Embed Size (px)
Citation preview

Perturbation of SphingolipidMetabolism and Ceramide Production
in HIV-DementiaNorman J. Haughey, PhD,1,2 Roy G. Cutler, MA,2 Anita Tamara, BSc,2 Justin C. McArthur, MD,1
Diana L. Vargas, MD,1 Carlos A. Pardo, MD,1,3 Jadwiga Turchan, PhD,1
Avindra Nath, MD,1 and Mark P. Mattson, PhD2,4
Infection by the human immunodeficiency virus type 1 (HIV-1) often results in neurological dysfunction including HIVdementia (HIVD). Alterations in cytokine and redox balance are thought to play important roles in the pathogenesis ofHIVD, but the specific mechanisms underlying neuronal dysfunction and death are unknown. Activation of cytokinereceptors and oxidative stress can induce the production of ceramide from membrane sphingomyelin, and recent findingssuggest that ceramide is an important mediator of a form of programmed cell death called apoptosis. We now report thatlevels of ceramide, sphingomyelin, and hydroxynonenal (HNE) are significantly increased in brain tissues and cerebro-spinal fluid of HIVD patients. Exposure of cultured neurons to the neurotoxic HIV proteins gp120 and Tat resulted inincreased cellular levels of sphingomyelin, ceramide, and HNE. The ceramide precursor palmitoyl-CoA sensitized neu-rons to Tat and gp120 toxicity, whereas an inhibitor of ceramide production reduced Tat and gp120-induced increasesof ceramide and HNE and protected the neurons from Tat and gp120-induced death. These results suggest that HIV-1infection may promote a lipid imbalance in neural cells, resulting in an overproduction of ceramide and consequentcellular dysfunction and death.
Ann Neurol 2004;55:257–267
In addition to a structural role for sphingolipids in eu-karoytic cell membranes, products of sphingolipid me-tabolism serve important signaling functions and mod-ulate a wide variety of inter cellular and intracellularevents. Sphingomyelin is the most abundant sphingo-lipid in brain and is an important constituent of spe-cialized signaling domains known as lipid rafts.1 Spin-gomyelin is metabolized by sphingomyelinases togenerate phosphocholine and a potent biomodulatorcalled ceramide. Ceramide is a ubiquitous second mes-senger that modulates cell differentiation, proliferation,survival, and apoptosis.2,3 There are two primary typesof sphingomyelinase with distinct subcellular localiza-tion. Acidic sphingomyelinase cleaves sphingomyelin inthe endoplasmic reticulum and in lysosmes.4 Neutralsphingomyelinase colocalizes with sphingomyelin inlipid rafts and primarily cleaves sphingomyelin locatedin the plasma membrane.5 Both forms of sphingomy-
elinase are present in brain where increases of ceramidecan sensitize neurons to excitotoxic damage and pro-mote apoptosis.6–8 There is considerable evidence thatlinks the production of reactive oxygen species (ROS)with ceramide generation. Pharmacological inhibitionof ROS with the antioxidants N-acetlycysteine (NAC),pyrrolidine dithiocarbamate (PDTC), or �-tocopherolprevent the generation of ceramide and rescue cellsfrom death induced by tumor necrosis factor–�, Fasligand, hypoxia, and ultraviolet radiation.9–13 Rela-tively little is known of the mechanisms that link ROSto ceramide production, although recent findings sug-gest potential roles for glutathione peroxidase and src-like tyrosine kinase p53/p56.12,14
Patients with human immunodeficiency virus de-mentia (HIVD) exhibit evidence of oxidative damagein their brains including increased amounts of per-oxynitrite, 4-hydroxynonenal (HNE), and protein car-
From the 1Department of Neurology, Johns Hopkins UniversitySchool of Medicine; 2Laboratory of Neurosciences, National Insti-tute on Aging Gerontology Research Center; Departments of 3Pa-thology and 4Neuroscience, Johns Hopkins University School ofMedicine, Baltimore, MD.
Received Apr 8, 2003, and in revised form Aug 27. Accepted forpublication Sep 2, 2003.
Address correspondence to Dr Haughey, Department of Neurology,Johns Hopkins University School of Medicine, Meyer 6-109, 600North Wolfe Street, Baltimore, MD 21287.E-mail: [email protected]
© 2004 American Neurological Association 257Published by Wiley-Liss, Inc., through Wiley Subscription Services

bonyls.15,16 The neurotoxic HIV-1 proteins gp120 andTat have been shown to increase oxidative stress, andantioxidants can protect cells from the damaging effectsof these proteins, suggesting an important role for ox-idative damage in the pathogenesis of HIVD.17,18 In-flammatory cytokines that are known to be increasedin the brains of HIVD dementia patients (TNF-�, in-terleukin [IL]–1, IFN-�, and Fas/FasL)19 can induceceramide production in nonneural cells by increasingthe activity of sphingomyelinases.20,21 There is a bidi-rectional relationship between cytokine balance andceramide. For instance, TNF�, IL-1, and Fas/FasL arepotent inducers of ceramide production and ceramidecan, in turn, stimulate the production of IL-2 andIL-6.22–24 In HIV infection, central nervous system(CNS) inflammation is thought to be initiated after theinflux of HIV-infected monocytes and macrophagesinto the brain parenchyma. The subsequent release ofmacrophage and HIV products then may promote acytokine imbalance.25,26 Prominent increases in theproinflammatory cytokines TNF�, IL-1, IL-2, IL-6,and Fas/FasL have been reported in brain and cerebro-spinal fluid (CSF) of HIVD patients.27 Successful an-tiretroviral treatment can drastically slow cognitive de-cline and restore the cytokine balance, suggesting thatinflammatory products play important roles in HIV-associated CNS dysfunction.28
Disruption of sphingolipid homeostasis has beendocumented in studies of human patients with, andanimal models of, several neurodegenerative conditionsincluding Alzheimer’s disease Parkinson’s disease,stroke, and amyotrophic lateral sclerosis.29–32 BecauseCNS inflammation and loss of redox balance arethought to play critical roles in the initiation and pro-gression of HIVD, we sought to determine whethersphingolipid metabolism is dysregulated in HIVD. Inbrain tissues from patients with well-established clinicalneuropathological documentation of HIVD, we foundevidence of increased oxidative stress and accumulationof sphingomyelin and ceramides. Studies of culturedneurons suggest a key role for ceramide production inthe cell death cascade induced by the HIV-1 proteinsgp120 and Tat. Our findings suggest that sphingolipidimbalance plays an important role in neuronal dysfunc-tion and death in HIVD and identifies a potentialtherapeutic target for neuroprotective intervention.
Materials and MethodsHuman Cerebrospinal Fluid Samples andBrain TissueCSF samples and brain tissues (pre-1996; pre–highly activeantiretroviral therapy) were obtained from the Johns Hop-kins AIDS brain bank. All patients were homosexual menwith no history of intravenous drug use and were similar inage (uninfected, 42 � 4; HIV-1 infected, 38 � 5). Brainswere predetermined to be free of opportunistic infections,
and HIVD patients were selected for indicators of encepha-litis using positive identification of macrophage infiltrationand/or multinucleated giant cells. HIVD patients were cate-gorized as either no dementia (HIVND) mild (HIVD-m) ormoderate-severe dementia (HIVD-MS) based on MemorialSloan-Kettering (MSK) staging score; MSK 0.5 to 1 �HIVD-m and MSK 2 to 3 � HIVD-MS.
