Embed Size (px)
Citation preview

Selective Prostacyclin Receptor Agonism AugmentsGlucocorticoid-Induced Gene Expression in Human BronchialEpithelial Cells1
Sylvia M. Wilson,* Pamela Shen,2* Christopher F. Rider,† Suzanne L. Traves,* David Proud,*Robert Newton,† and Mark A. Giembycz3*
Prostacyclin receptor (IP-receptor) agonists display anti-inflammatory and antiviral activity in cell-based assays and in preclinicalmodels of asthma and chronic obstructive pulmonary disease. In this study, we have extended these observations by demonstrating thatIP-receptor activation also can enhance the ability of glucocorticoids to induce genes with anti-inflammatory activity. BEAS-2B bron-chial epithelial cells stably transfected with a glucocorticoid response element (GRE) luciferase reporter were activated in a concen-tration-dependent manner by the glucocorticoid dexamethasone. An IP-receptor agonist, taprostene, increased cAMP in these cells andaugmented luciferase expression at all concentrations of dexamethasone examined. Analysis of the concentration-response relationshipthat described this effect showed that taprostene increased the magnitude of transcription without affecting the potency of dexameth-asone and was, thus, steroid-sparing in this simple system. RO3244794, an IP-receptor antagonist, and oligonucleotides that selectivelysilenced the IP-receptor gene, PTGIR, abolished these effects of taprostene. Infection of BEAS-2B GRE reporter cells with an adenovirusvector encoding a highly selective inhibitor of cAMP-dependent protein kinase (PKA) also prevented taprostene from enhancing GRE-dependent transcription. In BEAS-2B cells and primary cultures of human airway epithelial cells, taprostene and dexamethasoneinteracted either additively or cooperatively in the expression of three glucocorticoid-inducible genes (GILZ, MKP-1, and p57kip2) thathave anti-inflammatory potential. Collectively, these data show that IP-receptor agonists can augment the ability of glucocorticoids toinduce anti-inflammatory genes in human airway epithelial cells by activating a cAMP/PKA-dependent mechanism. This observationmay have clinical relevance in the treatment of airway inflammatory diseases that are either refractory or respond suboptimally toglucocorticoids. The Journal of Immunology, 2009, 183: 6788–6799.
P rostacyclin (PGI2)4 is a labile eicosanoid derived from ar-achidonic acid following the sequential action of cycloox-ygenase and PGI2 synthase (1). The biological actions of
PGI2 are mediated primarily through the PGI2 receptor (IP-recep-
tor), which typically couples to Gs for the activation of adenylylcyclase (2). Although the role of PGI2 in the regulation of vascularhomeostasis has, for many years, been a primary research focus, itis now appreciated that PGI2 and its cognate receptor may playimportant (patho)physiological roles in a variety of other pro-cesses, including airway inflammatory diseases (2).
The IP-receptor subtype is expressed on many immune andproinflammatory cells, including the monocyte (3), T lymphocyte(4, 5), dendritic cell (6), epithelial cell (7), and airway (8) andpulmonary vascular smooth muscle cells (9). Moreover, accumu-lated evidence suggests that IP-receptor agonism in the lung mayexert anti-inflammatory and/or antiviral activity. For example, theIP-receptor agonists cicaprost and iloprost attenuate cytokine pro-duction from anti-CD3-stimulated, murine CD4� T lymphocytescultured under conditions that cause Th1 or Th2 cell polarization(5). Cytokine secretion from murine bone marrow-derived den-dritic cells in response to LPS is also inhibited by iloprost (6, 10).A central role for the IP-receptor in regulating allergic inflamma-tory responses in vivo similarly has been documented. Thus, IP-receptor gene deficiency in allergen-challenged, sensitized mice isassociated with a phenotype characterized by exaggerated pulmo-nary inflammation and airway hyper-responsiveness when com-pared with wild-type animals (11, 12). These data are consistentwith the finding that PGI2 attenuates allergen-induced inflamma-tion in mice by limiting pulmonary CD4� T cell recruitment (4,13), which may be due to an inhibitory effect on dendritic cellfunction (14). Activation of the IP-receptor may also be beneficialin chronic obstructive pulmonary disease (COPD). Indeed, tapro-stene, an IP-receptor agonist, suppressed the generation of theCD8� T cell chemoattractants CXCL9 and CXCL10 from human
*Department of Physiology and Pharmacology and †Department of Cell Biologyand Anatomy, Airways Inflammation Research Group, Institute of Infection, Im-munity and Inflammation, Faculty of Medicine, University of Calgary, Calgary,Alberta, Canada
Received for publication August 19, 2009. Accepted for publication September21, 2009.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This study was funded in part by the Canadian Institutes of Health Research (CIHR)and the GlaxoSmithKline/Collaborative Innovation Research Fund. R.N. is a CIHRNew Investigator and an Alberta Heritage Foundation for Medical Research(AHFMR) Scholar. M.A.G. is an AHFMR Senior Scholar. D.P. holds a CanadianResearch Chair in Inflammatory Lung Diseases.2 Current Address: Department of Pathology and Molecular Medicine, Centre forGene Therapeutics, McMaster University, Hamilton, Ontario, Canada.3 Address correspondence and reprint requests to Dr. Mark A. Giembycz, Departmentof Physiology and Pharmacology, Airways Inflammation Research Group, Institute ofInfection, Immunity, and Inflammation, 3280 Hospital Drive Northwest, Calgary,Alberta, Canada T2N 4N1. E-mail address: [email protected] Abbreviations used in this paper: PGI2, prostacyclin; COPD, chronic obstructivepulmonary disease; GILZ, glucocorticoid-inducible leucine zipper; GR, glucocorti-coid receptor; GRE, glucocorticoid response element; E/[A], agonist concentrationeffect; HEK, human embryonic kidney; HpAEC, human primary airway epithelialcell; ICS, inhaled glucocorticoid; IP-receptor, PGI2 receptor; MKP, MAPK phospha-tase; MOI, multiplicity of infection; p57kip2, kinase inhibitor protein 2 of 57 kDa;PDE, phosphodiesterase; PKA, cAMP-dependent protein kinase A; PKI, PKA inhib-itor; PPAR, peroxisome proliferator-activated receptor; SFM, serum-free medium;siRNA, small interfering RNA; TX, thromboxane.
Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00
The Journal of Immunology
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0902738

airway epithelial cells in vitro (7). Moreover, another IP-recep-tor agonist, beraprost, protected rats against the development ofexperimental cigarette smoke-induced emphysema, possibly bymeans of a concerted inhibitory action on alveolar epithelialcell apoptosis, oxidative burden, matrix metalloproteinase ex-pression, and proinflammatory cytokine generation (15). Fi-nally, there is evidence from animal studies that IP-receptoragonists protect against the deleterious effects of respiratorysyncytial virus infection (16). Potentially, this is an importantobservation because viral infections are believed to precipitateexacerbation in many individuals with asthma and COPD (17).Collectively, therefore, these findings suggest that selective IP-receptor agonists could be exploited to therapeutic advantage inthe treatment of airway diseases where chronic inflammationwith or without associated parenchymal destruction is a defin-ing characteristic (11, 14).
Unlike asthma, COPD is an example of a chronic inflammatorydisease that is relatively insensitive to inhaled glucocorticoids(ICSs) (18). Clearly, therefore, a drug that can boost or reveallatent sensitivity to ICSs while having intrinsic anti-inflammatoryand/or antiviral activity could be a significant pharmacotherapeuticadvance. Repression of inflammatory gene expression by ICSs isbelieved to occur by at least two general mechanisms. The clas-sical repressive mode of glucocorticoid action is termed trans-repression, in which the activity of key proinflammatory transcrip-tion factors, such as NF-�B and AP-1, is inhibited via directinteractions with the ligand-bound glucocorticoid receptor (GR)(19). However, in simple model systems, glucocorticoids oftenare relatively weak (partial) inhibitors of inflammatory genetranscription, implying that processes in addition to trans-re-pression must be operative to account for the anti-inflammatoryeffects seen in bona fide models of inflammation (20 –23). Inthis respect, the induction (trans-activation) by glucocorticoidsof anti-inflammatory genes that then repress proinflammatoryprocesses is now believed to be a major mechanism of glu-cocorticoid action (20 –23). Moreover, in the context of thepresent study, cAMP-elevating agents can, in gene expressionstudies, interact positively with glucocorticoids (20, 24). Ac-cordingly, we have tested the hypothesis, using human bron-chial epithelial cells as a model system, that selective agonismof the IP-receptor will enhance the anti-inflammatory activity ofa glucocorticoid to a level that cannot be achieved by the glu-cocorticoid alone. We submit that the demonstration of such aphenomenon could have clinical utility in the treatment of air-way inflammatory diseases, including COPD, that are eitherrefractory or respond suboptimally to glucocorticoids. Such afinding would also provide a precedent for the idea that novelglucocorticoid combination therapies could be developed inwhich the cAMP-elevating drug is tailored to the inflammatorydisease of interest.
Materials and MethodsCulture of BEAS-2B cells
Cells were cultured for 2 days under a 5% CO2/air atmosphere at 37°C in6- or 24-well plastic plates containing DMEM/F12 (Invitrogen) supple-mented with 10% FBS (Invitrogen), L-glutamine (2.5 mM), and 0.15%(v/v) sodium bicarbonate. The cells were then growth-arrested for 24 h inserum-free medium (SFM). At this time, cultures were tightly confluentand were processed for biochemical and functional measurements as de-scribed below.
Culture of human primary airway epithelial cells (HpAECs)
Cells were obtained by proteinase digestion of nontransplanted normal hu-man lung (International Institute for the Advancement of Medicine, Edi-son, NJ), as previously described (25). Cells were seeded in 12-well plates(Corning Life Sciences) containing bronchial epithelial cell growth me-dium (Lonza) supplemented with penicillin (50 �g/ml) and streptomycin(10 �g/ml), cultured under a 5% CO2/air atmosphere at 37°C until con-fluent (typically 14 days; medium was changed every 3 to 4 days), growtharrested for 24 h in supplement-free, bronchial epithelial cell basal medium(Lonza), and processed for biochemical and functional measurements asdescribed below. Ethics approval for the use of human tissues has beengranted by the Conjoint Health Research Ethics Board of the University ofCalgary (Calgary, Alberta, Canada).
Culture of human embryonic kidney (HEK)-293 Epstein-Barrnuclear Ag cells
HEK-293 Epstein-Barr nuclear Ag cells expressing the human recombinantD prostanoid 1-receptor (DP1R-HEK), E prostanoid 2-receptor (EP2R-HEK), E prostanoid 4-receptor (EP4R-HEK), or IP-receptor (IPR-HEK)subtype were cultured for 2 days under a 5% CO2/air atmosphere at 37°Cin 24-well plastic plates containing DMEM supplemented with 10% (v/v)FBS, geneticin (200 �g/ml), and hygromycin B (200 �g/ml). Confluentcells were then growth-arrested in DMEM for 24 h under a 5% CO2 at-mosphere at 37°C in the absence of serum and antibiotics before processingfor cAMP experiments as described below.
