Embed Size (px)
Citation preview

Proangiogenic Role of Tumor-Activated HepaticStellate Cells in Experimental Melanoma Metastasis
Elvira Olaso,1,4 Clarisa Salado,2 Eider Egilegor,1 Virginia Gutierrez,1 Aitor Santisteban,1 Pau Sancho-Bru,3
Scott L. Friedman,4 and Fernando Vidal-Vanaclocha1,2
Myofibroblasts infiltrate malignant liver tumors, although their pathogenic implications areunclear. Immunohistochemical detection of �-smooth muscle actin, glial fibrillary acidic protein(GFAP), and CD31 and CD34 expression was used to analyze the contribution of myofibroblaststo angiogenesis in hepatic metastasis produced by intrasplenically-injected B16 melanoma(B16M). Because activated hepatic stellate cells (HSCs) are oxygen-sensing myofibroblasts pro-ducing vascular endothelial growth factor (VEGF), the effect of B16M and human A375 mela-noma supernatants on VEGF production by immortalized rat HSC line T6 and primary culturedhuman HSCs also was studied under an hypoxic atmosphere mimicking a tumor microenviron-ment. Myofibroblast infiltration preceded endothelium recruitment in avascular micrometasta-sis and generated specific stroma for sinusoidal-type and portal-type angiogeneses. Thereafter,myofibroblasts and endothelial cells colocalized within both angiogenic patterns and their nu-merical densities correlated with metastasis development. Myofibroblasts often were GFAP-positive, suggesting an HSC origin. Melanoma supernatants stimulated VEGF messenger RNAand protein synthesis by HSCs. These effects were potentiated by hypoxia. VEGF up-regulationwas accompanied by increased expression of cyclooxygenase type 2 (COX-2) and PGE2 synthe-sis. HSC production of VEGF decreased under COX-2 inhibition, whereas it was increased byexogenous PGE2. The high VEGF expression in HSCs induced by melanoma factors and hyp-oxia resulted in mitogenic, antiapoptotic, and motogenic stimulation of both murine hepaticsinusoidal endothelium and human umbilical vein endothelium. In conclusion, temporal andpositional relationships evolve between myofibroblast and endothelium recruitment during me-tastasis development. Mechanistically, hypoxic induction of VEGF in tumor-activated HSCsmay create a proangiogenic microenvironment, facilitating endothelial cell recruitment andsurvival during hepatic metastasis transition from an avascular to a vascular stage. (HEPATOLOGY
2003;37:674-685.)
Myofibroblast-like cells have been identifiedin primary and secondary malignant tu-mors of the liver.1-3 These cells may result
from a transdifferentiation process of quiescent hepaticstellate cells (HSCs) and other perivascular fibroblasts,induced by tumor cells4-6 and tumor-activated sinusoidalcells.7 Tumor-activated HSCs are responsible for the re-modeling and deposition of tumor-associated extracellu-lar matrix8-10 and have been involved in the migration andgrowth of hepatoma and metastatic cells.4-6,11,12 HSCsalso synthesize vascular endothelial growth factor (VEGF)upon in vitro activation and in response to hypoxia,13
suggesting their additional contribution to the angiogenicneeds of developing hepatic tumors. However, this hypo-thetical role is still unclear and, to date, has been suggestedonly by the morphologic appearance of intratumoral ves-sels lined by sinusoidal cells14 and by the encirclement ofhepatic metastasis by myofibroblast-like cells during an-giogenic development.15
Abbreviations: HSC, hepatic stellate cell; VEGF, vascular endothelial growth factor;B16M, B16 melanoma; A375M, A375 melanoma; COX-2, cyclooxygenase type 2;HUVEC, human umbilical vein endothelial cells; ASMA, alpha smooth muscle actin;GFAP, glial fibrillary acidic protein; DMEM, Dulbecco’s modifed Eagle medium; FCS,fetal calf serum; HSE, hepatic sinusoidal endothelium; CM, conditioned media; cDNA,complementary DNA; PCR, polymerase chain reaction; PGE2, prostaglandin E2;ELISA, enzyme-linked immunosorbent assay; TUNEL, terminal deoxynucleotidyltrans-ferase-mediated dUTP nick end labeling; mRNA, messenger RNA.
From the 1University of the Basque Country, School of Medicine and Dentistry,Bizkaia; 2Pharmakine Ltd, Zamudio Technology Park, Bizkaia; the 3HospitalClinic I Provincial, University of Barcelona School of Medicine, Barcelona, Spain;and the 4Mount Sinai School of Medicine, New York, NY.
Received February 14, 2002; accepted November 16, 2002.Supported in part by grants from the University of the Basque Country, Leioa
(13641/2001), the Plan Nacional de I�D de la Comision Interministerial deCiencia y Tecnologıa (SAF99-0042) to F.V.-V., and the NIH grant (DK56621) toS.L.F.; E.O. and E.E. were supported by fellowship from the Department of Edu-cation, University, and Research of the Basque Government and V.G. was supportedby a fellowship from the University of the Basque Country.
Address reprint requests to: Fernando Vidal-Vanaclocha, M.D., University ofthe Basque Country, School of Medicine and Dentistry, Department of CellularBiology and Morphological Sciences, Leioa 48940-Vizcaya, Spain. E-mail:[email protected]; fax: (34) 94-464-8966.
Copyright © 2003 by the American Association for the Study of Liver Diseases.0270-9139/03/3703-0024$30.00/0doi:10.1053/jhep.2003.50068
674

In the present work, we analyzed myofibroblast-likecell recruitment and their correspondence with differentangiogenic patterns associated with metastases,15 basedon sites of intrahepatic growth of intrasplenically-injectedmurine B16 melanoma (B16M) cells.16,17 In vitro, westudied the effect of B16M and human A375 melanoma(A375M) cell–conditioned media and hypoxia on VEGFproduction by an immortalized rat hepatic stellate cell lineHSC-T6 and primary cultured human HSCs, respec-tively. Because cyclooxygenase type-2 (COX-2) contrib-utes to VEGF synthesis during tumor development,18,19
its expression and contribution to VEGF production byHSCs also was studied. Next, the effect of HSC-derivedVEGF on the migration, survival and proliferation ofprimary cultured hepatic sinusoidal endothelial cells(HSE) and human umbilical vein endothelial cells(HUVECs) was tested. Our results point to a key proan-giogenic role of tumor-activated HSCs during hepaticcolonization of melanoma cells.
Materials and Methods
Histochemistry. Hepatic metastases were producedby intrasplenic injection of 3 � 105 B16M cells into anes-thetized male C57BL/6J mice (IFFA Credo, L’Arbreole,France). Mice were killed 5 to 10 days later. Five micron–thick paraffin sections from zinc-fixed livers20 were pro-cessed for hematoxylin/eosin and reticulin stain (Gordon-Sweets silver impregnation technique), or reacted with1:50 dilutions of antihuman � smooth muscle actin(ASMA) monoclonal antibody (Sygnet Pathology Sys-tems, Dedham, MA) or rat anti-mouse CD31 polyclonalantibody (BD Pharmingen, San Diego, CA). Immunola-beled cells were detected with an avidin-biotin-peroxidase(Vectastain ABC-AP kit; Vector Laboratories, Burlin-game, CA) according to the manufacturer’s instructions.Sections (n � 60, from 10 metastasized livers) were sortedunder microscopic observation according to their averagediameter with a reference calibration scale, and the in-trametastatic densities of ASMA-expressing cells andCD31-positive capillar cross-sections were determined athigh magnification by 2 independent observers. Ten mi-cron–thick liver sections fixed in cold acetone were re-acted with 1:300 dilution of a rabbit anti-bovine glialfibrillary acidic protein (GFAP) antibody (Dako,Glostrup, Denmark), 1:25 dilution of the rat anti-mouseCD31 antibody, and/or 1:100 dilution of a rat anti-mouse CD34 polyclonal antibody (BD Pharmingen) fol-lowed by the appropriate fluorescent secondaryantibodies (Dako). Some sections then were incubatedwith the anti-ASMA monoclonal antibody and its appro-priate fluorescent secondary antibody. Tissue sections
were preincubated with a F�ab fraction of goat anti-mouseimmunoglobulin G when appropriated. Pimonidazolewas used as a marker of hepatic tissue oxygenation, aspreviously described.21 Pimonidazole (120 mg/kg; NPI,Belmont, MA) was administered intravenously 3 hoursbefore mouse killing. Pimonidazole adducts were de-tected in paraffin sections with a specific monoclonal an-tibody (NPI) and the avidin-biotin-peroxidase technique.
