Embed Size (px)
Citation preview

ELSEVIER PIh S0016-2361(97)00134-8
Fuel Vol. 76, No. 12, pp. 1105-1115, 1997 © 1997 Elsevier Science Ltd. All rights reserved
Printed in Great Britain 0016-2361/97 $17.00+0.00
Reaction of methane with coal
Kezhan Yang a, Barry D. Batts a'*, Michael A. Wilson *b, Martin L. Gorbaty *c, Peter S. Maa c, Mervyn A. Long *d, Simon X. J. He d and Moetaz I. Attalla e aScho01 of Chemistry, Macquarie University, Macquarie, NSW 2109, Australia bChemistry Department, University of Technology, Sydney, PO Box 123, Broadway, NSW 2007, Australia CExxon Research and Engineering Company, PO Box 998, Annandale, NJ 08801-0998, USA dScho01 of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia eCSlRO Division of Coal and Energy Technology, PO Box 136, North Ryde, NSW 2113, Australia (Received 17 December 1996; revised 11 April 1997)
The reactivities of Australian coals and one American coal with methane or methane-hydrogen mixtures, in the range 350-400°C and a range of pressures (6.0-8.3 MPa, cold) have been examined. The effects of aluminophosphates (A1PO) or zeolite catalysts, with and without exchanged metals, on reactivity have also been examined. Yields of dichloromethane extractable material are increased by using a methane rather than a nitrogen atmosphere and different catalysts assist dissolution to various extents. It appears that surface exchanged catalysts are effective, but incorporating metals during AIPO lattice formation is detrimental. Aluminium phosphate catalysts are unstable to water produced during coal conversion, but are still able to increase extraction yields. For the American coal, under methane-hydrogen and a copper exchanged zeolite, 51.5% conversion was obtained, with a product selectivity close to that obtained under hydrogen alone, and with only 2% hydrogen consumption. The conversion under methane-hydrogen was close to that obtained under hydrogen alone, while a linear dependence of conversion on proportion of methane would predict a 43% conversion under methane- hydrogen. This illustrates a synergistic effect of the methane-hydrogen atmosphere for coal liquefaction using this catalyst system. © 1997 Elsevier Science Ltd.
(Keywords: coal; methane; zeolites; aluminophosphates)
INTRODUCTION
Forty to fifty per cent of the costs of direct coal liquefaction are in hydrogen generation. Thus, if coal can be liquefied by using an alternative, less expensive gas, savings up to 50% might be achievable. Methane is the major component of natural gas and, until recently, has been used solely for heating purposes by combustion. However, developments in catalyst design have made it useful for the formation of methanol via synthesis gas 1-5 ( C H 4 + H 2 0 ~ 3H2 + CO and then CO + 2H2 ~ CH3OH). It is believed that methane activation is promoted by oxide ions which are instrumental in the formation of methyl or methylene radicals as a first step in the overall process. This catalytic activation of methane does not operate at the typical low temperatures (400°C) which might be employed for selective thermal decomposition of the organic materials present in coal.
Recently, we have been able to demonstrate that micro- porous aluminophosphate (A1PO) catalysts can activate elemental hydrogen 6'7. The catalytic activity was enhanced by the incorporation of common metals such as lead and bismuth, and we were able to show that these catalysts can methylate toluene with methanol and convert methanol to dimethylether. A United States patent has appeared 8 which describes the activation of methane by A1PO
* Author to whom correspondence should be addressed
molecular sieves, A1PO4-5 and A1PO4-35. These were used with cobalt, manganese or iron as components of the catalyst. Between 400 and 500°C and at 1 atm pressure, the rate of methane conversion increased steadily to produce ethylene and higher hydrocarbons with efficiencies as high as 50% at 500°C. Since the mechanism by which these catalysts react with methane is different from that of the rare earth oxides, the purpose of this research was to explore the possibility of using these catalysts for low temperature (350-400°C) activation of methane toward reaction with coals. We have already re~o2rted activity with simple coal- related model compounds -
Once methane is incorporated, there may be other beneficial flow-on effects. We have previously demon- strated 13,14 that methyl groups on coal macromolecules play a crucial role in capping radicals generated by pyrolysis, and thus are key entities in forming liquid products. Thus, methyl groups in coal or from other reagents assist in promoting oil yields from coal. Highly reactive CH3 radicals actively promote liquefaction. Tetramethyl or tetraethyl lead 15 was found to be a superb additive for increasing yields of oil at low hydrogen pressures.
There also appears to be mounting evidence that methane can react directly with coal at coal liquefaction tempera- tures. Sundaram et al. 16, in another United States patent, have shown that in the temperatures range 350-450°C at 6.9-17.2 MPa pressure more coal is converted to volatile
Fuel 1997 Volume 76 Number 12 1105

Reaction of methane with coal: K. Yang et al.
products under methane than under nitrogen. A hydrogen donor solvent was used in these experiments, typically with a coal-to-solvent ratio of 3:1. Yields of residues before and after extraction with organic solvents were also found to be lower under methane than under helium 17. In a fairly intensive study, Egiebor and Gray 18 were able to show in 1:1 coal-to-tetralin ratio experiments that conversions to gaseous and liquid products were higher under methane than under hydrogen, and during pyrolysis without tetralin, conversions with methane were higher than under argon, but lower than under hydrogen. It was demonstrated that methane was consumed and liquefaction with methane produced more than seven times as much C2-C5 gases as with hydrogen and eleven times the amount produced with argon. Moreover, the hydrogen-to-carbon ratios of the toluene soluble products from the methane experi- ments were higher than those from the argon experiments. When the solvent was examined, the amounts of methyl- naphthalenes formed were found to be greater under methane than under hydrogen or argon. This appears to be definitive proof for the involvement of methane in reactions during coal liquefaction, even when catalyst is absent. Further support for this observation comes from studies of asphalt degradation 19. Ozawa and coworkers 19 have shown that the composition of the toluene soluble fraction obtained by treating asphalts with molten tin was dependent on the charge gas, and different results were obtained with methane from those with nitrogen. Indeed labelled model compound studies at 300°C with benzene, cyclopentene, methylenecyclopentane and simple alkenes showed that these compounds incorporated methane over nickel sup- ported catalysts containing carbon. The process is not just a simple methylation, since much of the carbon incorporated comes from the catalyst and not from the methane 2°'zl. Label has also shown that hydrogen in methane participates in heavy oil upgrading in a methane atmosphere 2z.
Pore size and mineral matter distribution can also be important in preventing agglomeration and enhancing
23 liquefaction . Phosphate and alumina have an important role in this regard z4. In this paper the effects of A1PO and zeolites on coal pyrolysis in the presence of methane or methane-hydrogen mixtures are examined. The effects of coal type, both in the presence and absence of methane, were studied, and the percentage conversion to gaseous and liquid products noted. Although the results are less convincing than those obtained with model compounds, there is evidence for methane capture and a synergistic effect is observed with methane-hydrogen atmospheres.
EXPERIMENTAL
Materials Coals. Details of the coals used are given in Table 1.
Most of the coals used were of Australian origin but one United States coal, Illinois No. 6 was used for comparative purposes.