Immunohistochemical AnalysesImmunohistochemistry was performed in paraffin-embeddedsections using the avidin-biotin-peroxidase complex method aspreviously described.33 Both glial fibrillary acidic protein(GFAP) and human-lucosite-associated (HLA)DR antibodieswere used at a dilution of 1 to 100 and biotinylated anti–rabbit and anti–mouse, respectively, secondary antibodies at 1to 200 (Vector Laboratories, Burlingame, CA). We quantifiedthe amount of immunoreactivity for GFAP and HLADR us-ing the fractional area method as described previously.33
Lipid Extraction and Measurements of Sphingolipids,Phospholipids, and Lipid PeroxidesTotal lipids from brain and CSF samples were prepared ac-cording to a modified Bligh and Dyer procedure.34 Snap-frozen brain tissues and premortem CSF were obtained fromthe same brains used for HLADR immunoreactivity studies.Electrospray ionization tandem mass spectrometry (ESI/MS/MS) analyses were performed using methods similar to thoseused in our previous studies.7
Cell Culture and Experimental TreatmentsHippocampal neuronal cultures were prepared from embry-onic day 18 Sprague Dawley rats using methods similar tothose described previously.35 Neurons were plated at a den-sity of 100,000 cells/ml on 25mm diameter poly-S-lysine–coated glass coverslips for calcium imaging, and at a densityof 200,000 cells/ml on 12mm diameter coverslips for apo-ptosis (survival) analysis in serum-free Neurobasal mediumcontaining 1% B-27 supplement (Gibco, Grand Island, NY).Immunofluorescent staining for MAP-2 showed that hip-pocampal cultures were greater than 98% neurons; the re-mainder of cells were predominantly astrocytes. Hippocam-pal cultures were used between 10 and 14 days in vitro.
Cultures were exposed to palmitoyl-CoA (0.1–1�M;Sigma, St. Louis, MMO) for 48 hours to increase the syn-thesis of sphingolipids. Cells then were exposed to Tat1-72
(100–500nM) or gp120IIIB (100–500pM) for 24 hours be-fore quantification of cell survival. The sphingomyelin inhib-itor GW4869 (1�M; Calbiochem, San Diego, CA) and theserine palmitoyl CoA-transferase inhibitor ISP-1 (10�M;Sigma) were applied to cultures for 30 minutes before Tat orgp120. At designated time points after treatment, the cellswere harvested for analysis of sphingomyelin and ceramidelevels or were used to quantify cell survival.
Cell Survival AnalysisQuantification of cell survival was performed in cells stainedwith the fluorescent DNA binding dye Hoechst 33342 usingmethods described previously.35 Nuclei were visualized andphotographed under epifluorescence illumination (340nmexcitation and 510nm barrier filter) using a �40 oil immer-
258 Annals of Neurology Vol 55 No 2 February 2004
sion objective. Two hundred cells from three fields in threeseparate cultures per experimental condition were countedwithout knowledge of experimental condition. Cells in whichnuclear staining was diffuse were considered viable and cellswhere nuclear staining was condensed or fragmented wereconsidered “apoptotic.”
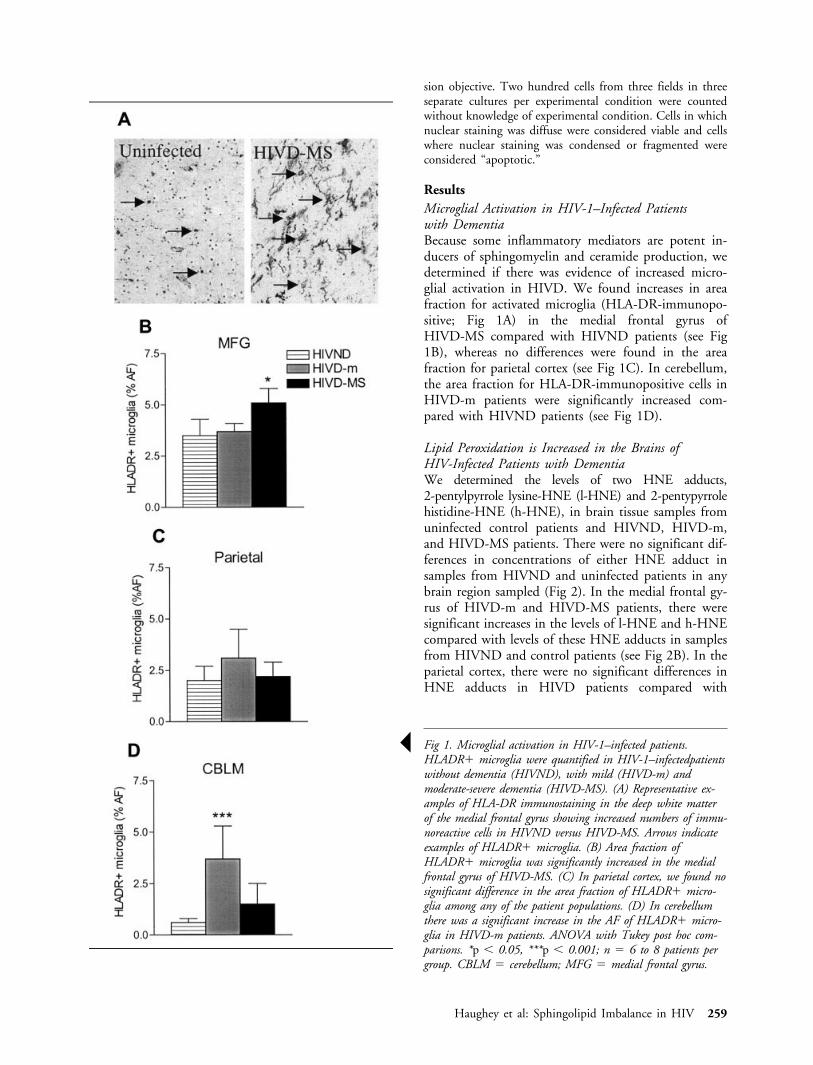
ResultsMicroglial Activation in HIV-1–Infected Patientswith DementiaBecause some inflammatory mediators are potent in-ducers of sphingomyelin and ceramide production, wedetermined if there was evidence of increased micro-glial activation in HIVD. We found increases in areafraction for activated microglia (HLA-DR-immunopo-sitive; Fig 1A) in the medial frontal gyrus ofHIVD-MS compared with HIVND patients (see Fig1B), whereas no differences were found in the areafraction for parietal cortex (see Fig 1C). In cerebellum,the area fraction for HLA-DR-immunopositive cells inHIVD-m patients were significantly increased com-pared with HIVND patients (see Fig 1D).
Lipid Peroxidation is Increased in the Brains ofHIV-Infected Patients with DementiaWe determined the levels of two HNE adducts,2-pentylpyrrole lysine-HNE (l-HNE) and 2-pentypyrrolehistidine-HNE (h-HNE), in brain tissue samples fromuninfected control patients and HIVND, HIVD-m,and HIVD-MS patients. There were no significant dif-ferences in concentrations of either HNE adduct insamples from HIVND and uninfected patients in anybrain region sampled (Fig 2). In the medial frontal gy-rus of HIVD-m and HIVD-MS patients, there weresignificant increases in the levels of l-HNE and h-HNEcompared with levels of these HNE adducts in samplesfrom HIVND and control patients (see Fig 2B). In theparietal cortex, there were no significant differences inHNE adducts in HIVD patients compared with
Š Fig 1. Microglial activation in HIV-1–infected patients.HLADR� microglia were quantified in HIV-1–infectedpatientswithout dementia (HIVND), with mild (HIVD-m) andmoderate-severe dementia (HIVD-MS). (A) Representative ex-amples of HLA-DR immunostaining in the deep white matterof the medial frontal gyrus showing increased numbers of immu-noreactive cells in HIVND versus HIVD-MS. Arrows indicateexamples of HLADR� microglia. (B) Area fraction ofHLADR� microglia was significantly increased in the medialfrontal gyrus of HIVD-MS. (C) In parietal cortex, we found nosignificant difference in the area fraction of HLADR� micro-glia among any of the patient populations. (D) In cerebellumthere was a significant increase in the AF of HLADR� micro-glia in HIVD-m patients. ANOVA with Tukey post hoc com-parisons. *p � 0.05, ***p � 0.001; n � 6 to 8 patients pergroup. CBLM � cerebellum; MFG � medial frontal gyrus.