Isolation and Primary Culture of Hepatic Sinusoi-dal Cells. Adult male Sprague-Dawley rats andC57BL/6J mice (IFFA Credo) were used. Sinusoidal cellswere separated by enzymatic perfusion followed by metri-zamide gradient, as previously described,7 and culturedon 0.001% type I collagen-coated wells (0.5 � 106 cells/cm2) in Dulbecco’s modified Eagle medium (DMEM)supplemented with 10% fetal calf serum (FCS) (bothfrom Sigma Chemicals, St Louis, MO). After 2 hours,cells were washed and HSE cell purity assessed by ovalbu-min endocytosis (�95%). Primary cultured humanHSCs were obtained in their activated phenotype by out-growth from explants of nontumoral tissue22 and culturedin noncoated wells (0.5 � 104 HSCs/cm2 ) in DMEMculture medium supplemented with 10% FCS. Cell iso-lates were characterized by a positive staining for ASMAand used at passage 2-6.
Cell Lines. The rat hepatic stellate cell line HSC-T6has been described previously.23 Murine B16 melanomacells (B16-F10 subline) and human A375 human mela-noma cells (parental cell line) also were used. Cell lineswere cultured in noncoated wells in DMEM supple-mented with 10% FCS. Conditioned media from sub-confluent melanoma cell cultures were obtained on thetwenty-fourth hour of culture under serum-free condi-tions and diluted 1:1 with fresh DMEM before use.HUVEC cells (American Type Culture Collection, Ma-nassas, VA) were cultured in gelatin-coated wells inKaigh�s medium supplemented with 10% FCS, 0.1mg/mL heparin and 0.03 mg/mL endothelial growth sup-plement (both from Sigma Chemicals).
HSC Treatments. Subconfluent HSC-T6 cells andhuman HSCs were maintained overnight in DMEM plus0.5% FCS and then incubated in a 1% O2 hypoxia cham-ber (Billups-Rothemberg, Inc. Del Mar, CA) or normoxia(21% O2) in serum-free DMEM or melanoma-condi-tioned media (CM). NS-398 (0 to 100 �mol/L; Biomol,Plymouth Meeting, PA) was added to some culture wells30 minutes before addition of the tumor-conditioned me-dia and then maintained until the experiment had ended.HSC-T6-CM was obtained on the twenty-fourth hour oftreatment, and human HSC-CM was obtained after an18-hour pretreatment followed by a 6-hour culture inserum-free DMEM with or without NS-398 unless oth-
HEPATOLOGY, Vol. 37, No. 3, 2003 OLASO ET AL. 675

erwise stated. In some experiments 10 �g/mL of blockingantibody against human or murine VEGF or its correspond-ing irrelevant immunoglobulin G (all from R&D Systems,Minneapolis, MN) were added to HSC-CM 30 minutesbefore their addition to HSE or HUVEC cultures.
Quantitative Reverse Transcription-PolymeraseChain Reaction. Total RNA from HSC-T6 cells wasextracted with Trizol reagent (Gibco BRL; Life Technol-ogies, Grand Island, NY) according to the manufacturer�sinstructions. First-strand complementary DNA (cDNA)was generated from 5 �g of total RNA with the Moloneymurine leukemia virus reverse transcriptase (Gibco BRL),as previously described.7 Quantitative real-time polymer-ase chain reaction (PCR) was performed in triplicate in 25�L with the Sybr Green PCR Core Reagent kit and theGene Amp 5700 sequence detection system (all from PEApplied Biosytems, Foster City, CA) as previously de-scribed7 with the following primers for murine VEGF:5�GAGACCCTGGTGGACATC3� and 5�TTTCTTT-GGTCTGCATTC3� (this set of primers amplified theconserved region of all VEGF splicing forms); for murine�-actin, 5�GCCTTCCTTCTTGGGTATGG3� and 5�-ACGCAGCTCAGTAACAGTCC3�. Each PCR run in-cluded 5 points of the standard curve (titrated dilutions ofplasmid containing the specific fragment to evaluate), ano-template control, the calibrator cDNA, and cDNAobtained from HSC-T6 cells. The average relative effi-ciencies of the VEGF and �-actin PCR reactions wereapproximately equal, as shown by a slope of the CT(VEGF–�-actin) versus log of input cDNA lower than0.1. The target messenger was analyzed by measuring Ct,which represents the fractional cycle number at which thefluorescence generated by cleavage of the probe (SYBR-Green dye) passes a fixed threshold above baseline. Theratio CtVEGF/Ct�-actin was calculated for each value andrelated to that obtained for HSC-T6 cells cultured undernormoxia in basal DMEM. The standard curve was usedto determine the target messenger quantity.
VEGF and COX-2 Protein Measurement. Onehundred-fold concentrated HSCT6-CM (Biomax, cutoff5K; Millipore, Bedford, MA) was subjected to 18% so-dium dodecyl sulfate polyacrylamide gel electrophoresis.Proteins were transferred to nitrocellulose membranesand incubated with a rabbit polyclonal antibody (SantaCruz Biotechnology, Santa Cruz, CA) that recognizes theVEGF121, VEGF165, and VEGF189 splice variants. HSCcells lysed in RIPA buffer24 plus protease inhibitors weresubjected to 8% sodium dodecyl sulfate polyacrylamidegel electrophoresis and transferred to nitrocellulose mem-branes. Blots were incubated with a mouse anti-humanmonoclonal antibody (Oxford Biomedical Research, Ox-ford, MI). Equal protein loading was confirmed by im-
munoblotting for �-tubulin expression. Bands werevisualized with the Super Signal West Dura Substrate(Pierce, Rockford, IL). VEGF amounts in nonconcen-trated HSC supernatants were analyzed with a commer-cial enzyme-linked immunosorbent assay (ELISA) (R&DSystems).
Determination of COX-2 Activity. PGE2 produc-tion was analyzed with a commercial enzyme immunoas-say (Amersham Pharmacia Biotek, Uppsala, Sweden).
Cell Migration Assay. Cell migration was analyzedwith modified Boyden chambers.24 HSE cells (2.5 �105), HSC-T6 cells (5 � 104) or HUVEC cells (5 � 104)were shed onto 0.001% type I collagen-coated insertswith 8 �m pores and placed on top of 2-cm2 wells (BDBiosciences, San Jose, CA) containing DMEM or condi-tioned media, plus 0.5% FCS. After 48 hours (for HSEcells) or 6 hours (for HSC-T6 and HUVEC cells), mi-grated cells were fixed, stained with 0.2% crystal violet,and counted in 10 high-power fields per membrane. Datawas expressed relative to migration of cells incubated inDMEM plus 0.5% FCS.
Detection of Apoptotic Cells. The terminal de-oxynucleotidyltransferase-mediated dUTP nick end la-beling (TUNEL)-based ApoAlert DNA FragmentationAssay (Clontech, Palo Alto, CA) was used according tothe manufacturer’s instructions. Results were expressed asthe ratio of TUNEL-positive cells versus propidium io-dide positive nuclei per high-power field.
Cell Growth Assay. HSC-T6 and HSE cells were cul-tured for 24 and 48 hours, respectively, in serum-freebasal media. DNA synthesis was determined by additionof 3[H]-thymidine (10 �Ci per mL of culture media) forthe last 12 hours of the assay. The radioactivity incorpo-rated in the nuclei of homogenized cells was analyzed witha beta counter, as previously reported.4
Statistical Results. Statistical results refer tomean � SD. Statistical analysis was performed by SPPSstatistical software for Microsoft Windows, release 6.0(Professional Statistic, Chicago, IL). Individual com-parisons were made with Student’s 2-tailed, unpaired ttest (program Statview 512, 1986; Abacus ConceptsInc, for Macintosh Berkely, CA). The criterion for sig-nificance was P � .05 for all comparisons. In vitro experi-ments were performed at least in duplicate with independentsamples.