Catalysts. Several catalyst types were used with various metal loadings. Catalysts used in this study are listed in Table 2 and details of the methods by which they were prepared are outlined below. The nomenclature for A1PO catalysts is somewhat complex and, hence, is first dealt with here. The simple AIPO catalysts are described as A1PO4-5 because they contain phosphate tetrahedra. If the tetrahedral structure of phosphate is incomplete, it is described as APO-5 (the subscript 4 and also the 1 are dropped). Thus silicon substituted catalyst is described as SAPO-5. If an additional metal is incorporated into the synthesis the metal is included first (e.g. PbAPO-5), but if the additional metal is exchanged on to the catalyst, the symbol for the metal is followed by a stroke. Thus Pb/A1PO4-5 is a catalyst with lead exchanged on to the catalyst after synth- esis, but there is no lead in the lattice which is strictly AIPO4-5.
Catalyst 1:ALP04-5 aluminophosphate molecular sieve. In a typical preparation, phosphoric acid (Analytical Reagent grade (AR), 85%) was diluted with distilled water in a glass vessel suitable for autoclave experiments and then pseudoboehmite (15.70 g) was gradually added with strong agitation until the reactants were well mixed. Tetraethylammonium hydroxide (TEAOH) (42.13 g AR, 20% solution in water) was then added to the reaction mix- ture while stirring, to produce a white gel. The mixture within the glass vessel was sealed in a stainless steel reactor (autoclave) and placed in an oven at 150-200°C for 24 h for hydrothermal crystallization (HTC). After cooling to room temperature in air, solids were recovered by filtration. They were washed with hot water to pH = 6-7 and dried at 100°C overnight.
The material was then calcined by the following procedure. Under compressed air blowing at 5 ml/min, the sample was heated to 100°C at 35°C/h, then kept at 100°C for 0.5 h followed by a second temperature programmed step of 35°C/h until 750°C was reached. Subsequently, the sample was kept at 750°C for 1 h, before cooling to room temperature.
Catalyst 2:ALP04-5 with lead incorporation on surface, (Pb/AIPO-5). AIPO4-5 was synthesized as above. Lead nitrate (2.69 g) was dissolved in distilled water and
Table 1 Ultimate, aromaticity, moisture and ash analysis of coals
Coal Elemental analysis (% w/w daf) a
C H N S O
Moisture Ash
(% w/w) (% w/w)
Illinois No. 6 75.8 5.4 1.5 4.9
Millmerran 77.8 6.5 I. 1 0.7
Leigh Creek 70.5 4.0 1.2 0.5
Collie 72.5 3.8 1.1 0.8
Wintana 70.9 4.3 1.9 3.3
Metropolitan 89.6 4.6 1.5 0.4
12.4 10.1 10.4 0.71 c
13.9 5.9 14.0 0.59
23.8 24.3 10.3 0.62
21.8 20.6 6.4 0.75
19.6 15.7 20.0 0.62
3.9 1.0 9.8 0.82
" Oxygen by difference b Aromaticity determined by solid state L3C NMR using c Ref25
1 ms contact time and cross polarization
1106 Fuel 1997 Volume 76 Number 12

Reaction of methane with coal: K. Yang et al.
Table 2 Structural information on catalysts
Catalyst % of AIPO4-5 Molar composition b phase"
Surface metal Surface % coordination as metal oxide c area d of AI which is
tetrahedral e
No. Type MeO2 SiO2 AlOe PO2 % w/w m2/g
1 AIPO4-5 > 99 0 0 0.50
2 Pb! AIPO-5 c > 99 0 0 0.50
3 PbAPO-5 c 95.5 0.06 0 0.48
4 NiAPO-5 > 98 0.06 0 0.48
5 NiAPO-5 f N/D 0 0 0.50
6 SAPO-5 > 95 0 0.08 0.49
7 CuSAPO-5 g > 98 0.06 0.05 0.46
8 Cu/SAPO-5 g > 98 0 0.05 0.46
9 CuAPO-5 > 98 0.06 0 0.48
10 Cu/ZSM5 N/A 0 0.98 0.02
11 Cu/Beta N/A 0 1.00 0
0.50 0 395 100
0.50 40.0 44 100
0.46 0 310 100
0.46 0 349 100
0.50 1.16 386 100
0.43 0 442 100
0.44 0 311 100
0.44 2.80 371 100
0.46 0 290 100
0 2.18 N/D N/A
0 0.49 N/D N/A
a Determined by X-ray diffraction b The molar compositions were determined by ICPAES before surface exchange c The percentage of metal in lattice as MeO2. Ignores surface adsorbed metal d Measured by BET method e By solid state NMR Y This is the reduced form of the catalyst, ie Ni metal (see experimental section)
These pairs of catalysts are distinguished by the way the metal was introduced into the catalysts as described in text N/A not applicable, N/D not determined
the solution was then heated to 30°C. A1PO4-5 (3.0 g) was added to the solution to form a suspension. The suspension was then stirred at 30°C for 3 h and then dried on a rotary evaporator to yield a fine powder. The dried powder was calcined under a stream of air in a similar manner to that used for AIPO4-5, except that the sample was heated to 500°C at 35°C/h and held at 500°C for 6 h. The catalyst prepared by this method contained lead oxide carried on the surface of the microporous A1PO4-5.
Catalyst 3:ALP04-5 with lead oxide incorporation during synthesis, PbAPO-5. Phosphoric acid (8.09g, 85%) was diluted with distilled water (6.05 g) in a glass vessel as above. A mixture of lead nitrate (1.09g) and pseudoboehmite (4.6 g) was gradually added to the diluted phosphoric acid solution with strong agitation until the reactants were well mixed. TEAOH, (25.7 g; 20% solution in water) was then added to the reaction mixture with stirring. The mixture was sealed within the glass vessel in a stainless steel reactor and allowed to stand for 30 min before being placed in an oven at 150-200°C for 24 h for HTC. After HTC, the reactor was cooled rapidly and the product was recovered by filtration. After washing with hot water to pH = 6 -7 and drying at 100°C overnight, the sample was calcined using a stream of air (5 ml/min) and following the temperature program as for catalyst 1 except that the sample was held at the upper limit of 750°C for only 5 min.
Catalyst 4:ALP04-5 catalysts with nickel incorporation, NiAPO-5. Nickel sulphate hexahydrate (1.74 g) was dis- solved in distilled water (22g) and phosphoric acid (39.3 ml; AR; 85%) was added. Psuedoboehmite (21.6 g), TEAOH (15 g; 20% solution in water) were added as described above. However, in this synthesis the mixture was placed in an autoclave with a Teflon liner at 150- 200°C for 24 h. After cooling, washing with hot water to pH = 6 -7 and drying overnight at 100°C, the sample was calcined as described for catalyst 1, except the upper temperature limit was 700°C.
Catalyst 5. The reduced form of NiAPO-5 catalyst was obtained by passing a stream of H2 through catalyst 4 at a flow rate of about 5 ml/min, for 6 h at 400°C.