Haughey et al: Sphingolipid Imbalance in HIV 259
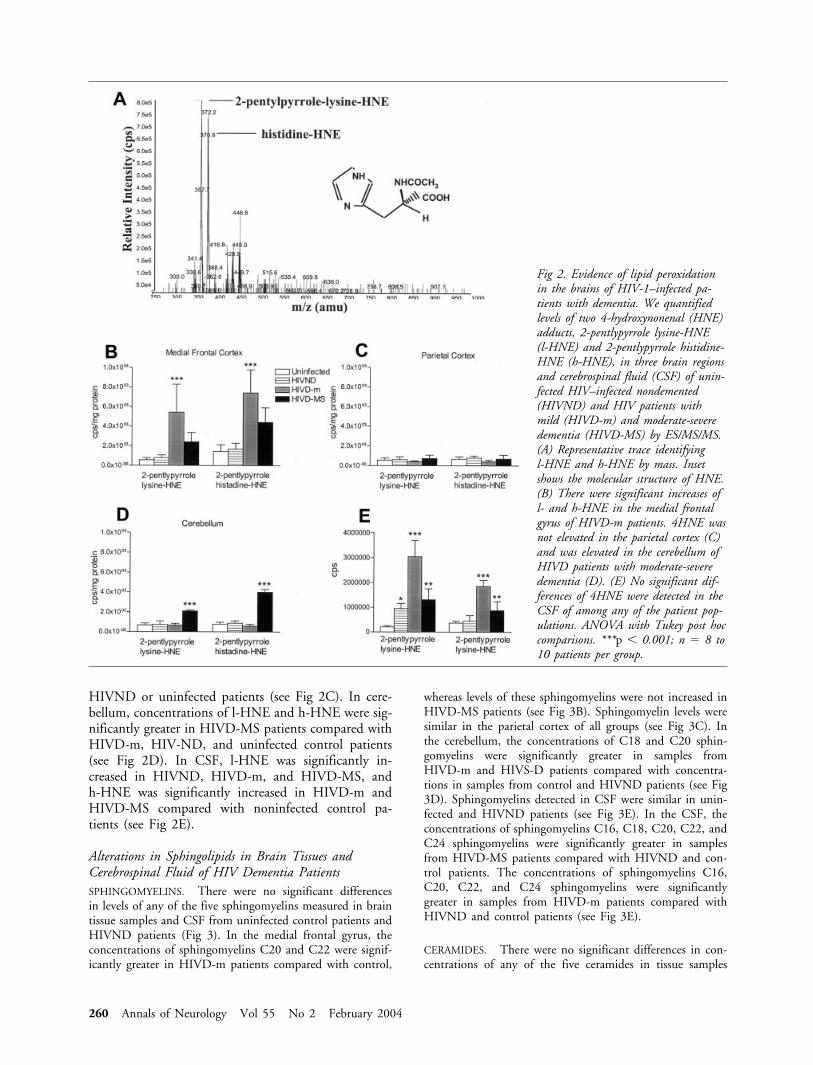
HIVND or uninfected patients (see Fig 2C). In cere-bellum, concentrations of l-HNE and h-HNE were sig-nificantly greater in HIVD-MS patients compared withHIVD-m, HIV-ND, and uninfected control patients(see Fig 2D). In CSF, l-HNE was significantly in-creased in HIVND, HIVD-m, and HIVD-MS, andh-HNE was significantly increased in HIVD-m andHIVD-MS compared with noninfected control pa-tients (see Fig 2E).
Alterations in Sphingolipids in Brain Tissues andCerebrospinal Fluid of HIV Dementia PatientsSPHINGOMYELINS. There were no significant differencesin levels of any of the five sphingomyelins measured in braintissue samples and CSF from uninfected control patients andHIVND patients (Fig 3). In the medial frontal gyrus, theconcentrations of sphingomyelins C20 and C22 were signif-icantly greater in HIVD-m patients compared with control,
whereas levels of these sphingomyelins were not increased inHIVD-MS patients (see Fig 3B). Sphingomyelin levels weresimilar in the parietal cortex of all groups (see Fig 3C). Inthe cerebellum, the concentrations of C18 and C20 sphin-gomyelins were significantly greater in samples fromHIVD-m and HIVS-D patients compared with concentra-tions in samples from control and HIVND patients (see Fig3D). Sphingomyelins detected in CSF were similar in unin-fected and HIVND patients (see Fig 3E). In the CSF, theconcentrations of sphingomyelins C16, C18, C20, C22, andC24 sphingomyelins were significantly greater in samplesfrom HIVD-MS patients compared with HIVND and con-trol patients. The concentrations of sphingomyelins C16,C20, C22, and C24 sphingomyelins were significantlygreater in samples from HIVD-m patients compared withHIVND and control patients (see Fig 3E).
CERAMIDES. There were no significant differences in con-centrations of any of the five ceramides in tissue samples
Fig 2. Evidence of lipid peroxidationin the brains of HIV-1–infected pa-tients with dementia. We quantifiedlevels of two 4-hydroxynonenal (HNE)adducts, 2-pentlypyrrole lysine-HNE(l-HNE) and 2-pentlypyrrole histidine-HNE (h-HNE), in three brain regionsand cerebrospinal fluid (CSF) of unin-fected HIV–infected nondemented(HIVND) and HIV patients withmild (HIVD-m) and moderate-severedementia (HIVD-MS) by ES/MS/MS.(A) Representative trace identifyingl-HNE and h-HNE by mass. Insetshows the molecular structure of HNE.(B) There were significant increases ofl- and h-HNE in the medial frontalgyrus of HIVD-m patients. 4HNE wasnot elevated in the parietal cortex (C)and was elevated in the cerebellum ofHIVD patients with moderate-severedementia (D). (E) No significant dif-ferences of 4HNE were detected in theCSF of among any of the patient pop-ulations. ANOVA with Tukey post hoccomparisons. ***p � 0.001; n � 8 to10 patients per group.