Results
Myofibroblastic Support of Sinusoidal-Type andPortal-Type Hepatic Metastases. Micrometastases rang-ing from 50 to 200 �m in diameter were evident by thefifth to seventh day after B16M cell injection at the pe-
676 OLASO ET AL. HEPATOLOGY, March 2003
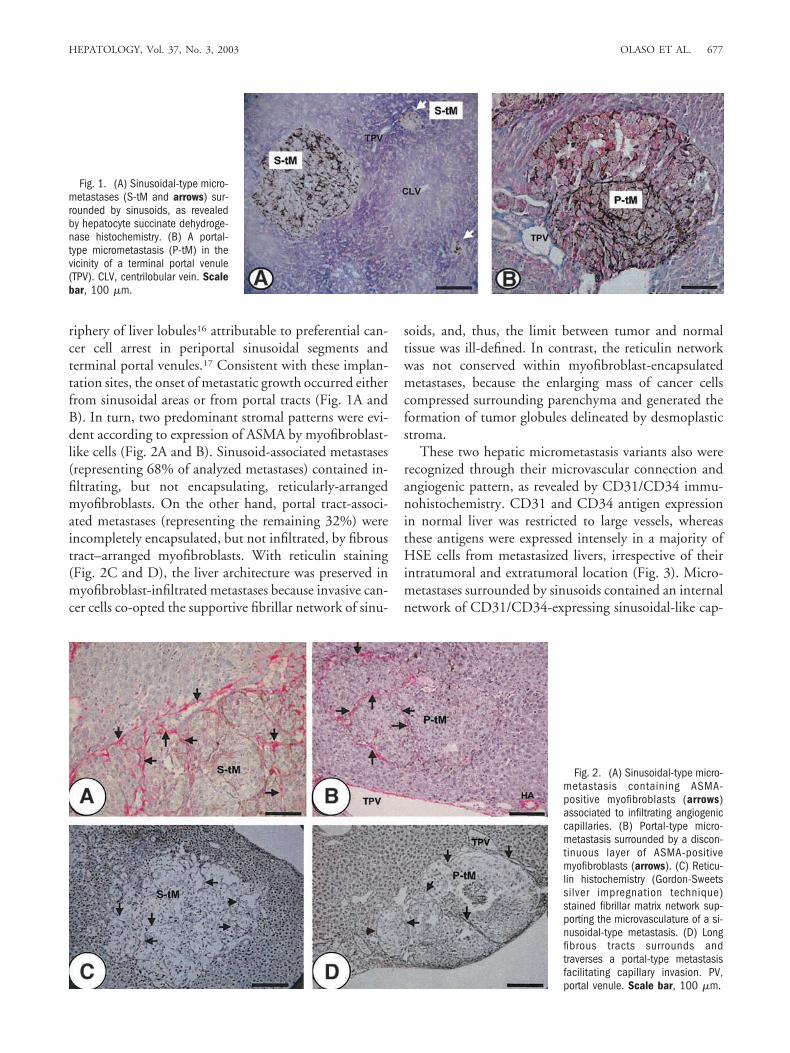
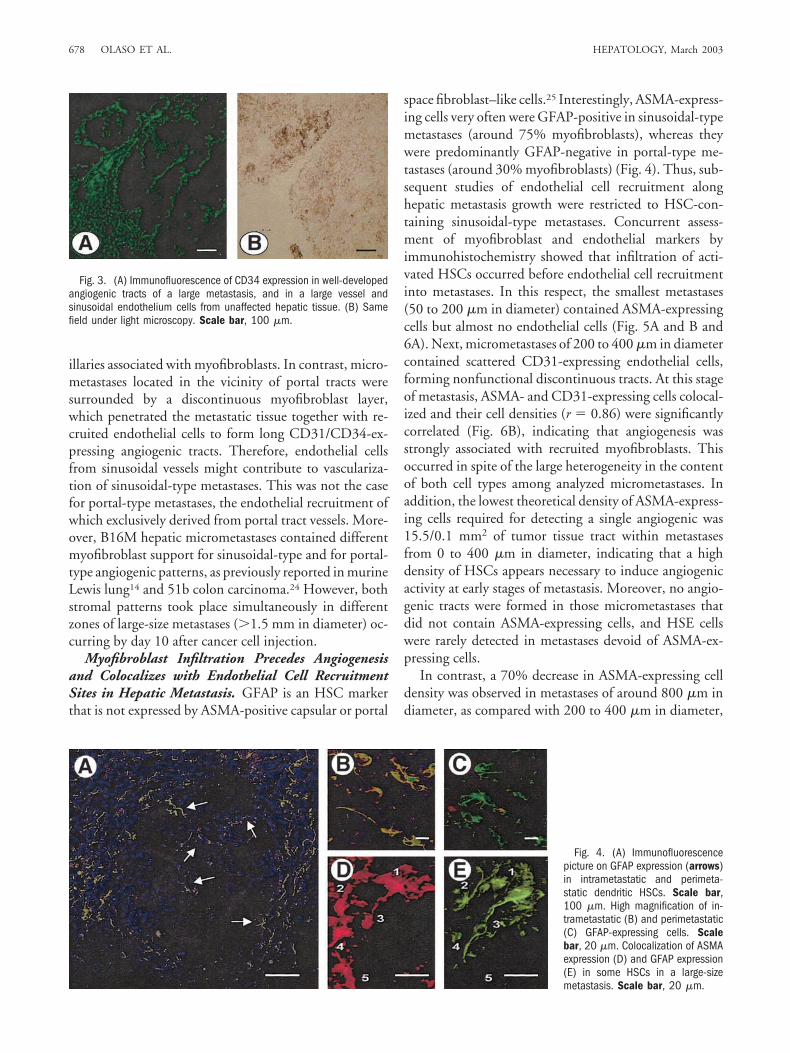
riphery of liver lobules16 attributable to preferential can-cer cell arrest in periportal sinusoidal segments andterminal portal venules.17 Consistent with these implan-tation sites, the onset of metastatic growth occurred eitherfrom sinusoidal areas or from portal tracts (Fig. 1A andB). In turn, two predominant stromal patterns were evi-dent according to expression of ASMA by myofibroblast-like cells (Fig. 2A and B). Sinusoid-associated metastases(representing 68% of analyzed metastases) contained in-filtrating, but not encapsulating, reticularly-arrangedmyofibroblasts. On the other hand, portal tract-associ-ated metastases (representing the remaining 32%) wereincompletely encapsulated, but not infiltrated, by fibroustract–arranged myofibroblasts. With reticulin staining(Fig. 2C and D), the liver architecture was preserved inmyofibroblast-infiltrated metastases because invasive can-cer cells co-opted the supportive fibrillar network of sinu-
soids, and, thus, the limit between tumor and normaltissue was ill-defined. In contrast, the reticulin networkwas not conserved within myofibroblast-encapsulatedmetastases, because the enlarging mass of cancer cellscompressed surrounding parenchyma and generated theformation of tumor globules delineated by desmoplasticstroma.
These two hepatic micrometastasis variants also wererecognized through their microvascular connection andangiogenic pattern, as revealed by CD31/CD34 immu-nohistochemistry. CD31 and CD34 antigen expressionin normal liver was restricted to large vessels, whereasthese antigens were expressed intensely in a majority ofHSE cells from metastasized livers, irrespective of theirintratumoral and extratumoral location (Fig. 3). Micro-metastases surrounded by sinusoids contained an internalnetwork of CD31/CD34-expressing sinusoidal-like cap-
Fig. 1. (A) Sinusoidal-type micro-metastases (S-tM and arrows) sur-rounded by sinusoids, as revealedby hepatocyte succinate dehydroge-nase histochemistry. (B) A portal-type micrometastasis (P-tM) in thevicinity of a terminal portal venule(TPV). CLV, centrilobular vein. Scalebar, 100 �m.
Fig. 2. (A) Sinusoidal-type micro-metastasis containing ASMA-positive myofibroblasts (arrows)associated to infiltrating angiogeniccapillaries. (B) Portal-type micro-metastasis surrounded by a discon-tinuous layer of ASMA-positivemyofibroblasts (arrows). (C) Reticu-lin histochemistry (Gordon-Sweetssilver impregnation technique)stained fibrillar matrix network sup-porting the microvasculature of a si-nusoidal-type metastasis. (D) Longfibrous tracts surrounds andtraverses a portal-type metastasisfacilitating capillary invasion. PV,portal venule. Scale bar, 100 �m.
HEPATOLOGY, Vol. 37, No. 3, 2003 OLASO ET AL. 677
illaries associated with myofibroblasts. In contrast, micro-metastases located in the vicinity of portal tracts weresurrounded by a discontinuous myofibroblast layer,which penetrated the metastatic tissue together with re-cruited endothelial cells to form long CD31/CD34-ex-pressing angiogenic tracts. Therefore, endothelial cellsfrom sinusoidal vessels might contribute to vasculariza-tion of sinusoidal-type metastases. This was not the casefor portal-type metastases, the endothelial recruitment ofwhich exclusively derived from portal tract vessels. More-over, B16M hepatic micrometastases contained differentmyofibroblast support for sinusoidal-type and for portal-type angiogenic patterns, as previously reported in murineLewis lung14 and 51b colon carcinoma.24 However, bothstromal patterns took place simultaneously in differentzones of large-size metastases (�1.5 mm in diameter) oc-curring by day 10 after cancer cell injection.
