Embed Size (px)
Citation preview

Mycobacterial Gene Induction in Macrophages • JID 2004:190 (15 July) • 341
M A J O R A R T I C L E
Role of Cellular Activation and Tumor NecrosisFactor–a in the Early Expressionof Mycobacterium tuberculosis 85B mRNAin Human Alveolar Macrophages
Najmul Islam,1 Andrew R. Kanost,1 Luciella Teixeira,1 John Johnson,1 Rana Hejal,1 Htin Aung,1 Robert J. Wilkinson,3
Christina S. Hirsch,1 and Zahra Toossi 1,2
1Division of Infectious Disease, Department of Medicine, Case Western Reserve University, and 2Veterans Affairs Medical Center Cleveland, Ohio;3Wellcome Center for Clinical Tropical Medicine, Imperial College of Science, Technology, and Medicine, London, United Kingdom
Background. Infection of alveolar macrophages (AMs), which constitute the first line of defense against My-cobacterium tuberculosis, initiates an intense interaction between the host’s innate immune response and mycobacteriathat may assist in the successful intracellular parasitism of M. tuberculosis.
Methods. Expression of tumor necrosis factor (TNF)–a and M. tuberculosis 85B mRNA was studied in M.tuberculosis–infected AMs, to better delineate the role of macrophages in the early events in initiation of infection.
Results. Both TNF-a mRNA and M. tuberculosis 85B were induced in AMs; at 24 h, the time point ofmaximum TNF-a induction, the mRNA levels for TNF-a and M. tuberculosis 85B correlated with one another,and induction of either gene correlated strongly with their protein levels. Inhibition of endogenous TNF-a bysoluble (s) TNF receptor (R) I and sTNFRII reduced expression of both TNF-a and M. tuberculosis 85B. Theactivation of nuclear factor–kB was found to underlie expression of both TNF-a and M. tuberculosis 85B. ExogenousTNF-a was slightly more potent than interleukin (IL)–6 and granulocyte-macrophage colony-stimulating factorand was significantly stronger than IL-1 in inducing expression of M. tuberculosis 85B. Interestingly, inhibition ofbactericidal mediators, reactive oxygen intermediates (ROIs) and reactive nitrogen intermediates (RNIs), reducedexpression of TNF-a and M. tuberculosis 85B genes in M. tuberculosis–infected AMs.
Conclusion. Activation of AMs by M. tuberculosis initiates a cascade of events whereby TNF-a, ROI, and RNIenhance the expression of the M. tuberculosis 85B gene.
Although Mycobacterium tuberculosis infects at least
one-third of the world’s population, a complete un-
derstanding of the mechanisms of its pathogenicity in
humans is still lacking. The virulence of M. tuberculosis
likely entails both its ability to survive the intracellular
environment of host alveolar macrophages (AMs), which
constitute the first line of defense against the pathogen,
Received 24 September 2003; accepted 6 February 2004; electronically published18 June 2004.
Financial support: National Institutes of Health (grants AI-18472, AI-45244, AI-95383, and AI-01514); Center for AIDS Research at Case Western ReserveUniversity (grant AI-36219); Wellcome Trust Advanced Fellowship in ClinicalScience (grant 064261 to R.W.).
Reprints or correspondence: Dr. Zahra Toossi, Div. of Infectious Disease, Dept.of Medicine, Case Western Reserve University, 11100 Euclid Ave., Cleveland, OH44106 ([email protected]).
The Journal of Infectious Diseases 2004; 190:341–51� 2004 by the Infectious Diseases Society of America. All rights reserved.0022-1899/2004/19002-0019$15.00
and its capacity to withstand the intense cytokine mi-
croenvironment generated after infection. Knowledge
of the role of the host’s innate immune responses in
the pathogenesis of M. tuberculosis infection may allow
better targeting of therapies and vaccines to eradicate
tuberculosis.
The initial interaction of M. tuberculosis with mono-
nuclear phagocytes gives rise to a cytokine profile that
is dominated by the proinflammatory macrophage-ac-
tivating molecule tumor necrosis factor (TNF)–a. TNF-
a is induced not only by phagocytic and nonphagocytic
interaction of M. tuberculosis bacilli with mononuclear
phagocytes [1, 2] but also by mycobacterial protein and
nonprotein components [3]. TNF-a is present at sites
of active M. tuberculosis infection in humans, regardless
of the stage of mycobacterial infection [4–7]. In mice
infected with mycobacteria, TNF-a is involved in the
development of microbicidal granulomas [8], and dis-
ruption of TNF-a responses is associated with myco-
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from

342 • JID 2004:190 (15 July) • Islam et al.
bacterial overgrowth [9, 10]. In subjects with latent M. tuber-
culosis infection, therapies that neutralize TNF-a have been
associated with reactivation of tuberculosis [11]. However, it is
not clear whether TNF-a contributes to protective immunity
during all stages of M. tuberculosis infection, because it has been
ascribed an immunopathological role in both animal models
of M. tuberculosis infection and recent human studies [12, 13].
In human macrophages, TNF-a is only modest in its anti–M.
tuberculosis activity [2], and its role in the formation of gran-
ulomas is unclear. In vitro studies indicate that, although AMs
are initially more susceptible to M. tuberculosis infection and
sustain higher M. tuberculosis growth, they ultimately contain
M. tuberculosis better than do their blood precursors, mono-
cytes [14]. The superiority of AMs in containment of growth
of M. tuberculosis has been ascribed to their expanded capacity
to produce TNF-a [14]. Nonetheless, other studies have shown
that production of TNF-a either promotes [15] or is associated
with [16] the growth of M. tuberculosis in human monocytes/
macrophages.
Cellular signaling by TNF-a is mediated mainly through ac-
tivation of NF-kB [17]. However, NF-kB–independent pathways
of cellular activation by TNF-a have been described [18, 19]. In
turn, activation of NF-kB and other pathways sustains TNF-a
activity [17]. Downstream effector pathways activated by TNF-
a in host tissues include activation of reactive oxygen interme-
diates (ROIs) and reactive nitrogen intermediates (RNIs) [20].
Phagocytosis of microbes and bacterial components, such as li-
popolysaccharide (LPS), also activates ROIs [21] and indirectly,
through oxygen radicals, activates inducible nitric oxide synthase
(iNOS) and, therefore, RNIs in macrophages [22, 23].
Of the many M. tuberculosis products that induce production
of TNF-a, antigen 85B is of note. Along with the other 2
proteins in the M. tuberculosis 85 complex (85A and 85C), with
which it has 70%–80% homology [24], 85B is abundantly se-
creted by M. tuberculosis [25], binds fibronectin (fn) [26], and
is involved in cell-wall biogenesis [27]. Interestingly, M. tuber-
culosis 85B is immunodominant [26] and potently induces TNF-
a when complexed to fn in mononuclear phagocytes [28]. It is
possible that, through sustaining TNF-a activity, the abundant
release of M. tuberculosis 85B in situ contributes to the patho-
genesis of M. tuberculosis infection. Recently, the expression of
M. tuberculosis 85B mRNA has been found to be increased early
during infection of monocytes [29] and cultured macrophages
[30]. The expression of 85B in M. tuberculosis–infected mono-
cytes correlates positively with both the amount of secreted
TNF-a and subsequent intracellular mycobacterial growth [29].
Therefore, the interaction of M. tuberculosis 85B and host TNF-
a may create a vicious circle in which each maintains the pro-
duction of the other.
The present study was conducted to examine the interaction
of M. tuberculosis 85B and TNF-a early after infection of AMs
with M. tuberculosis. To fully delineate the contribution of the
innate immune response to the pathogenesis of M. tuberculosis
infection within the cellular context of the lungs, the roles that
ROIs and RNIs, induction of TNF-a, and macrophage acti-
vation play in expression of M. tuberculosis 85B were examined.