260 Annals of Neurology Vol 55 No 2 February 2004
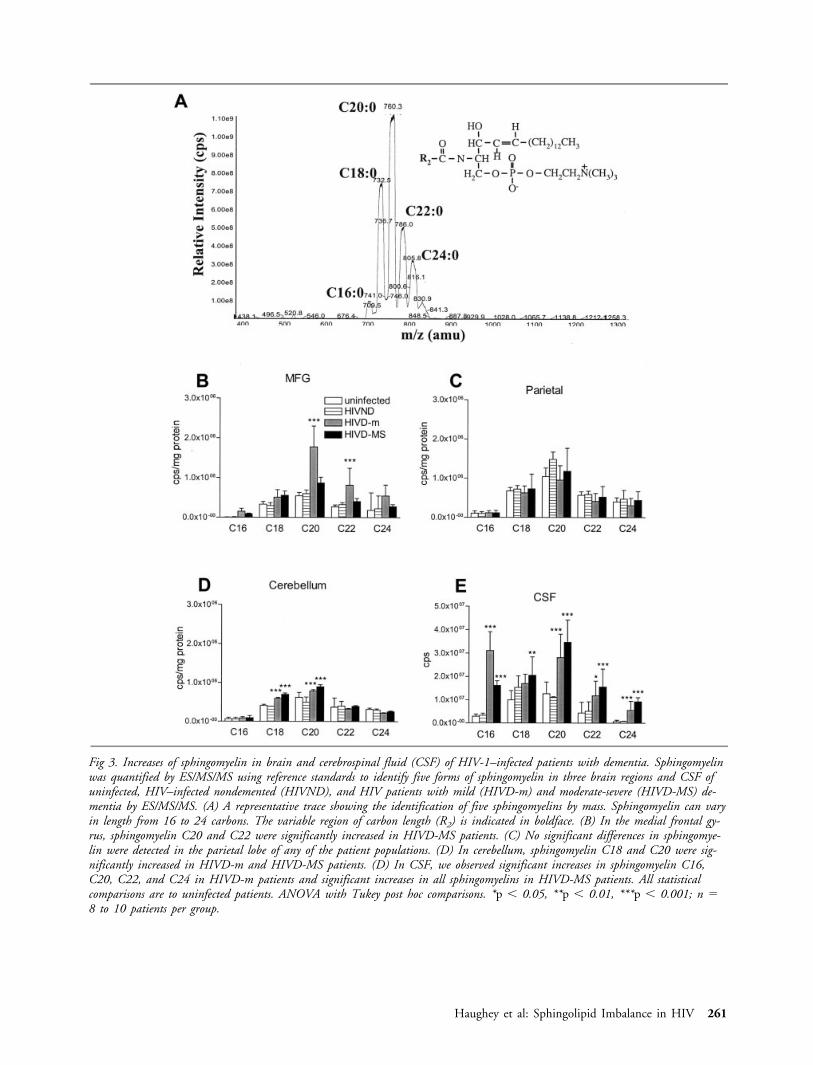
Fig 3. Increases of sphingomyelin in brain and cerebrospinal fluid (CSF) of HIV-1–infected patients with dementia. Sphingomyelinwas quantified by ES/MS/MS using reference standards to identify five forms of sphingomyelin in three brain regions and CSF ofuninfected, HIV–infected nondemented (HIVND), and HIV patients with mild (HIVD-m) and moderate-severe (HIVD-MS) de-mentia by ES/MS/MS. (A) A representative trace showing the identification of five sphingomyelins by mass. Sphingomyelin can varyin length from 16 to 24 carbons. The variable region of carbon length (R2) is indicated in boldface. (B) In the medial frontal gy-rus, sphingomyelin C20 and C22 were significantly increased in HIVD-MS patients. (C) No significant differences in sphingomye-lin were detected in the parietal lobe of any of the patient populations. (D) In cerebellum, sphingomyelin C18 and C20 were sig-nificantly increased in HIVD-m and HIVD-MS patients. (D) In CSF, we observed significant increases in sphingomyelin C16,C20, C22, and C24 in HIVD-m patients and significant increases in all sphingomyelins in HIVD-MS patients. All statisticalcomparisons are to uninfected patients. ANOVA with Tukey post hoc comparisons. *p � 0.05, **p � 0.01, ***p � 0.001; n �8 to 10 patients per group.
Haughey et al: Sphingolipid Imbalance in HIV 261
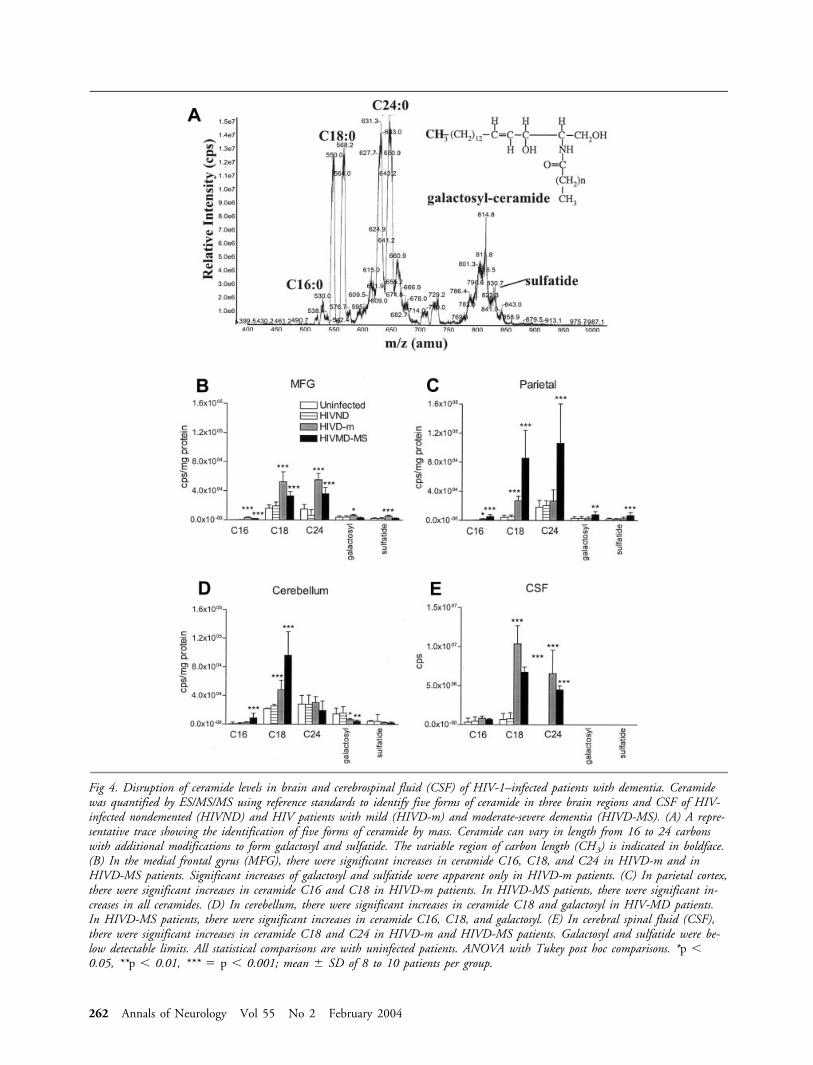
Fig 4. Disruption of ceramide levels in brain and cerebrospinal fluid (CSF) of HIV-1–infected patients with dementia. Ceramidewas quantified by ES/MS/MS using reference standards to identify five forms of ceramide in three brain regions and CSF of HIV-infected nondemented (HIVND) and HIV patients with mild (HIVD-m) and moderate-severe dementia (HIVD-MS). (A) A repre-sentative trace showing the identification of five forms of ceramide by mass. Ceramide can vary in length from 16 to 24 carbonswith additional modifications to form galactosyl and sulfatide. The variable region of carbon length (CH3) is indicated in boldface.(B) In the medial frontal gyrus (MFG), there were significant increases in ceramide C16, C18, and C24 in HIVD-m and inHIVD-MS patients. Significant increases of galactosyl and sulfatide were apparent only in HIVD-m patients. (C) In parietal cortex,there were significant increases in ceramide C16 and C18 in HIVD-m patients. In HIVD-MS patients, there were significant in-creases in all ceramides. (D) In cerebellum, there were significant increases in ceramide C18 and galactosyl in HIV-MD patients.In HIVD-MS patients, there were significant increases in ceramide C16, C18, and galactosyl. (E) In cerebral spinal fluid (CSF),there were significant increases in ceramide C18 and C24 in HIVD-m and HIVD-MS patients. Galactosyl and sulfatide were be-low detectable limits. All statistical comparisons are with uninfected patients. ANOVA with Tukey post hoc comparisons. *p �0.05, **p � 0.01, *** � p � 0.001; mean � SD of 8 to 10 patients per group.