Generation of a glucocorticoid response element (GRE)reporter
Stable transfection was used to generate a GRE reporter cell line as de-scribed previously (26). The construct, pGL3.neo.TATA.2GRE, containstwo copies of a consensus simple GRE site (sense strand, 5�-TGTACAGGATGTTCT-3�) positioned upstream of a minimal �-globin promoterdriving a luciferase gene and a separate neomycin gene to confer resistanceto geneticin (26). BEAS-2B cells at �70% confluence in T162 flasks weretransfected with 8 �g of plasmid DNA and 20 �l of Lipofectamine 2000(Invitrogen). After 24 h, geneticin (100 �g/ml) was added until foci ofstable transfectants appeared that were harvested to create heterogeneouspopulations of cells in which the site of integration was randomized.
Transfection of BEAS-2B GRE reporter cells with siRNAs
RNAiMax (Invitrogen) and the small interfering RNA (siRNA) of interest(20 nM) (see Table I for siRNA oligonucleotide sequences) were diluted to2� the desired final concentration with antibiotic-free, serum-free DMEM:F12 (Invitrogen) combined in a 1:1 ratio and left at room temperature for30 min. Subconfluent (�70%) BEAS-2B GRE reporter cells in 24-wellplates were exposed to the siRNA/lipid mix for 6 h at 37°C under a 5%CO2/air atmosphere. The medium was then replaced with fresh DMEM/
Table I. siRNA oligonucleotide sequences
Gene Oligonucleotides Accession No. Source
PTGIRa 5�-CATCCATCTCATTGTCTAAtt-3� NM_000960.3 Qiagen3�-ggGTAGGTAGAGTAACAGATT-5�
PTGIRb 5�-GGGCGACAGGAGCCAGAAAtt-3� NM_000960.3 Qiagen3�-agCCCGCTGTCCTCGGTCTTT-5�
GFP 5�-GGCAAGCTGACCCTGAAGTTCtt-3� U57609 Dharmacon3�-ttCCGTTCGACTGGGACTTCAAG-5�
GAPDH 5�-GAGCCACATCGCTCAGACAtt-3� NM_002046 Qiagen3�-ggCTCGGTGTAGCGAGTCTGT-5�
a Refers to PTGIR-1 siRNA in the text.b Refers to PTGIR-2 siRNA in the text.
6789The Journal of Immunology

F12 supplemented with 1% FBS (v/v) and left for 42 h. Cells were thengrowth arrested in SFM for a further 48 h before beginning the experiment.
Treatment of GRE BEAS-2B reporter cells and measurement ofluciferase
Confluent, growth-arrested cells were treated with dexamethasone, tapro-stene, PGE2, and the IP-receptor antagonist RO3244794 (27) as indicatedin the text and figure legends. Unless stated otherwise, cells were incubatedat 37°C under a 5% CO2 atmosphere and harvested 5 h later in 1� reporterlysis buffer (Promega). Luciferase activity was then measured using aMonolight luminometer (BD Biosciences) according to the manufacturer’sinstructions. Data are expressed as the fold induction of luciferase relativeto that of unstimulated cells.
Infection of BEAS-2B Cells with Ad5.CMV.PKI�
Subconfluent (�70%) BEAS-2B GRE reporter cells were infected (multi-plicity of infection (MOI) � 40) with an E1�/E3� replication-deficientadenovirus vector (Ad5.CMV.PKI�) containing a 251-base pair DNA frag-ment encoding the complete amino acid sequence of the �-isoform ofcAMP-dependent protein kinase (PKA) inhibitor � (PKI�) downstream ofthe constitutively active CMV immediate early promoter (28). After 48 h,cells were processed for the assessment of GRE-dependent transcription asdescribed above. To control for biological effects of the virus per se, thevector Ad5.CMV.Null, expressing no transgene, was used in parallel. Us-ing this experimental protocol, we have previously reported that �90% ofBEAS-2B cells are infected with Ad5.CMV.PKI�, resulting in the expres-sion of a completely functional transgene with no adverse effect on cellviability (28).
cAMP mass determination
BEAS-2B and HEK-293 cells in 24-well plates were growth arrested for24 h and then pretreated (30 min) with the phosphodiesterase (PDE) in-hibitors rolipram (10 �M) and siguazodan (10 �M). Prostanoid receptoragonists and antagonists were then added at the concentrations indicated inthe text and figure legends in the continued presence of PDE inhibitors.After 45 min of incubation at 37°C under 5% CO2, cells were lysed withHCl (100 mM) and cAMP mass was measured by enzyme immunoassay(Cayman Chemical) according to the manufacturer’s instructions.
Measurement of CXCL10
Growth-arrested BEAS-2B cells were pretreated (30 min) with GW9662 (1�M), a peroxisome proliferator-activated receptor (PPAR) � antagonist(29), or its vehicle followed by a PAR� agonist, rosiglitazone (1 �M), fora further 30 min. IFN-� (100 ng/ml; p[A]90) was then added and the cellsincubated at 37°C under a 5% CO2 atmosphere for 24 h. The amount ofCXCL10 released into the culture supernatant was quantified by sandwichELISA (Human DuoSet development system; R&D Systems) according tothe manufacturer’s instructions (see Ref. 7 for details).
RNA isolation, reverse transcription, and real-time quantitativePCR
Total RNA was extracted from BEAS-2B cells and HpAECs that had beentreated with dexamethasone (1 �M) and taprostene (1 �M) or forskolin (10�M) alone or in combination using RNeasy mini kits (Qiagen) and wasreverse transcribed to cDNA as described previously (26). Real-time quan-
titative PCR analysis of cDNA using the primer sequences shown in TableII (designed using Primer Express software; Applied Biosystems) encodingglucocorticoid-induced leucine zipper (GILZ; also known as TGF-�-stim-ulated clone 22, domain family member 3 or TSC22D3), MAPK phospha-tase (MKP)-1 (also known as dual-specificity phosphatase 1 or DUSP-1),and kinase inhibitor protein 2 of 57 kDa (p57kip2; also known as cyclin-dependent kinase inhibitor 1C or CDKN1C) was performed using an ABI7900HT instrument (Applied Biosystems) on 2.5 �l of cDNA in 20-�lreactions using SYBR GreenER chemistry (Invitrogen) according to themanufacturer’s guidelines. Relative cDNA concentrations were determinedfrom a cDNA standard curve that was analyzed simultaneously with thetest samples. Amplification conditions were as follows: 50°C for 2 min and95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1min. Dissociation (melt) curves (95°C for 15 s and 60°C for 20 s withramping to 95°C over 20 min and then 95°C for 15 s) were constructed toconfirm primer specificity.
Curve fitting
Monophasic agonist concentration effect (E/[A]) curves were fitted byleast-squares, nonlinear iterative regression to the following form of theHill equation (Prism 4; GraphPad Software) shown in Equation 1,
E � Emin ��Emax � Emin
1 � 10�pA�50 � pA�n (1)
where E is the effect, Emin and Emax are the lower and upper asymptotes(i.e., the basal response and maximum agonist-induced response, respec-tively), p[A] the log molar concentration of agonist, p[A]50 a location pa-rameter equal to the log molar concentration of agonist producing Emax/2,and n the gradient of the E/[A] curve at the p[A]50 level.
The antagonism of taprostene-induced responses by RO3244794 wasevaluated by least-squares, nonlinear regression using a modification of theHill and Gaddum/Schild equations derived by Waud et al. (30). Each fam-ily of E/[A] curves (i.e., the control E/[A] curve and E/[A] curves con-structed in the presence of increasing concentrations of RO3244794) werefitted simultaneously to Equation 2. Thus,
E � Emin � ��Emax � Emin
1 � �10pA�501 � �B�/10�pA2S�
A� �n� (2)
where [A] and [B] are the molar concentrations of taprostene andRO3244794 respectively, S is the Schild slope factor, which indicates thenature of antagonism, and pA2 is the affinity of the antagonist when S � 1,which is equivalent to the pKb. To determine whether S deviated signifi-cantly from unity, the entire family of E/[A] curves that made up an indi-vidual experiment was fitted globally to Equation 2 under two conditions:one where S was constrained to a constant equal to 1 and the other whereit was a shared value for all data sets. The F test was applied to determinewhich equation gave the best fit, which was then used for the analysis.
Assessment of cell viability
Cell viability was evaluated colorimetrically by measuring the reduction ofthe tetrazolium salt, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide, to formazan by mitochondrial dehydrogenases.
Drugs, Abs, and analytical reagents
ONO-AE1–259 ((Z)-7-[(1R,2S,3R,5R)-5-chloro-3-hydroxy-2-[(E,4S)-4-hydroxy-4-(1-prop-2-enylcyclo butyl)but-1-enyl]cyclopentyl]hept-5-enoicacid) and ONO-AE1–329 (2-[3-[(1R,2S,3R)-3-hydroxy-2-[(E,3S)-3-hydroxy-5-[2-(methoxymethyl)phenyl]pent-1-enyl]-5-oxo-cyclopentyl]sulfanylpropylsulfanyl]acetic acid) were from ONO Pharmaceuticals. RO3244794 ((R-3-(4-fluoro phenyl)-2-[5-(4-fluorophenyl)-benzofuran-2-ylmethoxycarbonylami-no]propionic acid) was provided by Roche Pharmaceuticals. Iloprost, PGD2,PGE2, BW245C (7-[3-(3-cyclohexyl-3-hydroxy-propyl)-2,5-dioxo-imidazoli-din-4-yl]heptanoic acid), rosiglitazone, and GW9662 (2-chloro-5-nitrobenza-nilide) were obtained from Cayman Chemical and rolipram and siguazodanwere purchased from Calbiochem (EMD Biosciences). Goat anti-human PKI�(code 1944) and goat anti-human �-actin (code 1615) were from AutogenBioclear. Taprostene, forskolin, and all other reagents were from Sigma-Aldrich. Drugs were dissolved in DMSO and diluted to the desired workingconcentration in the appropriate culture medium.
Definitions and statistics
In the text, the term “additivity” refers to two drugs that, when combined,produce an effect that is the sum of their individual components. In con-trast, the term “positive cooperativity” is used when the biological response
Table II. Primer pairs for real-time qPCRa
Gene Oligonucleotide Accession No.
MKP-1 (DUSP1)Forward 5�-GCTCAGCCTTCCCCTGAGTA-3� NM_004417Reverse 5�-GATACGCACTGCCCAGGTACA-3�
p57kip2 (CDKN1C)Forward 5�-CGGCGATCAAGAAGCTGTC-3� NM_000076Reverse 5�-GGCTCTAAATTGGCTCACCG-3�
GILZ (TSC22D3)Forward 5�-GGCCATAGACAACAAGATCG-3� NM_001015881Reverse 5�-ACTTACACCGCAGAACCACCA-3�
GAPDHForward 5�-TTCACCACCATGGAGAAGGC-3� NM_002046Reverse 5�-AGGAGGCATTGCTGATGATCT-3�
a Forward and reverse primers for each gene are listed. Common genes names areshown and gene symbols appear in brackets.