MATERIALS AND METHODS
Reagents. Recombinant (r) cytokines, soluble (s) TNF receptor
(R) I (sTNFRI), and sTNFRII were purchased from R&D Sys-
tems. NMMA (NG-monomethyl-l-arginine-monoacetate), which
specifically inhibits the iNOS required for production of RNI,
and NAC (N-acetyl cysteine), which scavenges oxygen radicals,
were purchased from Calbiochem and Sigma, respectively. In
some experiments, oxidized ATP (oATP; Sigma), which inhibits
the purinergic receptor P2X7 and reduces both RNI and ROI
[31], was used. NADPH and H2O2 (Sigma), nonoate-9 (NOC-
9) (Calbiochem), sodium nitroprusside (SNP; Sigma), and SN50
or its analogue SN50/M (Biomol) were used in some experi-
ments. The endotoxin content of all reagents, as assessed by use
of a lymulus amebocyte lysate assay (BioWhittaker), was �0.01
ng/mL. In preliminary dose-response experiments, the optimal
amount of each reagent was determined. The cytotoxicity of all
reagents was assessed by use of Trypan blue exclusion. Reagents
were added to cells and assessed either immediately or after 24
h of culture. None of the reagents was toxic to AMs.
Preparation of mycobacteria. Avirulent and virulent labo-
ratory-adapted M. tuberculosis (H37Ra and H37Rv) were grown
in Middlebrook 7H9 broth (Difco Laboratories) at 37�C in 5%
CO2. Midlogarithmic mycobacterial cultures (14 days) were har-
vested and quantified by use of a colony-forming unit assay, as
described elsewhere [2]. Aliquots of the stock were kept at �70�C.
The viability of the stock remained 199% at 1 year.
Study subjects. Healthy, nonsmoking volunteers, 20–45
years of age, were recruited for bronchoscopy and bronchoal-
veolar lavage (BAL). All study subjects fulfilled the following
criteria: not receiving medication, no history of heart or lung
disease, and no upper respiratory tract infection within 6 months
before the study. Furthermore, all subjects were tuberculin skin
test negative, HIV uninfected, and had not received vaccination
with bacille Calmette-Guerin. Informed, written consent was ob-
tained from all participants before undergoing bronchoscopy.
The protocol for this study was reviewed and approved by the
Institutional Review Board for Human Investigation at University
Hospitals of Cleveland.
Preparation of bronchoalveolar cells (BACs). BAL was
performed as described elsewhere [14]. In brief, after anesthe-
tizing the upper airway with topical 2% lidocaine, a flexible
bronchoscope (BF type 4B2 bronchoscope; Olympus Optical)
was wedged into the right middle lobe. Then, 360 mL of sterile
0.9% saline was instilled into 2 segments of the middle lobe,
and BAL fluid was harvested. BACs were suspended in complete
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from

Mycobacterial Gene Induction in Macrophages • JID 2004:190 (15 July) • 343
Table 1. Sequences of primers and probes for quantification of tumor necrosis factor (TNF)–a and Mycobacterium tuberculosis 85B mRNA in infected alveolar macrophages.
Target, strain, primer PCR primer sequence, 5′r3′ Taqman probe sequence, 5′
r3′
HumanTNF-a CCAGAGGGAAGAGTTCCCCAGGGAC
RT GGTTTCTACAACAForward AGGCGGTGCTTGTTCCTCAReverse GTTCGAGAAGATGATCTGACTGCC
R18 ACCGGCGCAAGACGGACCAGART GACGGTATCTGATCForward CGCCGCTAGAGGTGAAATTCReverse CATTCTTGGCAAATGCTTTC
M. tuberculosis85B TCGAGTGACCCGGCATGGGAGCG
RT TGTTGTTTGCGAForward TCAGGGGATGGGGCCTAGCCReverse GCTTGGGGATCTGCTGCGTA
16S AGCACCGGCCAACTACGTGCCAGRT CCCAGTAATTCCForward TTCTCTCGGATTGACGGTAGGTReverse CGCTCGCACCCTACGTATTAC
NOTE. PCR, polymerase chain reaction; RT, reverse transcription.
medium (RPMI 1640 medium containing 2 mol/L l-glutamine,
25 mmol/L HEPES [Gibco Laboratories], and no antibiotics)
and kept on ice. BACs were 90%–95% nonspecific-esterase
positive and, therefore, comprised predominantly AMs and
contained 5%–10% lymphocytes and !1% granulocytes, as as-
sessed by Wright staining.
Cell culture. The a-chymotryptic fragment (120 kDa) of
human fn (Life Sciences) was used to obtain adherent cells. In
brief, 0.5 mL of complete medium with fn (1 mg/mL) was placed
in 12-well tissue-culture plates (Costar) and incubated for 20
min at 37�C in 5% CO2; then the plates were washed extensively
to remove unbound fn. BACs were added to wells ( 60.5 � 10
cells/mL), and the plates were incubated for 1–2 h at 37�C in
5% CO2 and then were washed 4 times with medium to remove
nonadherent cells. In preliminary experiments, we found that
the purity of adherent AMs, as determined by cytostaining, was
100%. To maintain cell attachment, adherent AMs were cultured
in RPMI 1640 medium supplemented with 1% fn. Then the cells
were rested overnight at 37�C in 5% CO2. Before infection, the
plates were washed twice with RPMI 1640 medium.
Infection of AMs. Using conditions that maximize the
opsonic uptake of M. tuberculosis [14] in preliminary experi-
ments, we found an MOI of 1:1 (bacteria:cell) to be necessary
for quantification of intracellular M. tuberculosis mRNA by use
of real-time polymerase chain reaction (PCR). Thus, AMs were
infected with M. tuberculosis at an MOI of 1:1 in 30% autol-
ogous unheated serum for 90 min at 37�C in 5% CO2. There-
after, the infected monolayers were washed 4 times with complete
medium. Cells harvested at this time point were considered as
time zero (t0) after infection. Other cultures received RPMI 1640
medium with 2% autologous serum. At different time points,
cultures were harvested, and cells were lysed in 0.5 mL of TRIZOL
Reagent (Invitrogen). The cell-free culture supernatants were
kept at �70�C.
RNA extraction and quantitative reverse-transcription (RT)–
PCR. Cell lysates were transferred to FastRNA blue tubes and
agitated in a Fast Prep FP120 BIO 101 SAVANT vibrator/cell-
wall disrupter (Bio 101) at full speed, as described elsewhere [29].
RNA preparation and DNAase treatment were as before.
We used quantitative real-time RT-PCR with internal fluo-
rescent hybridization probes in ABI Prism 7700 Detection Sys-
tem (ABI/PerkinElmer Biosystems), which allows the sensitive
and specific quantification of individual host, as well as M.
tuberculosis RNA, transcripts [29]. A target-specific RT primer,
PCR primers, and probes for each assay were either designed
according to specifications recommended by ABI/PerkinElmer
Biosystems using Primer Express software or were as described
elsewhere [29, 32] (table 1). All probes were dually labelled
with FAM (5-carbofluorescein) at the 5′ end and TAMRA
(N,N,N′,N′-tetramethyl-6-carborhodamine) at the 3′ end. The
proximity of the dye (FAM) and the quencher (TAMRA) on
the intact probe prevents detection of any fluorescence. How-
ever, degradation of the probe during the course of PCR allows
the release and detection of FAM [33]. The PCRs for all am-
plifications were similar: 5 mL of each cDNA, 20 mL of Taqman
Universal PCR Master Mix (PE Biosystems), which contains
optimal amounts of AmpliTaq Gold DNA polymerase (which
protects against amplicon carryover) and of dNTPs, and op-
timal amounts of probe and primers calibrated to allow mea-
surement of the targets. First, cDNA was synthesized in the
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from

344 • JID 2004:190 (15 July) • Islam et al.
