.V. All rights reserved.
doi:10.1016/j.sleep.2006.03.011
* PRELUDE, Pramipexole for RLS: efficacy and tolerability of the usage of dopamine agonists.* Corresponding author. Address: Skogby Sleep Clinic, Rinnekoti Research Centre, Kumputie 3, FI-02980 Espoo, Finland. Tel.:C358 9 8551345; fax:
C358 9 8551375.
E-mail address: [email protected] (M. Partinen).1 Dr Partinen has received a grant from the Rinnekoti Research Foundation for his studies on restless legs syndrome (RLS), and he has received honoraria,
totaling less than USD10,000 per year, for his presentations in postgraduate symposia on RLS.2 The author has no conflict of interest related to this article.

M. Partinen et al. / Sleep Medicine 7 (2006) 407–417408
1. Introduction
Restless legs syndrome (RLS) is a sensorimotor disorder
affecting approximately 5–10% of the population [1–3]. Its
prevalence increases with advancing age and is higher in
women than in men [2,4,5]. Other risk factors have also
been identified, including positive family history, preg-
nancy, iron deficiency, and renal disease [1,6–8].
The primary clinical manifestations are a strong urge to
move the legs, and also abnormal sensations in the legs,
which become worse at night or during periods of inactivity
and are relieved by movement [9]. Periodic limb move-
ments during sleep (PLMS) occur in 70–80% of cases and
are defined as repetitive movements, usually extension of
the big toe and/or flexion of the ankle, knee and hip, of
which patients are often unaware [10,11]. Not surprisingly,
these night time symptoms often cause disruption of sleep,
insomnia, and sleepiness during the day, and it is often
sleep-related complaints that bring patients to the attention
of a physician [12]. RLS is often missed or misdiagnosed,
and its symptoms can be disabling [11,12]. Fortunately,
when properly diagnosed and managed, RLS is typically
very responsive to dopaminergic treatment [13].
The pathophysiology of RLS is not well understood, but
ongoing research suggests that dysregulation of dopamine
function plays a role [10,14]. Consistent with this
hypothesis, medications that enhance dopamine function
are considered to be appropriate treatments for the disorder.
For patients with intermittent symptoms, the dopamine
precursor levodopa may be utilized, although its pharma-
cokinetic and pharmacodynamic characteristics (e.g. short
half-life and augmentation) limit its use in patients with
regular symptoms [13]. In patients with daily symptoms,
dopamine agonists have been suggested as the treatment of
choice [13] and, in fact, are considered first-line treatment in
the guidelines established by the American Academy of
Sleep Medicine (AASM) [15]. Other, nondopaminergic
medications are also used clinically, but they carry risks of
abuse, dependence, or daytime somnolence.
Pramipexole, a second-generation, non-ergot dopamine
agonist with selectivity for the D2/D3 receptor [16], has
been marketed for several years for treatment of all stages of
Parkinson’s disease (PD). Meanwhile, some small studies
have shown that pramipexole can reduce the symptoms of
RLS. In a double-blind, placebo-controlled, 10-week,
crossover trial (4 weeks of treatment, 2 weeks of washout,
and 4 weeks of alternate treatment), Montplaisir and
colleagues [17] examined the effects of pramipexole
(0.375–1.5 mg/d salt) in 10 patients with RLS. Pramipexole
produced an 84% reduction in subjective leg restlessness
relative to placebo, compared with a 39% reduction for
levodopa in a historical control group. PLMS was reduced
by 98%, and limb movements during wakefulness were
reduced by more than 80%. The investigators followed
seven patients for up to 8 months and found persistent
benefit [18]. In addition, two open-label studies have shown
that pramipexole reduces RLS symptoms, with effects of a
similar magnitude to those observed in the Montplaisir
study [19–21]. A recently published open-label trial by
Stiasny-Kolster and Oertel [22] reports that low doses of
pramipexole reduced polysomnographic (PSG) measures of
RLS symptomatology, even in patients whose symptoms
were inadequately controlled by levodopa.
The present trial was designed to replicate and extend
earlier findings by exploring the effects of different doses of
pramipexole in a larger group of patients after short-term
treatment.
2. Methods
2.1. Patients
The study population consisted of 28 male and 79 female
patients, aged 27–76 years, who were experiencing mainly
moderate to severe idiopathic RLS, according to criteria of
the International Restless Legs Syndrome Study Group
(IRLSSG), with scores of at least 15 on the International
RLS Study Group Rating Scale (IRLS) [9,23]. All
participants were required to have PLMS at least five
times per hour, as documented by baseline polysomno-
graphy, and also weekly RLS symptoms that had disrupted
sleep within the previous 3 months. Females of childbearing
potential and males were required to use adequate contra-
ception and females who were pregnant or breast-feeding
were excluded. Potential participants were also excluded for
medical contraindications to use of pramipexole, for
medical conditions or prescriptions that might influence
disease course (including but not limited to diabetes
mellitus, anemia, renal or hepatic disease), and for comorbid
conditions that may cause or complicate symptoms of RLS.
Patients who were currently (within the previous week)
receiving treatment for RLS were not enrolled, nor were
those who had been in an investigational drug study within
the previous 60 days. All patients provided written informed
consent prior to participating, and the study was approved
by the Ethics Committee of the Helsinki and Uusimaa
Hospital District.