Myofibroblast Infiltration Precedes Angiogenesisand Colocalizes with Endothelial Cell RecruitmentSites in Hepatic Metastasis. GFAP is an HSC markerthat is not expressed by ASMA-positive capsular or portal
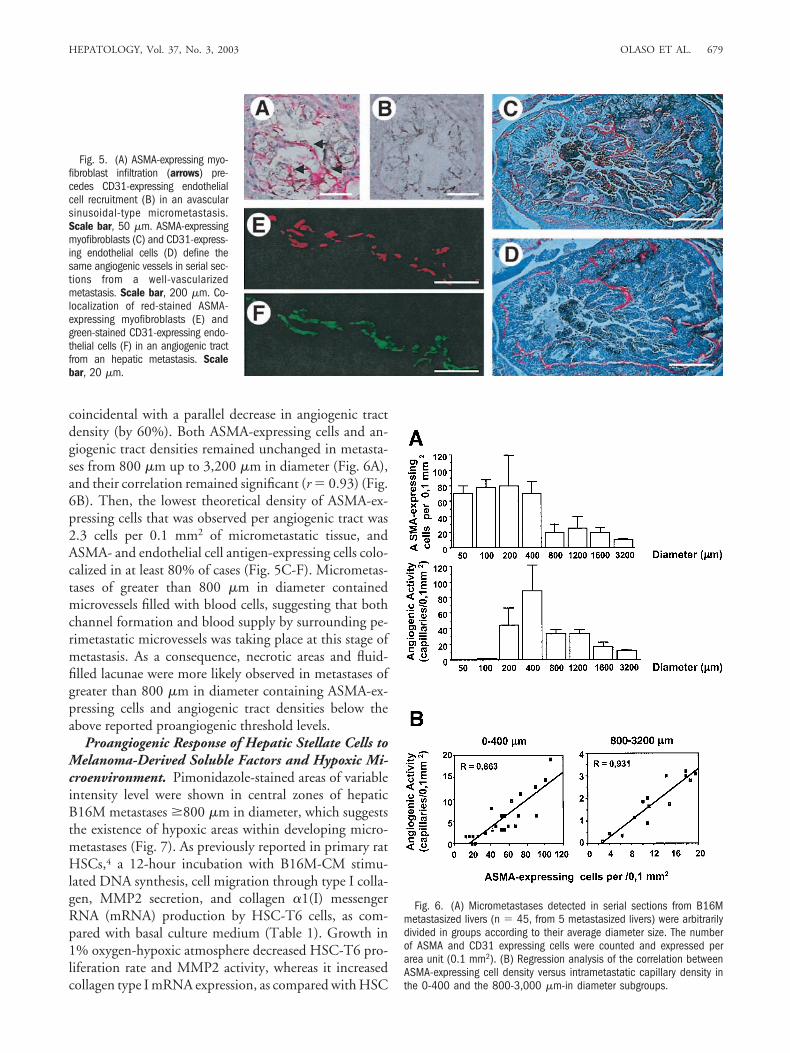
space fibroblast–like cells.25 Interestingly, ASMA-express-ing cells very often were GFAP-positive in sinusoidal-typemetastases (around 75% myofibroblasts), whereas theywere predominantly GFAP-negative in portal-type me-tastases (around 30% myofibroblasts) (Fig. 4). Thus, sub-sequent studies of endothelial cell recruitment alonghepatic metastasis growth were restricted to HSC-con-taining sinusoidal-type metastases. Concurrent assess-ment of myofibroblast and endothelial markers byimmunohistochemistry showed that infiltration of acti-vated HSCs occurred before endothelial cell recruitmentinto metastases. In this respect, the smallest metastases(50 to 200 �m in diameter) contained ASMA-expressingcells but almost no endothelial cells (Fig. 5A and B and6A). Next, micrometastases of 200 to 400 �m in diametercontained scattered CD31-expressing endothelial cells,forming nonfunctional discontinuous tracts. At this stageof metastasis, ASMA- and CD31-expressing cells colocal-ized and their cell densities (r � 0.86) were significantlycorrelated (Fig. 6B), indicating that angiogenesis wasstrongly associated with recruited myofibroblasts. Thisoccurred in spite of the large heterogeneity in the contentof both cell types among analyzed micrometastases. Inaddition, the lowest theoretical density of ASMA-express-ing cells required for detecting a single angiogenic was15.5/0.1 mm2 of tumor tissue tract within metastasesfrom 0 to 400 �m in diameter, indicating that a highdensity of HSCs appears necessary to induce angiogenicactivity at early stages of metastasis. Moreover, no angio-genic tracts were formed in those micrometastases thatdid not contain ASMA-expressing cells, and HSE cellswere rarely detected in metastases devoid of ASMA-ex-pressing cells.
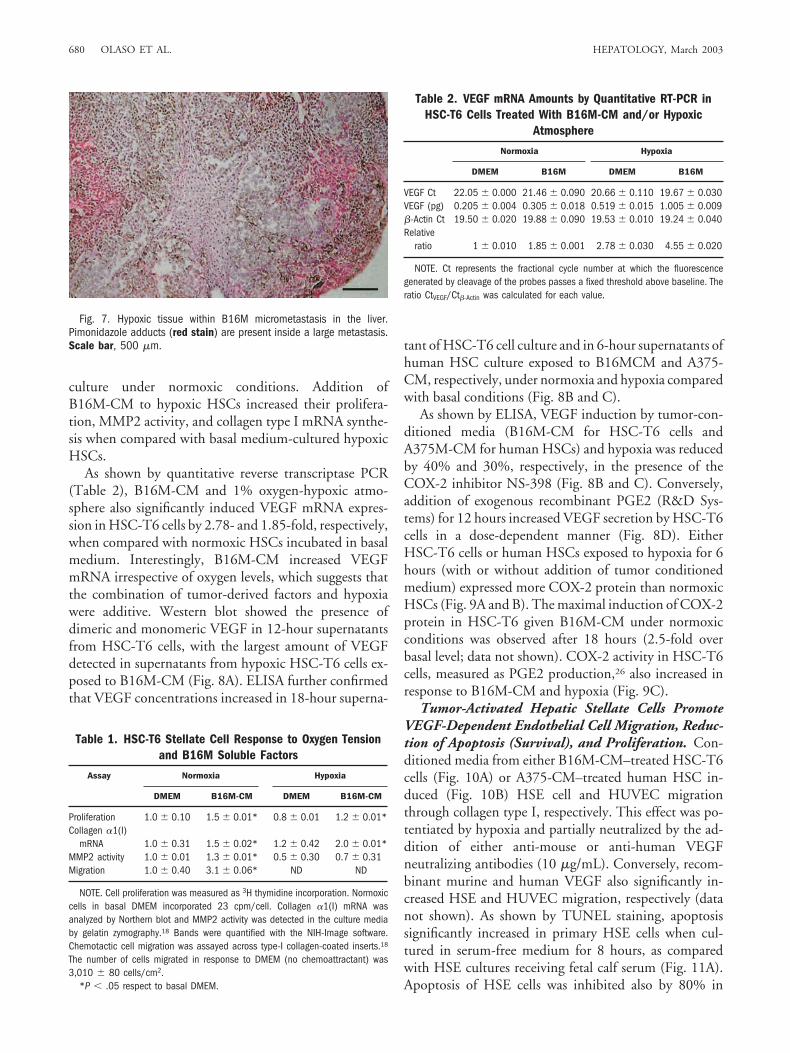
In contrast, a 70% decrease in ASMA-expressing celldensity was observed in metastases of around 800 �m indiameter, as compared with 200 to 400 �m in diameter,
Fig. 3. (A) Immunofluorescence of CD34 expression in well-developedangiogenic tracts of a large metastasis, and in a large vessel andsinusoidal endothelium cells from unaffected hepatic tissue. (B) Samefield under light microscopy. Scale bar, 100 �m.
Fig. 4. (A) Immunofluorescencepicture on GFAP expression (arrows)in intrametastatic and perimeta-static dendritic HSCs. Scale bar,100 �m. High magnification of in-trametastatic (B) and perimetastatic(C) GFAP-expressing cells. Scalebar, 20 �m. Colocalization of ASMAexpression (D) and GFAP expression(E) in some HSCs in a large-sizemetastasis. Scale bar, 20 �m.
678 OLASO ET AL. HEPATOLOGY, March 2003
coincidental with a parallel decrease in angiogenic tractdensity (by 60%). Both ASMA-expressing cells and an-giogenic tract densities remained unchanged in metasta-ses from 800 �m up to 3,200 �m in diameter (Fig. 6A),and their correlation remained significant (r � 0.93) (Fig.6B). Then, the lowest theoretical density of ASMA-ex-pressing cells that was observed per angiogenic tract was2.3 cells per 0.1 mm2 of micrometastatic tissue, andASMA- and endothelial cell antigen-expressing cells colo-calized in at least 80% of cases (Fig. 5C-F). Micrometas-tases of greater than 800 �m in diameter containedmicrovessels filled with blood cells, suggesting that bothchannel formation and blood supply by surrounding pe-rimetastatic microvessels was taking place at this stage ofmetastasis. As a consequence, necrotic areas and fluid-filled lacunae were more likely observed in metastases ofgreater than 800 �m in diameter containing ASMA-ex-pressing cells and angiogenic tract densities below theabove reported proangiogenic threshold levels.