Figure 1. Kinetics of simultaneous expression of tumor necrosis factor (TNF)–a and Mycobacterium tuberculosis 85B in human alveolar macrophages(AMs). AMs were infected with M. tuberculosis H37Ra (1:1 bacteria/cell) and cultured for 0–120 h in vitro. Total RNA was extracted and assessedfor TNF-a (A) and M. tuberculosis 85B (B). Expression of TNF-a was corrected to host 18S rRNA and expressed as copies of TNF-a in 1010 copiesof R18 (equivalent to cells). M. tuberculosis 85B was corrected to mycobacterial 16S rRNA and expressed as 85B:16S. Data are mean + SEM61 � 10of 5 experiments.
presence of 0.5 mL of murine leukemia virus enzyme (Invitro-
gen)/reaction and 10 mmol/L each RT primer, dNTPs, and other
substrate. Conditions for PCR were similar for all products (1
cycle of 2 min at 50�C and 1 cycle of 10 min at 95�C and then
40 cycles of 15 s at 95�C and 1 min at 60�C). The cycle threshold
for each sample was compared with the cycle threshold values
of known amounts of a standard DNA constructed for each
target and amplified simultaneously. To assure lack of DNA
contamination in the RNA samples, in some experiments, a
duplicate tube of sample with no RT enzyme was included as
control. DNA contamination remained negligible. Expression
of TNF-a mRNA was corrected to host 18S rRNA in the same
sample and was expressed as copies of TNF-a in 1010 copies
of R18 (equivalent to cells). M. tuberculosis 85B mRNA61 � 10
was corrected to mycobacterial 16S rRNA in the same sample
and expressed as 85B:16S.
Measurement of TNF-a and M. tuberculosis 85 complex
protein by ELISA. The TNF-a content of culture superna-
tants was determined by use of a commercial ELISA (R&D
Systems), according to the manufacturer’s specifications. The
lower limit of sensitivity was 4.4 pg/mL.
An ELISA for detection of the mycobacterial 85 complex
(includes M. tuberculosis 85A, B, and C proteins) was devel-
oped using the monoclonal antibody to mycobacterial 85
complex (CS-90; available from Colorado State University,
Fort Collins) as coating and a rabbit anti–Mycobacterium bovis
antibody (DAKO) as detection reagent [34]. Purified 85 com-
plex antigen (also available from Colorado State University)
was used as a standard. This assay has a lower limit of sen-
sitivity of 50 pg/mL and does not detect other M. tuberculosis
products (purified M. tuberculosis 19-kDa and 45-kDa anti-
gens, or lipoarabinomannan).
Statistical analysis. Results were analyzed by use of Mann-
Whitney rank sum and paired t tests, and linear correlation
and regression analysis were performed where needed. P! .05
was considered to be significant.
RESULTS
Expression of TNF-a and M. tuberculosis mRNA in M. tuber-
culosis–infected AMs. First, we investigated the kinetics (0–
120 h) of expression of TNF-a and M. tuberculosis 85B mRNA
in M. tuberculosis H37Ra–infected AMs. TNF-a mRNA was
corrected to host 18S rRNA in the same sample, and M. tu-
berculosis 85B mRNA was expressed as 85B:16S. Expression of
TNF-a was maximal at 24 h ( ) and thereafter decreasedP ! .05
(figure 1A). Expression of M. tuberculosis 85B continued to
increase up to 120 h (figure 1B); at 4 h, M. tuberculosis 85B:
16S was 3-fold higher ( ), and, at 24 h, it was 5-foldP ! .05
higher ( ), compared with levels in t0 cultures. Further-P ! .01
more, between 4 and 24 h, M. tuberculosis 85B:16S increased
significantly ( ). In contrast to the increase in intracellularP ! .01
expression of M. tuberculosis 85B:16S, it did not increase when
bacteria (104–107 cfu) were cultured for up to 120 h in medium
alone (in the absence of cells). Of importance, the early levels
of expression of TNF-a mRNA and M. tuberculosis 85B:16S
(at t0 and 4 h) correlated with one another ( , P! .001).2r p 0.6
In some experiments ( ), the intracellular growth of M.n p 4
tuberculosis was assessed in duplicate wells of M. tuberculosis–
infected AMs (by the colony-forming unit assay) at t0 and at
4 days. The increase in intracellular growth of M. tuberculosis
at 4 days correlated with intracellular M. tuberculosis 85B:16S
at 24 h ( ; ).2r p 0.93 P ! .05
To ascertain that the intracellular changes in expression of
M. tuberculosis 85B were not attributable to any extracellular
bacteria left after extensive washing of cultures or released from
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from

Mycobacterial Gene Induction in Macrophages • JID 2004:190 (15 July) • 345
Figure 2. Effect of neutralization of tumor necrosis factor (TNF)–a by soluble (s) TNF receptors (Rs) on expression of TNF-a and Mycobacteriumtuberculosis 85B. Alveolar macrophages were infected with M. tuberculosis H37Ra (1:1 bacteria/cell). Cultures then received 10 ng/mL sTNFRI (stripedbars) or sTNRFII (hatched bars) or media alone (black bars) for 4 and 24 h. Total RNA was extracted and assessed for TNF-a mRNA (A) and 85B:16S (B). Data are of 3 experiments. * , vs. M. tuberculosis alone; ** , vs. M. tuberculosis alone; *** , vs. M.mean � SEM P ! .05 P ! .01 P ! .001tuberculosis alone.
cells during 24 h, in some experiments, culture supernatants
of M. tuberculosis H37Ra–infected AMs (at t0 and 4 and 24 h)
also were collected and centrifuged in the presence of 10%
polyethylene glycol 8000 (Sigma), and M. tuberculosis RNA was
extracted in the presence of excess yeast RNA (1 mg). Expression
of M. tuberculosis 85B mRNA in these samples was negligible.
Next, culture supernatants from M. tuberculosis–infected AMs
at 24 h were assessed for TNF-a and antigen 85 complex by use
of ELISA. Expression of TNF-a mRNA at 4 h correlated with
that of TNF-a protein at 24 h ( ; ). Similarly,2r p 0.73 P ! .001
expression of M. tuberculosis 85B at 4 h correlated with im-
munoreactivity of 85 complex in 24-h culture supernatants
( ; ). The concentration of M. tu-2r p 0.5 P ! .05 mean � SEM
berculosis 85 complex in 24-h culture supernatants was 592 �
pg/mL ( ). As noted elsewhere [34], M. tuberculosis182 n p 12
85B is a component of M. tuberculosis antigen 85 complex, and
changes in the 85 complex immunoreactivity in supernatants
likely reflect changes in 85B.
To ascertain that expression of the M. tuberculosis 85B gene
was not divergent in H37Rv- and H37Ra-infected AMs, we
compared expression of TNF-a and M. tuberculosis 85B in the
same experiment, using AMs infected with either strain of M.
tuberculosis, from 3 donors. Again, despite differences in levels
of expression of TNF-a and M. tuberculosis 85B, the patterns
of gene expression were similar. At 4 h, the level of TNF-a
mRNA induced by H37Rv was 12–20 times higher than that
induced by H37Ra, and the level of M. tuberculosis 85B induced
by H37Rv was 16 times higher than that induced by H37Ra.
At 24 h, levels of both TNF-a and M. tuberculosis 85B induced
by H37Rv were higher than those induced by H37Ra. Thus,
virulent M. tuberculosis strains induce higher levels of both
TNF-a and M. tuberculosis 85B in AMs.