2.2. Study design
The study was a 3week, double-blind, placebo-con-
trolled, parallel-group, fixed-dose trial designed to evaluate
the dose effects of pramipexole salt (0.125, 0.25, 0.50, and
0.75 mg/d, where 0.125 mg salt is equivalent to 0.088 mg
base) on objective and subjective ratings of RLS sympto-
matology. After completing baseline assessments, patients
were randomly assigned to 1 of 4 dose levels of pramipexole
or to placebo in a 1:1:1:1:1 ratio. All participants
randomized to active drug were started on 0.125 mg/d and
titrated up to their assigned dose in 4-day intervals. They
stayed on their assigned dose until the end of week 3. Doses

M. Partinen et al. / Sleep Medicine 7 (2006) 407–417 409
were taken once daily 2–3 h before bedtime. The primary
endpoint was change from baseline in the periodic limb
movements during time in bed index (PLMI). Secondary
assessments included additional PSG measures (described
in the following paragraph), along with changes from
baseline in subjective ratings on the IRLS and on clinician-
rated (clinical global impressions (CGI)) and patient-rated
(patient global impression (PGI)) scales. Another secondary
objective was to assess the effect of pramipexole on quality
of sleep and daytime well-being, as evaluated by self-
reported ratings of sleep quality, daytime somnolence, and
quality of life (QOL).
2.3. PSG measures
As an objective measure of periodic limb movements
during time in bed (asleep or awake) PLMI was assessed
by polysomnography, according to AASM guidelines, at
days 0 and 21. The procedure was performed in single
sound- and light-isolated bedrooms. Recordings were
started at the time of ‘lights off.’ After 8 h, the patients
were awakened (if asleep), and the recording was
stopped (‘lights on’). Several PSG measures were
evaluated as secondary efficacy endpoints, namely the
periodic limb movements during sleep index (PLMSI),
the periodic limb movements during wakefulness index
(PLMWI), the periodic limb movements during sleep
with arousal index (PLMAI), the total number of periodic
limb movements (PLM), the total number of PLMS, the
total number of periodic limb movements during sleep
with arousal (PLMSA), the total number of awakening-
s/arousals during sleep, and also sleep latency (SL), sleep
efficiency (SE), total sleeping time (TST), percentage of
delta sleep, and percentage of stage rapid eye movement
(sREM) sleep.
2.4. Subjective assessments
2.4.1. RLS Symptoms
By interview, patients completed the IRLS, formulated
by the IRLSSG [23], at baseline and at week 3. The
instrument is a 10-item self-reported assessment of the
severity of RLS symptoms over the preceding week in
5 degrees (e.g. ‘none’ to ‘very severe’); the maximum score
is 40. Change from baseline to the end of treatment was
taken as a measure of efficacy.
2.4.2. Sleepiness and sleep quality
The Epworth sleepiness scale (ESS) [24] is a self-
reported measure of daytime sleepiness. At baseline and at
the end of the 3 weeks of treatment, this instrument was used
to assess the likelihood of dozing off or falling asleep in
eight different situations, using a 4-point verbal rating scale
(‘no chance’ to ‘high chance’); items were summed to yield
a total score (range 0–24). Patients were asked to respond to
items with respect to the preceding week. Participants were
also asked to estimate their sleep quality, using the
subjective sleep quality scale (SSQ), (during the previous
night), on a scale of 1–10 in which 1 signifies ‘very poor
sleep quality’ and 10 ‘excellent sleep quality,’ and their
tiredness after waking up in the morning, on a scale from 1
to 10 in which 1 represents ‘very tired’ and 10 denotes ‘fully
awake’.
2.4.3. Quality of life
Health-related quality of life (QOL) was assessed by the
Short Form 36 Health Survey questionnaire (SF-36) [25] as
change from baseline to week 3. The SF-36 consists of four
physical domains (i.e. physical functioning, role physical,
bodily pain, and general health) and four nonphysical
domains (i.e. vitality, social functioning, role emotional,
and mental health index). A global evaluation of health is
also included. Each dimension is transformed to a scale
ranging from 0 to 100, with higher scores indicating better
health. The number of response alternatives per item varies
from 2 to 6. The SF-36 is designed to assess QOL during the
day and does not include parameters assessing well-being
during the night.
2.4.4. PGI and CGI scales
Patient and clinician ratings of improvement were
obtained by PGI and CGI [26]. In seven categories, these
instruments assess a patient’s overall condition at a given
timepoint in relation to baseline on a scale ranging from
‘very much better/improved’ to ‘very much worse’.
2.5. Adverse events
Adverse events (AEs) were assessed by non-probing
questions at each study visit and also by examination of
clinical test results (vital signs, electrocardiogram, blood
count and/or chemistry, and urinalysis) at regular intervals.
Events were recorded whether or not they were believed to
be causally related to the study drug. A serious adverse
event (SAE) was defined as any AE that resulted in death,
was immediately life-threatening, resulted in persistent or
significant disability/incapacity, required or prolonged
patient hospitalization, was a congenital anomaly/birth
defect, or was deemed serious for any other reason
representing a significant hazard. Sudden onset of sleep
(SOOS) during daily activities was considered to be a
significant AE.
2.6. Data analysis
The intent-to-treat principle was used to include as many
patients as possible in the analyses, according to protocol.
The analyses therefore encompassed all randomized
patients with at least baseline data and post-treatment
polysomnography. Continuous or discrete parameters were
analyzed by descriptive statistics or by frequency tables,
respectively.

M. Partinen et al. / Sleep Medicine 7 (2006) 407–417410
The primary analysis was planned and performed using
an analysis of covariance (ANCOVA) model to compare the
efficacy of different doses of pramipexole for reducing the
PLMI after 3 weeks of randomized treatment. For
application of ANCOVA, the distribution of PLMI indices
required log transformation. Therefore, the final PSG
assessment was taken as the response parameter (instead
of change from baseline), and the log-transformed baseline
PLMI score was used as the covariate. Hierarchical testing
was utilized, sequentially comparing placebo with the
highest to lowest pramipexole dose. All tests were
conducted as 2-tailed at the 5% level of significance.