Proangiogenic Response of Hepatic Stellate Cells toMelanoma-Derived Soluble Factors and Hypoxic Mi-croenvironment. Pimonidazole-stained areas of variableintensity level were shown in central zones of hepaticB16M metastases �800 �m in diameter, which suggeststhe existence of hypoxic areas within developing micro-metastases (Fig. 7). As previously reported in primary ratHSCs,4 a 12-hour incubation with B16M-CM stimu-lated DNA synthesis, cell migration through type I colla-gen, MMP2 secretion, and collagen �1(I) messengerRNA (mRNA) production by HSC-T6 cells, as com-pared with basal culture medium (Table 1). Growth in1% oxygen-hypoxic atmosphere decreased HSC-T6 pro-liferation rate and MMP2 activity, whereas it increasedcollagen type I mRNA expression, as compared with HSC
Fig. 5. (A) ASMA-expressing myo-fibroblast infiltration (arrows) pre-cedes CD31-expressing endothelialcell recruitment (B) in an avascularsinusoidal-type micrometastasis.Scale bar, 50 �m. ASMA-expressingmyofibroblasts (C) and CD31-express-ing endothelial cells (D) define thesame angiogenic vessels in serial sec-tions from a well-vascularizedmetastasis. Scale bar, 200 �m. Co-localization of red-stained ASMA-expressing myofibroblasts (E) andgreen-stained CD31-expressing endo-thelial cells (F) in an angiogenic tractfrom an hepatic metastasis. Scalebar, 20 �m.
Fig. 6. (A) Micrometastases detected in serial sections from B16Mmetastasized livers (n � 45, from 5 metastasized livers) were arbitrarilydivided in groups according to their average diameter size. The numberof ASMA and CD31 expressing cells were counted and expressed perarea unit (0.1 mm2). (B) Regression analysis of the correlation betweenASMA-expressing cell density versus intrametastatic capillary density inthe 0-400 and the 800-3,000 �m-in diameter subgroups.
HEPATOLOGY, Vol. 37, No. 3, 2003 OLASO ET AL. 679
culture under normoxic conditions. Addition ofB16M-CM to hypoxic HSCs increased their prolifera-tion, MMP2 activity, and collagen type I mRNA synthe-sis when compared with basal medium-cultured hypoxicHSCs.
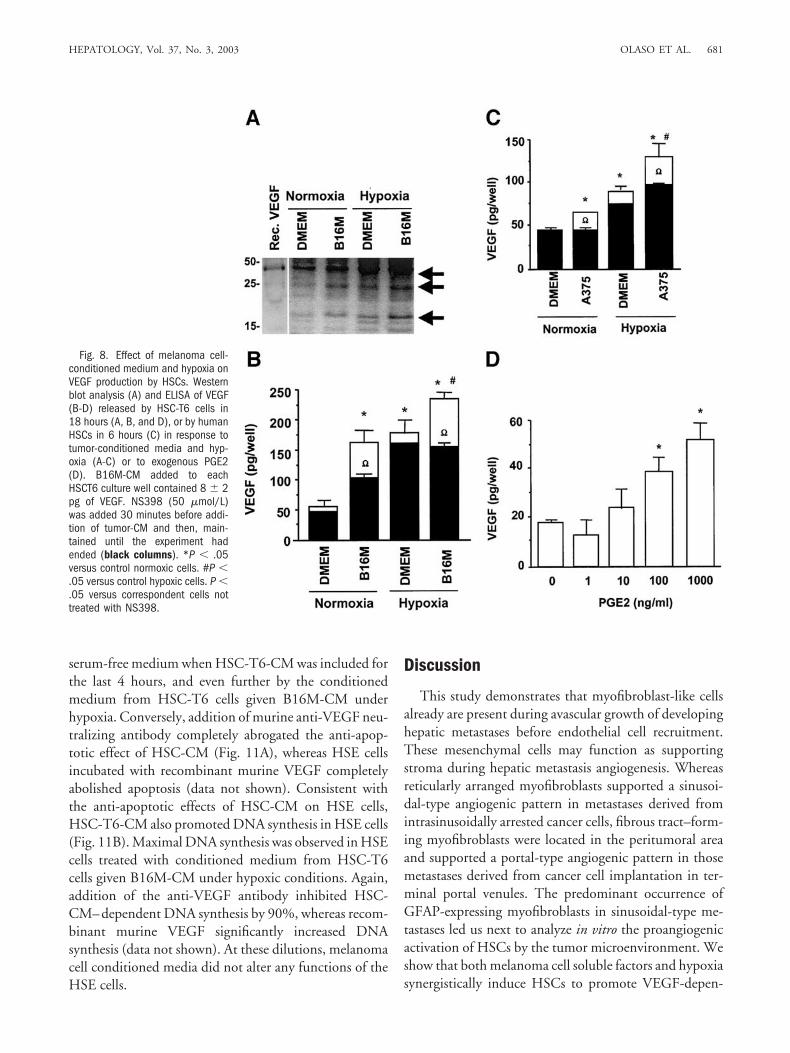
As shown by quantitative reverse transcriptase PCR(Table 2), B16M-CM and 1% oxygen-hypoxic atmo-sphere also significantly induced VEGF mRNA expres-sion in HSC-T6 cells by 2.78- and 1.85-fold, respectively,when compared with normoxic HSCs incubated in basalmedium. Interestingly, B16M-CM increased VEGFmRNA irrespective of oxygen levels, which suggests thatthe combination of tumor-derived factors and hypoxiawere additive. Western blot showed the presence ofdimeric and monomeric VEGF in 12-hour supernatantsfrom HSC-T6 cells, with the largest amount of VEGFdetected in supernatants from hypoxic HSC-T6 cells ex-posed to B16M-CM (Fig. 8A). ELISA further confirmedthat VEGF concentrations increased in 18-hour superna-
tant of HSC-T6 cell culture and in 6-hour supernatants ofhuman HSC culture exposed to B16MCM and A375-CM, respectively, under normoxia and hypoxia comparedwith basal conditions (Fig. 8B and C).
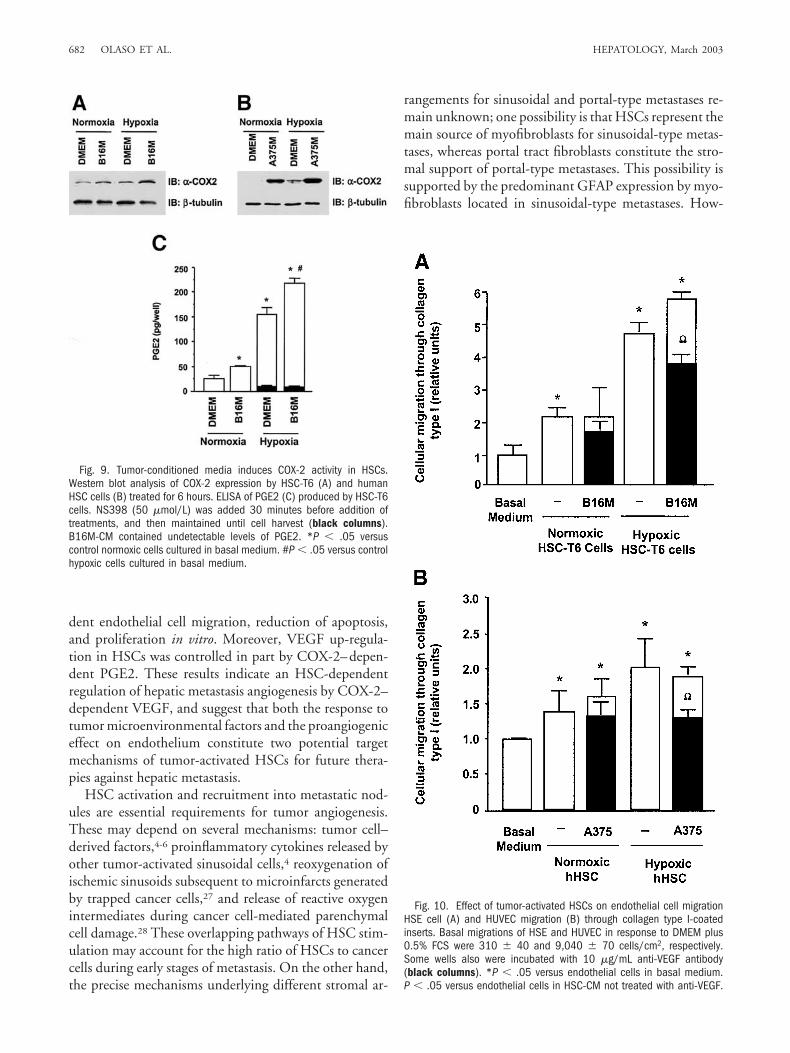
As shown by ELISA, VEGF induction by tumor-con-ditioned media (B16M-CM for HSC-T6 cells andA375M-CM for human HSCs) and hypoxia was reducedby 40% and 30%, respectively, in the presence of theCOX-2 inhibitor NS-398 (Fig. 8B and C). Conversely,addition of exogenous recombinant PGE2 (R&D Sys-tems) for 12 hours increased VEGF secretion by HSC-T6cells in a dose-dependent manner (Fig. 8D). EitherHSC-T6 cells or human HSCs exposed to hypoxia for 6hours (with or without addition of tumor conditionedmedium) expressed more COX-2 protein than normoxicHSCs (Fig. 9A and B). The maximal induction of COX-2protein in HSC-T6 given B16M-CM under normoxicconditions was observed after 18 hours (2.5-fold overbasal level; data not shown). COX-2 activity in HSC-T6cells, measured as PGE2 production,26 also increased inresponse to B16M-CM and hypoxia (Fig. 9C).