Modulation by sTNFRs of expression of TNF-a and M.
tuberculosis 85B gene in M. tuberculosis–infected AMs. On
the basis of results presented in the previous section, we hy-
pothesized that endogenous M. tuberculosis–induced TNF-a in
cultures of AMs may contribute to modulation of expression
of the M. tuberculosis 85B gene during the initial 24 h after
infection. To assess the effect of TNF-a on expression of M.
tuberculosis 85B in AMs early after M. tuberculosis infection, we
inhibited TNF-a signaling by use of sTNFRI and sTNFRII. In
preliminary experiments, we defined the optimal dose of each
reagent to inhibit TNF-a. H37Ra-infected AMs received either
sTNFR1 (10 ng/mL), sTNFRII (10 ng/mL), or medium alone
for 4 and 24 h. M. tuberculosis–induced TNF-a mRNA was
down-regulated significantly in AMs by sTNFRI and sTNFRII at
4 h ( for both) and at 24 h ( for both) (figure 2).P ! .001 P ! .01
Both soluble receptors also significantly decreased expression of
M. tuberculosis 85B in AMs ( at 4 h and at 24 h,P ! .05 P ! .02
for both). Despite the fact that sTNFRI appeared to be stronger
than sTNFRII in inhibition of expression of TNF-a and M.
tuberculosis 85B gene in AMs, these differences were not signif-
icant. In addition, in a few experiments, we found that the co-
presence of the 2 sTNFRs was not synergistic in inhibition of
expression of TNF-a or 85B:16S (data not shown).
Because the results presented in the previous section indi-
cated that H37Rv was stronger in induction of both TNF-a
and 85B mRNA, we also examined the effect of neutralization
of TNF-a (by sTNFRI or sTNFRII) on expression of either in
AMs infected with H37Rv. Neutralization of TNF-a (by s-
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from
346 • JID 2004:190 (15 July) • Islam et al.
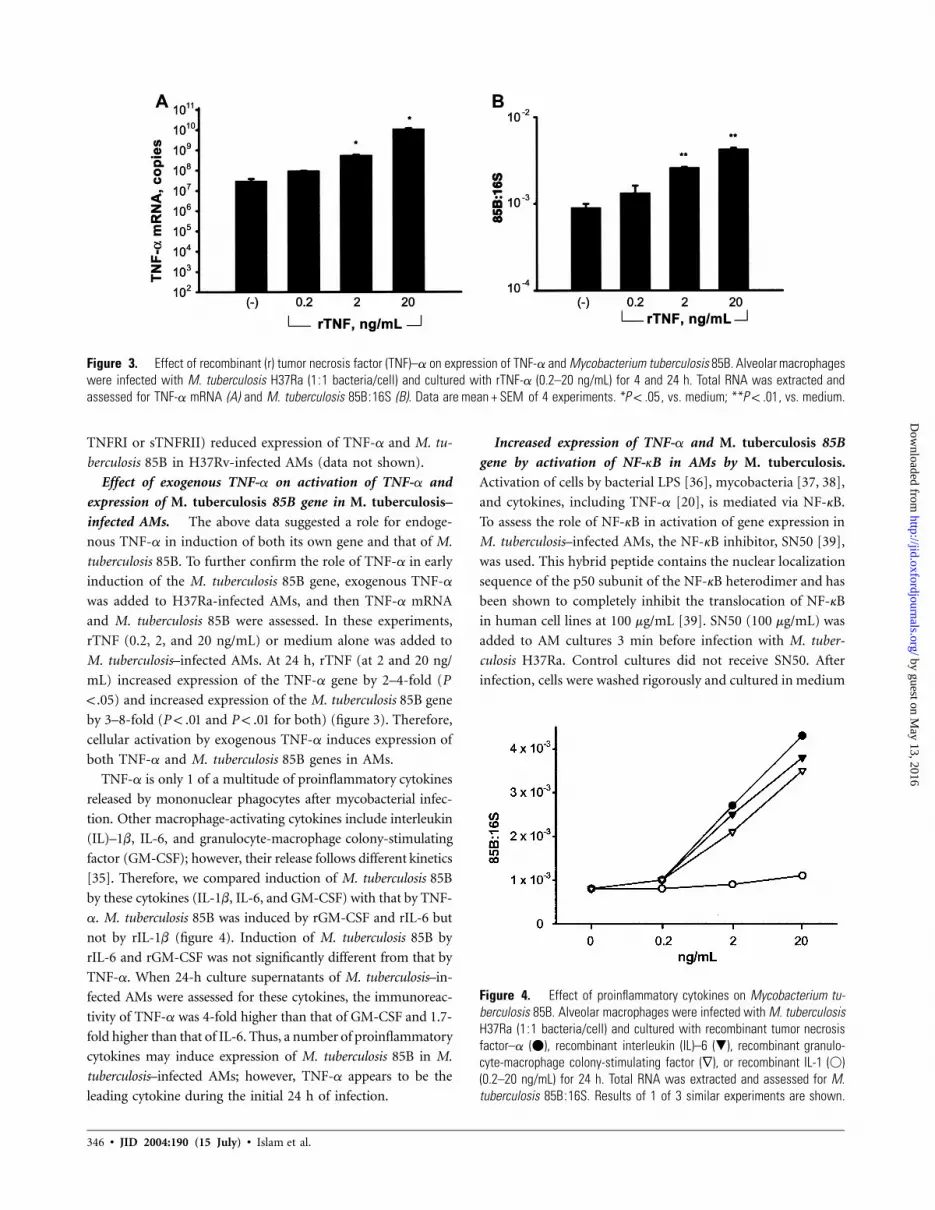
Figure 3. Effect of recombinant (r) tumor necrosis factor (TNF)–a on expression of TNF-a and Mycobacterium tuberculosis 85B. Alveolar macrophageswere infected with M. tuberculosis H37Ra (1:1 bacteria/cell) and cultured with rTNF-a (0.2–20 ng/mL) for 4 and 24 h. Total RNA was extracted andassessed for TNF-a mRNA (A) and M. tuberculosis 85B:16S (B). Data are of 4 experiments. * , vs. medium; ** , vs. medium.mean + SEM P ! .05 P ! .01
Figure 4. Effect of proinflammatory cytokines on Mycobacterium tu-berculosis 85B. Alveolar macrophages were infected with M. tuberculosisH37Ra (1:1 bacteria/cell) and cultured with recombinant tumor necrosisfactor–a (�), recombinant interleukin (IL)–6 (�), recombinant granulo-cyte-macrophage colony-stimulating factor (∇), or recombinant IL-1 (�)(0.2–20 ng/mL) for 24 h. Total RNA was extracted and assessed for M.tuberculosis 85B:16S. Results of 1 of 3 similar experiments are shown.
TNFRI or sTNFRII) reduced expression of TNF-a and M. tu-
berculosis 85B in H37Rv-infected AMs (data not shown).
Effect of exogenous TNF-a on activation of TNF-a and
expression of M. tuberculosis 85B gene in M. tuberculosis–
infected AMs. The above data suggested a role for endoge-
nous TNF-a in induction of both its own gene and that of M.
tuberculosis 85B. To further confirm the role of TNF-a in early
induction of the M. tuberculosis 85B gene, exogenous TNF-a
was added to H37Ra-infected AMs, and then TNF-a mRNA
and M. tuberculosis 85B were assessed. In these experiments,
rTNF (0.2, 2, and 20 ng/mL) or medium alone was added to
M. tuberculosis–infected AMs. At 24 h, rTNF (at 2 and 20 ng/
mL) increased expression of the TNF-a gene by 2–4-fold (P
! .05) and increased expression of the M. tuberculosis 85B gene
by 3–8-fold ( and for both) (figure 3). Therefore,P ! .01 P ! .01
cellular activation by exogenous TNF-a induces expression of
both TNF-a and M. tuberculosis 85B genes in AMs.
TNF-a is only 1 of a multitude of proinflammatory cytokines
released by mononuclear phagocytes after mycobacterial infec-
tion. Other macrophage-activating cytokines include interleukin
(IL)–1b, IL-6, and granulocyte-macrophage colony-stimulating
factor (GM-CSF); however, their release follows different kinetics
[35]. Therefore, we compared induction of M. tuberculosis 85B
by these cytokines (IL-1b, IL-6, and GM-CSF) with that by TNF-
a. M. tuberculosis 85B was induced by rGM-CSF and rIL-6 but
not by rIL-1b (figure 4). Induction of M. tuberculosis 85B by
rIL-6 and rGM-CSF was not significantly different from that by
TNF-a. When 24-h culture supernatants of M. tuberculosis–in-
fected AMs were assessed for these cytokines, the immunoreac-
tivity of TNF-a was 4-fold higher than that of GM-CSF and 1.7-
fold higher than that of IL-6. Thus, a number of proinflammatory
cytokines may induce expression of M. tuberculosis 85B in M.
tuberculosis–infected AMs; however, TNF-a appears to be the
leading cytokine during the initial 24 h of infection.