262 Annals of Neurology Vol 55 No 2 February 2004

from HIVND compared with uninfected patients in anybrain region sampled (Fig 4). The concentrations of C1,C18, and C24 ceramides and of galactosyl ceramide and sul-fatide were significantly increased in samples from the medialfrontal gyrus of HIVD-m patients compared with uninfectedcontrol and HIVND patients (see Fig 4B). The concentra-tions of C16, C18, and C24 ceramides were significantly in-creased in the medial frontal gyrus of HIVD-MS patientscompared with control and HIVND patients. In the parietalcortex, concentrations of each of the five ceramides examinedwere significantly greater in samples from HIVD-MS pa-tients compared with control and HIVND patients (see Fig4C). The concentration of C18 ceramide was significantlygreater in parietal cortex of HIVD-m patients as comparedwith control and HIVND patients. The concentrations ofC16 and C18 ceramides were significantly greater in the cer-ebellum of HIVD-MS patients compared with control andHIVND patients, and the C18 ceramide concentration wasalso significantly elevated in the cerebellum of HIVD-m pa-tients (see Fig 4D). In contrast, the concentration of galac-tosyl ceramide was significantly lower in the cerebellum ofHIVD-m and HIVD-MS compared with control andHIVND patients. The concentrations of C18 and C24 cer-amides were greatly elevated in the CSF of HIVD-m andHIVD-MS patients compared with control and HIVND pa-tients, whereas the CSF concentration of CSF C16 ceramidewas unchanged in HIV dementia patients (see Fig 4E).Galactosyl ceramide and sulfatide were not detectable in CSFsamples from any of the patients.
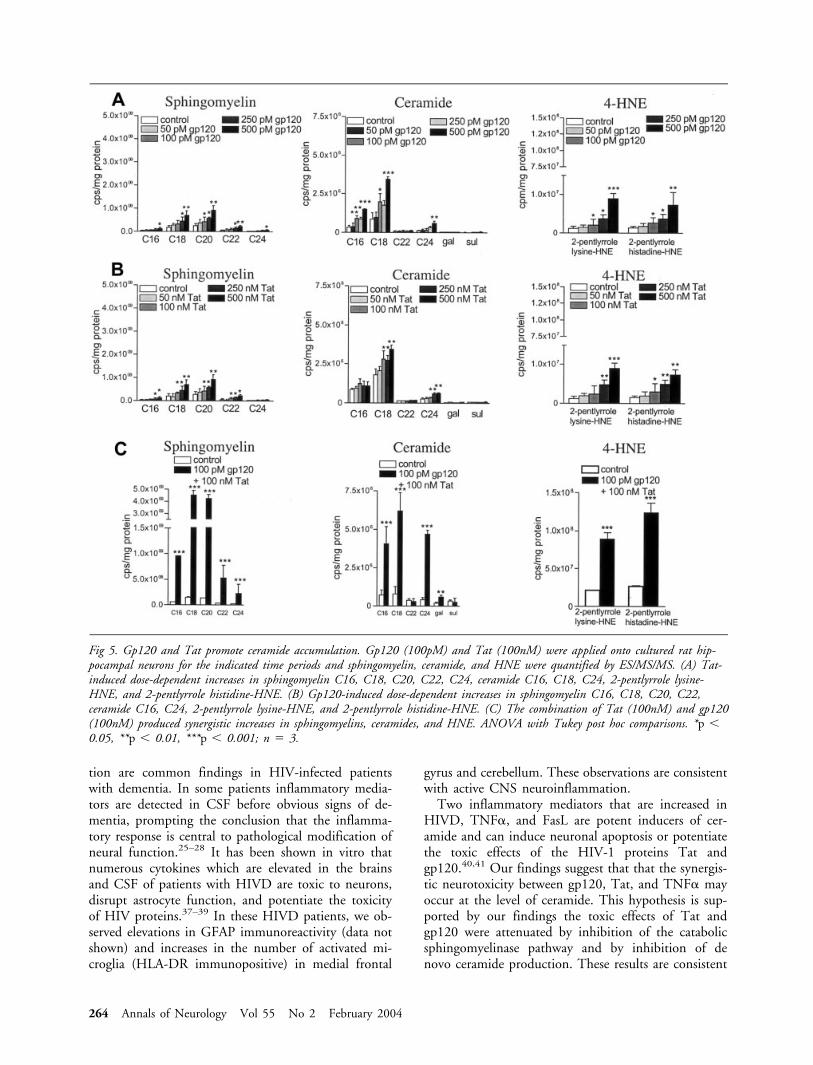
Evidence that Perturbed Sphingolipid MetabolismMediates the Neurotoxicity of HIV-1 ProteinsWe exposed cultured hippocampal neurons to gp120,Tat, or gp120 � Tat and determined sphingomyelin,ceramide, and HNE levels by ES/MS/MS. We present,based on findings that stimulation with gp120 or Tatresulted in peak sphingomyelin levels at 6 hours, cer-amide at 12 hours and HNE after 24 hours, only thedata corresponding to these time points. When neu-rons were exposed to a dose range of gp120, concen-trations in the range of 250 to 500pM gp120 signifi-cantly increased sphingomyelin C16, C18, C20, C22,and C24. Ceramide C16, C18, and C24 were signifi-cantly increased by gp120 concentrations in the rangeof 100 to 500pM. The HNE adducts, 2-pentlypyrrolelysine-HNE and 2-pentlyrrole histidine-HNE were sig-nificantly increased by gp120 concentrations equal orgreater than 100pM (Fig 5A). When neurons were ex-posed to a dose range of Tat, concentrations of Tat inthe range of 250 to 500nM significantly increasedsphingomyelin C16, C18, C20, and C22. CeramideC18 and C24 were significantly increased by concen-trations of Tat in the range of 100 to 500nM. TheHNE adducts 2-pentlypyrrole lysine-HNE and2-pentlyrrole histidine-HNE were significantly in-creased by 100 to 500nM Tat (see Fig 5B). The com-bination of gp120 (100pM) and Tat (100nM) re-sulted in synergistic increases of sphingomyelin C16,
C18, C20, C22, and C24, ceramide C16, C18, C24,and galactosyl, and 2-pentlypyrrole lysine-HNE and2-pentlyrrole histidine-HNE (see Fig 5C).