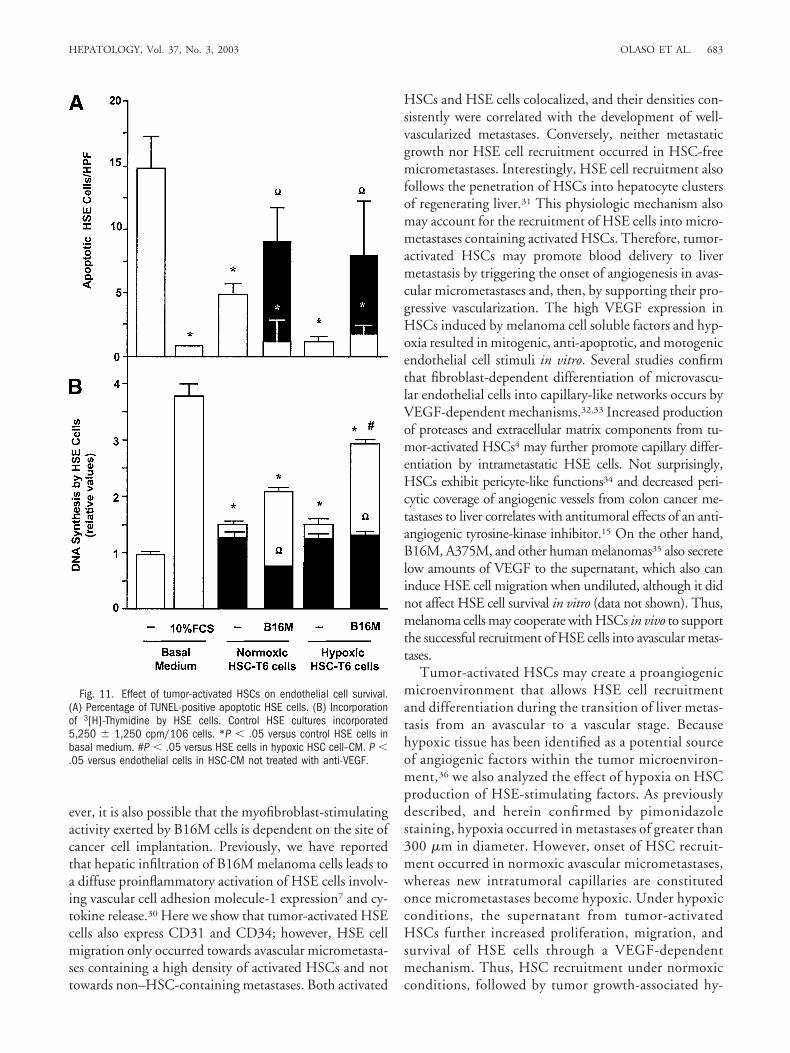
Tumor-Activated Hepatic Stellate Cells PromoteVEGF-Dependent Endothelial Cell Migration, Reduc-tion of Apoptosis (Survival), and Proliferation. Con-ditioned media from either B16M-CM–treated HSC-T6cells (Fig. 10A) or A375-CM–treated human HSC in-duced (Fig. 10B) HSE cell and HUVEC migrationthrough collagen type I, respectively. This effect was po-tentiated by hypoxia and partially neutralized by the ad-dition of either anti-mouse or anti-human VEGFneutralizing antibodies (10 �g/mL). Conversely, recom-binant murine and human VEGF also significantly in-creased HSE and HUVEC migration, respectively (datanot shown). As shown by TUNEL staining, apoptosissignificantly increased in primary HSE cells when cul-tured in serum-free medium for 8 hours, as comparedwith HSE cultures receiving fetal calf serum (Fig. 11A).Apoptosis of HSE cells was inhibited also by 80% in
Fig. 7. Hypoxic tissue within B16M micrometastasis in the liver.Pimonidazole adducts (red stain) are present inside a large metastasis.Scale bar, 500 �m.
Table 1. HSC-T6 Stellate Cell Response to Oxygen Tensionand B16M Soluble Factors
Assay Normoxia Hypoxia
DMEM B16M-CM DMEM B16M-CM
Proliferation 1.0 � 0.10 1.5 � 0.01* 0.8 � 0.01 1.2 � 0.01*Collagen �1(I)
mRNA 1.0 � 0.31 1.5 � 0.02* 1.2 � 0.42 2.0 � 0.01*MMP2 activity 1.0 � 0.01 1.3 � 0.01* 0.5 � 0.30 0.7 � 0.31Migration 1.0 � 0.40 3.1 � 0.06* ND ND
NOTE. Cell proliferation was measured as 3H thymidine incorporation. Normoxiccells in basal DMEM incorporated 23 cpm/cell. Collagen �1(I) mRNA wasanalyzed by Northern blot and MMP2 activity was detected in the culture mediaby gelatin zymography.18 Bands were quantified with the NIH-Image software.Chemotactic cell migration was assayed across type-I collagen-coated inserts.18
The number of cells migrated in response to DMEM (no chemoattractant) was3,010 � 80 cells/cm2.
*P � .05 respect to basal DMEM.
Table 2. VEGF mRNA Amounts by Quantitative RT-PCR inHSC-T6 Cells Treated With B16M-CM and/or Hypoxic
Atmosphere
Normoxia Hypoxia
DMEM B16M DMEM B16M
VEGF Ct 22.05 � 0.000 21.46 � 0.090 20.66 � 0.110 19.67 � 0.030VEGF (pg) 0.205 � 0.004 0.305 � 0.018 0.519 � 0.015 1.005 � 0.009�-Actin Ct 19.50 � 0.020 19.88 � 0.090 19.53 � 0.010 19.24 � 0.040Relative
ratio 1 � 0.010 1.85 � 0.001 2.78 � 0.030 4.55 � 0.020
NOTE. Ct represents the fractional cycle number at which the fluorescencegenerated by cleavage of the probes passes a fixed threshold above baseline. Theratio CtVEGF/Ct�-Actin was calculated for each value.
680 OLASO ET AL. HEPATOLOGY, March 2003
serum-free medium when HSC-T6-CM was included forthe last 4 hours, and even further by the conditionedmedium from HSC-T6 cells given B16M-CM underhypoxia. Conversely, addition of murine anti-VEGF neu-tralizing antibody completely abrogated the anti-apop-totic effect of HSC-CM (Fig. 11A), whereas HSE cellsincubated with recombinant murine VEGF completelyabolished apoptosis (data not shown). Consistent withthe anti-apoptotic effects of HSC-CM on HSE cells,HSC-T6-CM also promoted DNA synthesis in HSE cells(Fig. 11B). Maximal DNA synthesis was observed in HSEcells treated with conditioned medium from HSC-T6cells given B16M-CM under hypoxic conditions. Again,addition of the anti-VEGF antibody inhibited HSC-CM–dependent DNA synthesis by 90%, whereas recom-binant murine VEGF significantly increased DNAsynthesis (data not shown). At these dilutions, melanomacell conditioned media did not alter any functions of theHSE cells.
Discussion
This study demonstrates that myofibroblast-like cellsalready are present during avascular growth of developinghepatic metastases before endothelial cell recruitment.These mesenchymal cells may function as supportingstroma during hepatic metastasis angiogenesis. Whereasreticularly arranged myofibroblasts supported a sinusoi-dal-type angiogenic pattern in metastases derived fromintrasinusoidally arrested cancer cells, fibrous tract–form-ing myofibroblasts were located in the peritumoral areaand supported a portal-type angiogenic pattern in thosemetastases derived from cancer cell implantation in ter-minal portal venules. The predominant occurrence ofGFAP-expressing myofibroblasts in sinusoidal-type me-tastases led us next to analyze in vitro the proangiogenicactivation of HSCs by the tumor microenvironment. Weshow that both melanoma cell soluble factors and hypoxiasynergistically induce HSCs to promote VEGF-depen-
Fig. 8. Effect of melanoma cell-conditioned medium and hypoxia onVEGF production by HSCs. Westernblot analysis (A) and ELISA of VEGF(B-D) released by HSC-T6 cells in18 hours (A, B, and D), or by humanHSCs in 6 hours (C) in response totumor-conditioned media and hyp-oxia (A-C) or to exogenous PGE2(D). B16M-CM added to eachHSCT6 culture well contained 8 � 2pg of VEGF. NS398 (50 �mol/L)was added 30 minutes before addi-tion of tumor-CM and then, main-tained until the experiment hadended (black columns). *P � .05versus control normoxic cells. #P �.05 versus control hypoxic cells. P �.05 versus correspondent cells nottreated with NS398.