Increased expression of TNF-a and M. tuberculosis 85B
gene by activation of NF-kB in AMs by M. tuberculosis.
Activation of cells by bacterial LPS [36], mycobacteria [37, 38],
and cytokines, including TNF-a [20], is mediated via NF-kB.
To assess the role of NF-kB in activation of gene expression in
M. tuberculosis–infected AMs, the NF-kB inhibitor, SN50 [39],
was used. This hybrid peptide contains the nuclear localization
sequence of the p50 subunit of the NF-kB heterodimer and has
been shown to completely inhibit the translocation of NF-kB
in human cell lines at 100 mg/mL [39]. SN50 (100 mg/mL) was
added to AM cultures 3 min before infection with M. tuber-
culosis H37Ra. Control cultures did not receive SN50. After
infection, cells were washed rigorously and cultured in medium
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from

Mycobacterial Gene Induction in Macrophages • JID 2004:190 (15 July) • 347
Figure 5. Effect of inhibition of NF-kB on expression of tumor necrosis factor (TNF)–a and Mycobacterium tuberculosis 85B. Alveolar macrophageswere infected with M. tuberculosis H37Ra (1:1 bacteria/cell) in the presence or absence (�) of SN50 (100 mg/mL). Total RNA was extracted at 4 hand was assessed for expression of TNF-a (A) and M. tuberculosis 85B:16S (B). Data are of 3 experiments. * , vs. medium;mean � SEM P ! .05** , vs. medium.P ! .01
alone for 4 h; SN50 reduced expression of both TNF-a mRNA
( ) and M. tuberculosis 85B:16S ( ) in AMs (figureP ! .005 P ! .05
5). To assure that cellular inhibition was not nonspecific, in
some experiments, we compared the effect of SN50 (100 mg/
mL) with its inactive analogue, SN50/M (100 mg/mL). SN50/
M did not affect expression of either TNF-a or M. tuberculosis
85B (data not shown). Therefore, the increased expression of
85B and TNF-a mRNA in M. tuberculosis–infected AMs is me-
diated via activation of NF-kB.
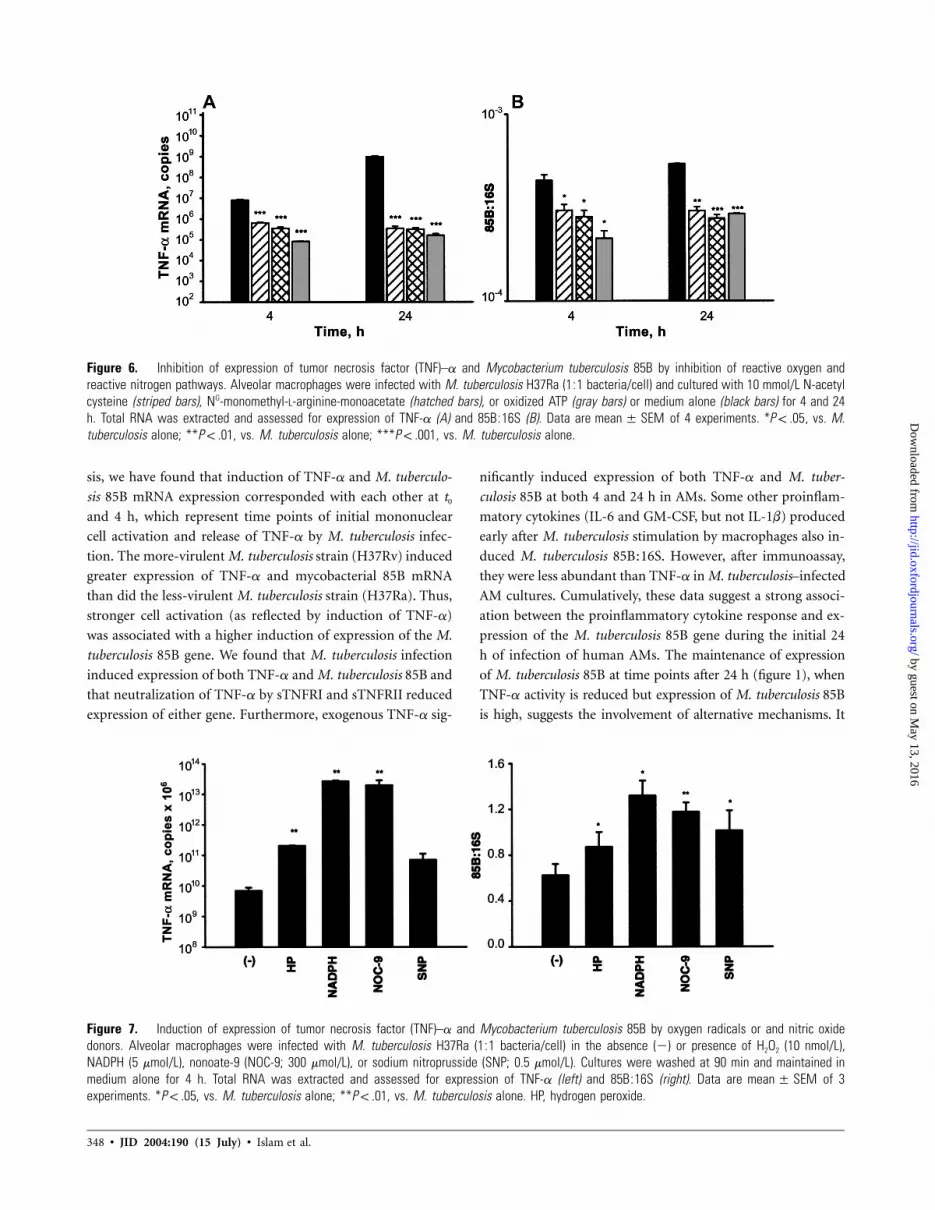
Role of RNI and ROI in expression of TNF-a and M. tu-
berculosis mRNA in M. tuberculosis–infected AMs. After
stimulation of mononuclear phagocytes by a variety of sub-
stances, including microbes and their products, ROI and RNI
pathways are produced. The activation of RNI and ROI path-
ways are further potentiated by macrophage-activating cyto-
kines [40, 41]. To assess the effect of activation of ROI and
RNI on expression of the M. tuberculosis 85B gene, we used
specific inhibitors of each pathway, NAC and NMMA. In some
experiments, oATP, which inhibits both pathways [31], was
used. After infection of AMs with M. tuberculosis H37Ra, NAC
(10 nmol/L), NMMA (10 nmol/L), or oATP (10 nmol/L) was
added to cultures. Control cultures received medium alone.
NAC, NMMA, and oATP significantly reduced expression of
both TNF-a mRNA and M. tuberculosis 85B at both 4 and 24
h (figure 6). At 4 h, expression of TNF-a was reduced by 1–
2 logs ( ). At 24 h, reduction in expression of TNF-aP ! .001
by NAC, NMMA, or oATP was comparable (∼3 logs; P! .001
for all). NAC, NMMA, and oATP also reduced expression of
M. tuberculosis 85B at both 4 ( for all) and 24 h (P ! .05 P !
for NAC; for NMMA and oATP). In a single ex-.01 P ! .001
periment, we ascertained that the reduction in expression of
TNF-a and M. tuberculosis 85B by these inhibitors was also
demonstrable in AMs infected with H37Rv.