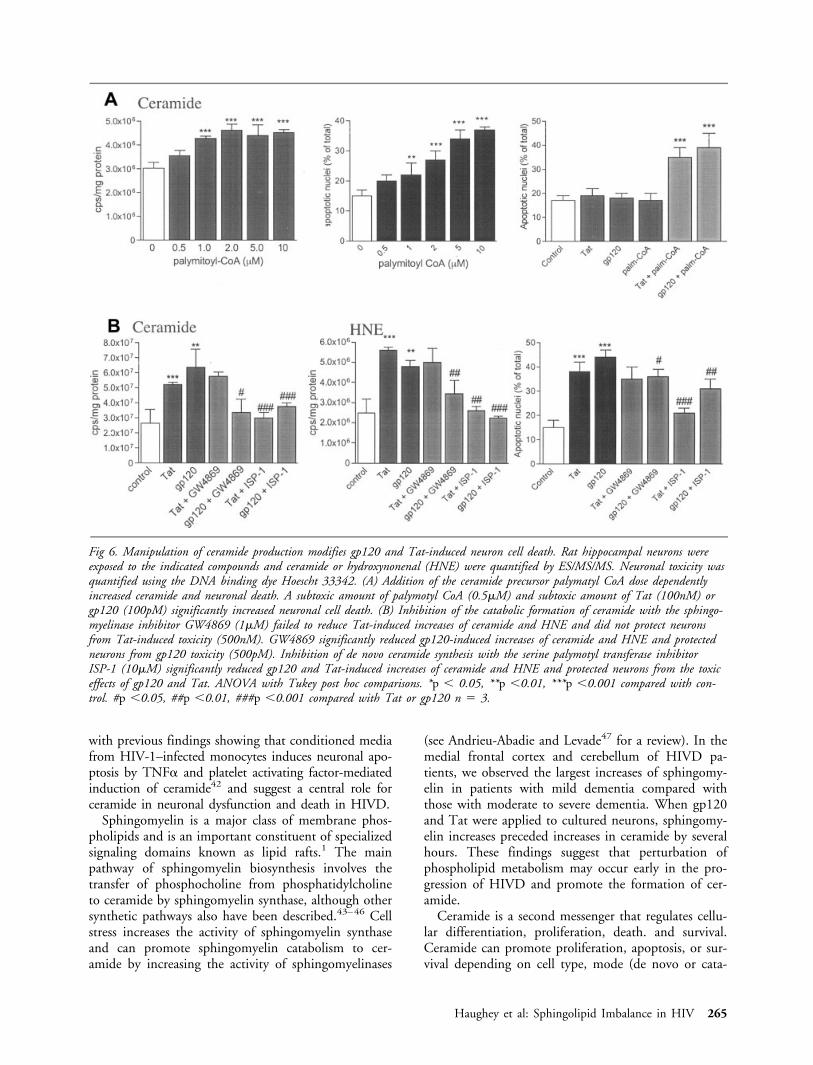
To determine whether the alterations in sphingolip-ids induced by gp120 and Tat played a role in the celldeath process, we treated hippocampal neurons withthe sphingomyelin/ceramide precursor palmitoyl-CoAto increase the amount of sphingomyelins and cer-amide in the cells. Palmitoyl-CoA increased ceramideand induced neuronal death in a concentration-dependent manner (Fig 6A), suggesting that increasedceramide levels is sufficient to induce neuronal death.When neurons were exposed to a sublethal concentra-tion of palmitoyl-CoA, in combination with sublethalconcentrations of Tat or gp120, neuronal death wassignificantly increased (see Fig 6A). To determinewhether increased levels of sphingomyelin and cer-amide were required for the neurotoxic action ofgp120 and Tat, we treated neurons with the serinepalmitoyl CoA-transferase inhibitor ISP-1 (myriocin),an agent previously shown to deplete sphingomyelinand ceramide levels in cultured neurons.7 Neuronstreated with ISP-1 were significantly more resistant tobeing killed by gp120 or Tat and showed significantlylower concentrations of ceramide and HNE comparedwith neurons in cultures treated with gp120 or Tat (seeFig 6B). Treatment of neurons with GW4869, asphingomyelinase inhibitor,36 significantly protectedneurons against the toxicity of gp120 and significantlylowered Tat and gp120-induced increases of sphingo-myelin, ceramide, and HNE levels (see Fig 6B).GW4869 did not protect against Tat-induced neuronaldeath and failed to reduced ceramide and HNE levels(see Fig 6B). At the concentrations tested, ISP-1 andGW4869 were not toxic to neurons and did not sig-nificantly alter sphingomyelin or ceramide concentra-tions after a 24-hour exposure (data not shown).
DiscussionIn patients with HIVD and pathological evidence ofencephalitis, we found significant increases in sphingo-myelins and ceramides in brain tissue and CSF. Thesechanges were associated with the accumulation HNEin the medial frontal cortex and cerebellum. The neu-rotoxic HIV-1 proteins gp120 and Tat significantly in-creased sphingomyelin and ceramide concentrations incultured hippocampal neurons, suggesting a possiblemechanism whereby HIV-1 induces alterations insphingolipid and ceramide metabolism in the brains ofHIVD patients. The ability of an inhibitor of serinepalmitoyl transferase, the rate limiting step in ceramidesynthesis, to protect neurons from being killed bygp120 and Tat suggests that sphingolipid imbalancemay play an important role in the neuronal dysfunc-tion and death that occurs in HIVD.
CNS inflammation, astroglial, and microglial activa-
Haughey et al: Sphingolipid Imbalance in HIV 263
tion are common findings in HIV-infected patientswith dementia. In some patients inflammatory media-tors are detected in CSF before obvious signs of de-mentia, prompting the conclusion that the inflamma-tory response is central to pathological modification ofneural function.25–28 It has been shown in vitro thatnumerous cytokines which are elevated in the brainsand CSF of patients with HIVD are toxic to neurons,disrupt astrocyte function, and potentiate the toxicityof HIV proteins.37–39 In these HIVD patients, we ob-served elevations in GFAP immunoreactivity (data notshown) and increases in the number of activated mi-croglia (HLA-DR immunopositive) in medial frontal
gyrus and cerebellum. These observations are consistentwith active CNS neuroinflammation.
Two inflammatory mediators that are increased inHIVD, TNF�, and FasL are potent inducers of cer-amide and can induce neuronal apoptosis or potentiatethe toxic effects of the HIV-1 proteins Tat andgp120.40,41 Our findings suggest that that the synergis-tic neurotoxicity between gp120, Tat, and TNF� mayoccur at the level of ceramide. This hypothesis is sup-ported by our findings the toxic effects of Tat andgp120 were attenuated by inhibition of the catabolicsphingomyelinase pathway and by inhibition of denovo ceramide production. These results are consistent
Fig 5. Gp120 and Tat promote ceramide accumulation. Gp120 (100pM) and Tat (100nM) were applied onto cultured rat hip-pocampal neurons for the indicated time periods and sphingomyelin, ceramide, and HNE were quantified by ES/MS/MS. (A) Tat-induced dose-dependent increases in sphingomyelin C16, C18, C20, C22, C24, ceramide C16, C18, C24, 2-pentlyrrole lysine-HNE, and 2-pentlyrrole histidine-HNE. (B) Gp120-induced dose-dependent increases in sphingomyelin C16, C18, C20, C22,ceramide C16, C24, 2-pentlyrrole lysine-HNE, and 2-pentlyrrole histidine-HNE. (C) The combination of Tat (100nM) and gp120(100nM) produced synergistic increases in sphingomyelins, ceramides, and HNE. ANOVA with Tukey post hoc comparisons. *p �0.05, **p � 0.01, ***p � 0.001; n � 3.
264 Annals of Neurology Vol 55 No 2 February 2004
with previous findings showing that conditioned mediafrom HIV-1–infected monocytes induces neuronal apo-ptosis by TNF� and platelet activating factor-mediatedinduction of ceramide42 and suggest a central role forceramide in neuronal dysfunction and death in HIVD.
Sphingomyelin is a major class of membrane phos-pholipids and is an important constituent of specializedsignaling domains known as lipid rafts.1 The mainpathway of sphingomyelin biosynthesis involves thetransfer of phosphocholine from phosphatidylcholineto ceramide by sphingomyelin synthase, although othersynthetic pathways also have been described.43–46 Cellstress increases the activity of sphingomyelin synthaseand can promote sphingomyelin catabolism to cer-amide by increasing the activity of sphingomyelinases
(see Andrieu-Abadie and Levade47 for a review). In themedial frontal cortex and cerebellum of HIVD pa-tients, we observed the largest increases of sphingomy-elin in patients with mild dementia compared withthose with moderate to severe dementia. When gp120and Tat were applied to cultured neurons, sphingomy-elin increases preceded increases in ceramide by severalhours. These findings suggest that perturbation ofphospholipid metabolism may occur early in the pro-gression of HIVD and promote the formation of cer-amide.
Ceramide is a second messenger that regulates cellu-lar differentiation, proliferation, death. and survival.Ceramide can promote proliferation, apoptosis, or sur-vival depending on cell type, mode (de novo or cata-
Fig 6. Manipulation of ceramide production modifies gp120 and Tat-induced neuron cell death. Rat hippocampal neurons wereexposed to the indicated compounds and ceramide or hydroxynonenal (HNE) were quantified by ES/MS/MS. Neuronal toxicity wasquantified using the DNA binding dye Hoescht 33342. (A) Addition of the ceramide precursor palymatyl CoA dose dependentlyincreased ceramide and neuronal death. A subtoxic amount of palymotyl CoA (0.5�M) and subtoxic amount of Tat (100nM) orgp120 (100pM) significantly increased neuronal cell death. (B) Inhibition of the catabolic formation of ceramide with the sphingo-myelinase inhibitor GW4869 (1�M) failed to reduce Tat-induced increases of ceramide and HNE and did not protect neuronsfrom Tat-induced toxicity (500nM). GW4869 significantly reduced gp120-induced increases of ceramide and HNE and protectedneurons from gp120 toxicity (500pM). Inhibition of de novo ceramide synthesis with the serine palymotyl transferase inhibitorISP-1 (10�M) significantly reduced gp120 and Tat-induced increases of ceramide and HNE and protected neurons from the toxiceffects of gp120 and Tat. ANOVA with Tukey post hoc comparisons. *p � 0.05, **p �0.01, ***p �0.001 compared with con-trol. #p �0.05, ##p �0.01, ###p �0.001 compared with Tat or gp120 n � 3.