6790 TAPROSTENE AUGMENTS GRE-DEPENDENT TRANSCRIPTION
of two drugs given in combination is greater than the sum of their indi-vidual effects.
Data points and values in the text and figure legends represent themean � SEM of N independent determinations. Data were analyzed byStudent’s t test or ANOVA (one-way or two-way as indicated) followed,when appropriate, by Bonferroni’s multiple comparison test. The null hy-pothesis was rejected when p 0.05.
ResultsNone of the compounds or their vehicles used in the experimentsdescribed herein significantly affected cell viability.
Selection of taprostene as an IP-receptor agonist
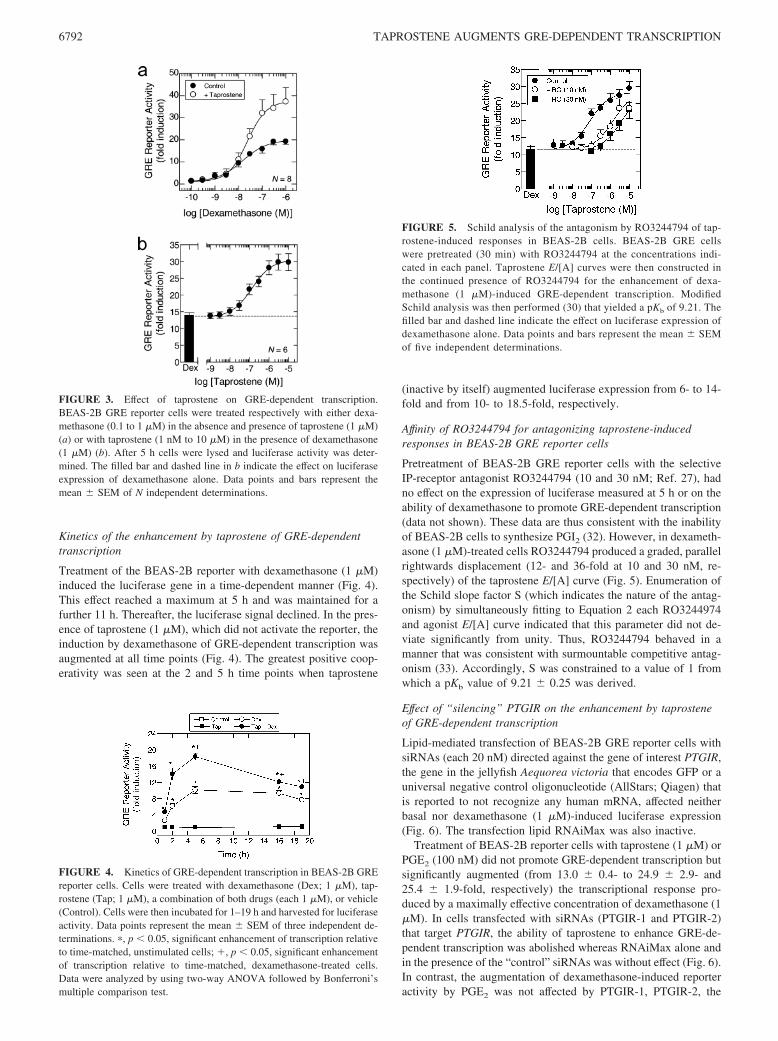
The explicit classification of responses mediated by IP-receptors ishindered by a paucity of suitable pharmacological tools. In thepresent study, we established that a synthetic PGI2 analog, tapro-stene (31), is a highly selectively agonist suitable for examiningthe (patho)physiological role of the IP-receptor subtype (Fig. 1).Thus, in IPR-HEK cells, taprostene increased the cAMP content ina concentration-dependent manner with a p[A]50 of 12.7 � 0.2(Fig. 2a). Relative to iloprost, taprostene was a full agonist in thesecells (intrinsic activity (�) � 1.01; Fig. 2a). In contrast, taprostenefailed to increase cAMP mass in HEK-293 cells expressing therecombinant DP1-, EP2-, and EP4-receptor subtypes (all adenylylcyclase coupled) at a concentration (1 nM) that maximally in-creased the cAMP level in IPR-HEK cells (Fig. 2, b–d). Indeed,even at a concentration of 10 �M, taprostene was weak, elicitinga cAMP response () that was 13% of that produced by PGD2 inDP1R-HEK cells and 4.9 and 10% of that produced by PGE2
in EP2R-HEK and EP4R-HEK cells, respectively (Fig. 2, b–d). Incontrast, at a concentration of 100 nM, BW245C (DP1-selective),ONO-AE1–259 (EP2-selective), and ONO-AE1–329 (EP4-selec-tive) were full agonists in DP1R-HEK, EP2R-HEK, and EP4R-HEK cells, respectively (Fig. 2, b–d).
Effect of taprostene on GRE-dependent transcription
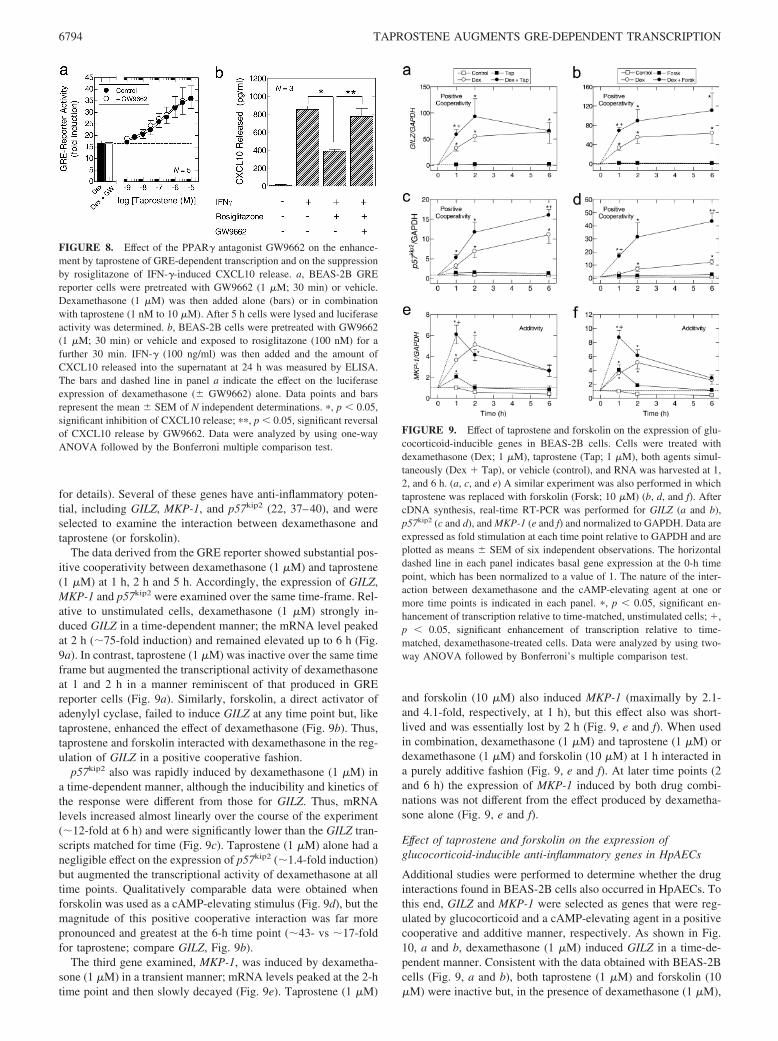
Treatment of BEAS-2B reporter cells with dexamethasone (0.1nM to 1 �M) for 5 h induced GRE-dependent transcription in aconcentration-dependent manner with a p[A]50 and Emax of7.96 � 0.11 and 19.3 � 3.8-fold, respectively (Fig. 3a). Tap-rostene (1 �M) alone was without effect on the GRE reporterbut, when added to cells concurrently with dexamethasone, sig-nificantly augmented GRE-dependent transcription above thatproduced by the glucocorticoid alone (Fig. 3a). Thus, the in-teraction of taprostene and dexamethasone was one of positivecooperativity. Analysis of the E/[A] relationship that describedthis effect showed that taprostene enhanced the magnitude(Emax) of transcription (from 19.3 � 3.8- to 37.3 � 5.5-fold)without affecting the potency of dexamethasone (p[A]50 �7.73 � 0.2) (Fig. 3a). Further studies established that in thepresence of a maximally effective concentration of dexameth-asone (1 �M), the ability of taprostene to augment GRE-de-pendent transcription was concentration-dependent, with ap[A]50 of 6.82 � 0.12 (Fig. 3b).
FIGURE 1. Chemical structure of PGI2, taprostene, and RO3244794.Asterisk (�) denotes chiral center. Optically active isomers are indicated.
FIGURE 2. Effect of taprostene on cAMP accumulation in HEK-293cells stably expressing adenylyl cyclase-coupled prostanoid receptor sub-types. Cells expressing the human recombinant IP-(IPR-HEK) (a), DP1-(DP1R-HEK) (b), EP2-(EP2R-HEK) (c), and EP4-receptor (EP4R-HEK) (d)were pretreated (30 min) with the PDE3 and PDE4 inhibitors and sigua-zodan (S; 10 �M) and rolipram (R; 10 �M), respectively, to prevent cAMPhydrolysis. E/[A] curves to taprostene were then constructed (IP: from 100aM to 10 nM; DP1, EP2, and EP4: from 100 pM to 10 �M) using cAMPand measured after 45 min as a biochemical output. The open and filled/hatched bars in each panel represent the cAMP response in cells treatedwith the PDE inhibitors in the absence or presence of the natural prosta-glandin and/or synthetic agonist, respectively, as indicated (each at 100nM). The intrinsic activity (�) of taprostene in IPR-HEK relative to iloprostis indicated in a. In b–d, the cAMP response () of taprostene (10 �M) isshown relative to that of PGD2 (DP1) and PGE2 (EP2 and EP4), which areused as reference full agonists. The dashed line in each panel indicates thebaseline cAMP level. Data points and bars represent the mean � SEM ofN independent determinations.
6791The Journal of Immunology
Kinetics of the enhancement by taprostene of GRE-dependenttranscription
Treatment of the BEAS-2B reporter with dexamethasone (1 �M)induced the luciferase gene in a time-dependent manner (Fig. 4).This effect reached a maximum at 5 h and was maintained for afurther 11 h. Thereafter, the luciferase signal declined. In the pres-ence of taprostene (1 �M), which did not activate the reporter, theinduction by dexamethasone of GRE-dependent transcription wasaugmented at all time points (Fig. 4). The greatest positive coop-erativity was seen at the 2 and 5 h time points when taprostene
(inactive by itself) augmented luciferase expression from 6- to 14-fold and from 10- to 18.5-fold, respectively.