Catalyst 6: silicon substituted A1PO, SAPO-5. Phos- phoric acid (5.27 g; 85%) was diluted with distilled water. Pseudoboehmite (3.74 g) was then gradually added to the diluted H3PO4 solution with strong agitation until the reac- tants were well mixed. TEAOH (16.83 g; 20% solution in water) was added to the reaction mixture with stirring. Tetraethyl orthosilicate Si(OC2H5)4 (0.52 g) was finally added and the mixture was stirred for about 10 min and then sealed into a stainless steel reactor with glass liner. The mixture was allowed to stand at room temperature for 30 min before being placed in an oven at 150-200°C for 24 h for HTC. After cooling, the product was recovered as described above and dried overnight at 100°C. Calcina- tion was carried out under a stream of air using the same temperature program as that for catalyst 3.
Catalyst 7: copper and silicon co-substituted A1PO, CuSAPO-5. This was synthesized in a manner similar to that for catalyst 6 but cupric nitrate (Cu(NO3)2.3H20) was added to the dilute phosphoric acid. The amounts of the components in the sample were adjusted so the molar relationship (Si + P) = (Cu + A1) was main- tained. The material is a green solid and is designated CuSAPO-5.
Catalyst 8: copper exchanged SAPO-5, Cu/SAPO-5. The preparation involved two steps. SAPO-5 (catalyst 6) was first synthesized and then copper was exchanged on to the SAPO-5 rather than incorporated into the thermal synthesis. A cupric nitrate solution in water (30 ml; 3 M) was warmed to 55°C. SAPO-5 (1.5 g) was then added with stirring. The suspension was further stirred for 3 h at 55°C and then filtered. The solids were washed thoroughly with distilled water and dried in air, and then dried in an oven at 100°C overnight to yield a fine blue solid.
Fuel 1997 Volume 76 Number 12 1107

Reaction of methane with coal: K. Yang et al.
Table 3 Stabilities of A1PO4-5 and CuSAPO-5 in presence of water
Expt. No. CH4 Temp. Time H20/Catalyst AIPO4-5 phase (MPa) (°C) (h) (wt/wt) after experiment"
AIPO4-5
1 atmospheric 15 24 0.52 > 99%
2 atmospheric 15 24 1.50 > 99%
3 atmospheric 15 24 1.97 > 99%
4 6.9 400 1 1.00 0%
5 6.9 400 1 0.25 90%
6 6.9 400 1 0.10 90%
CuSAPO-5
7 6.9 400 1 0.15 45%
8 6.9 400 1 0.05 70%
9 6.9 400 1 0.00 100%
a by XRD analysis
Catalyst 9:ALP04-5 catalysts with copper incorpora- tion, CuAPO-5. The procedure for the preparation of this catalyst was the same as that described above for catalyst 7 except that tetraethyl orthosilicate [Si(OC;Hs)4] was not added. The final material was green in colour.
Catalyst 10: copper surface exchanged zeolite, Cu/ZSM-5. A slurry of Zeolite HZSM-5 (100 g) in a cupric nitrate solu- tion, prepared by dissolving 90 g of Cu(NO3)2.3H20 in 400 mL water, was stirred for 5 h at 65°C. The suspension was filtered and the precipitate again stirred with a similar, but fresh, copper nitrate solution under the same condition and again filtered. The stirring and filtering procedure was repeated once more, and the final precipitate washed thoroughly with distilled water (1.5L) and dried in a vacuum oven at 100°C for 20 h. After cooling to room tem- perature in a desiccator, the dried solid was gently ground and passed through a 710/~m screen. The fraction below 710/~m was used as a catalyst.
Catalyst 11: zeolite beta, Cu/beta. The procedure for copper ion exchange was similar to that used to Cu/ ZSM-5, except that 30 g of acid washed Beta (Si/A1 = 100) zeolite and 135 g of Cu(NO3)2.3H20 in 200mL water were used. The dried solid was gently ground and passed through a 710/zm screen.
CATALYST CHARACTERIZATION AND STABILITY
The catalysts were characterized by X-ray diffraction (XRD), nuclear magnetic resonance and surface area measurements. Table 2 gives a brief description of the various physical properties of the catalysts. Further details are available elsewhere 26'27.
Although the A1PO catalysts used in this work were found to be thermally stable, and stable to hydrolysis by water at room temperature, they were found to be unstable to water at 400°C. The reaction can be followed by X-ray diffraction or infrared spectroscopy. For example XRD showed that 45% of the CuSAPO-5 catalyst was undecom- posed at 400°C in the presence of 15% by weight of water (Table 3). Typical infrared spectra are shown in Figure l(a) and Figure l(b). The characteristic A1PO absorption at 550cm -1 is present in untreated or room temperature- treated catalyst (Figure la). This absorption is greatly reduced in the thermally treated product (Experiment 7, Table 3 and Figure lb).
¢q
t-
@
0 . 8 ¸
0.6
04 =
0.2
400
550 cmt
30100 I 2000 10100
0.2!
@
0.1
400 3000 2000 1000 Wavenumber (cm ~)
Figure 1 Infra red spectra to show the thermal instability of aluminophosphate catalysts at 400°C in the presence of water. (a) CuSAPO-5 treatment for 24 h at room temperature with 15% by weight water; (b) treatment at 400°C with 15% by weight water f o r l h
Thus, it is likely that the AIPO materials would not be stable at coal liquefaction conditions, because the reactions usually take place at temperatures of 400°C and above, and because water is usually a by-product of the liquefaction. On the other hand, the zeolite-based catalysts were found to be hydrolytically stable at these temperatures.
EXPERIMENTS WITH AUSTRALIAN COALS
Initial experiments were carried out in small rocking tubular reactors heated in a jacket. These are recorded in Table 4. In later experiments (Tables 5 and Table 6) coal
1108 Fuel 1997 Volume 76 Number 12
Reaction of methane with coal: K. Yang et al.