Haughey et al: Sphingolipid Imbalance in HIV 265

bolic reaction), and subcellular site of ceramide forma-tion.46 In neurons, there is an intimate relationshipbetween oxidative stress, ceramide, and apoptosis.48–50
In this study, we observed increases in levels of cer-amides in HIVD patients in each brain regions sam-pled. We found biphasic increases in medial frontal gy-rus with peak ceramide in patients of mild dementiathat declined with more severe forms of dementia. Incontrast, ceramide in parietal cortex and cerebellum in-creased with the severity of dementia. These observa-tions are consistent with the progressive involvement ofmedial frontal gyrus, parietal cortex, and cerebellumduring disease progression. The decline in ceramidelevels in the medial frontal cortex of patients with moresevere dementia may be the result of neuronal death inthis brain region early in disease progression when cer-amide levels were at their peak. This hypothesis is sup-ported by previous studies of nonneuronal cells inwhich ceramide levels decreased after irreversible initi-ation of apoptotic signaling,51 a result similar to thetransient increases in sphingomyelins and ceramidesthat occurred in hippocampal neurons exposed togp120 and Tat. Thus, irreversible initiation of neuro-nal damage may occur early in disease progression. Be-cause both cytokine and HIV protein–induced neuro-nal death can be prevented by inhibition of ceramidesynthesis, this point of convergence is an attractivetherapeutic target.
Free radical species such as hydroxyl radical and per-oxynitrite can bind covalently to cellular macromoleculesand promote lipid peroxidation of cellular membraneswith the resultant formation of very reactive aldehydessuch as HNE. These aldehydes play an active role inneuronal death by modifying sulfhydryl groups and dis-rupting protein functions. Because some protein thiolsare essential components of the molecular arrangementresponsible for the calcium transport across cellularmembranes, loss of such thiols can affect the calciumsequestration activity of subcellular compartments anddiminish the capacity of mitochondria and microsomesto regulate the cytosolic calcium levels, thereby promot-ing cellular dysfunction and death. We observed in-creases in two HNE adducts in the medial frontal cortexand cerebellum of HIVD patients. The greatest increasesof HNE in the medial frontal cortex were in patientswith mild dementia that decreased slightly in patientswith more severe dementia. We interpret these findingto indicate that HNE may perturb neuronal functionearly in the pathogenesis of HIVD. This interpretationis supported by observations that suggest HIV-1protein–induced neural dysfunction involves disruptionsin calcium flux, mitochondria function, and redox bal-ance17,18,19,52,53 and, in vivo, findings in which increasesof HNE and oxidized proteins were greatest in HIVD-mpatients compared with HIVD-MS patients.15,16
Channel blockers have proved to be minimally pro-
tective in HIVD and have considerable side effects.Our data suggest that metabolic pathways leading tothe overproduction of ceramide may present two addi-tional targets for therapeutic intervention. Blocking ormodifying these points of convergence in inflammatoryand HIV protein–induced apoptotic pathways are at-tractive approaches for the treatment of HIVD.
References1. Dobrowsky RT. Sphingolipid signalling domains floating on
rafts or buried in caves? Cell Signal 2000;12:81–90.2. Billis W, Fuks Z, Kolesnick R. Signaling in and regulation of
ionizing radiation induced apoptosis in endothelial cells. RecentProg Horm Res 1998;53:85–92.
3. Andrieu-Abadie N, Gouaze V, Salvayre R, Levade T. Ceramidein apoptosis signaling: relationship with oxidative stress. FreeRadic Biol Med 2001;31:717–728.
4. Monney L, Olivier R, Otter I, et al. Role of an acidic compart-ment in tumor-necrosis-factor-alpha-induced production of cer-amide, activation of caspase-3 and apoptosis. Eur J Biochem1998;15:295–303.
5. Veldman RJ, Maestre N, Aduib OM, et al. A neutral sphingo-myelinase resides in sphingolipid-enriched microdomains and isinhibited by the caveolin-scaffolding domain: potential implica-tions in tumour necrosis factor signalling. Biochem J 2001;355:859–868.
6. Hofmann K, Tomiuk S, Wolff G, Stoffel W. Cloning andcharacterization of the mammalian brain-specific, Mg2�-dependent neutral sphingomyelinase. Proc Natl Acad Sci USA2000;97:5895–5900.
7. Cutler RG, Pedersen WA, Camandola S, et al. Evidence thataccumulation of ceramides and cholesterol esters mediates oxi-dative stress-induced death of motor neurons in amyotrophiclateral sclerosis. Ann Neurol 2002;52:448–457.
8. Movsesyan VA, Yakovlev AG, Dabaghyan EA, et al. Ceramideinduces neuronal apoptosis through the caspase-9/caspase-3pathway. Biochem Biophys Res Commun 2002;299:201–207.
9. Singh I, Kishimoto Y. Ceramide synthesis in rat brain: charac-terization of the synthesis requiring pyridine nucleotide. ArchBiochem Biophys 1980;202:93–100.
10. Liu B, Andrieu-Abadie N, Levade T, et al. Glutathione regula-tion of neutral sphingomyelinase in tumor necrosis factor-alpha-induced cell death. J Biol Chem 1998;273:11313–11320.
11. Yoshimura S, Banno Y, Nakashima S, et al. Inhibition of neu-tral sphingomyelinase activation and ceramide formation byglutathione in hypoxic PC12 cell death. J Neurochem 1999;73:675–683.
12. Maziere C, Meignotte A, Dantin F, et al. Oxidized LDL in-duces an oxidative stress and activates the tumor suppressor p53in MRC5 human fibroblasts. Biochem Biophys Res Commun2000;276:718–723.
13. Bezombes C, Plo I, Mansat-De Mas V, et al. Oxidative stress-induced activation of Lyn recruits sphingomyelinase and is req-uisite for its stimulation by Ara-C. FASEB J 2001;15:1583–1585.
14. Gouaze V, Mirault ME, Carpentier S, et al. Glutathioneperoxidase-1 overexpression prevents ceramide production andpartially inhibits apoptosis in doxorubicin-treated human breastcarcinoma cells. Mol Pharmacol 2001;60:488–496.
15. Boven LA, Gomes L, Hery C, et al. Increased peroxynitrite ac-tivity in AIDS dementia complex: implications for the neuro-pathogenesis of HIV-1 infection. J Immunol 1999;162:4319–4327.
266 Annals of Neurology Vol 55 No 2 February 2004

16. Turchan J, Pocernich CB, Gairola C, et. al. Oxidative stress inHIV demented patients and protection ex vivo with novel an-tioxidants. Neurology 2003;28:307–314.
17. Foga IO, Nath A, Hasinoff BB, Geiger JD. Antioxidants anddipyridamole inhibit HIV-1 gp120-induced free radical-basedoxidative damage to human monocytoid cells. J Acquir Im-mune Defic Syndr Hum Retrovirol 1997;16:223–229.
18. Kruman II, Nath A, Mattson MP.HIV-1 protein Tat inducesapoptosis of hippocampal neurons by a mechanism involvingcaspase activation, calcium overload, and oxidative stress. ExpNeurol 1998;154:276–288.