Affinity of RO3244794 for antagonizing taprostene-inducedresponses in BEAS-2B GRE reporter cells
Pretreatment of BEAS-2B GRE reporter cells with the selectiveIP-receptor antagonist RO3244794 (10 and 30 nM; Ref. 27), hadno effect on the expression of luciferase measured at 5 h or on theability of dexamethasone to promote GRE-dependent transcription(data not shown). These data are thus consistent with the inabilityof BEAS-2B cells to synthesize PGI2 (32). However, in dexameth-asone (1 �M)-treated cells RO3244794 produced a graded, parallelrightwards displacement (12- and 36-fold at 10 and 30 nM, re-spectively) of the taprostene E/[A] curve (Fig. 5). Enumeration ofthe Schild slope factor S (which indicates the nature of the antag-onism) by simultaneously fitting to Equation 2 each RO3244974and agonist E/[A] curve indicated that this parameter did not de-viate significantly from unity. Thus, RO3244794 behaved in amanner that was consistent with surmountable competitive antag-onism (33). Accordingly, S was constrained to a value of 1 fromwhich a pKb value of 9.21 � 0.25 was derived.
Effect of “silencing” PTGIR on the enhancement by taprosteneof GRE-dependent transcription
Lipid-mediated transfection of BEAS-2B GRE reporter cells withsiRNAs (each 20 nM) directed against the gene of interest PTGIR,the gene in the jellyfish Aequorea victoria that encodes GFP or auniversal negative control oligonucleotide (AllStars; Qiagen) thatis reported to not recognize any human mRNA, affected neitherbasal nor dexamethasone (1 �M)-induced luciferase expression(Fig. 6). The transfection lipid RNAiMax was also inactive.
Treatment of BEAS-2B reporter cells with taprostene (1 �M) orPGE2 (100 nM) did not promote GRE-dependent transcription butsignificantly augmented (from 13.0 � 0.4- to 24.9 � 2.9- and25.4 � 1.9-fold, respectively) the transcriptional response pro-duced by a maximally effective concentration of dexamethasone (1�M). In cells transfected with siRNAs (PTGIR-1 and PTGIR-2)that target PTGIR, the ability of taprostene to enhance GRE-de-pendent transcription was abolished whereas RNAiMax alone andin the presence of the “control” siRNAs was without effect (Fig. 6).In contrast, the augmentation of dexamethasone-induced reporteractivity by PGE2 was not affected by PTGIR-1, PTGIR-2, the
FIGURE 3. Effect of taprostene on GRE-dependent transcription.BEAS-2B GRE reporter cells were treated respectively with either dexa-methasone (0.1 to 1 �M) in the absence and presence of taprostene (1 �M)(a) or with taprostene (1 nM to 10 �M) in the presence of dexamethasone(1 �M) (b). After 5 h cells were lysed and luciferase activity was deter-mined. The filled bar and dashed line in b indicate the effect on luciferaseexpression of dexamethasone alone. Data points and bars represent themean � SEM of N independent determinations.
FIGURE 4. Kinetics of GRE-dependent transcription in BEAS-2B GREreporter cells. Cells were treated with dexamethasone (Dex; 1 �M), tap-rostene (Tap; 1 �M), a combination of both drugs (each 1 �M), or vehicle(Control). Cells were then incubated for 1–19 h and harvested for luciferaseactivity. Data points represent the mean � SEM of three independent de-terminations. �, p 0.05, significant enhancement of transcription relativeto time-matched, unstimulated cells; �, p 0.05, significant enhancementof transcription relative to time-matched, dexamethasone-treated cells.Data were analyzed by using two-way ANOVA followed by Bonferroni’smultiple comparison test.
FIGURE 5. Schild analysis of the antagonism by RO3244794 of tap-rostene-induced responses in BEAS-2B cells. BEAS-2B GRE cellswere pretreated (30 min) with RO3244794 at the concentrations indi-cated in each panel. Taprostene E/[A] curves were then constructed inthe continued presence of RO3244794 for the enhancement of dexa-methasone (1 �M)-induced GRE-dependent transcription. ModifiedSchild analysis was then performed (30) that yielded a pKb of 9.21. Thefilled bar and dashed line indicate the effect on luciferase expression ofdexamethasone alone. Data points and bars represent the mean � SEMof five independent determinations.
6792 TAPROSTENE AUGMENTS GRE-DEPENDENT TRANSCRIPTION

“control” siRNAs, or RNAiMax (Fig. 6), thereby confirming theselectivity of the knockdown.
Role of the cAMP/PKA pathway in the enhancement bytaprostene of GRE-dependent transcription
The IP-receptor couples typically to Gs for the stimulation of ad-enylyl cyclase, although in some systems it can also promote phos-pholipase C-dependent inositol phosphate production (34, 35). Toestablish the mechanism by which activation of the IP-receptorenhanced GRE-dependent transcription, BEAS-3B cells were ex-posed to taprostene and cAMP mass was determined. As shown inFig. 7a, taprostene increased cAMP in a concentration-dependentmanner with a p[A]50 of 8.01 � 0.01. In subsequent experiments,BEAS-2B GRE reporter cells were infected with an adenovirusvector, Ad5.CMV.PKI�, which directs overexpression of PKI�, ahighly selective inhibitor of PKA (28). In uninfected cells, PKI�was not detected by Western blotting in any experiment. However,48 h after infection of BEAS-2B cells with Ad5.CMV.PKI�(MOI � 40), a single peptide was labeled by the anti-PKI� Ab thatmigrated as a 12-kDa band on SDS-polyacrylamide gels (Fig. 7c).As shown in Fig. 7b, taprostene enhanced dexamethasone (1 �M)-induced GRE-dependent transcription in a concentration-depen-dent manner (p[A]50 � 6.91 � 0.14; Emax � 24.0 � 0.9-fold),which was prevented in cells expressing the PKI� transgene (Fig.7b). In contrast, cells infected with a null virus, Ad5.CMV.Null,responded to taprostene in a manner that was not significantlydifferent from uninfected cells (p[A]50 � 6.86 � 0.08; Emax �26.1 � 1.6-fold; Fig. 8b). Forskolin, a direct activator of adenylylcyclase, also augmented GRE-dependent transcription by 2.78-fold (from 25.0 � 5.8 to 69.5 � 11.3; n � 5), and this effect alsowas abolished in cells expressing the PKI� transgene (data notshown).
Role of PPAR� in the enhancement by taprostene ofGRE-dependent transcription
Agonism of the human IP-receptor subtype is reported to activatethe nuclear hormone receptor PPAR� (36). Accordingly, using
GW9662 as a selective antagonist (29), studies were performedto establish whether PPAR� mediated the augmentation by tap-rostene of dexamethasone-induced GRE-dependent transcrip-tion. GW9662 (1 �M) had no effect on basal or dexamethasone-induced luciferase expression (Fig. 8a). Similarly, GW9662 didnot affect the E/[A] relationship that described the enhancementby taprostene of GRE-dependent transcription (Fig. 8a). Indeed,neither the p[A]50 (6.68 � 0.07) nor Emax (38.2 � 5.5-fold)were altered by GW9662 (p[A]50 � 6.81 � 0.19; Emax �39.3 � 6.3-fold). In contrast, GW9662 (1 �M) abolished theinhibitory effect of rosiglitazone, a PPAR� agonist, on IFN-�-induced CXCL10 output from BEAS-2B cells under similar ex-perimental conditions (Fig. 8b).
Effect of taprostene and forskolin on the expression ofglucocorticoid-inducible anti-inflammatory genes in BEAS-2Bcells
The data presented in the preceding sections indicate that tapro-stene enhanced dexamethasone-induced transcription from a con-ventional simple GRE reporter. To determine whether this findingis applicable to real genes, we took advantage of data derived froma prior microarray analysis in which dexamethasone-induciblegenes were identified in pulmonary type II A549 cells (see Ref. 20
FIGURE 6. Effect of silencing PTGIR by using siRNA on the enhance-ment by taprostene of GRE-dependent transcription. Subconfluent (70%)BEAS-2B GRE reporter cells were transfected (6 h) using RNAiMax withsiRNAs (each 20 nM) directed against PTGIR, GFP, and a universal neg-ative control oligonucleotide. Cells were washed, incubated for 18 h inDMEM/F12 supplemented with 1% (v/v) FBS and then growth arrested for48 h in SFM. The effect of dexamethasone (Dex: 1 �M) alone and in thepresence of taprostene (Tap; 1 �M) and PGE2 (100 nM) on GRE-de-pendent luciferase expression at 5 h was then determined. The dashedline indicates the effect on luciferase expression of dexamethasonealone. Bars represent the mean � SEM of seven independent determi-nations. See Materials and Methods for further details. NS, No stimu-lation representing basal luciferase expression; �, p 0.05, significantinhibition of the enhancement by taprostene of GRE-dependent tran-scription relative to cells transfected with a universal control oligonu-cleotide. Data were analyzed by using one-way ANOVA followed byBonferroni’s multiple comparison test.
FIGURE 7. Effect of taprostene on the cAMP/PKA cascade in BEAS-2Bcells. a, BEAS-2B cells were pretreated (30 min) with the PDE3 and PDE4inhibitors and siguazodan (S; 10 �M) and rolipram (R; 10 �M), re-spectively, to prevent cAMP hydrolysis and then exposed to taprostene(0.1 nM to 10 �M) for 45 min. Cells were lysed and cAMP mass wasmeasured by enzyme immunoassay according to the manufacturer’s in-structions (Cayman Chemical). b, Subconfluent cells were infected withAd5.CMV.PKI�, Ad5.CMV.Null (both at MOI � 40), or left untreatedfor 48 h. Cells were then treated with dexamethasone (Dex; 1 �M)alone (filled bar) or taprostene (1 nM to 10 �M) in the presence ofdexamethasone (1 �M). After 5 h cells were lysed and luciferase ac-tivity was determined. c, Western blot, representative of three experi-ments, of the expression of the PKI� transgene in BEAS-2B cells 48 hafter infection with Ad5.CMV.PKI�. The filled bar and dashed line inb indicate the effect on luciferase expression of dexamethasone alone.Data points and bars represent the mean � SEM of N independentdeterminations. Lane 1, Naive cells; lane 2, Ad5.CMV.Null-infectedcells; lane 3, Ad5.CMV.PKI�-infected cells.
6793The Journal of Immunology
for details). Several of these genes have anti-inflammatory poten-tial, including GILZ, MKP-1, and p57kip2 (22, 37–40), and wereselected to examine the interaction between dexamethasone andtaprostene (or forskolin).