Nonparametric analyses (Wilcoxon–Mann–Whitney tests)
of the original data were performed as well. The hierarchical
testing allows inferences to be made on all primary endpoint
comparisons, while preserving an overall type I error
probability of 0.05.
Every effort was made to collect PSG and other data at
each visit. However, PSG and other data were collected
prematurely for patients wishing to drop out of double-blind
treatment. Randomly missing data (due, for instance, to a
mechanical failure) were estimated either by linear
interpolation of adjacent data or, if no subsequent data
were available, by a last observation carried forward
(LOCF) approach. For patients discontinuing due to
unexpected worsening of RLS, the missing data were
imputed by the least favorable data prior to discontinuation.
For patients who missed a visit due to other reasons, the
missing data were estimated by LOCF. The evaluability of
patients with protocol deviations likely to confound
treatment response was either predefined by the protocol
or decided before unblinding. For secondary endpoints,
there were no multiplicity adjustments.
All subjects who received at least one dose of study
medication were included in the analysis of safety
(NZ109). The occurrence of AEs was analyzed both by
randomized dose group (to permit a comparison of the
Fig. 1. Patient d
different pramipexole target doses) and by ‘treatment at
onset’ (to elucidate the AE pattern at the different
pramipexole dose levels).
3. Results
3.1. Patients and treatment
One hundred and forty-one patients were recruited for
the study. Among them, 32 patients were screening failures
and were not entered, leaving 109 patients who were
randomized to active treatment or placebo (Fig. 1). Two of
them withdrew prematurely: one patient in the placebo
group was lost to follow-up, without providing a second
PSG assessment, and one patient in the 0.125 mg group was
withdrawn due to an AE. For inclusion in the intent-to-treat
population, patients had to provide two PSG assessments (at
baseline and at the end of 3 weeks of treatment). One patient
from the 0.75 mg group was assessed 48 h after discontinu-
ation of treatment.
All 107 patients included in the intent-to-treat population
were Caucasian, and most (73.8%) were female. The gender
distribution was similar among treatment groups, although
the 0.75 mg group had fewer females (61.9%) and the
placebo and 0.50 mg groups had more females (81.0 and
81.8%, respectively) than the overall study population did.
Patients were a mean age of 56.2 years (standard deviation
(SD)Z10.9), except that the 0.125 mg group was slightly
but not significantly older (60.0 years, SDZ10.1). Patient
groups did not differ with respect to body mass index or
proportion previously treated (vs de novo), except in the
0.50 mg group, where 59.1% of participants had received
prior treatment (vs 41.1% overall). Table 1 summarizes
demographic and disease-related characteristics of the study
participants.
isposition.

Table 1
Demographic and disease-related characteristics in the intent-to-treat population
PBO N (%) PPX 0.125 mg N
(%)
PPX 0.25 mg N
(%)
PPX 0.50 mg N
(%)
PPX 0.75 mg
N (%)
Total N (%)
Number of patients 21 21 22 22 21 107
Gender
Male 4 (19.0) 6 (28.6) 6 (27.3) 4 (18.2) 8 (38.1) 28 (26.2)
Age (Mean [SD] years) 53.3 [11.1] 60.0 [10.1] 54.8 [10.9] 58.4 [9.5] 54.5 [12.2] 56.2 [10.9]
RLS duration (Mean [SD] years) 2.7 [10.1] 5.1 [11.1] 6.1 [10.6] 5.3 [10.6] 4.5 [10.3] 4.8 [10.4]
Previously treated for RLS 7 (33.3) 7 (33.3) 8 (36.4) 13 (59.1) 9 (42.9) 44 (41.1)
IRLS score (Mean, [SD]) 22.9 [4.2] 22.4 [4.7] 23.0 [3.4] 23.6 [3.7] 21.7 [4.6] 22.7 [4.1]
PLMI (median) 51.75 63.10 36.85 35.90 40.30
PLMS (median) 42.85 22.30 29.40 24.75 29.68 28.60
Total sleeping time (median, h) 6.4 5.7 6.8 6.1 6.6 6.3
Sleep efficiency (median, %) 84.55 84.00 89.65 82.55 88.20 85.10
PBO, placebo; PPX, pramipexole; SD, standard deviation; IRLS, International RLS Study Group Rating Scale; PLMI, periodic limb movements during time in
bed index; PLMS, periodic limb movements during sleep.
M. Partinen et al. / Sleep Medicine 7 (2006) 407–417 411
3.2. Objective assessments
Primary and secondary objective measures of efficacy
showed pramipexole to be superior to placebo. In particular,
the ANCOVA analysis of log-transformed PLMI showed
that the adjusted means at the end of week 3 were
significantly smaller in all active-dose groups than in the
placebo group (P!0.0001). In adjusted mean differences
from placebo, the largest effects (log-transformed data)
were in the 0.25 and 0.50 mg groups (Table 2). Nonpara-
metric analyses of PLMI changes from baseline (original
data) confirmed the ANCOVA results with P!0.001 for all
comparisons vs placebo. Pramipexole dose-independently
reduced PLMI at endpoint; the median change in each
treatment group was K3.00 for placebo, K52.70 for
Table 2
Median changes from baseline in PLM parameters
Parameter PBO PPX
0.125 mg
P
0
Number of patients 20 21
PLM during time in
bed index (PLMI)K3.00 K52.70b
PLM during sleep
index (PLMSI)K3.45 K20.90b
PLM during
wakefulness
index (PLMWI)
K11.00 K41.20a
PLM during sleep
with arousal
index (PLMAI)
K1.85 5.20
Total number of
awakenings/arousalsK22.50 8.00
Total number of PLM K24.00 K404.000b KTotal number of PLM
during sleepK19.50 K126.00b K
Total number of
PLM during sleep
with arousal
K9.00 K30.00
PLM, periodic limb movements; PBO, placebo; PPX, pramipexole.a P for difference from placebo !0.05 by Wilcoxon–Mann–Whitney test.b P for difference from placebo !0.01 by Wilcoxon–Mann–Whitney test.c P for difference from placebo !0.001 by Wilcoxon–Mann–Whitney test.