To confirm the above observations, in some experiments
( ), classic agents that increase formation of oxygen rad-n p 3
icals (NADPH or H2O2), or the nitric oxide (NO) donors (SNP
or NOC-9), were added to H37Ra-infected AMs during and
after infection. Control AM cultures received oxygen radicals
or NO donors but were not infected. The oxygen radicals and
NO donors significantly increased the expression of TNF-a and
M. tuberculosis 85B in M. tuberculosis–infected AMs (figure 7).
Oxygen radicals and NO donors also increased TNF-a mRNA
in uninfected AMs, but to low levels (103–104 copies/sample)
(data not shown).
DISCUSSION
The initial interaction of M. tuberculosis and host mononuclear
phagocytes at sites of M. tuberculosis infection is critical to
intracellular parasitism of M. tuberculosis. Intense cellular ac-
tivation and proinflammatory cytokine release, predominated
by TNF-a, occurs and may affect the pathogenesis of M. tu-
berculosis. Of the ∼12 M. tuberculosis genes shown to be up-
regulated after M. tuberculosis infection of human macrophages,
most of which are essential components of bacterial metabolism
and stress adaptation, M. tuberculosis 85B was expressed most
frequently [30]. However, the role of 85B in the pathogenesis
of M. tuberculosis in humans is not clear. Here, the relationship
between expression of TNF-a and M. tuberculosis 85B mRNA
during the first 24 h of infection of AMs with M. tuberculosis
has been examined.
On the basis of results of previous studies examining kinetics
of induction of expression of TNF-a mRNA and levels of TNF-
a [2, 14], we hypothesized that this cytokine may play a major
role in the innate immune response early after M. tuberculosis
infection of human macrophages. In support of this hypothe-
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from
348 • JID 2004:190 (15 July) • Islam et al.
Figure 6. Inhibition of expression of tumor necrosis factor (TNF)–a and Mycobacterium tuberculosis 85B by inhibition of reactive oxygen andreactive nitrogen pathways. Alveolar macrophages were infected with M. tuberculosis H37Ra (1:1 bacteria/cell) and cultured with 10 mmol/L N-acetylcysteine (striped bars), NG-monomethyl-L-arginine-monoacetate (hatched bars), or oxidized ATP (gray bars) or medium alone (black bars) for 4 and 24h. Total RNA was extracted and assessed for expression of TNF-a (A) and 85B:16S (B). Data are of 4 experiments. * , vs. M.mean � SEM P ! .05tuberculosis alone; ** , vs. M. tuberculosis alone; *** , vs. M. tuberculosis alone.P ! .01 P ! .001
Figure 7. Induction of expression of tumor necrosis factor (TNF)–a and Mycobacterium tuberculosis 85B by oxygen radicals or and nitric oxidedonors. Alveolar macrophages were infected with M. tuberculosis H37Ra (1:1 bacteria/cell) in the absence (�) or presence of H2O2 (10 nmol/L),NADPH (5 mmol/L), nonoate-9 (NOC-9; 300 mmol/L), or sodium nitroprusside (SNP; 0.5 mmol/L). Cultures were washed at 90 min and maintained inmedium alone for 4 h. Total RNA was extracted and assessed for expression of TNF-a (left) and 85B:16S (right). Data are of 3mean � SEMexperiments. * , vs. M. tuberculosis alone; ** , vs. M. tuberculosis alone. HP, hydrogen peroxide.P ! .05 P ! .01
sis, we have found that induction of TNF-a and M. tuberculo-
sis 85B mRNA expression corresponded with each other at t0
and 4 h, which represent time points of initial mononuclear
cell activation and release of TNF-a by M. tuberculosis infec-
tion. The more-virulent M. tuberculosis strain (H37Rv) induced
greater expression of TNF-a and mycobacterial 85B mRNA
than did the less-virulent M. tuberculosis strain (H37Ra). Thus,
stronger cell activation (as reflected by induction of TNF-a)
was associated with a higher induction of expression of the M.
tuberculosis 85B gene. We found that M. tuberculosis infection
induced expression of both TNF-a and M. tuberculosis 85B and
that neutralization of TNF-a by sTNFRI and sTNFRII reduced
expression of either gene. Furthermore, exogenous TNF-a sig-
nificantly induced expression of both TNF-a and M. tuber-
culosis 85B at both 4 and 24 h in AMs. Some other proinflam-
matory cytokines (IL-6 and GM-CSF, but not IL-1b) produced
early after M. tuberculosis stimulation by macrophages also in-
duced M. tuberculosis 85B:16S. However, after immunoassay,
they were less abundant than TNF-a in M. tuberculosis–infected
AM cultures. Cumulatively, these data suggest a strong associ-
ation between the proinflammatory cytokine response and ex-
pression of the M. tuberculosis 85B gene during the initial 24
h of infection of human AMs. The maintenance of expression
of M. tuberculosis 85B at time points after 24 h (figure 1), when
TNF-a activity is reduced but expression of M. tuberculosis 85B
is high, suggests the involvement of alternative mechanisms. It
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from

Mycobacterial Gene Induction in Macrophages • JID 2004:190 (15 July) • 349
has to be noted that expression of the TNF-a gene and pro-
duction of its protein are under strong intracellular control mech-
anisms. Expression of the M. tuberculosis 85B gene likely follows
the unrestrained intracellular growth of M. tuberculosis. However,
an effect of cytokines released by M. tuberculosis–infected AMs
on intracellular or even extracellular (as macrophages undergo
lysis) expression of M. tuberculosis gene throughout infection of
mononuclear phagocytes needs to be considered.
Induction of expression of TNF-a and M. tuberculosis 85B
in M. tuberculosis–infected AMs was mediated through acti-
vation of NF-kB, because both mRNAs were suppressed when
SN50, an inhibitor of NF-kB, was present in cultures. The an-
alogue of SN50 did not have any effect. Thus, cellular activation
is associated with augmentation of expression of both TNF-a
and M. tuberculosis 85B in M. tuberculosis–infected AMs.
After phagocytosis of microbes and cellular activation, ROIs
are activated [21]. One product of the ROI pathway, H2O2,
activates expression of iNOS and production of NO [22]. On
the other hand, both ROI and RNI are downstream mediators
of macrophage-activating cytokines and are thought to be mi-
crobicidal. Activation of iNOS and production of NO may be
important in the final containment of M. tuberculosis by mac-
rophages [42]. However, M. tuberculosis has evolved resistance
mechanisms against both ROI [43, 44] and RNI [45, 46]. Here,
when the activation of RNI and ROI was inhibited by NMMA,
NAC, or oATP, the expression of both TNF-a and M. tuber-
culosis 85B was significantly lowered in M. tuberculosis–infected
AMs at 24 h. We had previously found that these treatments
had no effect on M. tuberculosis–infected monocytes [29]. We
confirmed these observations by using agents that release ox-
ygen radicals (NADPH and H2O2) or NO (SNP or NOC-9).
In M. tuberculosis–infected AMs, these reagents increased ex-
pression of both TNF-a and M. tuberculosis 85B. Thus, both
RNI and ROI, induced early after M. tuberculosis infection of
AMs, activate expression of M. tuberculosis gene in turn. Mod-
erate-to-high production of NO has been reported in AMs from
2–3 healthy donors after stimulation with M. tuberculosis [47],
and ROI activates NF-kB [22].