The data derived from the GRE reporter showed substantial pos-itive cooperativity between dexamethasone (1 �M) and taprostene(1 �M) at 1 h, 2 h and 5 h. Accordingly, the expression of GILZ,MKP-1 and p57kip2 were examined over the same time-frame. Rel-ative to unstimulated cells, dexamethasone (1 �M) strongly in-duced GILZ in a time-dependent manner; the mRNA level peakedat 2 h (�75-fold induction) and remained elevated up to 6 h (Fig.9a). In contrast, taprostene (1 �M) was inactive over the same timeframe but augmented the transcriptional activity of dexamethasoneat 1 and 2 h in a manner reminiscent of that produced in GREreporter cells (Fig. 9a). Similarly, forskolin, a direct activator ofadenylyl cyclase, failed to induce GILZ at any time point but, liketaprostene, enhanced the effect of dexamethasone (Fig. 9b). Thus,taprostene and forskolin interacted with dexamethasone in the reg-ulation of GILZ in a positive cooperative fashion.
p57kip2 also was rapidly induced by dexamethasone (1 �M) ina time-dependent manner, although the inducibility and kinetics ofthe response were different from those for GILZ. Thus, mRNAlevels increased almost linearly over the course of the experiment(�12-fold at 6 h) and were significantly lower than the GILZ tran-scripts matched for time (Fig. 9c). Taprostene (1 �M) alone had anegligible effect on the expression of p57kip2 (�1.4-fold induction)but augmented the transcriptional activity of dexamethasone at alltime points. Qualitatively comparable data were obtained whenforskolin was used as a cAMP-elevating stimulus (Fig. 9d), but themagnitude of this positive cooperative interaction was far morepronounced and greatest at the 6-h time point (�43- vs �17-foldfor taprostene; compare GILZ, Fig. 9b).
The third gene examined, MKP-1, was induced by dexametha-sone (1 �M) in a transient manner; mRNA levels peaked at the 2-htime point and then slowly decayed (Fig. 9e). Taprostene (1 �M)
and forskolin (10 �M) also induced MKP-1 (maximally by 2.1-and 4.1-fold, respectively, at 1 h), but this effect also was short-lived and was essentially lost by 2 h (Fig. 9, e and f). When usedin combination, dexamethasone (1 �M) and taprostene (1 �M) ordexamethasone (1 �M) and forskolin (10 �M) at 1 h interacted ina purely additive fashion (Fig. 9, e and f). At later time points (2and 6 h) the expression of MKP-1 induced by both drug combi-nations was not different from the effect produced by dexametha-sone alone (Fig. 9, e and f).
Effect of taprostene and forskolin on the expression ofglucocorticoid-inducible anti-inflammatory genes in HpAECs
Additional studies were performed to determine whether the druginteractions found in BEAS-2B cells also occurred in HpAECs. Tothis end, GILZ and MKP-1 were selected as genes that were reg-ulated by glucocorticoid and a cAMP-elevating agent in a positivecooperative and additive manner, respectively. As shown in Fig.10, a and b, dexamethasone (1 �M) induced GILZ in a time-de-pendent manner. Consistent with the data obtained with BEAS-2Bcells (Fig. 9, a and b), both taprostene (1 �M) and forskolin (10�M) were inactive but, in the presence of dexamethasone (1 �M),
FIGURE 8. Effect of the PPAR� antagonist GW9662 on the enhance-ment by taprostene of GRE-dependent transcription and on the suppressionby rosiglitazone of IFN-�-induced CXCL10 release. a, BEAS-2B GREreporter cells were pretreated with GW9662 (1 �M; 30 min) or vehicle.Dexamethasone (1 �M) was then added alone (bars) or in combinationwith taprostene (1 nM to 10 �M). After 5 h cells were lysed and luciferaseactivity was determined. b, BEAS-2B cells were pretreated with GW9662(1 �M; 30 min) or vehicle and exposed to rosiglitazone (100 nM) for afurther 30 min. IFN-� (100 ng/ml) was then added and the amount ofCXCL10 released into the supernatant at 24 h was measured by ELISA.The bars and dashed line in panel a indicate the effect on the luciferaseexpression of dexamethasone (� GW9662) alone. Data points and barsrepresent the mean � SEM of N independent determinations. �, p 0.05,significant inhibition of CXCL10 release; ��, p 0.05, significant reversalof CXCL10 release by GW9662. Data were analyzed by using one-wayANOVA followed by the Bonferroni multiple comparison test.
FIGURE 9. Effect of taprostene and forskolin on the expression of glu-cocorticoid-inducible genes in BEAS-2B cells. Cells were treated withdexamethasone (Dex; 1 �M), taprostene (Tap; 1 �M), both agents simul-taneously (Dex � Tap), or vehicle (control), and RNA was harvested at 1,2, and 6 h. (a, c, and e) A similar experiment was also performed in whichtaprostene was replaced with forskolin (Forsk; 10 �M) (b, d, and f). AftercDNA synthesis, real-time RT-PCR was performed for GILZ (a and b),p57kip2 (c and d), and MKP-1 (e and f) and normalized to GAPDH. Data areexpressed as fold stimulation at each time point relative to GAPDH and areplotted as means � SEM of six independent observations. The horizontaldashed line in each panel indicates basal gene expression at the 0-h timepoint, which has been normalized to a value of 1. The nature of the inter-action between dexamethasone and the cAMP-elevating agent at one ormore time points is indicated in each panel. �, p 0.05, significant en-hancement of transcription relative to time-matched, unstimulated cells; �,p 0.05, significant enhancement of transcription relative to time-matched, dexamethasone-treated cells. Data were analyzed by using two-way ANOVA followed by Bonferroni’s multiple comparison test.
6794 TAPROSTENE AUGMENTS GRE-DEPENDENT TRANSCRIPTION

augmented (markedly at 6 h) the expression of GILZ. However,several notable differences were apparent. First, GILZ was at leastan order of magnitude less inducible in HpAECs at all time points(compare Fig. 10a with Fig. 9a). Second, although taprostene anddexamethasone interacted cooperatively in HpAECs, this was seenonly at the 6-h time point whereas cooperativity was seen at alltime points when forskolin and dexamethasone were combined(compare Fig. 10, a and b with Fig. 9, a and b). Third, the kineticsof GILZ induction in response to dexamethasone in combinationwith taprostene or forskolin appeared to be slower in HpAECswhen compared with BEAS-2B cells (compare Fig. 10, a and bwith Fig. 9, a and b).
In HpAECs the MKP-1 gene was also induced by dexametha-sone (1 �M) and, additionally, by taprostene (1 �M) and forskolin(10 �M; Fig. 10, c and d). For all three stimuli, MKP-1 mRNAappeared very transiently (more so than in BEAS-2B cells);peak levels were achieved at 1 h and had declined to baseline at2 h. In combination, taprostene and dexamethasone, or forsko-lin and dexamethasone interacted additively on MKP-1 expres-sion with similarly transient kinetics (Fig. 10, c and d). In con-trast to GILZ, the inducibility of MKP-1 in HpAECs was similarto that found in BEAS-2B cells (compare Fig. 10, c and d withFig. 9, e and f).
DiscussionThe results of the present study show that the selective agonismof the IP-receptor on the human bronchial epithelial cell lineBEAS-2B with taprostene enhanced dexamethasone-inducedtranscription of a simple GRE reporter by activating the cAMP/PKA cascade. Similarly, taprostene augmented the expression
of certain glucocorticoid-inducible genes in BEAS-2B cells andHpAECs, including GILZ and MKP-1, which have potential anti-inflammatory activity. These findings support and extend theexisting body of knowledge, by demonstrating that IP-receptoragonists may have utility in treating chronic airway inflamma-tion by boosting the clinical efficacy of glucocorticoids as partof an IP-receptor agonist/ICS combination therapy.
Selection of taprostene as an IP-receptor agonist
A major problem in classifying responses mediated by IP-recep-tors has been a paucity of suitable pharmacological tools. Indeed,PGI2 and many PGI2 analogs and mimetics, including iloprost,carbacyclin, AFP-07, and TEI-9063, are not sufficiently selectivefor their biological actions to be diagnostic of IP-receptor agonism(41, 42). Even cicaprost, which is often the agonist of choice instudies examining IP-receptor pharmacology, must be used withcaution at it does not definitively discriminate IP-, EP4-, and, to alesser extent, EP3-receptor-mediated responses (41, 42). To over-come these problems, we used, in the present study, a syntheticPGI2 analog, taprostene (31). Two sets of data led us to select thisligand. First, taprostene showed high selectivity for the humanrecombinant IP-receptor expressed in HEK-293 cells relative tothe other prostanoid receptors that couple positively to adenylylcyclase. Although absolute selectivity ratios cannot be calculatedfrom these data, taprostene at its �p[A]95 (i.e., 100 pM) was in-active at the DP1-, EP2-, and EP4-subtypes (Fig. 2). Second, theability of taprostene to enhance GRE-dependent transcription inBEAS-2B reporter cells was antagonized in a competitive mannerby RO3244794, an IP-receptor-blocking drug, with an affinity(pKb � 9.21) consistent with an interaction at IP-receptors (27,43). Collectively, these results strongly suggest that in BEAS-2Bcells, taprostene augments GRE-dependent transcription by an IP-receptor-mediated mechanism. This conclusion was confirmed ingene silencing studies where, under stringently controlled condi-tions, two siRNAs that target PTGIR abolished the ability of tap-rostene to enhance GRE-dependent transcription, whereas thesame response evoked by PGE2 was preserved.
Interaction of taprostene and dexamethasone
Using a simple GRE-reporter construct stably transfected intoBEAS-2B cells, taprostene augmented the ability of dexameth-asone to promote the induction of the luciferase gene in a time-and concentration-dependent manner. Inspection of the E/[A]curves showed that taprostene increased the ability of dexa-methasone to “drive” the luciferase gene (i.e., Emax was in-creased) without significantly affecting its potency (i.e., p[A]50
was unchanged). Of potential clinical significance is that thisinteraction is one of positive cooperativity, because taprostenealone did not promote GRE-dependent transcription (see be-low). Moreover, taprostene was “steroid-sparing” in this modelsystem. For example, dexamethasone, at its p[A]95, produced a17.4-fold induction of the luciferase gene. However, in the pres-ence of taprostene (1 �M), which was inactive by itself, thesame degree of gene induction was achieved at a concentrationof dexamethasone that was significantly lower (7.3-fold if thep[A]95 of the glucocorticoid response alone is taken as refer-ence) (see Fig. 11).
Prior microarray analysis of genes induced by dexamethasone inA549 cells identified several candidates for subsequent analysisthat are predicted to exert anti-inflammatory activity. The threegenes studied here were found to be regulated in an additive orpositive cooperative manner by glucocorticoid and taprostene incombination. Thus, GILZ and p57kip2 were induced in a positivecooperative fashion in BEAS-2B, and a similar effect was seen
FIGURE 10. Effect of taprostene (Tap) and forskolin (Forsk) on theexpression of glucocorticoid-inducible genes in HpAECs. Cells weretreated and cDNA prepared as described in the legend to Fig. 9. Real-timeRT-PCR was performed for GILZ (a and b) and MKP-1 (c and d) andnormalized to GAPDH. Data are expressed as fold stimulation at each timepoint relative to GAPDH and are plotted as means � SEM of three inde-pendent observations. The horizontal dashed line in each panel indicatesbasal gene expression at the 0-h time point, which has been normalized toa value of 1. The nature of the interaction between dexamethasone (Dex)and a cAMP-elevating agent at one or more time points is indicated in eachpanel. �, p 0.05, significant enhancement of transcription relative totime-matched, unstimulated cells; �, p 0.05, significant enhancement oftranscription relative to time-matched, dexamethasone-treated cells. Datawere analyzed by using two-way ANOVA followed by Bonferroni’s mul-tiple comparison test.