HEPATOLOGY, Vol. 37, No. 3, 2003 OLASO ET AL. 681
dent endothelial cell migration, reduction of apoptosis,and proliferation in vitro. Moreover, VEGF up-regula-tion in HSCs was controlled in part by COX-2–depen-dent PGE2. These results indicate an HSC-dependentregulation of hepatic metastasis angiogenesis by COX-2–dependent VEGF, and suggest that both the response totumor microenvironmental factors and the proangiogeniceffect on endothelium constitute two potential targetmechanisms of tumor-activated HSCs for future thera-pies against hepatic metastasis.
HSC activation and recruitment into metastatic nod-ules are essential requirements for tumor angiogenesis.These may depend on several mechanisms: tumor cell–derived factors,4-6 proinflammatory cytokines released byother tumor-activated sinusoidal cells,4 reoxygenation ofischemic sinusoids subsequent to microinfarcts generatedby trapped cancer cells,27 and release of reactive oxygenintermediates during cancer cell-mediated parenchymalcell damage.28 These overlapping pathways of HSC stim-ulation may account for the high ratio of HSCs to cancercells during early stages of metastasis. On the other hand,the precise mechanisms underlying different stromal ar-
rangements for sinusoidal and portal-type metastases re-main unknown; one possibility is that HSCs represent themain source of myofibroblasts for sinusoidal-type metas-tases, whereas portal tract fibroblasts constitute the stro-mal support of portal-type metastases. This possibility issupported by the predominant GFAP expression by myo-fibroblasts located in sinusoidal-type metastases. How-
Fig. 10. Effect of tumor-activated HSCs on endothelial cell migrationHSE cell (A) and HUVEC migration (B) through collagen type I-coatedinserts. Basal migrations of HSE and HUVEC in response to DMEM plus0.5% FCS were 310 � 40 and 9,040 � 70 cells/cm2, respectively.Some wells also were incubated with 10 �g/mL anti-VEGF antibody(black columns). *P � .05 versus endothelial cells in basal medium.P � .05 versus endothelial cells in HSC-CM not treated with anti-VEGF.
Fig. 9. Tumor-conditioned media induces COX-2 activity in HSCs.Western blot analysis of COX-2 expression by HSC-T6 (A) and humanHSC cells (B) treated for 6 hours. ELISA of PGE2 (C) produced by HSC-T6cells. NS398 (50 �mol/L) was added 30 minutes before addition oftreatments, and then maintained until cell harvest (black columns).B16M-CM contained undetectable levels of PGE2. *P � .05 versuscontrol normoxic cells cultured in basal medium. #P � .05 versus controlhypoxic cells cultured in basal medium.
682 OLASO ET AL. HEPATOLOGY, March 2003
ever, it is also possible that the myofibroblast-stimulatingactivity exerted by B16M cells is dependent on the site ofcancer cell implantation. Previously, we have reportedthat hepatic infiltration of B16M melanoma cells leads toa diffuse proinflammatory activation of HSE cells involv-ing vascular cell adhesion molecule-1 expression7 and cy-tokine release.30 Here we show that tumor-activated HSEcells also express CD31 and CD34; however, HSE cellmigration only occurred towards avascular micrometasta-ses containing a high density of activated HSCs and nottowards non–HSC-containing metastases. Both activated
HSCs and HSE cells colocalized, and their densities con-sistently were correlated with the development of well-vascularized metastases. Conversely, neither metastaticgrowth nor HSE cell recruitment occurred in HSC-freemicrometastases. Interestingly, HSE cell recruitment alsofollows the penetration of HSCs into hepatocyte clustersof regenerating liver.31 This physiologic mechanism alsomay account for the recruitment of HSE cells into micro-metastases containing activated HSCs. Therefore, tumor-activated HSCs may promote blood delivery to livermetastasis by triggering the onset of angiogenesis in avas-cular micrometastases and, then, by supporting their pro-gressive vascularization. The high VEGF expression inHSCs induced by melanoma cell soluble factors and hyp-oxia resulted in mitogenic, anti-apoptotic, and motogenicendothelial cell stimuli in vitro. Several studies confirmthat fibroblast-dependent differentiation of microvascu-lar endothelial cells into capillary-like networks occurs byVEGF-dependent mechanisms.32,33 Increased productionof proteases and extracellular matrix components from tu-mor-activated HSCs4 may further promote capillary differ-entiation by intrametastatic HSE cells. Not surprisingly,HSCs exhibit pericyte-like functions34 and decreased peri-cytic coverage of angiogenic vessels from colon cancer me-tastases to liver correlates with antitumoral effects of an anti-angiogenic tyrosine-kinase inhibitor.15 On the other hand,B16M, A375M, and other human melanomas35 also secretelow amounts of VEGF to the supernatant, which also caninduce HSE cell migration when undiluted, although it didnot affect HSE cell survival in vitro (data not shown). Thus,melanoma cells may cooperate with HSCs in vivo to supportthe successful recruitment of HSE cells into avascular metas-tases.
Tumor-activated HSCs may create a proangiogenicmicroenvironment that allows HSE cell recruitmentand differentiation during the transition of liver metas-tasis from an avascular to a vascular stage. Becausehypoxic tissue has been identified as a potential sourceof angiogenic factors within the tumor microenviron-ment,36 we also analyzed the effect of hypoxia on HSCproduction of HSE-stimulating factors. As previouslydescribed, and herein confirmed by pimonidazolestaining, hypoxia occurred in metastases of greater than300 �m in diameter. However, onset of HSC recruit-ment occurred in normoxic avascular micrometastases,whereas new intratumoral capillaries are constitutedonce micrometastases become hypoxic. Under hypoxicconditions, the supernatant from tumor-activatedHSCs further increased proliferation, migration, andsurvival of HSE cells through a VEGF-dependentmechanism. Thus, HSC recruitment under normoxicconditions, followed by tumor growth-associated hy-
Fig. 11. Effect of tumor-activated HSCs on endothelial cell survival.(A) Percentage of TUNEL-positive apoptotic HSE cells. (B) Incorporationof 3[H]-Thymidine by HSE cells. Control HSE cultures incorporated5,250 � 1,250 cpm/106 cells. *P � .05 versus control HSE cells inbasal medium. #P � .05 versus HSE cells in hypoxic HSC cell–CM. P �.05 versus endothelial cells in HSC-CM not treated with anti-VEGF.
HEPATOLOGY, Vol. 37, No. 3, 2003 OLASO ET AL. 683

poxia, may constitute two synergistic stimuli towardsintratumoral migration and survival of HSE cells.
Finally, B16M-dependent HSC production of VEGFwas regulated in part by a COX-2–dependent mecha-nism, suggesting that COX-2 inhibitors may interferewith a relevant proangiogenic activity of tumor-activatedHSC. Interestingly, the majority of hepatic metastases inthe present model are interleukin-1–dependent.30 Inter-leukin 1� up-regulates COX-2 activity in many cellularsystems including HSCs.37 Therefore, it is possible thatangiogenesis of interleukin-1–dependent metastases isregulated in part by COX-2 activity.
In summary, our results show positional and temporalrelationships between HSC and HSE cell recruitmentduring melanoma metastasis development in the liver.Mechanistically, hypoxia and tumor-derived soluble fac-tors promote production of VEGF by recruited HSCsthrough a COX-2–dependent mechanism. In turn,VEGF increases HSE cell migration, proliferation, andsurvival, which may contribute to angiogenic develop-ment within metastases.
Acknowledgment: The authors thank Marco de Lucaand Itziar Otxotorena for their skillful technical assis-tance.
References1. Dvorak HF. Tumors: wounds that do not heal. Similarities between
tumor stroma reaction and wound healing. N Engl J Med 1986;315:1650-1659.
2. Gregoire M, Lieubeau B. The role of fibroblasts in tumor behaviour. Can-cer Metast Rev 1995;14:339-350.
3. Johnson SJ, Burr AW, Toole K, Dack CL, Mathew J, Burt AD. Macro-phage and hepatic stellate cell responses during experimental hepatocarci-nogenesis. J Gastroenterol Hepatol 1998;13:145-151.