0.125 mg, K31.05 for 0.25 mg, K26.55 for 0.50 mg, and
K30.00 for 0.75 mg.
Data distributions for all other PSG parameters were
checked for normality. Since, all parameters except ‘SL’
and ‘percentage of time spent in sREM sleep’ showed a non-
normal distribution (and hence failed to meet the
assumption of normality required for a linear model such
as ANCOVA), natural logarithmic transformations were
used. In consequence, data were lost for a small number of
patients where baseline or on-treatment measurements were
zero. For all ANCOVA analyses, age was included as a
covariate. However, age was not found to have a significant
influence as a covariate for any of the parameters.
Pramipexole was significantly superior to placebo for
values of PLMI, PLMSI, PLMWI, total PLM, and total
PX
.25 mg
PPX
0.50 mg
PPX
0.75 mg
22 22 21
K31.05c K26.55c K30.00c
K26.65c K22.45b K17.00b
K36.50a K38.45a K21.30
K2.35 K3.25 K2.90
4.00a K4.00 1.00
244.00c K212.50b K180.00b
147.00c K135.50b K111.00c
K16.00 K21.50 K18.00
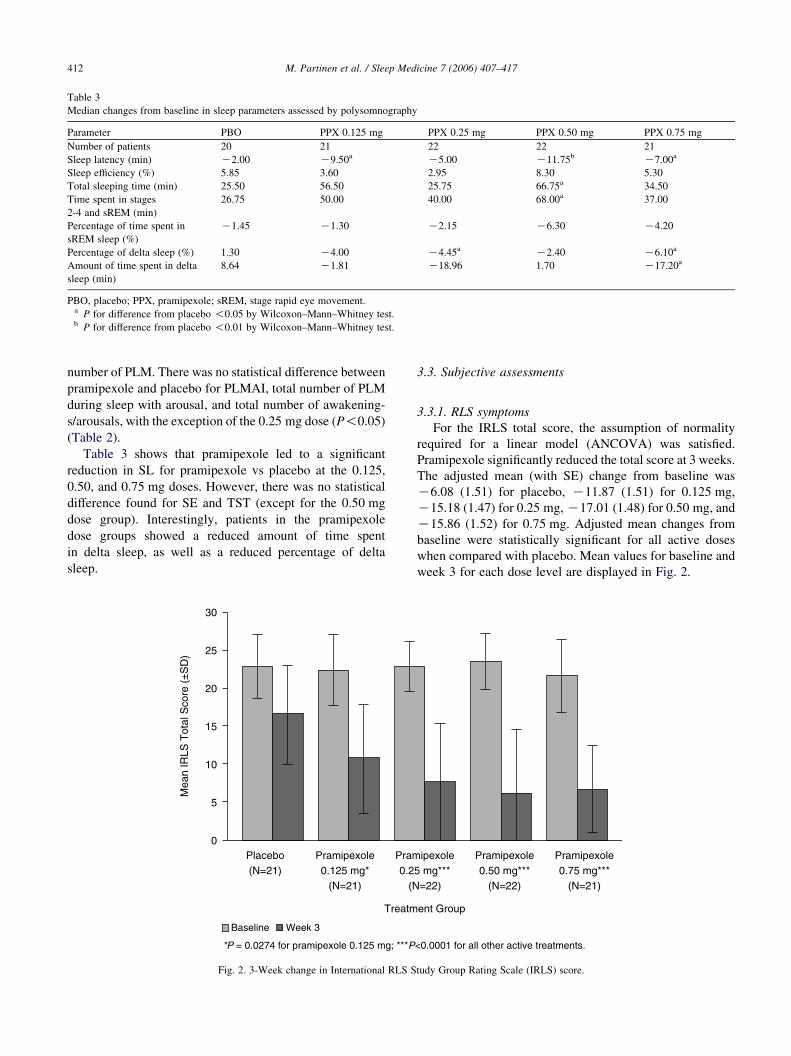
Table 3
Median changes from baseline in sleep parameters assessed by polysomnography
Parameter PBO PPX 0.125 mg PPX 0.25 mg PPX 0.50 mg PPX 0.75 mg
Number of patients 20 21 22 22 21
Sleep latency (min) K2.00 K9.50a K5.00 K11.75b K7.00a
Sleep efficiency (%) 5.85 3.60 2.95 8.30 5.30
Total sleeping time (min) 25.50 56.50 25.75 66.75a 34.50
Time spent in stages
2-4 and sREM (min)
26.75 50.00 40.00 68.00a 37.00
Percentage of time spent in
sREM sleep (%)
K1.45 K1.30 K2.15 K6.30 K4.20
Percentage of delta sleep (%) 1.30 K4.00 K4.45a K2.40 K6.10a
Amount of time spent in delta
sleep (min)
8.64 K1.81 K18.96 1.70 K17.20a
PBO, placebo; PPX, pramipexole; sREM, stage rapid eye movement.a P for difference from placebo !0.05 by Wilcoxon–Mann–Whitney test.b P for difference from placebo !0.01 by Wilcoxon–Mann–Whitney test.