6795The Journal of Immunology

with GILZ in HpAECs. Although there were differences in themagnitude and kinetics of gene induction between BEAS-2B cellsand HpAECs, both cell types responded at certain time points in amanner that resembled activation of the GRE reporter. Indeed, afunctional conventional simple GRE is present in the p57kip2 andGILZ promoters (44, 45), which implies that taprostene and dexa-methasone in combination can induce in a positive cooperativemanner real anti-inflammatory genes. GILZ has clear anti-inflammatory potential through its ability to attenuate the activa-tion of NF-�B and/or AP-1, which are critical transcription factorsthat control the expression of many proinflammatory genes (37,38). Similarly, p57kip2 encodes a cell cycle kinase inhibitor thatregulates the anti-proliferative effects of glucocorticoids (39).Given that airway remodeling in COPD could be explained, inpart, by the increase in airway wall volume occupied by bronchialepithelial cells as well as smooth muscle (46), it is possible that anIP-receptor agonist/ICS combination therapy could be diseasemodifying through its ability to promote the expression of p57kip2.The p57kip2 gene product also blocks JNK (47) suggesting that itmay suppress proinflammatory events known to be regulated bythis MAPK.
The third gene examined was MKP-1. This gene encodes anenzyme that dephosphorylates (inactivates) the three core mam-malian MAPKs (p38 MAPK, ERK, and JNK) that are central tothe induction of many proinflammatory genes (e.g., growth-relatedoncogene-�) (Ref. 40). As shown in Figs. 9 and 10, the regulationof MKP-1 was distinct from that of p57kip2 and GILZ. Thus,MKP-1 was induced by both taprostene and dexamethasone, and incombination the interaction of these stimuli was transient andpurely additive. These data indicate that that unlike p57kip2 andGILZ, the glucocorticoid-dependent regulation of MKP-1 is notprimarily mediated via simple GREs, which is consistent with thefinding that the promoter of this and many other glucocorticoid-inducible genes do not feature a conventional GRE consensus se-quence (i.e., sense strand, 5�-TGTACAGGATGTTCT-3�) (48).
It is noteworthy that the transcription of glucocorticoid-induc-ible genes that mediate adverse effects may also be augmented by
an IP-receptor agonist, thereby limiting the clinical efficacy of sucha combination therapy. For example, the expression of metabolicgenes such as phosphoenol pyruvate carboxykinase and glucose-6-phosphastase, which regulate gluconeogenesis, are induced bycAMP-elevating stimuli and glucocorticoids and, in combination,act at least additively (49–51). Clearly, therefore, the pharmaco-kinetics of a glucocorticoid will dictate the propensity to whichsuch adverse effect genes may be induced. In respiratory diseases,a glucocorticoid should be delivered directly to the lung and havelow oral absorption, high plasma protein binding to limit systemicexposure, and high first-pass hepatic metabolism. Under these con-ditions, the ability of an IP-receptor agonist to enhance the tran-scriptional activity of that glucocorticoid should be retained pre-dominantly in the lung, allowing the superior clinical efficacy ofthe combination therapy to be achieved.
Mechanism of action
It has been reported that PGI2 analogs, including carbacyclin andtreprostinil, are PPAR� agonists (36). However, no evidence forsuch a mechanism was obtained in the present study based on theinability of GW9662, a PPAR� antagonist (29), to inhibit tapro-stene-induced transcriptional responses. Indeed, our data suggestthat the mechanistic basis for the enhancement by taprostene ofGRE-reporter activity involves the activation of the classicalcAMP/PKA pathway. Thus, this effect was mimicked by forskolinand abolished in cells infected with Ad5.CMV.PKI�. Similarly,the ability of forskolin to interact either additively or cooperativelywith dexamethasone on the expression of GILZ, p57kip2, andMKP-1 in BEAS-2B cells and HpAECs confirms that cAMP can,in some way, augment the transcription of real glucocorticoid-inducible genes.
We have reported previously that salmeterol, a �2-adreno-ceptor agonist, and forskolin also augment GRE-dependenttranscription in BEAS-2B cells, but only a certain, undefinedpopulation of glucocorticoid-inducible genes are affected (20).Indeed, genes that encode tristetraprolin, aminopeptidase N,and plasminogen activator inhibitor-1 are not regulated in anadditive or positive cooperative manner by either of thesecAMP-elevating agents (20). Current data indicate that �2-ad-renoceptor agonists and forskolin display an identical, qualita-tive profile of activity on a panel of glucocorticoid-induciblegenes (20). Whether taprostene shares this defined activity ordisplays a distinct or overlapping profile is unknown. Althoughthe present study was undertaken to establish the concept thatan IP-receptor agonist can augment GRE-dependent trans-acti-vation, this is nevertheless an important objective for futureresearch. Indeed, it is conceivable that different cAMP-elevat-ing drugs may enhance the transcription of distinct populationsof glucocorticoid-inducible genes. Clearly, this raises the pos-sibility of selectively regulating the expression of certain genes,which would have clear therapeutic potential.
The identity of the targets downstream of PKA that moredirectly up-regulate GRE-dependent transcription is vague. Ithas been proposed that cAMP-elevating agents enhance thetranslocation of the GR from the cytosol to the nucleus and thatthis boosts transcription (52–54). Indeed, the cAMP-elevatingdrug formoterol augments the ability of budesonide to promotethe binding of the GR to GREs on target genes above the levelproduced by the glucocorticoid alone (55). Such an effect wouldbe consistent with the enhanced GR:DNA binding seen in cellsoverexpressing PKA (56). Another possibility is that cAMP-elevating agents increase the expression of functional GR (57)and/or stabilize the interaction of glucocorticoid-bound GRwith DNA through activation of PKA (58). Although these are
FIGURE 11. Interaction between taprostene and dexamethasone onGRE-dependent transcription. The graph in Fig. 3a has been redrawn toillustrate that taprostene augments dexamethasone-induced GRE reporteractivity in a steroid-sparing manner. BEAS-2B cells stably expressing aGRE-reporter were treated with taprostene (1 �M) in the absence otr pres-ence of dexamethasone (100 pM to 1 �M). After 6 h, cells were harvestedfor luciferase assay. The data show that the effect of taprostene and dexa-methasone in combination promotes GRE-dependent transcription abovethe maximum effect achieved by the glucocorticoid alone (1.93-fold at theEmax). Taprostene was also glucocorticoid sparing in this model. Thus,dexamethasone at a concentration that evoked 95% of the maximum re-sponse produced a 19.3-fold induction of the luciferase gene. However, inthe presence of taprostene (1 �M), which was inactive by itself, the samedegree of gene induction was achieved at a concentration of glucocorticoidthat was significantly (7.3-fold) lower.
6796 TAPROSTENE AUGMENTS GRE-DEPENDENT TRANSCRIPTION

plausible ideas, both mechanisms necessarily imply that thetranscription of all glucocorticoid-inducible genes would be en-hanced by a cAMP-elevating drug, which is not the case (20).To account for this, we propose that taprostene and othercAMP-elevating agents may act predominantly within the nu-cleus by regulating the activity and/or recruitment of specificcofactors that affect only the transcription of a specific subset ofglucocorticoid-inducible genes (59 – 61). Although this pro-posal remains to be investigated, it would confer promoter spec-ificity and explain why the expression of some glucocorticoid-inducible genes is augmented in a positive cooperative fashionby cAMP-elevating agents whereas others are not.
Targeting the IP-receptor in COPD
COPD is a multifaceted disorder. Typically, neutrophilic inflam-mation of the small airways and lungs is a defining pathologicalfeature that is associated with chronic airflow limitation (60).Many individuals with COPD also have systemic inflammation(62) that is positively related to disease severity (63). Pulmonaryvascular remodeling leading to impaired gas exchange and/or pul-monary hypertension is also frequently seen. In many cases, suchabnormalities occur in conjunction with right-side heart failure(64, 65) and platelet hyper-reactivity, rendering afflicted individ-uals at increased risk of developing thromboses (66–68). Unlikeasthma, stable COPD is an example of a chronic inflammatorydisease that is relatively insensitive to ICSs (18). Conceivably, an“add-on” therapy that is known to effectively target the cardiovas-cular pathologies and concurrently augment the anti-inflammatoryeffect of an ICS could provide an effective and potentially disease-modifying treatment. We submit that a selective IP-receptor ago-nist could fulfill this role for several tangible reasons. In particular,the expression of PGI2 synthase and the level of 6-keto-PGF1�, theprimary metabolite of PGI2, are reduced in whole lung lysatesprepared from subjects with emphysema (69). Conversely, theurinary excretion of 11-dehydro-thromboxane (TX) B2, the ma-jor enzymatic metabolite of TXA2 in subjects with COPD, issignificantly enhanced relative to that in healthy individualsmatched for age and gender (70). Thus, there appears to be aPGI2 deficiency in emphysematous COPD and a reciprocal in-crease in TXA2 generation with predicted undesirable effects onplatelet reactivity and pulmonary vascular smooth muscle func-tion. Clearly, a selective IP-receptor agonist could restore thisPGI2/TXA2 imbalance and, thereby, reduce the threshold forplatelet aggregation (71), lower pulmonary vascular resistance(72) (through smooth muscle relaxation and, long term, by in-hibiting mitogenesis), and even promote bronchodilatation (73).The results of this study indicate that an IP-receptor agonistcould also suppress airway and pulmonary vascular inflamma-tion by enhancing the clinical efficacy of an ICS. It is notewor-thy that IP-receptor agonists are effective in the treatment ofpulmonary arterial hypertension (74). However, there have beenfew controlled trials in pulmonary hypertension associated withCOPD (65), and the suitability of these drugs for treating car-diovascular defects in this pathological setting would need to betested empirically in well-designed clinical trials.
Recently, there has been a considerable effort in medicinalchemistry to develop new PGI2 mimetics for pulmonary arterialhypertension that are both stable and selective for the IP sub-type. For example, chemists at Nippon Shinyaku have discov-ered a number of nonprostanoid pro-drugs that are metabolizedin vivo into potent and selective IP-receptor agonists (75). Oneof these, NS-304, is an N-acylsulphonamide that is slowly hy-drolyzed by the liver to produce the corresponding carboxylicacid MRE-269, which has a t1/2 in blood of 7.9 h (75). In the
event that IP-receptor agonists are shown to be efficacious intreating the cardiovascular abnormalities in COPD, a drug ex-emplified by NS-304 could be suitable for twice a day dosingwith an existing ICS.
Conclusions
In the present study we report that a selective IP-receptor ag-onist, taprostene, enhances the ability of a glucocorticoid totranscribe genes, in airway epithelial cells, with potential anti-inflammatory activity. This novel mechanism of action has clearclinical relevance in diseases, such as COPD, that are generallyrefractory to glucocorticoids as a monotherapy. Moreover, thesedata complement a growing body of literature in which PGI2
analogs are reported to have both anti-inflammatory and anti-viral activity in preclinical models of airway inflammation (seeIntroduction). We submit that because activation of the IP-re-ceptor also elicits beneficial effects on platelet reactivity, pul-monary vascular smooth muscle tone. and, indirectly, cardiacfunction, ICS/IP-receptor agonist combination therapy couldprovide an effective treatment option in COPD that may beparticularly effective in subjects with hematological and car-diovascular dysfunction.