19. Shi B, Raina J, Lorenzo A, et al. Neuronal apoptosis induced byHIV-1 Tat protein and TNF-alpha: potentiation of neurotox-icity mediated by oxidative stress and implications for HIV-1dementia. J Neurovirol 1998;4:281–290.
20. Cifone MG, De Maria R, Roncaioli P, et al. Apoptotic signal-ing through CD95 (Fas/Apo-1) activates an acidic sphingomy-elinase. J Exp Med 1994;180:1547–1552.
21. Wiegmann K, Schutze S, Machleidt T, et al. Functional dichot-omy of neutral and acidic sphingomyelinases in tumor necrosisfactor signaling. Cell 1994;78:1005–1015.
22. Jarvis WD, Kolesnick RN, Fornari FA, et al. Induction of ap-optotic DNA damage and cell death by activation of the sphin-gomyelin pathway. Proc Natl Acad Sci USA 1994;91:73–77.
23. Obeid LM, Linardic CM, Karolak LA, Hannun YA. Pro-grammed cell death induced by ceramide. Science 1993;259:1769–1771.
24. Gulbins E, Bissonnette R, Mahboubi A, et al. FAS-induced ap-optosis is mediated via a ceramide-initiated RAS signaling path-way. Immunity 1995;2:341–351.
25. Fischer-Smith T, Croul S, Sverstiuk AE, et al. CNS invasion byCD14�/CD16� peripheral blood-derived monocytes in HIVdementia: perivascular accumulation and reservoir of HIV in-fection. J Neurovirol 2001;7:528–541.
26. Gartner S. 2000. HIV infection and dementia. Science 2000;287:602–604.
27. Wesselingh SL, Power C, Glass JD, et al. Intracerebral cytokinemessenger RNA expression in acquired immunodeficiency syn-drome dementia. Ann Neurol 1993;33:576–582.
28. McArthur JC, Haughey N, Gartner S, et al. HIV-associateddementia: an evolving disease. J Neurovirol 2003;9:205–221.
29. Cherayil GD, Cyrus AE Jr. The quantitative estimation ofglycolipids in Alzheimer’s disease. J Neurochem 1966;13:579–590.
30. France-Lanord V, Brugg B, Michel PP, et al. Mitochondrialfree radical signal in ceramide-dependent apoptosis: a putativemechanism for neuronal death in Parkinson’s disease. J Neuro-chem 1997;69:1612–1621.
31. Yu ZF, Nikolova-Karakashian M, Zhou D, et al. Pivotal rolefor acidic sphingomyelinase in cerebral ischemia-induced cer-amide and cytokine production, and neuronal apoptosis. J MolNeurosci 2000;15:85–97.
32. Pedersen WA, Fu W, Keller JN, et al. Protein modification bythe lipid peroxidation product 4-hydroxynonenal in the spinalcords of amyotrophic lateral sclerosis patients. Ann Neurol1998;44:819–824.
33. Haughey NJ, Nath A, Chan SL, et al. Disruption of neurogen-esis by amyloid beta-peptide, and perturbed neural progenitorcell homeostasis, in models of Alzheimer’s disease. J Neurochem2002;83:1509–1524.
34. Shaikh NA. Assessment of various techniques for the quantita-tive extraction of lysophospholipids from myocardial tissues.Anal Biochem 1994;216:313–321.
35. Haughey NJ, Nath A, Mattson MP, et al. HIV-1 Tat throughphosphorylation of NMDA receptors potentiates glutamate ex-citotoxicity. J Neurochem 2001;78:457–467.
36. Luberto C, Hassler DF, Signorelli P, et al. Inhibition of tumornecrosis factor-induced cell death in MCF7 by a novel inhibitorof neutral sphingomyelinase. J Biol Chem 2002;277:41128–41139.
37. Tornatore C, Nath A, Amemiya K, Major EO. Persistent hu-man immunodeficiency virus type 1 infection in human fetalglial cells reactivated by T-cell factor(s) or by the cytokines tu-mor necrosis factor alpha and interleukin-1 beta. J Virol 1991;65:6094–6100.
38. Gelbard HA, Dzenko KA, DiLoreto D, et al. Neurotoxic effectsof tumor necrosis factor alpha in primary human neuronal cul-tures are mediated by activation of the glutamate AMPA recep-tor subtype: implications for AIDS neuropathogenesis. DevNeurosci 1993;15:417–422.
39. Kelder W, McArthur JC, Nance-Sproson T, et al. Beta-chemokines MCP-1 and RANTES are selectively increased incerebrospinal fluid of patients with human immunodeficiencyvirus-associated dementia. Ann Neurol 1998;44:831–835.
40. Mayne M, Bratanich AC, Chen P, et al. HIV-1 tat moleculardiversity and induction of TNF-alpha: implications for HIV-induced neurological disease. Neuroimmunomodulation 1998;5:184–192.
41. Jeohn GH, Kong LY, Wilson B, et al. Synergistic neurotoxic ef-fects of combined treatments with cytokines in murine primarymixed neuron/glia cultures. J Neuroimmunol 1998;85:1–10.
42. Perry SW, Hamilton JA, Tjoelker LW, et al. Platelet-activatingfactor receptor activation. An initiator step in HIV-1 neuro-pathogenesis. J Biol Chem 1998;273:17660–17664.
43. Marggraf WD, Anderer FA, Kanfer JN, et al. The formation ofsphingomyelin from phosphatidylcholine in plasma membranepreparations from mouse fibroblasts. Biochim Biophys Acta1981;664:61–73.
44. Kallen KJ, Allan D, Whatmore J, Quinn P. Synthesis of surfacesphingomyelin in the plasma membrane recycling pathway ofBHK cells. Biochim Biophys Acta 1994;1191:52–58.
45. Malgat M, Maurice A, Baraud J, et al. Sphingomyelin andceramide-phosphoethanolamine synthesis by microsomes andplasma membranes from rat liver and brain. J Lipid Res 1986;27:251–260.
46. Diringer H, Marggraf WD, Koch MA, Anderer FA. Evidencefor a new biosynthetic pathway of sphingomyelin in SV 40transformed mouse cells. Biochem Biophys Res Commun 1972;47:1345–1352.
47. Andrieu-Abadie N, Levade T. Sphingomyelin hydrolysis duringapoptosis. Biochim Biophys Acta 2002;1585:126–314.
48. Arora AS, Jones BJ, Patel TC, et al. Ceramide induces hepato-cyte cell death through disruption of mitochondrial function inthe rat. Hepatology 1997;25:958–963.
49. Garcia-Ruiz C, Colell A, Mari M, et al. Direct effect of cer-amide on the mitochondrial electron transport chain leads togeneration of reactive oxygen species. Role of mitochondrialglutathione. J Biol Chem 1997;272:11369–11377.
50. Quillet-Mary A, Jaffrezou JP, Mansat V, et al. Implication ofmitochondrial hydrogen peroxide generation in ceramide-induced apoptosis. J Biol Chem 1997;272:21388–21395.
51. Herr I, Martin-Villalba A, Kurz E, et al. FK506 preventsstroke-induced generation of ceramide and apoptosis signaling.Brain Res 1999;826:210–219.
52. Haughey NJ, Holden CP, Nath A, Geiger JD. Involvement ofinositol 1,4,5-trisphosphate-regulated stores of intracellular cal-cium in calcium dysregulation and neuron cell death caused byHIV-1 protein tat. J Neurochem 1999;73:1363–1374.
53. Haughey NJ, Mattson MP. Calcium dysregulation and neuro-nal apoptosis by the HIV-1 proteins Tat and gp120. J AcquirImmune Defic Syndr 2002;31:S55–S61.
Haughey et al: Sphingolipid Imbalance in HIV 267





