4. Olaso E, Santisteban A, Bidaurrazaga J, Gressner AM, Rosenbaum J,Vidal-Vanaclocha F. Tumor-dependent activation of rodent hepatic stel-late cells during experimental melanoma metastasis. HEPATOLOGY 1997;26:634-642.
5. Faouzi S, Lepreux S, Bedin C, Dubuisson L, Balabaud C, Bioulac-Sage P,Desmouliere A, et al. Activation of cultured rat hepatic stellate cells bytumoral hepatocytes. Lab Invest 1999;79:485-493.
6. Shimizu S, Yamada N, Sawada T, Ikeda K, Kawada N, Seki S, KanedaK, et al. In vivo and in vitro interactions between human colon carci-noma cells and hepatic stellate cells. Jpn J Cancer Res 2000;91:1285-1295.
7. Mendoza L, Carrascal T, de Luca M, Fuentes AM, Salado C, Blanco J,Vidal-Vanaclocha F. Hydrogen peroxide mediates vascular cell adhesionmolecule-1 expression from interleukin-18-activated hepatic sinusoidalendothelium: implications for circulating cancer cell arrest in murine liver.HEPATOLOGY 2001;34:298-310.
8. Klieverik L, Fehres O, Griffini P, Van Noorden CJF, Frederiks W. Pro-motion of colon cancer metastasis in rat liver by fish oil diet is not due toreduced stromal formation. Clin Exp Met 2001;18:371-377.
9. Terada T, Makimoto K, Terayama N, Suzuki Y, Nakanuma Y. Alpha-smooth muscle actin-positive stromal cells in cholangiocarcinomas, hepa-tocellular carcinoma and metastatic liver carcinoma. J Hepatol 1996;24:706-712.
10. Hautekeete ML, Geerts A. The hepatic stellate (Ito) cell: its role in humanliver disease. Virchow Arch 1997;430:195-207.
11. Theret N, Musso O, Turlin B, Lotrian D, Bioulac-Sage P, Campion JP,Boudjema K, et al. Increased extracellular matrix remodeling is associatedwith tumor progresssion in human hepatocellular carcinoma. HEPATOL-OGY 2001;34:82-88.
12. Mouvoisin A, Bisson C, Si-Tayeb K, Balabaud C, Desmouliere A, Rosen-baum J. Involvement of matrix metalloproteinase type-3 in hepatocytegrowth factor induced invasion of human hepatocellular carcinoma cells.Int J Cancer 2002;97:157-162.
13. Ankoma-Sey V, Wang Y, Dai Z. Hypoxic stimulation of vascular endo-thelial growth factor expression in activated rat hepatic stellate cells. HEPA-TOLOGY 2000;31:141-148.
14. Paku S, Lapis K. Morphological aspects of angiogenesis in experimentalliver metastases. Am J Pathol 1993;143:926-936.
15. Shaheen RM, Tseng W, Davis DW, Liu W, Reinmuth N, Vellagas R,Wieczorek AA, et al. Tyrosine kinase inhibition of multiple angiogenicgrowth factors receptors improves survival in mice bearing colon cancerliver metastases by inhibition of endothelial cell survival mechanism. Can-cer Res 2001;61:1464-1468.
16. Barbera-Guillem E, Alonso-Varona A, Vidal-Vanaclocha F. Selective im-plantation and growth in rats and mice of experimental liver metastasis inacinar zone 1. Cancer Res 1989;49:4003-4010.
17. Anasagasti MJ, Martin JJ, Mendoza L, Obrador E, Estrela JM, McCuskeyRS, Vidal-Vanaclocha F. Glutathione protects metastatic melanoma cellsagainst oxidative stress in the murine hepatic microvasculature. HEPATOL-OGY 1998;27:1249-1256.
18. Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cycloox-ygenase regulates angiogenesis induced by colon cancer cells. Cell 1998;93:705-716.
19. Masferrer JL, Leahy KM, Koki AT, Zweifel BS, Settle SL, Woerner BM,Edwards DA, et al. Antiangiogenic and antitumor activities of cyclooxy-genase-2 inhibitors. Cancer Res 2000;60:1306-1311.
20. Solaun MS, Mendoza L, de Luca M, Gutierrez V, Lopez M-P, Olaso E,Sim BKL, et al. Endostatin inhibits murine colon carcinoma sinusoidal-type metastases by preferential targeting of hepatic sinusoidal endothe-lium. HEPATOLOGY 2002;35:1104-1116.
21. Arteel GE, Thurman RG, Yates JM, Raleigh JA. Evidence that hypoxiamarkers detect oxygen gradients in livers: pimonidazole and retrogradeperfusion in rat liver. Br J Cancer 1995;72:889-895.
22. Bataller R, Nicolas JM, Gines P, Esteve A, Nieves Gorbig M, Garcia-Ramallo E, Pinzani M, et al. Arginine vasopressin induces contraction andstimulation growth of cultures hepatic stellate cells. Gastroenterology1997;113:615-624.
23. Vogel S, Piantedosi R, Frank J, Lalazar A, Rockey DC, Friedman SL,Blaner WS. An immortalized rat liver stellate cell line (HSC-T6): a new cellmodel for the study of retinoid metabolism in vitro. J Lipid Res 2000;41:882-893.
24. Olaso E, Ikeda K, Eng FJ, Xu L, Wang LH, Lin HC, Friedman SL. DDR2receptor promotes MMP-2-mediated proliferation and invasion by hepaticstellate cells. J Clin Invest 2001;108:1369-1379.
25. Niki T, De Blesser PJ, Xu G, Van Den Berg K, Wisse E, Geerts A.Comparison of glial fibrillary acidic protein and desmin staining innormal and CCl4-induced fibrotic rat livers. HEPATOLOGY 1996;23:1538-1545.
26. Nieto N, Greenwell P, Friedman SL, Zhang F, Dannenberg AJ, Ceder-baum AI. Ethanol and arachydonic acid increase �2(I) collagen expressionin rat hepatic stellate cells overexpressing cytochrome P450 2E1. Role ofH2O2 and cyclooxygenase-2. J Biol Chem 2000;275:20136-20145.
27. Jessup J, Battle P, Waller H, Edmiston K, Stolz D, Watkins S, Locker J, etal. Reactive nitrogen and oxygen radicals formed during hepatic ischemia-reperfusion kill weakly metastatic colorectal cancer cells. Cancer Res 1999;59:1825-1829.
28. Weiss L. Biomechanical interactions of cancer cells with the microvasculatureduring hematogenous metastasis. Cancer Met Rev 1992;11:227-235.
29. Soares FA, Shaughnessy SG, MacLarkey WR, Orr FW. Quantification andmorphologic demonstration of reactive oxygen species produced by
684 OLASO ET AL. HEPATOLOGY, March 2003

Walker 256 tumor cells in vitro and during metastasis in vivo. Lab Invest1994;71:480-489.
30. Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, Fuentes A, Anasagasti M,Martın J, Carrascal T, et al. IL-18 regulates IL-1�-dependent hepaticmelanoma metastasis via vascular cell adhesion molecule-1. Proc Natl AcadSci U S A 2000;97:734-739.
31. Martinez-Hernandez A, Amenta PS. The extracellular matrix in hepaticregeneration. FASEB J 1995;9:1401-1410.
32. Montesano R, Pepper MS, Orci L. Paracrine induction of angiogenesis invitro by Swiss 3T3 fibroblasts. J Cell Sci 1993;105:1013-1024.
33. Omaida C, Velazquez U, Ruthanne Snyder U, Zhao-Jun Liu U, RonaldM, Fairman U, et al. Fibroblast-dependent differentiation of human mi-crovascular endothelial cells into capillary-like, three-dimensional net-works. FASEB J 2002;16:1316-1318.
34. Rockey DC. Hepatic blood flow regulation by stellate cells in normal andinjured liver. Semin Liver Dis 2001;21:337-349.
35. Graeven U, Rodeck U, Karpinski S, Jost M, Philippou S, Schmiegel W.Modulation of angiogenesis and tumorigenicity of human melanocyticcells by vascular endothelial growth factor and basic fibroblast growthfactor. Cancer Res 2001;61:7282-7290.
36. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factorinduced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature1992;59:843-845.
37. Efsen E, Bonacchi A, Pastacaldi S, Valente AJ, Wenzal UO, Tosti-GuerraC, Pinzani M, et al. Agonist-specific regulation of monocyte chemoattrac-tant protein-1 expression by cyclooxygenase metabolites in hepatic stellatecells. HEPATOLOGY 2001;33:713-721.
HEPATOLOGY, Vol. 37, No. 3, 2003 OLASO ET AL. 685





