DisclosuresThe authors have no financial conflict of interest.
References1. Vane, J. R., and R. M. Botting. 1995. Pharmacodynamic profile of prostacyclin.
Am. J. Cardiol. 75: 3A–10A.2. Hata, A. N., and R. M. Breyer. 2004. Pharmacology and signaling of prostaglan-
din receptors: multiple roles in inflammation and immune modulation. Pharma-col. Ther. 103: 147–166.
3. Meja, K. K., P. J. Barnes, and M. A. Giembycz. 1997. Characterization of theprostanoid receptor(s) on human blood monocytes at which prostaglandin E2
inhibits lipopolysaccharide-induced tumour necrosis factor-� generation.Br. J. Pharmacol. 122: 149–157.
4. Jaffar, Z., K. S. Wan, and K. Roberts. 2002. A key role for prostaglandin I2 inlimiting lung mucosal Th2, but not Th1, responses to inhaled allergen. J. Immu-nol. 169: 5997–6004.
5. Zhou, W., T. S. Blackwell, K. Goleniewska, J. F. O’Neal, G. A. FitzGerald,M. Lucitt, R. M. Breyer, and R. S. Peebles, Jr. 2006. Prostaglandin I2 analogsinhibit Th1 and Th2 effector cytokine production by CD4 T cells. J. LeukocyteBiol. 81: 809–887.
6. Zhou, W., K. Hashimoto, K. Goleniewska, J. F. O’Neal, S. Ji, T. S. Blackwell,G. A. FitzGerald, K. M. Egan, M. W. Geraci, and R. S. Peebles. 2007. Prosta-glandin I2 analogs inhibit proinflammatory cytokine production and T cell stim-ulatory function of dendritic cells. J. Immunol. 178: 702–710.
7. Ayer, L. M., S. M. Wilson, S. L. Traves, D. Proud, and M. A. Giembycz.2008. (4,5-Dihydro-1H-imidazol-2-yl)-[4-(4-isopropoxy-benzyl)-phenyl]-amine (RO1138452) is a selective, pseudo-irreversible orthosteric antagonistat the prostacyclin (IP)-receptor expressed by human airway epithelial cells:negative IP-receptor-mediated regulation of CXCL9 and CXCL10 release.J. Pharmacol. Exp. Ther. 324: 815– 826.
8. Clarke, D. L., M. G. Belvisi, S. J. Smith, E. Hardaker, M. H. Yacoub, K. K. Meja,R. Newton, D. M. Slater, and M. A. Giembycz. 2005. Prostanoid receptor ex-pression by human airway smooth muscle cells and regulation of the secretion ofgranulocyte colony-stimulating factor. Am. J. Physiol. 288: L238–L250.
9. Walch, L., C. Labat, J. P. Gascard, M. de, V., C. Brink, and X. Norel. 1999.Prostanoid receptors involved in the relaxation of human pulmonary vessels.Br. J. Pharmacol. 126: 859–866.
10. Jozefowski, S., M. Bobek, and J. Marcinkiewicz. 2003. Exogenous but not en-dogenous prostanoids regulate cytokine secretion from murine bone marrow den-dritic cells: EP2, DP, and IP but not EP1, EP3, and FP prostanoid receptors areinvolved. Int. Immunopharmacol. 3: 865–878.
11. Takahashi, Y., S. Tokuoka, T. Masuda, Y. Hirano, M. Nagao, H. Tanaka,N. Inagaki, S. Narumiya, and H. Nagai. 2002. Augmentation of allergic inflam-mation in prostanoid IP-receptor deficient mice. Br. J. Pharmacol. 137: 315–322.
12. Nagao, K., H. Tanaka, M. Komai, T. Masuda, S. Narumiya, and H. Nagai. 2003.Role of prostaglandin I2 in airway remodeling induced by repeated allergen chal-lenge in mice. Am. J. Respir. Cell Mol. Biol. 29: 314–320.
13. Jaffar, Z., M. E. Ferrini, M. C. Buford, G. A. FitzGerald, and K. Roberts. 2007.Prostaglandin I2-IP signaling blocks allergic pulmonary inflammation by pre-venting recruitment of CD4� Th2 cells into the airways in a mouse model ofasthma. J. Immunol. 179: 6193–6203.
14. Idzko, M., H. Hammad, M. van Nimwegen, M. Kool, N. Vos, H. C. Hoogsteden,and B. N. Lambrecht. 2007. Inhaled iloprost suppresses the cardinal features ofasthma via inhibition of airway dendritic cell function. J. Clin. Invest. 117:464–472.
6797The Journal of Immunology

15. Chen, Y., M. Hanaoka, P. Chen, Y. Droma, N. F. Voelkel, and K. Kubo. 2009.Protective effect of beraprost sodium, a stable prostacyclin analog, in the devel-opment of cigarette smoke extract-induced emphysema. Am. J. Physiol. 296:L648–L656.
16. Hashimoto, K., B. S. Graham, M. W. Geraci, G. A. FitzGerald, K. Egan,W. Zhou, K. Goleniewska, J. F. O’Neal, J. D. Morrow, R. K. Durbin, et al. 2004.Signaling through the prostaglandin I2 receptor IP protects against respiratorysyncytial virus-induced illness. J. Virol. 78: 10303–10309.
17. Traves, S. L., and D. Proud. 2007. Viral-associated exacerbations of asthma andCOPD. Curr. Opin. Pharmacol. 7: 252–258.
18. Suissa, S., R. McGhan, D. Niewoehner, and B. Make. 2007. Inhaled corticoste-roids in chronic obstructive pulmonary disease. Proc. Am. Thor. Soc. 4: 535–542.
19. De Bosscher, K., W. Vanden Berghe, and G. Haegeman. 2003. The interplaybetween the glucocorticoid receptor and nuclear factor-�B or activator protein-1:molecular mechanisms for gene repression. Endocr. Rev. 24: 488–522.
20. Kaur, M., J. E. Chivers, M. A. Giembycz, and R. Newton. 2008. Long-acting�2-adrenoceptor agonists synergistically enhance glucocorticoid-dependent tran-scription in human airway epithelial and smooth muscle cells. Mol. Pharmacol.73: 201–214.
21. Newton, R., and N. S. Holden. 2007. Separating transrepression and transacti-vation: A distressing divorce for the glucocorticoid receptor? Mol. Pharmacol.72: 799–809.
22. Clark, A. R., J. R. S. Martins, and C. R. Tchen. 2008. Role of dual specificityphosphatases in biological responses to glucocorticoids. J. Biol. Chem. 283:25765–25769.
23. Clark, A. R. 2007. Anti-inflammatory functions of glucocorticoid-induced genes.Mol. Cell. Endocrinol. 275: 79–97.
24. Nordeen, S. K., M. L. Moyer, and B. J. Bona. 1994. The coupling of multiplesignal transduction pathways with steroid response mechanisms. Endocrinology134: 1723–1732.
25. Churchill, L., F. H. Chilton, J. H. Resau, R. Bascom, W. C. Hubbard, andD. Proud. 1989. Cyclooxygenase metabolism of endogenous arachidonic acid bycultured human tracheal epithelial cells. Am. Rev. Respir. Dis. 140: 449–459.
26. Chivers, J. E., L. M. Cambridge, M. C. Catley, J. C. Mak, L. E. Donnelly,P. J. Barnes, and R. Newton. 2004. Differential effects of RU486 reveal distinctmechanisms for glucocorticoid repression of prostaglandin E release. Eur. J. Bio-chem. 271: 4042–4052.
27. Bley, K. R., A. Bhattacharya, D. V. Daniels, J. Gever, A. Jahangir, C. O’Yang,S. Smith, D. Srinivasan, A. P. Ford, and M. F. Jett. 2006. RO1138452 andRO3244794: characterization of structurally distinct, potent and selective IP(prostacyclin) receptor antagonists. Br. J. Pharmacol. 147: 335–345.
28. Meja, K., M. C. Catley, L. M. Cambridge, P. J. Barnes, H. Lum, R. Newton, andM. A. Giembycz. 2004. Adenovirus-mediated delivery and expression of acAMP-dependent protein kinase inhibitor gene to BEAS-2B epithelial cells abol-ishes the anti-inflammatory effects of rolipram, salbutamol and prostaglandin E2:a comparison with H-89. J. Pharmacol. Exp. Ther. 309: 833–844.
29. Leesnitzer, L. M., D. J. Parks, R. K. Bledsoe, J. E. Cobb, J. L. Collins,T. G. Consler, R. G. Davis, E. A. Hull-Ryde, J. M. Lenhard, L. Patel, et al. 2002.Functional consequences of cysteine modification in the ligand binding sites ofperoxisome proliferator activated receptors by GW9662. Biochemistry 41:6640–6650.
30. Waud, D. R., S. L. Son, and B. E. Waud. 1978. Kinetic and empirical analysis ofdose-response curves illustrated with a cardiac example. Life Sci. 22: 1275–1285.
31. Schneider, J., E. Friderichs, B. Kogel, U. Seipp, H.-J. Stahlberg, R. Terlinden,and K. Heintze. 1993. Taprostene sodium. Cardiovasc. Drug Rev. 11: 479–500.
32. Aksoy, M. O., X.-x. Li, M. Borenstein, Y. Yi, and S. G. Kelsen. 1999. Effects oftopical corticosteroids on inflammatory mediator-induced eicosanoid release byhuman airway epithelial cells. J. Allergy. Clin. Immunol. 103: 1081–1091.
33. Neubig, R. R., M. Spedding, T. Kenakin, and A. Christopoulos. 2003. Interna-tional Union of Pharmacology Committee on Receptor Nomenclature and DrugClassification. XXXVIII. Update on terms and symbols in quantitative pharma-cology. Pharmacol. Rev. 55: 597–606.
34. Wise, H., and R. L. Jones. 1996. Focus on prostacyclin and its novel mimetics.Trends Pharmacol. Sci. 17: 17–21.
35. Chow, K. B., R. L. Jones, and H. Wise. 2004. Agonists can discriminate betweencloned human and mouse prostacyclin receptors. Prostaglandins Leukot. Essent.Fatty Acids 70: 423–429.
36. Falcetti, E., D. M. Flavell, B. Staels, A. Tinker, S. G. Haworth, and L. H. Clapp.2007. IP-receptor-dependent activation of PPAR� by stable prostacyclin ana-logues. Biochem. Biophys. Res. Commun. 360: 821–827.
37. Mittelstadt, P. R., and J. D. Ashwell. 2001. Inhibition of AP-1 by the glucocor-ticoid-inducible protein GILZ. J. Biol. Chem. 276: 29603–29610.
38. Eddleston, J., J. Herschbach, A. L. Wagelie-Steffen, S. C. Christiansen, andB. L. Zuraw. 2007. The anti-inflammatory effect of glucocorticoids is mediatedby glucocorticoid-induced leucine zipper in epithelial cells. J. Allergy Clin. Im-munol. 119: 115–122.