M. Partinen et al. / Sleep Medicine 7 (2006) 407–417412
number of PLM. There was no statistical difference between
pramipexole and placebo for PLMAI, total number of PLM
during sleep with arousal, and total number of awakening-
s/arousals, with the exception of the 0.25 mg dose (P!0.05)
(Table 2).
Table 3 shows that pramipexole led to a significant
reduction in SL for pramipexole vs placebo at the 0.125,
0.50, and 0.75 mg doses. However, there was no statistical
difference found for SE and TST (except for the 0.50 mg
dose group). Interestingly, patients in the pramipexole
dose groups showed a reduced amount of time spent
in delta sleep, as well as a reduced percentage of delta
sleep.
Fig. 2. 3-Week change in International RLS S
3.3. Subjective assessments
3.3.1. RLS symptoms
For the IRLS total score, the assumption of normality
required for a linear model (ANCOVA) was satisfied.
Pramipexole significantly reduced the total score at 3 weeks.
The adjusted mean (with SE) change from baseline was
K6.08 (1.51) for placebo, K11.87 (1.51) for 0.125 mg,
K15.18 (1.47) for 0.25 mg,K17.01 (1.48) for 0.50 mg, and
K15.86 (1.52) for 0.75 mg. Adjusted mean changes from
baseline were statistically significant for all active doses
when compared with placebo. Mean values for baseline and
week 3 for each dose level are displayed in Fig. 2.
tudy Group Rating Scale (IRLS) score.

M. Partinen et al. / Sleep Medicine 7 (2006) 407–417 413
Data for IRLS ratings were also evaluated as proportion
of responders. In all but the 0.125 mg group, the proportion
of patients who experienced at least a 50% reduction in RLS
severity was significantly greater than in the placebo group.
Using categorical data-analysis techniques, pair-wise com-
parisons were performed for the number of responders in
each active-treatment group vs placebo. Using the Fisher’s
exact test method, statistically significant results were
obtained in the 0.25 mg group (PZ0.023), the 0.50 mg
group (PZ0.004), and the 0.75 mg group (PZ0.006). The
percentage of responders in each of the active-treatment
groups was 61.9% (0.125 mg group), 68.2% (0.25 mg
group), 77.3% (0.50 mg group), and 76.2% (0.75 mg
group). Thus, the higher-dose groups of 0.25 mg, 0.50 mg,
and 0.75 mg showed a larger clinical response than the
0.125 mg group.
3.3.2. Ratings of improvement and response to treatment
Responder rates in PGI (combined categories ‘much
better’ and ‘very much better’) were higher in the
pramipexole groups than in the placebo group. Using the
Fisher’s exact test method, statistically significant results
were obtained for the 0.50 mg group (PZ0.039) and the
0.75 mg group (PZ0.041). At week 3, the proportion of
patients who rated their current condition as ‘very much
better’ was 27.2% in the 0.50 mg group and 23.8% in the
0.75 mg group, compared with 4.8% in the placebo group.
Fifty percent of patients in the 0.50 mg group and 33.3% of
patients in the 0.75 mg group were classified as ‘much
better,’ compared with 33.3% in the placebo group. No
Fig. 3. Clinical global impressions scale (CG
patients in any treatment group assessed their condition as
‘much worse’ or ‘very much worse’.
Generally, the clinicians’ assessment of patients’ improve-
ment was similar to the patients’ own assessment. For CGI
ratings of global improvement (Fig. 3), more than 60% of
patients in all pramipexole dose groups were rated by the
investigator as being ‘much improved’ or ‘very much
improved’ after 3 weeks of therapy, compared with 42.9%
of patients in the placebo group. A comparison of the lowest
active dose (0.125 mg) with placebo was not statistically
significant (PO0.31); however, the comparison for the highest
active doses (0.25, 0.50, and 0.75 mg) did reach statistical
significance (PZ0.022, 0.001, and 0.008, respectively).
Similar results were obtained for CGI ratings of severity of
illness. At baseline, the majority of patients were considered
either ‘markedly ill’ (38.3%) or ‘moderately ill’ (34.6%). At
week 3, in stark contrast, a substantial proportion (overall,
30.8%) of patients were evaluated as ‘not at all ill,’ including
9.5% of the placebo group and 19.0% (0.125 mg), 36.4%
(0.25 mg), 54.5% (0.50 mg), and 33.3% (0.75 mg) of the
pramipexole groups. Fig. 4 shows the percentage of patients in
each treatment group who exhibited improvement in severity
ratings at the end of week 3.
Moreover, pramipexole increased the percentage of
patients rated by clinicians as showing a therapeutic effect.
For 47.6% of patients in the placebo group, a ‘marked’ or
‘moderate’ therapeutic effect was seen, compared with
66.7–90.5% of patients in the pramipexole groups. A
comparison of the 0.125 mg group with placebo was not
statistically significant (PZ0.162), but for the higher-dose
groups (0.25, 0.50, and 0.75 mg), the comparisons with
I): global improvement after 3 weeks.

Fig. 4. Clinical global impressions scale (CGI): severity of illness after 3 weeks.
M. Partinen et al. / Sleep Medicine 7 (2006) 407–417414
placebo were statistically significant (PZ0.0127, 0.002, and
0.004, respectively). Pramipexole did not affect the number
of clinician-rated side effects reported at the end of week 3.