Table 4 Small tubular reactor studies with Millmerran coal and methane using GC/MS for product yield determination
Expt. No. Coal Gas pressure Catalyst Temp. Time Oil weight Residue b Oil yield Coal conversion
(g) (MPa) Type Wt(%) (°C) (h) (g) (g) (%) (%)c
1 0.630 6.9" - - - - 400 4 0.0300 0. 4300 4.8 31.8
2 0.629 6.9 - - - - 400 4 0.0313 0.4160 5.0 33.9
3 0.630 6.9 Pb/A1PO-5 10.0 400 4 0.0393 N/D 6.2 N/D
4 0.635 6.9 Pb/AIPO-5 10.4 400 4 0.0420 0.4690 6.6 26.2
5 0.314 6.9 Pb/A1PO-5 48.9 400 4 0.0220 0.2170 7.0 30.9
6 0.632 8.3 Pb/AIPO-5 10.0 400 4 0.0391 0.4810 6.2 23.9
7 0.631 7.6 Cu/ZSM-5 10.1 400 4 0.0742 0.4350 11.8 3 I. 1
Under nitrogen b Excludes catalyst c Calculated as (100 × coal wt (as received) - residue (catalyst free))/coal wt (as received) N/D not determined
Table 5 Yield of liquids (determined by NMR) from pyrolysis of Millmerran coal with methane for 60 min a
Expt No. Coal Cat. No. Cat.wt CH 4 Temp. % oil Elemental composition (daf coal)
(mg) (mg) (MPa) (°C)
Proton distribution by NMR (%)b
%C %H %N Ar-CH2 Ar-CH3 Naph -CH 2 -CH 3 Har
l 2000 - - - - - - 25 4.5 82.1 9.5 0.40
2 2484 - - - - - - 350 9.0 82.6 11.9 0.99
3 2016 - - - - 6 350 13.2 81.5 8.3 < 0.2
4 2000 1 100 6 350 20.1 83.6 9.5 < 0.2
5 2002 1 102 6 400 17.6 77.7 7.9 1.5
6 2002 2 101 6 350 30.1 80.0 8.0 < 0.2
7 2008 3 115 6 350 13.6 75.6 8.3 0.57
8 2002 4 123 6 350 11.3 72.8 9.0 0.47
9 2033 5 123 6 350 12.7 83.5 10.5 0.66
10 2000 6 125 6 350 10.2 83.4 11.3 0.42
11 2009 7 119 6 350 8.7 71.8 7.6 0.77
12 1994 8 101 6 350 16.3 83.8 7.6 0.54
16 8 9 47 13 7
13 8 3 53 11 12
15 8 3 52 11 12
15 9 5 49 12 11
39 19 3 24 6 9
17 10 6 47 10 9
14 8 3 53 11 11
11 11 4 55 I1 8
15 10 5 55 10 8
N/D N/D N/D N/D N/D N/D
N/D N/D N/D N/D N/D N/D
N/D N/D N/D N/D N/D N/D
"Yields are from soxhlet extraction for 3 days bproton types as defined in text, Naph = naphthenics (cycloalkylaromatics), Ha, = proton aromaticity % N/D not determined
Table 6 Yield of liquids (determined by GC/MS) from pyrolysis of coal in methane"
Proton distribution by ~H NMR (%)c
Expt, No. Coal Catalyst b Temp. (°C) Ar-CH2 Ar-CH3 Naph -CH2 -CH3 H,r % oil (daf coal)
1 Wintana - - 350 15 5 7 48 11 14 0.81
2 Wintana 1 350 16 10 2 40 13 19 2.23
3 Wintana 2 400 20 8 7 31 6 27 1.44
4 Collie - - 350 16 9 3 38 9 26 3.04
5 Collie I 350 15 12 3 40 9 21 1.85
6 Collie 2 400 20 12 3 22 3 40 1.37
7 Leigh Creek - - 350 18 9 4 40 10 19 1.34
8 Leigh Creek 1 350 12 13 2 47 11 16 2.22
9 Leigh Creek 2 400 56 6 5 21 2 I I 1.53
10 Metropolitan - - 350 22 5 4 27 10 33 1.07
11 Metropolitan 1 350 29 5 6 20 5 34 0.90
12 Metropolitan 2 400 67 21 1 3 1 7 1.53
13 Millmerran - - 350 15 9 3 45 13 15 5.59
14 Millmerran 1 350 14 8 3 47 12 16 6.46
15 Millmerran 2 400 29 9 4 37 9 13 8.66
OReaction conditions: coal (2 g), catalyst t00 mg, CH4, 5 MPa cold pressure, reaction time bCatalysts see Table 1 "See text, Naph = naphthenics (cycloalkylaromatics), H~r = proton aromaticity %
1 h. Yields are from soxhlet extraction for 3 days
Fuel 1997 Volume 76 Number 12 1109

Reaction of methane with coal: K. Yang et al.
Table 7 Products from hydrogen-methane mixture experiments with Illinois No. 6
Gas Methane Methane/hydrogen Hydrogen
Conversion a (wt% dry ash-free coal basis) 26.8 51.5 60.1 Yield C2-C4 gases 3.1 4.6 5.5 H2 consumption - 0.2 b 1.8 3.6 CH4 consumption 1.3 - 2.1 b - 3.9 b Product IBP-204°C c 7.7 19.2 21.1 Product 204-343°C c 10.4 14.3 18.2 Product 343-535°C c - 7.7 d - 5.8 d - 3.4 d Product 535°C plus c 73.2 48.5 39.9 COxx = 1,2 c 5.7 5.3 6.1 H2S c 2.2 2.4 2.6 H20 c 6.5 11.2 9.8 CHdC 2H6 ratio e 27.0 12.0 1.4
aCalculated by mass balance blndicates production rather than consumption cSelectivities. Yield = conversion x selectivity dlndicates solvent in this temperature boiling range consumed eThe methane-to-ethane ratio in the product gas, expressed as a ratio is dimensionless. The ratio is based on the wt% of each gas in the product, calculated on a
dry ash-free coal basis IBP = initial boiling point
and catalyst (5 or 10%) were sealed in a 100 ml autoclave and then charged with methane at 6.9 MPa (or other pressure) at room temperature. The autoclave was then heated to 400°C for 4 h before being cooled to room temperature. Rocking of the autoclave accompanied the whole process. The autoclave was cooled in ice before being vented. For the experiments recorded in Tables 4-6, the oil was then extracted from the residue with dichloromethane for 3 days.
Two methods of determining oil yields were used. Gas chromatography mass spectrometry (GC/MS) was used initially, and quantitation was achieved by means of an internal standard. Details of these experiments are shown in Tables 4, and 6. This method has the drawback that materials which are not volatile under the GC conditions are not detected. In other experiments ~H NMR was used (Table 5). In this method, a known weight of dioxane (D) is added and the fraction of protons measured relative to the number of protons in dioxane. Since there are 8 g protons per 88 g of dioxane then the weight of hydrogen in the dioxane is dioxane wt × 8/88. Then the wt of hydrogen in the sample is the weight of hydrogen in dioxane X sample signal (Ss)/dioxane signal (SD), i.e. 8DSs/88SD. By defini- tion the weight of oil sample is 100 X wt hydrogen in sample/%hydrogen in sample. From this, the weight of sample is wt hydrogen in sample x 100/% hydrogen in sample (H) = (8DSs/88SD) X (100/H) = 800DSs/88SDH. This allows a more accurate estimate of the oil as a percentage of the dry ash-free coal to be determined.
EXPERIMENTS WITH ILLINOIS NO. 6 (MONTEREY) COAL
All experiments were performed in a 380cm 3 stirred autoclave. The autoclave was operated in a batch mode and charged with 35 g of vacuum dried Monterey coal, 28 g of tetralin, 28 g of coal-derived vacuum gas oil (VGO) and 10% of catalyst (either Cu/beta or Cu/ZSM5) based on the weight of coal. The VGO was produced by distillation of coal hydroliquefaction products 28 and had an atmos- pheric pressure equivalent boiling range of about 250- 535°C. The treat gas was either CH4, equimolar CH4/H2,
or H2. When CH4 was used, 6.9 MPa was initially charged at ambient temperature, after which the autoclave was heated to the desired temperature in about 35 min, and maintained at that temperature for 60 min. In order to maintain the system at about 16.6 MPa, additional CH4 was charged to a pressure of 17 MPa if the system pressure dropped below 16.3 MPa. When hydrogen was used, the initial pressure charge was 8 MPa. After heating to 450°C, the system pressure was 16.6 MPa and was maintained at that pressure by addition of hydrogen on demand. For the CH4/H2 mixture, 3.5 MPa of methane was charged first, then 3.5 MPa of hydrogen was charged to get a 7.0 MPa pressure at room temperature. At reaction temperature, hydrogen was added as needed to maintain the system pressure at 16.6 MPa. After the reaction, the reactor was cooled to room temperature, the gas was collected in a sampling bag, the volume determined, and a small sample was analysed by mass spectrometry. The slurry was collected in a 250 ml distillation pot and distilled by an ASTM standard vacuum distillation procedure 29. The liquids boiling below an atmospheric pressure equivalent temperature of 535°C were combined and analysed by a gas chromatoKraphic simulated distillation (GC-SIMDIS)
..90 procedure to determine the product boiling point distri- butions (Table 7). The GC-SIMDIS procedure used for these analyses was modified from ASTM D2887 and similar methods by using calibrations derived from coal liquid samples. Hydrogen and methane consumption (or produc- tion) were calculated from the difference between the amounts charged (determined by pressure) and the amounts recovered (determined by gas volume and gas chromato- graphy and mass spectrometry of the gas). Other gases were measured in a similar way. Water is collected in the IBP- 204°C fraction and physically collected and weighed. Conversion was calculated by mass balance and the selectivities to each product fraction recorded (Table 7).