39. Samuelsson, M. K., A. Pazirandeh, B. Davani, and S. Okret. 1999. p57Kip2, aglucocorticoid-induced inhibitor of cell cycle progression in HeLa cells. Mol.Endocrinol. 13: 1811–1822.
40. Issa, R., S. Xie, N. Khorasani, M. Sukkar, I. M. Adcock, K. Y. Lee, andK. F. Chung. 2007. Corticosteroid inhibition of growth-related oncogene pro-tein-� via mitogen-activated kinase phosphatase-1 in airway smooth muscle cells.J. Immunol. 178: 7366–7375.
41. Abramovitz, M., M. Adam, Y. Boie, M. Carriere, D. Denis, C. Godbout,S. Lamontagne, C. Rochette, N. Sawyer, N. M. Tremblay, et al. 2000. The use ofrecombinant prostanoid receptors to determine the affinities and selectivities ofprostaglandins and related analogs. Biochim. Biophys. Acta. 1483: 285–293.
42. Wise, H., and R. L. Jones. 2000. Prostacyclin and Its Receptors. Kluwer Aca-demic Publishers, Hingham, MA.
43. Jones, R. L., M. A. Giembycz, and D. F. Woodward. 2009. Prostanoid receptorantagonists: development and therapeutic applications. Br. J. Pharmacol. 158:104–145.
44. Alheim, K., J. Corness, M. K. Samuelsson, L. G. Bladh, T. Murata, T. Nilsson,and S. Okret. 2003. Identification of a functional glucocorticoid response elementin the promoter of the cyclin-dependent kinase inhibitor p57Kip2. J. Mol. Endo-crinol. 30: 359–368.
45. van der Laan, S., R. A. Sarabdjitsingh, M. F. Van Batenburg, S. B. Lachize, H. Li,T. F. Dijkmans, E. Vreugdenhil, E. R. de Kloet, and O. C. Meijer. 2008. Chro-matin immunoprecipitation scanning identifies glucocorticoid receptor bindingregions in the proximal promoter of a ubiquitously expressed glucocorticoid tar-get gene in brain. J. Neurochem. 106: 2515–2523.
46. Hogg, J. C., F. Chu, S. Utokaparch, R. Woods, W. M. Elliott, L. Buzatu,R. M. Cherniack, R. M. Rogers, F. C. Sciurba, H. O. Coxson, and P. D. Pare.2004. The nature of small-airway obstruction in chronic obstructive pulmonarydisease. N. Engl. J. Med. 350: 2645–2653.
47. Chang, T. S., M. J. Kim, K. Ryoo, J. Park, S. J. Eom, J. Shim, K. I. Nakayama,K. Nakayama, M. Tomita, K. Takahashi, et al. 2003. p57KIP2 modulates stress-activated signaling by inhibiting c-Jun NH2-terminal kinase/stress-activated pro-tein Kinase. J. Biol. Chem. 278: 48092–48098.
48. So, A. Y., C. Chaivorapol, E. C. Bolton, H. Li, and K. R. Yamamoto. 2007.Determinants of cell- and gene-specific transcriptional regulation by the glu-cocorticoid receptor. PLoS. Genet. 3: e94.
49. Yeagley, D., and P. G. Quinn. 2005. 3�,5�-Cyclic adenosine monophosphate re-sponse element-binding protein and CCAAT enhancer-binding protein are dis-pensable for insulin inhibition of phosphoenolpyruvate carboxykinase transcrip-tion and for its synergistic induction by protein kinase A and glucocorticoids.Mol. Endocrinol. 19: 913–924.
50. Schmoll, D., C. Wasner, C. J. Hinds, B. B. Allan, R. Walther, and A. Burchell.1999. Identification of a cAMP response element within the glucose-6-phospha-tase hydrolytic subunit gene promoter which is involved in the transcriptionalregulation by cAMP and glucocorticoids in H4IIE hepatoma cells. Biochem. J.338: 457–463.
51. van Schaftingen, E., and I. Gerin. 2002. The glucose-6-phosphatase system. Bio-chem. J. 362: 513–532.
52. Eickelberg, O., M. Roth, R. Lorx, V. Bruce, J. Rudiger, M. Johnson, andL. H. Block. 1999. Ligand-independent activation of the glucocorticoid receptorby �2-adrenergic receptor agonists in primary human lung fibroblasts and vas-cular smooth muscle cells. J. Biol. Chem. 274: 1005–1010.
53. Usmani, O. S., K. Ito, K. Maneechotesuwan, M. Ito, M. Johnson, P. J. Barnes,and I. M. Adcock. 2005. Glucocorticoid receptor nuclear translocation in airwaycells after inhaled combination therapy. Am. J. Respir. Crit Care Med. 172:704–712.
54. Profita, M., R. Gagliardo, R. Di Giorgi, F. Pompeo, M. Gjomarkaj, G. Nicolini,J. Bousquet, and A. M. Vignola. 2005. Biochemical interaction between effects ofbeclomethasone dipropionate and salbutamol or formoterol in sputum cells frommild to moderate asthmatics. Allergy 60: 323–329.
55. Roth, M., P. R. Johnson, J. J. Rudiger, G. G. King, Q. Ge, J. K. Burgess,G. Anderson, M. Tamm, and J. L. Black. 2002. Interaction between glucocorti-coids and �2 agonists on bronchial airway smooth muscle cells through synchro-nised cellular signalling. Lancet 360: 1293–1299.
56. Rangarajan, P. N., K. Umesono, and R. M. Evans. 1992. Modulation of glu-cocorticoid receptor function by protein kinase A. Mol. Endocrinol. 6:1451–1457.
57. Oikarinen, J., L. Hamalainen, and A. Oikarinen. 1984. Modulation of glucocor-ticoid receptor activity by cyclic nucleotides and its implications on the regulationof human skin fibroblast growth and protein synthesis. Biochim. Biophys. Acta799: 158–165.
58. Espinas, M. L., J. Roux, R. Pictet, and T. Grange. 1995. Glucocorticoids andprotein kinase A coordinately modulate transcription factor recruitment at a glu-cocorticoid-responsive unit. Mol. Cell. Biol. 15: 5346–5354.
59. Rowan, B. G., N. Garrison, N. L. Weigel, and B. W. O’Malley. 2000. 8-Bromo-cyclic AMP induces phosphorylation of two sites in SRC-1 that facilitate ligand-independent activation of the chicken progesterone receptor and are critical forfunctional cooperation between SRC-1 and CREB binding protein. Mol. Cel.lBiol. 20: 8720–8730.
60. Fenne, I. S., T. Hoang, M. Hauglid, J. V. Sagen, E. A. Lien, and G. Mellgren.2008. Recruitment of coactivator glucocorticoid receptor interacting protein 1 toan estrogen receptor transcription complex is regulated by the 3�,5�-cyclic aden-osine 5�-monophosphate-dependent protein kinase. Endocrinology 149:4336–4345.
61. Hoang, T., I. S. Fenne, C. Cook, B. Borud, M. Bakke, E. A. Lien, andG. Mellgren. 2004. cAMP-dependent protein kinase regulates ubiquitin-protea-some-mediated degradation and subcellular localization of the nuclear receptorcoactivator GRIP1. J. Biol. Chem. 279: 49120–49130.
62. Barnes, P. J. 2008. Immunology of asthma and chronic obstructive pulmonarydisease. Nat. Rev. Immunol. 8: 183–192.
63. Sin, D. D., and S. F. Man. 2007. Do chronic inhaled steroids alone or in com-bination with a bronchodilator prolong life in chronic obstructive pulmonarydisease patients? Curr. Opin. Pulm. Med. 13: 90–97.
64. Peinado, V. I., S. Pizarro, and J. A. Barbera. 2008. Pulmonary vascular involve-ment in COPD. Chest 134: 808–814.
65. Chaouat, A., R. Naeije, and E. Weitzenblum. 2008. Pulmonary hypertension inCOPD. Eur. Respir. J. 32: 1371–1385.
6798 TAPROSTENE AUGMENTS GRE-DEPENDENT TRANSCRIPTION

66. Mitchell, R. S., G. W. Silvers, G. A. Dart, T. L. Petty, T. N. Vincent, S. F. Ryan,and G. F. Filley. 1968. Clinical and morphologic correlations in chronic airwayobstruction. Aspen. Emphysema. Conf. 9: 109–123.
67. Ferroni, P., S. Basili, F. Martini, M. Vieri, G. Labbadia, C. Cordova,C. Alessandri, and P. P. Gazzaniga. 2000. Soluble P-selectin as a marker ofplatelet hyperactivity in patients with chronic obstructive pulmonary disease.J. Investig. Med. 48: 21–27.
68. Cordova, C., A. Musca, F. Violi, C. Alessandri, A. Perrone, and F. Balsano. 1985.Platelet hyperfunction in patients with chronic airways obstruction. Eur.J. Respir. Dis. 66: 9–12.
69. Nana-Sinkam, S. P., J. D. Lee, S. Sotto-Santiago, R. S. Stearman, R. L. Keith,Q. Choudhury, C. Cool, J. Parr, M. D. Moore, T. M. Bull, et al. 2007. Prosta-cyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke.Am. J. Respir. Crit Care Med. 175: 676–685.
70. Davi, G., S. Basili, M. Vieri, F. Cipollone, S. Santarone, C. Alessandri,P. Gazzaniga, C. Cordova, and F. Violi. 1997. Enhanced thromboxane biosyn-thesis in patients with chronic obstructive pulmonary disease. The Chronic Ob-
structive Bronchitis and Haemostasis Study Group. Am. J. Respir. Crit Care Med.156: 1794–1799.
71. Beghetti, M., G. Reber, M. P. de, L. Vadas, A. Chiappe, I. Spahr-Schopfer, andP. C. Rimensberger. 2002. Aerosolized iloprost induces a mild but sustainedinhibition of platelet aggregation. Eur. Respir. J. 19: 518–524.
72. McLaughlin, V. V., A. Shillington, and S. Rich. 2002. Survival in primary pul-monary hypertension: the impact of epoprostenol therapy. Circulation 106:1477–1482.
73. Norel, X., L. Walch, C. Labat, J. P. Gascard, E. Dulmet, and C. Brink. 1999.Prostanoid receptors involved in the relaxation of human bronchial preparations.Br. J. Pharmacol. 126: 867–872.
74. Oudiz, R. J., and H. W. Farber. 2009. Dosing considerations in the use of intra-venous prostanoids in pulmonary arterial hypertension: an experience-based re-view. Am. Heart J. 157: 625–635.
75. Kuwano, K., A. Hashino, T. Asaki, T. Hamamoto, T. Yamada, K. Okubo, andK. Kuwabara. 2007. 2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl)acetamide (NS-304), an orally available and long-acting pros-tacyclin receptor agonist prodrug. J. Pharmacol. Exp. Ther. 322: 1181–1188.
6799The Journal of Immunology





