3.3.3. Sleep quality and sleepiness
Evaluated by ESS, pramipexole likewise showed no
effect on daytime sleepiness. All groups had mean baseline
ESS scores in the range of healthy persons and, after 3
weeks, all groups showed minimal change, with no
significant difference between placebo and any active
treatment (Table 4).
At baseline, subjective sleep quality (SSQ; median over 1
week) ranged from6.4 to 7.0 among the various groups. By the
end of week 3, it had improved in all groups, the improvement
being substantial in the higher-dose groups (1.01, 1.44, and
1.36 for the 0.25, 0.50, and 0.75 mg groups, respectively).
For placebo and for the 0.125 mg group, only small increases
(0.54 and 0.59, respectively) were observed.
For subjective tiredness after awakening, a baseline of
6.5–7.3 (median over 1 week) improved in all treatment
groups, with larger median increases for the 0.25 and
Table 4
Median [range] changes from baseline in Epworth Sleepiness Scale (ESS)
Parameter PBO PPX 0.125 mg
Number of patients 20 21
ESS median change, range K1.0 [K6, 3] 0.0 [K4, 3]
PBO, placebo; PPX, pramipexole.
0.75 mg groups (0.82 and 0.70, respectively) than for
placebo (0.50) (Table 5).
3.3.4. Quality of life
At baseline, the mean overall index for social
functioning (87.38G17.55) was comparable to a healthy
Finnish population (81.20G19.30) [27]. Changes in
health-related QOL, as measured by the generic SF-36
instrument, were not significantly different in the
pramipexole and placebo groups for 7 subscales, with
the exception of a positive effect of pramipexole on the
social functioning subscale. While the adjusted mean SF-
36 social functioning subscore decreased in the placebo
group, it increased in the pramipexole groups. Pair-wise
comparisons of pramipexole doses vs placebo showed
significant adjusted mean differences for the 0.125 mg
(PZ0.041), 0.50 mg (PZ0.031), and 0.75 mg groups
(PZ0.008).
3.3.5. Safety and tolerability
The overall incidence of AEs was similar between
placebo and pramipexole total (77.3 vs. 74.7%). Among
PPX 0.25 mg PPX 0.50 mg PPX 0.75 mg
22 22 21
0.0 [K5, 4] 0.0 [K4, 3] 1.0 [K13, 2]

Table 5
Subjective sleep quality assessment
PBO
(NZ21)
PPX 0.125 mg
(NZ21)
PPX 0.25 mg
(NZ22)
PPX 0.50 mg
(NZ22)
PPX 0.75 mg
(NZ21)
Total
(NZ107)
Sleep quality during the previous night
Baseline (visits 1–2)
median
7.00 6.43 6.62 6.50 6.71 6.67
Week 3 (visits 2–4,
maintenance) median
7.38 7.30 7.59 7.58 7.20 7.44
Change from baseline
median
0.54 0.59 1.01 1.44 1.36 0.77
Tiredness after waking up in the morning
Baseline (visits 1–2)
median
6.50 7.29 6.93 7.20 6.50 6.86
Week 3 (visits 2–4,
Maintenance) median
7.00 7.50 7.80 7.41 7.20 7.40
Change from baseline
median
0.50 0.22 0.82 0.18 0.70 0.50
PBO, placebo; PPX, pramipexole. For the evaluation of baseline, the last 7 days prior to randomization were used.
M. Partinen et al. / Sleep Medicine 7 (2006) 407–417 415
the different doses of pramipexole, the highest frequency
of AEs was reported for 0.125 mg (44.8%, compared
with 33.3% for 0.25 mg, 31.8% for 0.50 mg, and 27.3%
for 0.75 mg). Since all patients had started their
treatment at 0.125 mg/d, this pattern reflects a titration
of pramipexole during which, in general, the increasing
dosage led neither to new AEs nor to a worsening of
existing ones.
The most frequently reported AEs (O5%) more often
reported in the combined pramipexole groups than in the
placebo group were nausea (14.9 vs. 4.5%) and nasophar-
yngitis (6.9 vs. 0.0%). Conversely, fatigue (22.7 vs. 18.4%)
and headache (31.8 vs. 19.5%) were reported more
frequently in the placebo group than in the combined
pramipexole groups (Table 6).
Overall, 45.0% of patients reported AEs related to study
drug, as assessed by an investigator. In the placebo group,
the proportion was 50.0% of patients, while in the
Table 6
Adverse events occurring in O5% of the population
MedDRA system organ
class/preferred term
PBO DB PPX PPX
N (%) N (%) N
Total treated 22 100.0 87 100.0 87
Total with any adverse event 17 77.3 65 74.7 39
Nervous system disorders 8 36.4 30 34.5 14
Headache 7 31.8 17 19.5 7
Gastrointestinal disorders 2 9.1 21 24.1 11
Nausea 1 4.5 13 14.9 7
General disorders and
administration-site conditions
6 27.3 17 19.5 14
Fatigue 5 22.7 16 18.4 14
Infections and infestations 3 13.6 15 17.2 3
Nasopharyngitis 0 0.0 6 6.9 2
Musculoskeletal and connective
tissue disorders
2 9.1 4 4.6 2
Psychiatric disorders 3 13.6 3 3.4 2
PBO, placebo; DB, double-blind; PPX, pramipexole.
pramipexole groups it ranged between 31.8% (0.75 mg)
and 61.9% (0.125 mg). The most frequent study-drug-
related AEs were fatigue (overall, 16.5%), nausea (overall,
12.8%), and headache (overall, 5.5%). All other drug-
related AEs occurred in less than 5.0% of all patients.