M. tuberculosis 85B is a predominant protein [25] during
human M. tuberculosis infection; however, its role in the patho-
genesis of M. tuberculosis infection is not clear. At least with
regard to mycolyl transferase activity, which underlies cell-wall
biosynthesis, it appears that both M. tuberculosis 85A and 85B
are redundant [48]. In sputum from patients with tuberculosis,
levels of M. tuberculosis 85B protein and mRNA correlate with
M. tuberculosis growth, and maintenance of 85B levels correlates
with a lack of response to therapy [34, 49]. Studies using the
human myelo-monocytic cell line, THP-1, have shown that 85B
may be dispensable for intracellular growth of M. tuberculosis
[50]. However, the activation state of primary macrophages is
different from tumor cell lines [51], and, despite the fact that
THP-1 cells have been used in studies of macrophage cytokine
expression [52], their secretory capacities are different from pri-
mary macrophages (Z.T., unpublished data). Therefore, studies
of relevance of 85B to pathogenesis of M. tuberculosis need to
be extended to primary human macrophages. Interestingly, al-
though many mycobacterial components induce TNF-a in mono-
nuclear phagocytes [53–55], only the members of 85 complex
interact with host fn [26]. Moreover, binding of 85B to fn en-
hances the expression of TNF-a in monocytes [28]. Therefore,
the role of M. tuberculosis 85B in intracellular infection may be
the maintenance of an inflammatory response. It is likely that
other fn-binding proteins of M. tuberculosis increase proinflam-
matory cytokines as well. In addition, M. tuberculosis 85 complex
may act as an intermediary to synthesis of trehalose dimycolate,
which enhances the host inflammatory response [56]. A vicious
circle may exist in which expression of host inflammation and
mycobacterial products amplify one another.
In summary, M. tuberculosis infection of AMs leads to a
concomitant activation of TNF-a and expression of M. tuber-
culosis 85B gene. Although the intracellular induction of ex-
pression of M. tuberculosis 85B may be merely a marker for a
set of M. tuberculosis genes associated with initial mycobacterial
survival, an intense interaction of M. tuberculosis with host AMs
is suggested by our data. M. tuberculosis infection of lung mac-
rophages initiates a cascade of events whereby cellular activation
by TNF-a, RNI, and ROI enhances the expression of M. tu-
berculosis 85B in AMs. Further studies on the initial interplay
of human macrophages and M. tuberculosis will help delineate
the contribution of this early host-pathogen interaction to the
pathogenesis of tuberculosis.
Acknowledgments
We thank H. Boom and K. Eisenach, for careful reading of the manuscript.
References
1. Means TK, Jones BW, Schromm AB, et al. Differential effects of a Toll-like receptor antagonist on Mycobacterium tuberculosis–induced mac-rophage responses. J Immunol 2001; 166:4074–82.
2. Hirsch CS, Yoneda T, Averill L, Ellner JJ, Toossi Z. Enhancement ofintracellular growth of Mycobacterium tuberculosis in human mono-cytes by transforming growth factor–b1. J Infect Dis 1994; 170:1229–37.
3. Dahl KE, Shiratsuchi H, Hamilton BD, Ellner JJ, Toossi Z. Selectiveinduction of transforming growth factor b in human monocytes bylipoarabinomannan of Mycobacterium tuberculosis. Infect Immun 1996;64:399–405.
4. Schwander SK, Torres M, Carranza CC, et al. Pulmonary mononuclearcell responses to antigens of Mycobacterium tuberculosis in healthyhousehold contacts of patients with active tuberculosis and healthycontrols from the community. J Immunol 2000; 165:1479–85.
5. Barnes PF, Fong SJ, Brennan PJ, Twomey PE, Mazumder A, ModlinRL. Local production of tumor necrosis factor and IFN-g in tuber-culous pleuritis. J Immunol 1990; 145:149–54.
6. Hirsch CS, Toossi Z, Johnson JL, et al. Augmentation of apoptosis and
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from

350 • JID 2004:190 (15 July) • Islam et al.
interferon-g production at sites of active Mycobacterium tuberculosisinfection in human tuberculosis. J Infect Dis 2001; 183:779–88.
7. Condos R, Rom WN, Liu YM, Schluger NW. Local immune responsescorrelate with presentation and outcome in tuberculosis. Am J RespirCrit Care Med 1998; 157:729–35.
8. Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducingrole of tumor necrosis factor in the development of bactericidal gran-ulomas during BCG infection. Cell 1989; 56:731–40.
9. Jacobs M, Brown N, Allie N, Ryffel B. Fatal Mycobacterium bovis BCGinfection in TNF-LT-a–deficient mice. Clin Immunol 2000; 94:192–9.
10. Ehlers S, Benini J, Kutsch S, Endres R, Rietschel ET, Pfeffer K. Fatalgranuloma necrosis without exacerbated mycobacterial growth in tu-mor necrosis factor receptor p55 gene–deficient mice intravenouslyinfected with Mycobacterium avium. Infect Immun 1999; 67:3571–9.
11. Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with in-fliximab, a tumor necrosis factor a–neutralizing agent. N Engl J Med2001; 345:1098–104.
12. Tsenova L, Bergtold A, Freedman VH, Young RA, Kaplan G. Tumornecrosis factor a is a determinant of pathogenesis and disease pro-gression in mycobacterial infection in the central nervous system. ProcNatl Acad Sci USA 1999; 96:5657–62.
13. Bekker LG, Maartens G, Steyn L, Kaplan G. Selective increase in plasmatumor necrosis factor–a and concomitant clinical deterioration afterinitiating therapy in patients with severe tuberculosis. J Infect Dis 1998;178:580–4.
14. Hirsch CS, Ellner JJ, Russell DG, Rich EA. Complement receptor–medi-ated uptake and tumor necrosis factor–a–mediated growth inhibition ofMycobacterium tuberculosis by human alveolar macrophages. J Immunol1994; 152:743–53.
15. Byrd TF. Tumor necrosis factor a (TNFa) promotes growth of virulentMycobacterium tuberculosis in human monocytes: iron-mediated growthsuppression is correlated with decreased release of TNFa from iron-treated infected monocytes. J Clin Invest 1997; 99:2518–29.
16. Silver RF, Li Q, Ellner JJ. Expression of virulence of Mycobacteriumtuberculosis within human monocytes: virulence correlates with intra-cellular growth and induction of tumor necrosis factor a but not withevasion of lymphocyte-dependent monocyte effector functions. InfectImmun 1998; 66:1190–9.
17. Baeuerle PA, Baltimore D. NF-kB: ten years after. Cell 1996; 87:13–20.18. Cheng N, Chen J. Tumor necrosis factor–a induction of endothelial eph-
rin A1 expression is mediated by a p38 MAPK- and SAPK/JNK-depend-ent but nuclear factor-kB–independent mechanism. J Biol Chem 2001;276:13771–7.
19. Haddad JJ, Land SC. A non-hypoxic, ROS-sensitive pathway mediatesTNF-a–dependent regulation of HIF-1a. FEBS Lett 2001; 505:269–74.
20. Fan J, Frey RS, Rahman A, Malik AB. Role of neutrophil NADPH oxidasein the mechanism of TNFa-induced NF-kB activation and intracellularadhesion molecule–1 expression in endothelial cells. J Biol Chem 2002;277:3404–11.
21. Takao S, Smith EH, Wang D, Chan CK, Bulkley GB, Klein AS. Role ofreactive oxygen metabolites in murine peritoneal macrophage phago-cytosis and phagocytic killing. Am J Physiol 1996; 271:C1278–84.
22. Han YJ, Kwon YG, Chung HT, et al. Antioxidant enzymes suppressnitric oxide production through the inhibition of NF-kB activation:role of H2O2 and nitric oxide in inducible nitric oxide synthase ex-pression in macrophages. Nitric Oxide 2001; 5:504–13.
23. Bergamini S, Rota C, Canali R, et al. N-acetylcysteine inhibits in vivonitric oxide production by inducible nitric oxide synthase. Nitric Oxide2001; 5:349–60.
24. Content J, de la Cuvellerie A, De Wit L, Vincent-Levy-Frebault V, OomsJ, De Bruyn J. The genes coding for the antigen 85 complexes ofMycobacterium tuberculosis and Mycobacterium bovis BCG are membersof a gene family: cloning, sequence determination, and genomic or-ganization of the gene coding for antigen 85-C of M. tuberculosis. InfectImmun 1991; 59:3205–12.