NMR SPECTROSCOPY
Proton IH NMR spectroscopy was carried out on samples dissolved in carbon tetrachloride using a Varian XL-200 NMR Spectrometer operating at 200 MHz. A 90 ° pulse with
1110 Fuel 1997 Volume 76 Number 12

Reaction of methane with coal: K. Yang et al.
a pulse delay of 6 sec was used with a spectral width of 2600 Hz. Five hundred transients or less were collected depending on the sample concentration. The data were acquired into 16 K data points and Fourier transformed to a resolution of 0.1 Hz. Chemical shifts were measured relative to internal tetramethylsilane. The ~H NMR data can be used for comparative purposes to analyse the oil by subdividing the spectra into various regions. While the method is inaccurate for detailed assignments, since there is considerable overlap between protons of various different types, it can be used for comparative purposes. Spectra were subdivided on the basis of chemical shift. Structural groups were assigned to each region for this purpose as follows: aromatics (Ar) 8.3-6.6, olefin 5.5-4.5, naphthenes (Naph) 4.0-2.3, methylene attached to aromatic (Ar-CH2) 2.3-1.9, methyl attached to aromatic (Ar-CH3) 1.9-1.6, methylene in aliphatic (-CH2) 1.6-1.0, methyl in aliphatic (-CH3) 1.0-0.5 ppm. Data which distinguish between this range of different protons are shown in Table 5 for Millmerran coal and in Table 6 for Millmerran and other coals.
GAS CHROMATOGRAPHY MASS SPECTROMETRY
Gas chromatography mass spectrometry work was per- formed on a Carlo Erba Gas Chromatograph linked to a Kratos MS25 Mass Spectrometer. Data collection and processing were performed using a Teknivent Ventor/Two data acquisition system. The mass spectrometer was run with an ionization energy of 70eV and the source
temperature at 200°C. A 5 0 m SGE 50Q C3/BP1/0.5 column was used in the gas chromatograph which was programmed to run at an initial temperature of 30°C for 2 min and then heated at 4°/min to 300°C, which was maintained for a further 30 min. Quantitation was achieved using a known amount of n-C23 alkane as an internal standard. Helium was used as the carrier gas at a linear carrier velocity of 20 cm/s at 110°C.
INFRARED SPECTROSCOPY
FTIR spectra were obtained on a Digilabs FTS 20/80 instrument. Spectra were obtained using 1-2 mg of sample dispersed in potassium bromide discs. An acceptable signal-to-noise ratio was obtained using about 64 scans at 4 cm -1 resolution.
RESULTS AND DISCUSSION
Effect of catalysts Results of the experiments using GC/MS to determine
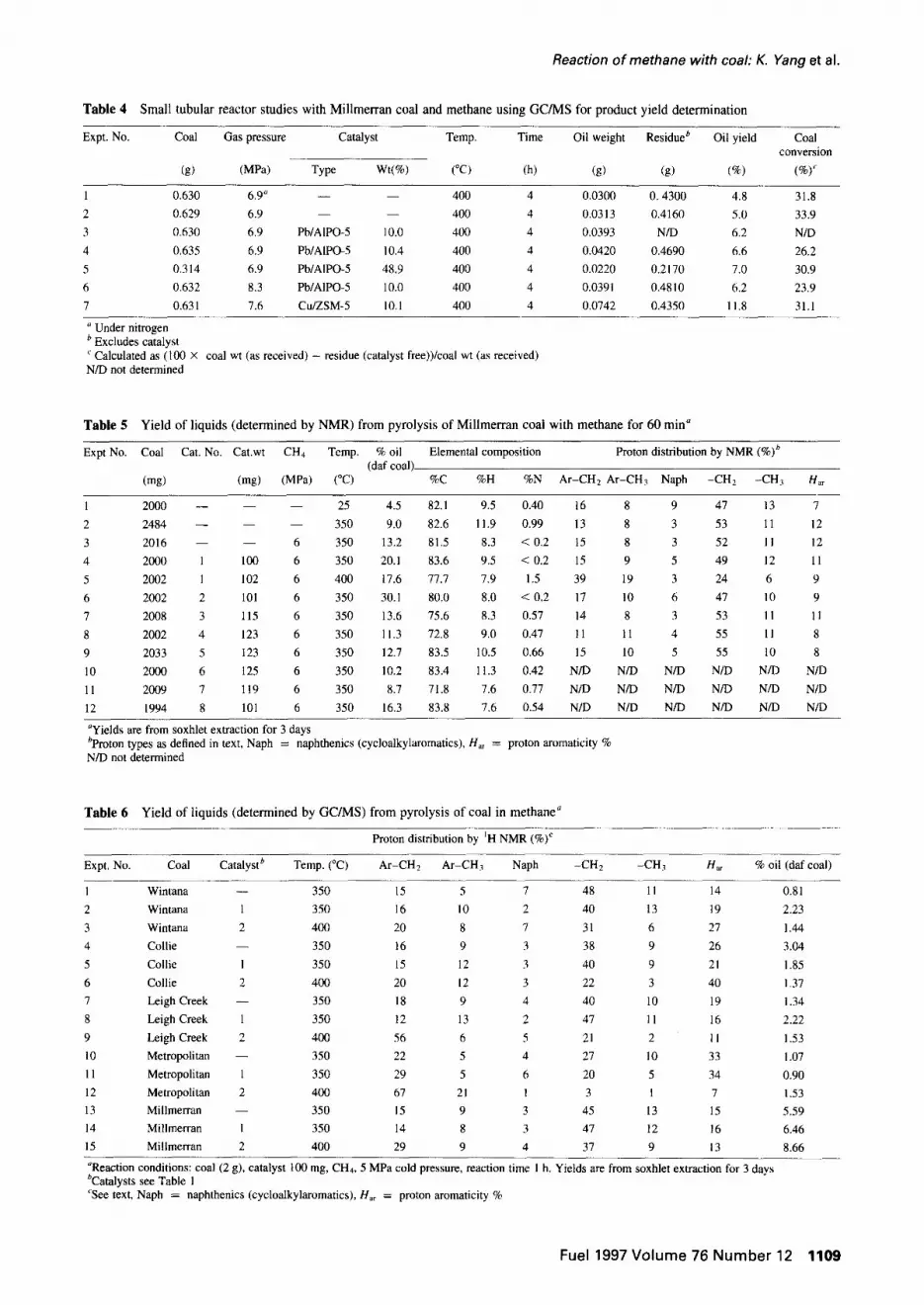
yields from the reaction of Millmerran coal with methane are shown in Table 4. Oil yields under methane are around 5% which is a little more than that obtained from the same experiment under nitrogen (compare experiments 1 and 2). These experiments also show that the lead-exchanged catalysts appear to increase oil yields slightly. The Cu/ZSM-5 catalyst is much better. Conversion, however, does not appear to be increased by catalyst. Indeed, it
%H
Metro Collie Leigh Wintan a Mill
Coals (increasing H/C) Figure 2 Effect of increasing (H/C) on hydrogen distributions for 350 °, no catalyst, 100 ml autoclave reactions: [] aryl-CH3; • aliphatic chain CH3; [] aromatic hydrogen
Fuel 1997 Vo lume 76 Number 12 1111
Reaction of methane with coal: K. Yang et al.