The majority of AEs had a maximum intensity of mild or
moderate; only three patients (3.4%) reported AEs with
severe intensity. Significant AEs that led to study
discontinuation or to a decrease in study drug dose were
reported for two patients receiving pramipexole and for no
patient in the placebo group. No patient in any group
experienced an SAE.
Aggravation of RLSwas observed in four patients (3.7%):
one at 0.125 mg, two at 0.25 mg, and one at 0.50 mg.
Somnolence was reported by three patients (2.8%), all at the
0.125 mg level.
No significant change in any laboratory parameter or
vital sign (blood pressure, pulse rate, or weight) was
0.125 mg PPX 0.25 mg PPX 0.50 mg PPX 0.75 mg
(%) N (%) N (%) N (%)
100.0 66 100.0 44 100.0 22 100.0
44.8 22 33.3 14 31.8 6 27.3
16.1 10 15.2 6 13.6 2 9.1
8.0 4 6.1 5 11.4 2 9.1
12.6 5 7.6 4 9.1 1 4.5
8.0 3 4.5 2 4.5 1 4.5
16.1 2 3.0 2 4.5 0 0.0
16.1 1 1.5 1 2.3 0 0.0
3.4 8 12.1 2 4.5 2 9.1
2.3 2 3.0 1 2.3 1 4.5
2.3 1 1.5 0 0.0 1 4.5
2.3 1 1.5 0 0.0 0 0.0

M. Partinen et al. / Sleep Medicine 7 (2006) 407–417416
observed in any pramipexole group, compared with the
placebo group. Only one patient reported symptomatic
orthostatic hypotension during the study. At screening, this
patient had exhibited asymptomatic orthostatic hypotension
(systolic change R20 mmHg in combination with a
diastolic change R10 mmHg from supine to standing).
4. Discussion
Overall, pramipexole was highly effective and safe
for key RLS parameters. In all active-treatment groups,
the objective measure of PLMI was significantly reduced
(P!0.001); the largest reduction was observed at a dosage
of 0.125 mg/d. This marked improvement in the primary
endpoint demonstrated that pramipexole rapidly controlled
RLS symptoms. Statistical improvement from baseline was
shown for pramipexole vs placebo for the total IRLS score
(P%0.001), the CGI global assessment (P!0.05), and the
CGI ‘severity of illness’ assessment (P!0.05) across all
dosing groups.
Secondary endpoints analyzed by polysomnography
evaluated statistical significant changes from baseline for
PLMSI, PLMWI, and total number of PLM with the
treatment of pramipexole. No statistical difference was
found for SE and TST. This is in line with findings
from PSG studies with other dopamine agonists [12,28].
Pramipexole numerically decreased the PLM with arousal
compared with placebo, but the difference did not reach
statistical difference. The total number of arousals
decreased with placebo and increased slightly in the
pramipexole groups. However, this is in contrast to the
improvements seen in the subjective sleep quality assess-
ment. The median change from baseline for ‘sleep quality
during the previous night’ and ‘tiredness after waking up in
the morning’, as assessed using the SSQ, favored
pramipexole over placebo for these endpoints.
A disconcordance of ‘objective sleep findings’ and the
subjective assessments was also reported with other
dopaminergic drugs [12,28]. There is no conclusive
explanation for this finding at the present time.
The safety and tolerability of pramipexole over 3 weeks
was comparable to placebo and comparable to that reported
for other agents in this class [28–30]. The incidence of new
AEs or aggravation of existing ones did not increase with
increasing doses of pramipexole. The rapid up-titration to
effective dosing was well tolerated.
Previous studies have shown that RLS has an impact on
QOL comparable to that of many other chronic diseases
[31,32]. In the present study, statistical improvement over
placebo in ‘social functioning’ was shown for pramipexole
using the SF-36 health questionnaire. The fact that the
SF-36 does not account for nighttime symptoms from RLS
bypasses an important aspect of this disorder.
In summary, the findings of the present trial support
previous, smaller-scale studies in showing that
pramipexole produced improvement in both objective
and subjective measures of RLS symptomatology at all
doses tested and was generally well tolerated. The effects
of pramipexole were robust and were apparent within 3
weeks of treatment.
The outcomes suggest that pramipexole has considerable
promise in patients with RLS in the dose range tested. The
lowest dose, 0.125 mg, was safe and effective and generally
indistinguishable from the higher doses, suggesting that it
may be a reasonable place to start when targeting maximum
dose for optimum efficacy.
Acknowledgements
This study was supported by Boehringer-Ingelheim
International GmbH. The authors wish to thank the main
research nurse, M. Halavaara, and the personnel of the
Haaga Neurological Research Centre in Helsinki, Finland,
for providing sleep laboratory facilities and help in the
polysomnographic recordings, and also L.H. Brauer, PhD,
for her help in the preparation of the article, and R. Morton
for his statistical contribution.
References
[1] Phillips B, Young T, Finn L, et al. Epidemiology of restless legs
symptoms in adults. Arch Intern Med 2000;160:2137–41.
[2] Rothdach AJ, Trenkwalder C, Haberstock J, et al. Prevalence and risk
factors of RLS in an elderly population: the MEMO study. Memory
and morbidity in augsburg elderly. Neurology 2000;54:1064–8.
[3] Hening W, Walters AS, Allen RP, et al. Impact, diagnosis and
treatment of restless legs syndrome (RLS) in a primary care
population: the REST (RLS epidemiology, symptoms, and treatment)
primary care study. Sleep Med 2004;5:237–46.
[4] Lavigne GJ, Montplaisir JY. Restless legs syndrome and sleep
bruxism: prevalence and association among Canadians. Sleep 1994;
17:739–43.