25. Salata RA, Sanson AJ, Malhotra IJ, et al. Purification and characteri-
zation of the 30,000 dalton native antigen of Mycobacterium tuberculosisand characterization of six monoclonal antibodies reactive with a majorepitope of this antigen. J Lab Clin Med 1991; 118:589–98.
26. Abou-Zeid C, Ratliff TL, Wiker HG, Harboe M, Bennedsen J, RookGA. Characterization of fibronectin-binding antigens released by My-cobacterium tuberculosis and Mycobacterium bovis BCG. Infect Immun1988; 56:3046–51.
27. Wiker HG, Harboe M. The antigen 85 complex: a major secretionproduct of Mycobacterium tuberculosis. Microbiol Rev 1992; 56:648–61.
28. Aung H, Toossi Z, Wisnieski JJ, et al. Induction of monocyte expressionof tumor necrosis factor a by the 30-kD a antigen of Mycobacteriumtuberculosis and synergism with fibronectin. J Clin Invest 1996; 98:1261–8.
29. Wilkinson RJ, Desjardin LE, Islam N, et al. An increase in expression ofa Mycobacterium tuberculosis mycolyl transferase gene (fbpB) occurs earlyafter infection of human monocytes. Mol Microbiol 2001; 39:813–21.
30. Graham JE, Clark-Curtiss JE. Identification of Mycobacterium tuber-culosis RNAs synthesized in response to phagocytosis by human mac-rophages by selective capture of transcribed sequences (SCOTS). ProcNatl Acad Sci USA 1999; 96:11554–9.
31. Sikora A, Liu J, Brosnan C, Buell G, Chessel I, Bloom BR. Cutting edge:purinergic signaling regulates radical-mediated bacterial killing mecha-nisms in macrophages through a P2X7-independent mechanism. J Im-munol 1999; 163:558–61.
32. Hartel C, Bein G, Kirchner H, Kluter H. A human whole-blood assayfor analysis of T-cell function by quantification of cytokine mRNA.Scand J Immunol 1999; 49:649–54.
33. Holland PM, Abramson RD, Watson R, Gelfand DH. Detection ofspecific polymerase chain reaction product by utilizing the 5r3′ exo-nuclease activity of Thermus aquaticus DNA polymerase. Proc NatlAcad Sci USA 1991; 88:7276–80.
34. Wallis RS, Perkins M, Phillips M, et al. Induction of the antigen 85complex of Mycobacterium tuberculosis in sputum: a determinant ofoutcome in pulmonary tuberculosis treatment. J Infect Dis 1998; 178:1115–21.
35. Shiratsuchi H, Toossi Z, Mettler MA, Ellner JJ. Colonial morphotypeas a determinant of cytokine expression by human monocytes infectedwith Mycobacterium avium. J Immunol 1993; 150:2945–54.
36. Yao J, Mackman N, Edgington TS, Fan ST. Lipopolysaccharide induc-tion of the tumor necrosis factor–a promoter in human monocyticcells: regulation by Egr-1, c-Jun, and NF-kB transcription factors. JBiol Chem 1997; 272:17795–801.
37. Toossi Z, Hamilton BD, Phillips MH, Averill LE, Ellner JJ, Salvekar A.Regulation of nuclear factor–kB and its inhibitor IkB- a/MAD-3 inmonocytes by Mycobacterium tuberculosis and during human tuber-culosis. J Immunol 1997; 159:4109–16.
38. Giri DK, Aggarwal BB. Constitutive activation of NF-kB causes resis-tance to apoptosis in human cutaneous T cell lymphoma HuT-78 cells:autocrine role of tumor necrosis factor and reactive oxygen interme-diates. J Biol Chem 1998; 273:14008–14.
39. Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition ofnuclear translocation of transcription factor NF-kB by a synthetic pep-tide containing a cell membrane-permeable motif and nuclear local-ization sequence. J Biol Chem 1995; 270:14255–8.
40. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of in-terferon-gamma as the lymphokine that activates human macrophageoxidative metabolism and antimicrobial activity. J Exp Med 1983; 158:670–89.
41. Rubin BY, Sekar V, Martimucci WA. Comparative antiproliferative ef-ficacies of human a and g interferons. J Gen Virol 1983; 64:1743–8.
42. MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, NathanCF. Identification of nitric oxide synthase as a protective locus againsttuberculosis. Proc Natl Acad Sci USA 1997; 94:5243–8.
43. Hillas PJ, del Alba FS, Oyarzabal J, Wilks A, Ortiz De Montellano PR.The AhpC and AhpD antioxidant defense system of Mycobacteriumtuberculosis. J Biol Chem 2000; 275:18801–9.
44. Manca C, Paul S, Barry CE 3rd, Freedman VH, Kaplan G. Mycobac-
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from

Mycobacterial Gene Induction in Macrophages • JID 2004:190 (15 July) • 351
terium tuberculosis catalase and peroxidase activities and resistance tooxidative killing in human monocytes in vitro. Infect Immun 1999;67:74–9.
45. St John G, Brot N, Ruan J, et al. Peptide methionine sulfoxide reductasefrom Escherichia coli and Mycobacterium tuberculosis protects bacteriaagainst oxidative damage from reactive nitrogen intermediates. ProcNatl Acad Sci USA 2001; 98:9901–6.
46. Ruan J, St John G, Ehrt S, Riley L, Nathan C. noxR3, a novel gene fromMycobacterium tuberculosis, protects Salmonella typhimurium from ni-trosative and oxidative stress. Infect Immun 1999; 67:3276–83.
47. Rich EA, Torres M, Sada E, Finegan CK, Hamilton BD, Toossi Z. My-cobacterium tuberculosis (MTB)–stimulated production of nitric oxide byhuman alveolar macrophages and relationship of nitric oxide productionto growth inhibition of MTB. Tuber Lung Dis 1997; 78:247–55.
48. Puech V, Guilhot C, Perez E, et al. Evidence for a partial redundancyof the fibronectin-binding proteins for the transfer of mycoloyl residuesonto the cell wall arabinogalactan termini of Mycobacterium tubercu-losis. Mol Microbiol 2002; 44:1109–22.
49. Desjardin LE, Perkins MD, Wolski K, et al. Measurement of sputumMycobacterium tuberculosis messenger RNA as a surrogate for responseto chemotherapy. Am J Respir Crit Care Med 1999; 160:203–10.
50. Armitige LY, Jagannath C, Wanger AR, Norris SJ. Disruption of the genesencoding antigen 85A and antigen 85B of Mycobacterium tuberculosisH37Rv: effect on growth in culture and in macrophages. Infect Immun2000; 68:767–78.
51. Goerdt S, Politz O, Schledzewski K, et al. Alternative versus classicalactivation of macrophages. Pathobiology 1999; 67:222–6.
52. Silver BJ, Hamilton BD, Toossi Z. Suppression of TNF-a gene ex-pression by hemin: implications for the role of iron homeostasis inhost inflammatory responses. J Leukoc Biol 1997; 62:547–52.
53. Wallis RS, Paranjape R, Phillips M. Identification by two-dimensionalgel electrophoresis of a 58-kilodalton tumor necrosis factor–inducingprotein of Mycobacterium tuberculosis. Infect Immun 1993; 61:627–32.
54. Barnes PF, Chatterjee D, Abrams JS, et al. Cytokine production inducedby Mycobacterium tuberculosis lipoarabinomannan: relationship to chem-ical structure. J Immunol 1992; 149:541–7.
55. Toossi Z. The inflammatory response in Mycobacterium tuberculosisinfection. Arch Immunol Ther Exp (Warsz) 2000; 48:513–9.
56. Lima VM, Bonato VL, Lima KM, et al. Role of trehalose dimycolatein recruitment of cells and modulation of production of cytokines andNO in tuberculosis. Infect Immun 2001; 69:5305–12.
by guest on May 13, 2016
http://jid.oxfordjournals.org/D
ownloaded from





