oil yield % (daO 9-
8-
7-
6-
5-
4-
3-
2-
1-
Metro Collie Leigh Wintana Mill
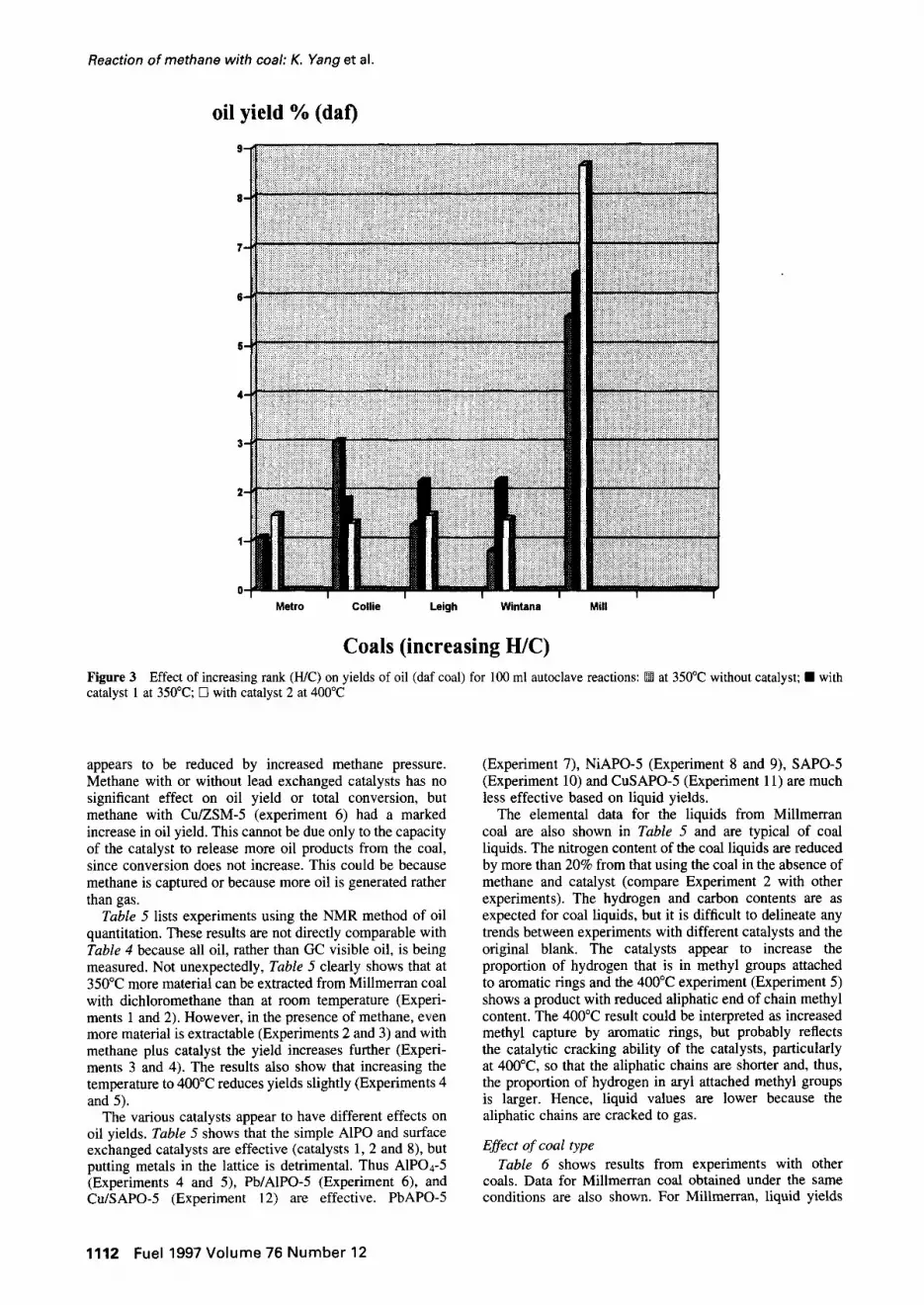
Coals (increasing H/C) Figure 3 Effect of increasing rank (H/C) on yields of oil (daf coal) for 100 ml autoclave reactions: [] at 350°C without catalyst; [] with catalyst 1 at 350°C; [] with catalyst 2 at 400°C
appears to be reduced by increased methane pressure. Methane with or without lead exchanged catalysts has no significant effect on oil yield or total conversion, but methane with Cu/ZSM-5 (experiment 6) had a marked increase in oil yield. This cannot be due only to the capacity of the catalyst to release more oil products from the coal, since conversion does not increase. This could be because methane is captured or because more oil is generated rather than gas.
Table 5 lists experiments using the NMR method of oil quantitation. These results are not directly comparable with Table 4 because all oil, rather than GC visible oil, is being measured. Not unexpectedly, Table 5 clearly shows that at 350°C more material can be extracted from Millmerran coal with dichloromethane than at room temperature (Experi- ments 1 and 2). However, in the presence of methane, even more material is extractable (Experiments 2 and 3) and with methane plus catalyst the yield increases further (Experi- ments 3 and 4). The results also show that increasing the temperature to 400°C reduces yields slightly (Experiments 4 and 5).
The various catalysts appear to have different effects on oil yields. Table 5 shows that the simple AIPO and surface exchanged catalysts are effective (catalysts 1, 2 and 8), but putting metals in the lattice is detrimental. Thus A1PO4-5 (Experiments 4 and 5), Pb/A1PO-5 (Experiment 6), and Cu/SAPO-5 (Experiment 12) are effective. PbAPO-5
(Experiment 7), NiAPO-5 (Experiment 8 and 9), SAPO-5 (Experiment 10) and CuSAPO-5 (Experiment 11) are much less effective based on liquid yields.