[5] Berger K, Luedemann J, Trenkwalder C, et al. Sex and the risk of
restless legs syndrome in the general population. Arch Intern Med
2004;164:196–202.
[6] Earley CJ, Allen RP, Beard JL, Connor JR. Insight into the
pathophysiology of restless legs syndrome. J Neurosci Res 2000;62:
623–8.
[7] Earley CJ. Clinical practice. Restless legs syndrome. N Engl J Med
2003;348:2103–9.
[8] Sonka K, Kemlink D. Restless legs syndrome in 2004. Prague Med
Rep 2004;105:337–56.
[9] Allen RP, Picchietti D, Hening WA, et al. Restless legs syndrome:
diagnostic criteria, special considerations, and epidemiology. A report
from the restless legs syndrome diagnosis and epidemiology work-
shop at the national institutes of health. Sleep Med 2003;4:101–19.
[10] Allen RP, Earley CJ. Restless legs syndrome: a review of clinical and
pathophysiologic features. J Clin Neurophysiol 2001;18:128–47.
[11] Stiasny-Kolster K, Benes H, Peglau I, Hornyak M, et al. Effective
cabergoline treatment in idiopathic restless legs syndrome. Neurology
2004;63:2272–9.
[12] Allen R, Becker PM, Bogan R, et al. Ropinirole decreases periodic leg
movements and improves sleep parameters in patients with restless
leg syndrome. Sleep 2004;27:907–14.

M. Partinen et al. / Sleep Medicine 7 (2006) 407–417 417
[13] Silber MH, Ehrenberg BL, Allen RP, et al. An algorithm for the
management of restless legs syndrome. Mayo Clin Proc 2004;79:
916–22.
[14] Michaud M, Dumont M, Selmaoui B, et al. Circadian rhythm of
restless legs syndrome: relationship with biological markers. Ann
Neurol 2004;55:372–80.
[15] Chesson Jr AL, Wise M, Davila D, et al. Practice parameters for the
treatment of restless legs syndrome and periodic limb movement
disorder. An American academy of sleep medicine report. Standards
of practice committee of the American academy of sleep medicine.
Sleep 1999;22:961–8.
[16] Deleu D, Northway MG, Hanssens Y. Clinical pharmacokinetic and
pharmacodynamic properties of drugs used in the treatment of
Parkinson’s disease. Clin Pharmacokinet 2002;41:261–309.
[17] Montplaisir J, Nicolas A, Denesle R, Gomez-Mancilla B. Restless legs
syndrome improved by pramipexole: a double-blind randomized trial.
Neurology 1999;52:938–43.
[18] Montplaisir J, Denesle R, Petit D. Pramipexole in the treatment of
restless legs syndrome: a follow-up study. Eur J Neurol 2000;7(Suppl.
1):27–31.
[19] Lin SC, Kaplan J, Burger CD, Fredrickson PA. Effect of pramipexole
in treatment of resistant restless legs syndrome. Mayo Clin Proc 1998;
73:497–500.
[20] Lin S, Kaplan J, Burger C, Fredrickson P. Long-term use of
pramipexole in the treatment of restless legs syndrome: a two-year
follow-up. The sixth international congress of Parkinson’s disease and
movement disorders, Barcelona, Spain, 11–15 June, 2000.
[21] Becker PM, Ondo W, Sharon D. Encouraging initial response of
restless legs syndrome to pramipexole. Neurology 1998;51:1221–3.
[22] Stiasny-Kolster K, Oertel WH. Low-dose pramipexole in the
management of restless legs syndrome. Neuropsychobiology 2004;
50:65–70.
[23] Walters AS, LeBrocq C, Dhar A, et al. For the international restless
legs syndrome study group. Validation of the international restless
legs syndrome study group rating scale for restless legs syndrome.
Sleep Med 2003;4:121–32.
[24] Johns MW. A new method for measuring daytime sleepiness: the
Epworth sleepiness scale. Sleep 1991;14:540–5.
[25] Jenkinson C, Coulter A, Wright L. Short form 36 (SF36) health survey
questionnaire: normative data for adults of working age. Br Med J
1993;306:1437–40.
[26] National Institute of Mental Health (NIMH). Early clinical drug
evaluation unit (ECDEU). Clinical global impressions. In: Guy W,
editor. ECDEU assessment manual for psychopharmacology. rev.
Rockville, MD: NIMH USA; 1976.
[27] Solovieva S, Santavirta N, Santavirta S, Konttinen YT. Assessing
quality of life in individuals with hereditary blood coagulation
disorders. Qual Life Res 2004;13:987–1000.
[28] Trenkwalder C, Hundemer HP, Lledo A, et al. Efficacy of pergolide in
treatment of restless legs syndrome: the PEARLS study. Neurology
2004;62:1391–7.
[29] Benes H, Heinrich CR, Ueberall MA, Kohnen R. Long-term safety
and efficacy of cabergoline for the treatment of idiopathic restless legs
syndrome: results from an open-label 6-month clinical trial. Sleep
2004;27:674–82.
[30] Adler CH, Hauser RA, Sethi K, et al. Ropinirole for restless legs
syndrome. Neurology 2004;62:1405–7.
[31] Abetz L, Allen R, Follet A, et al. Evaluating the quality of life
of patients with restless legs syndrome. Clin Ther 2004;26:
925–35.
[32] Allen RP, Walters AS, Montplaisir J, et al. Restless legs syndrome
prevalence and impact: REST general population study. Arch Intern
Med 2005;165:1286–92.





