The elemental data for the liquids from Millmerran coal are also shown in Table 5 and are typical of coal liquids. The nitrogen content of the coal liquids are reduced by more than 20% from that using the coal in the absence of methane and catalyst (compare Experiment 2 with other experiments). The hydrogen and carbon contents are as expected for coal liquids, but it is difficult to delineate any trends between experiments with different catalysts and the original blank. The catalysts appear to increase the proportion of hydrogen that is in methyl groups attached to aromatic rings and the 400°C experiment (Experiment 5) shows a product with reduced aliphatic end of chain methyl content. The 400°C result could be interpreted as increased methyl capture by aromatic rings, but probably reflects the catalytic cracking ability of the catalysts, particularly at 400°C, so that the aliphatic chains are shorter and, thus, the proportion of hydrogen in aryl attached methyl groups is larger. Hence, liquid values are lower because the aliphatic chains are cracked to gas.
Effect of coal type Table 6 shows results from experiments with other
coals. Data for Millmerran coal obtained under the same conditions are also shown. For Millmerran, liquid yields
1112 Fuel 1997 Volume 76 Number 12

Reaction of methane with coal: K. Yang et al.
%H
Metro Collie Leigh Wintana Mill
Coals (increasing H/C) Figure 4 Effect of catalysts and temperature on aliphatic chain methyl hydrogen distribution for 100 ml autoclave reactions: [] blank at 350°C; • catalyst 1 at 350°C; [] catalyst 2 at 400°C
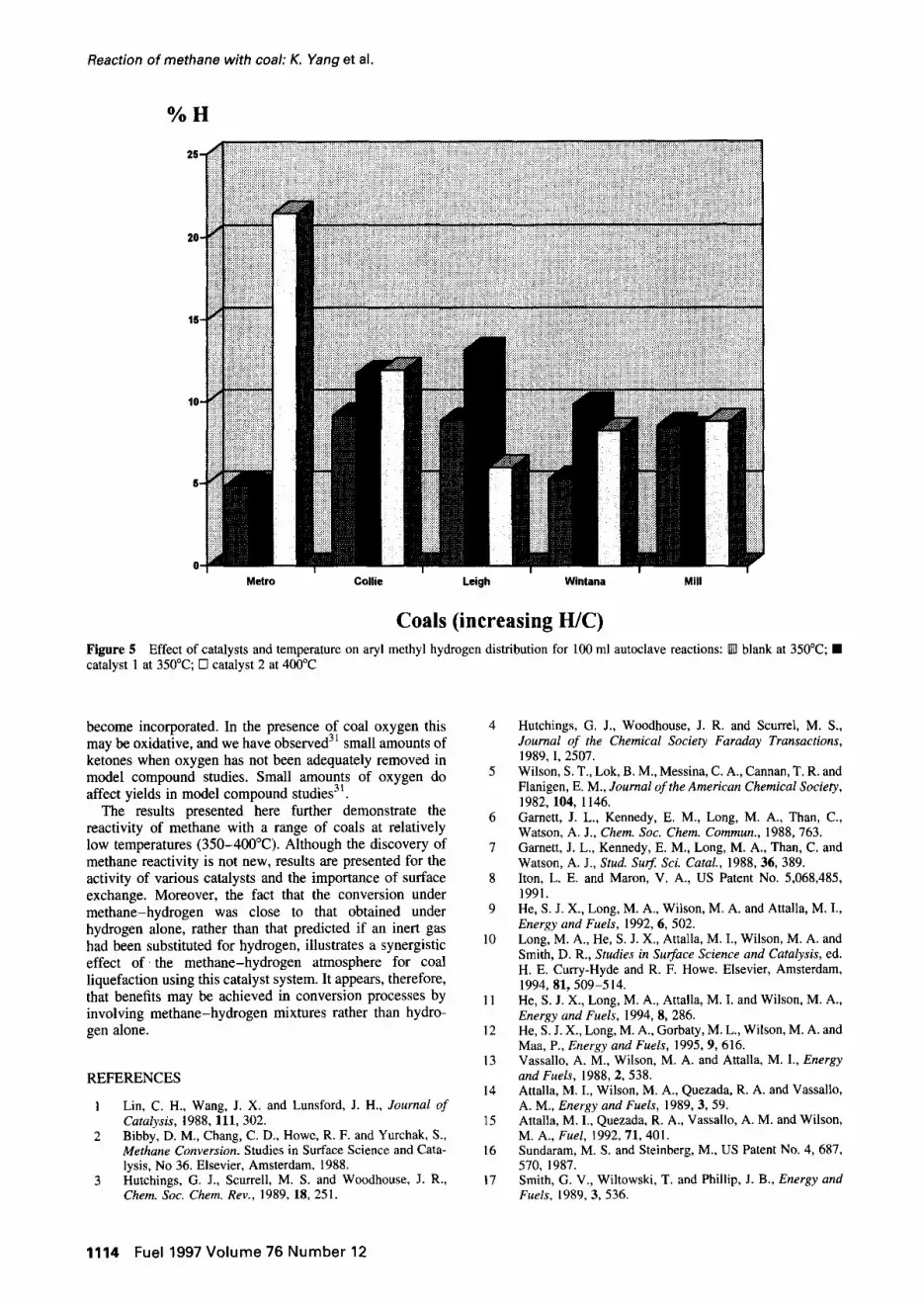
are lower than in Table 5 because of the different conditions, and method of determining oil yield, but trends are similar. The coal liquids obtained from all the coals do not show any clear trends in elemental composi- tions betweeen catalysts or coals. However, there is a clear decrease in proportion of hydrogen which is aromatic with an increase in hydrogen-to-carbon ratio (Figure 2), and the yield of liquids appears to increase with increase in hydrogen-to-carbon ratio (Figure 3). There is an increase in proportion of hydrogen which is methyl and attached to the end of aliphatic chains with increase in hydrogen-to- carbon ratio and this trend is increased with catalyst and increased temperature (Figure 4). Thus, it appears that increased yield with increasing rank is due to extraction of longer chain aliphatic material. There appears to be no clear increase in the proportion of aryl methyl groups (Figure 5) even in the presence of catalyst and increased tem- perature, so there is no evidence of increased cracking due to catalyst.
Effect of hydrogen mixing Elsewhere, we have shown hydrogen may assist in the
27 incorporation of methane in model systems . Table 7 displays the results of extending this to the liquefaction of Illinois No. 6 coal using the Cu/Beta catalyst. Table 7 shows that under methane only 27% of the coal is converted and the selectivity to liquids is low. Under hydrogen, a 60% conversion was obtained, with good selectivity to liquids while consuming 4 wt% hydrogen. Under methane-
hydrogen, 51.5% conversion was obtained, with a product selectivity close to that obtained under hydrogen alone, and with only 2% hydrogen consumption. The conversion under methane-hydrogen was close to that obtained under hydrogen alone, while a linear combination of conversions would predict a 43% conversion under methane-hydrogen. This illustrates a synergistic effect of the methane- hydrogen atmosphere for coal liquefaction using this catalyst system in the presence of a solvent. Table 7 shows a decrease in the boiling fraction 343-535°C. This fraction is solvent but not inert; additional hydrogenation and cracking take place just as well as on coal molecules. In fact, since the solvent is derived from coal itself, it is relatively high in molecular weight (i.e. its b.p.) and probably looks a little like its parent, more so than it looks like a product. The fact that the loss of this fraction decreases as hydrogen partial pressure increases does not mean less solvent is reacting; rather, it merely means that more solvent fraction is produced as conversions increase. The net is a smaller change in solvent consumption. More interesting is the fact that water production is greater in the presence of methane and hydrogen than hydrogen alone. This clearly demonstrates the beneficial effect of methane on the reaction.
Overview A little can be said concerning the mechanism of activity
of methane. Apart from acting as a hydrogen shuttle, it is clear from model compound studies 9- ~2,27 that methane can
Fuel 1997 Volume 76 Number 12 1113
Reaction of methane with coal: K. Yang et al.
% H
25-
20-
15-
10-
5 -
0-
4
i~iiiiii;
: . : . : . : . : - _
: : : : = . . . . . . . . . = . . . . . . . . . = . . . . . . . . = • . . . . . . . =
. ? i . ! . ? = . . . . . . . . . =
. . . . . . . . =
. . . . . . . . =
: : : : : . . . . . . . . . =
: : : : : : : : : i
iiiililill : : : : : : : : 2
. . . , . . . . ~ .
: : : : =
Metro Collie Leigh Wintana Mill
Coals (increasing H/C) Figure 5 Effect of catalysts and temperature on aryl methyl hydrogen distribution for 100 ml autoclave reactions: [] blank at 350°C; • catalyst 1 at 350°C; [] catalyst 2 at 400°C
become incorporated. In the presence of coal oxygen this may be oxidative, and we have observed 31 small amounts of ketones when oxygen has not been adequately removed in model compound studies. Small amounts of oxygen do affect yields in model compound studies 3~.
The results presented here further demonstrate the reactivity of methane with a range of coals at relatively low temperatures (350-400°C). Although the discovery of methane reactivity is not new, results are presented for the activity of various catalysts and the importance of surface exchange. Moreover, the fact that the conversion under methane-hydrogen was close to that obtained under hydrogen alone, rather than that predicted if an inert gas had been substituted for hydrogen, illustrates a synergistic effect of ~ the methane-hydrogen atmosphere for coal liquefaction using this catalyst system. It appears, therefore, that benefits may be achieved in conversion processes by involving methane-hydrogen mixtures rather than hydro- gen alone.
REFERENCES
1 Lin, C. H., Wang, J. X. and Lunsford, J. H., Journal of Catalysis, 1988, 111, 302.
2 Bibby, D. M., Chang, C. D., Howe, R. F. and Yurchak, S., Methane Conversion. Studies in Surface Science and Cata- lysis, No 36. Elsevier, Amsterdam, 1988.
3 Hutchings, G. J., Scurrell, M. S. and Woodhouse, J. R., Chem. Soc. Chem. Rev., 1989, 18, 251.
4 Hutchings, G. J., Woodhouse, J. R. and Scurrel, M. S., Journal of the Chemical Society Faraday Transactions, 1989, I, 2507.
5 Wilson, S. T., Lok, B. M., Messina, C. A., Cannan, T. R. and Flanigen, E. M., Journal of the American Chemical Society, 1982, 104, 1146.
6 Garnett, J. L., Kennedy, E. M., Long, M. A., Than, C., Watson, A. J., Chem. Soc. Chem. Commun., 1988, 763.
7 Garnett, J. L., Kennedy, E. M., Long, M. A., Than, C. and Watson, A. J., Stud. Su~ Sci. Catal., 1988, 36, 389.
8 Iton, L. E. and Maron, V. A., US Patent No. 5,068,485, 1991.
9 He, S. J. X., Long, M. A., Wilson, M. A. and Attalla, M. I., Energy and Fuels, 1992, 6, 502.
10 Long, M. A., He, S. J. X., Attalla, M. I., Wilson, M. A. and Smith, D. R., Studies in Surface Science and Catalysis, ed. H. E. Curry-Hyde and R. F. Howe. Elsevier, Amsterdam, 1994, 81, 509-514.
11 He, S. J. X., Long, M. A., Attalla, M. I. and Wilson, M. A., Energy and Fuels, 1994, 8, 286.
12 He, S. J. X., Long, M. A., Gorbaty, M. L., Wilson, M. A. and Maa, P., Energy and Fuels, 1995, 9, 616.
13 Vassallo, A. M., Wilson, M. A. and Attalla, M. I., Energy and Fuels, 1988, 2, 538.
14 Attalla, M. I., Wilson, M. A., Quezada, R. A. and Vassallo, A. M., Energy and Fuels, 1989, 3, 59.
15 Attalla, M. I., Quezada, R. A., Vassallo, A. M. and Wilson, M. A., Fuel, 1992, 71, 401.
16 Sundaram, M. S. and Steinberg, M., US Patent No. 4, 687, 570, 1987.
17 Smith, G. V., Wiltowski, T. and Phillip, J. B., Energy and Fuels, 1989, 3, 536.
1114 Fuel 1997 V o l u m e 76 N u m b e r 12

Reaction of methane with coal: K. Yang et al.
18 Egiebor, N. O. and Gray, M. R., Fuel, 1990, 69, 1276. 19 Ozawa, S., Ohsaki, H. and Ogino, Y., Fuel Proc. Technol.,
1987, 17, 187. 20 Loftier, I. D., Maier, W. F., Andrade, J. G., Thies, I.,
Schleyer, P. J., Chem. Soc. Chem. Commun., 1984, 1178. 21 Tanaka, K., Yaegashi, I., Aomura, K. J., Chem. Soc. Chem.
Commun., 1982, 938. 22 Ovalles, C., Hamana, A., Rojas, I. and Boliva, R. A., Fuel,
1995, 74, 1162. 23 Barron, P. F., Collin, P. J., Russell, N. J. and Wilson, M. A.,
Fuel Processing Technology, 1982, 6, 147. 24 Attalla, M. I., Bruce, L. A., Hodgson, S. I., Turner, T. W.,
Wilson, M. A. and Batts, B. D., Fuel, 1990, 69, 725. 25 Silbernagel, B. G.and Botto, R. E., in Magnetic Resonance
of Carbonaceous Solids, Advances in Chemistry Series 229, ed R. E. Botto and Y. Sanada. American Chemical Society, Washington DC, 1993, pp. 629-643.
26 He, J. X., Ph.D. thesis, University of New South Wales, Australia, 1995.
27 Pang, L. S. K., Wilson, M. A., Quezada, R. A., Prochazka, J. L., Long, M. A., He, S. J. X., Gorbaty, M. L. and Maa, P.S., Fuel, 1997, in press.
28 Stuntz, G. F., Premium products from direct coal liquefac- tion. Alternate Energy '91, Scottsdale, Az, 4/1/91.
29 ASTM D-5236, Standard Test Method for Distillation of Heavy Hydrocarbon Mixtures (Vacuum Potstill Method).
30 Butler, R. D., in Chromatography in Petroleum Analysis, ed. K. H. Altgelt and T. H. Gouw. Marcel Dekker, New York, 1979.
31 Pang, L. S. K., Quezada, R. A. and Wilson M. A., unpub- lished work.
Fuel 1997 Volume 76 Number 12 1115





























