Embed Size (px)
Citation preview

Shear Stress Regulates Angiotensin Type 1 ReceptorExpression in Endothelial Cells
Bhama Ramkhelawon, Jose Vilar, Daniel Rivas, Barend Mees, Rini de Crom,Alain Tedgui, Stephanie Lehoux
Rationale: Shear stress (SS) has an established role in atherosclerotic plaque localization, but how it exerts itsprotective effect is not fully understood.
Objective: To test the hypothesis that SS may downregulate angiotensin type 1 receptors (AT1Rs). Angiotensin IIhas been shown to be proinflammatory and to promote atherosclerosis.
Methods and Results: Using immunohistochemistry, we found a pronounced expression of AT1R in the inner,atheroprone regions of the aortic arch of C57BL/6 and endothelial NO synthase–deficient (eNOS�/�) mice butnot eNOS-overexpressing mice. In human umbilical vein endothelial cells (HUVECs), laminar SS (15 dyn/cm2)induced a biphasic decrease in AT1R protein expression characterized by a first reduction at 1 hour (31�4% ofstatic control, P<0.01), partial recovery at 3 hours (65�9%), and a second more prolonged decline at 6, 12, and24 hours (48�9%, 36�9%, 33�5%, respectively, P<0.05). One and 24 hours of SS significantly reducedfluorescent angiotensin binding compared to static HUVECs. Shear-induced downregulation of AT1R wasabolished by treatment with protein kinase A and G inhibitors or NG-nitro-L-arginine methyl ester (L-NAME).Fittingly, stimulating static HUVECs with an NO donor decreased AT1R protein levels. RT-PCR revealed asignificant (P<0.05) decrease of AT1R mRNA in HUVECs exposed to SS during 3 (6�2% of static control), 6(4�1%), 12 (4�1%), and 24 hours (15�4%), suggesting a transcriptional downregulation of AT1R at length.Finally, angiotensin-induced vascular cell adhesion molecule was abated in HUVECs exposed to SS and in theouter aortic arch of mice.
Conclusions: Our results demonstrate that SS may convey some of its atheroprotective effects through downregu-lation of AT1R in endothelial cells. (Circ Res. 2009;105:869-875.)
Key Words: shear stress � angiotensin � nitric oxide � endothelium � atherosclerosis
Hemodynamic forces are major determinants of athero-sclerotic plaque localization. Plaques tend to form at
arterial bifurcations, branch points, and curvatures whereblood flow is low and oscillatory, whereas blood vesselsexposed to high laminar shear stress (SS) remain compara-tively devoid of plaques.1–3 The protective effect of SS islargely attributable to the local release of nitric oxide (NO),an important vasoprotective agent, known to reduce endothe-lial permeability, leukocyte adhesion, vascular smooth mus-cle cell proliferation, and thrombosis, while favoring endo-thelial survival.4–6 In contrast, oscillatory blood flow or lowSS induces the expression of adhesion molecules (intracellu-lar adhesion molecule-1, vascular cell adhesion molecule-1),chemokines (monocyte chemoattractant protein-1, interleu-kins), and growth factors that contribute to leukocyte recruit-ment and infiltration,7,8 which constitute the early steps of
plaque formation. Appropriately, the knockout (KO) or phar-maceutical inhibition of endothelial NO synthase (eNOS) isassociated with enhanced lesion growth in animal models ofatherosclerosis.9,10
Opposite to the NO/NO synthase system, the renin–angio-tensin system, which participates in vasoconstriction andarterial remodeling, has rather proatherosclerotic effects inthe vascular wall.11 In animal models of atherosclerosis,angiotensin II (Ang II) perfusion exacerbates the develop-ment of atherosclerotic plaques.12 Conversely, angiotensintype 1 receptor (AT1R) antagonists show a beneficial effecton plaque regression,12 caused by a decrease in the inflam-matory properties of the plaque.13 The atherogenic effects ofAng II can be explained not only by its impact on bloodpressure but also by a direct inflammatory action of thehormone on vascular cells, inducing the expression of adhe-sion molecules and chemokines.14
Original received August 6, 2008; first resubmission received march 27, 2009; second resubmission received July 2, 2009; revised second resubmissionreceived September 3, 2009; accepted September 8, 2009.
From the Paris Cardiovascular Research Center (B.R., J.V., A.T., S.L.), Institut National de la Sante et de la Recherche Medicale U970, HopitalEuropeen Georges Pompidou, France; Lady Davis Institute for Medical Research (D.R., S.L.), McGill University, Montreal, Canada; and Department ofVascular Surgery (B.M., R.d.C.) and Cell Biology & Genetics (B.M.), Erasmus University medical Center, Rotterdam, The Netherlands.
Correspondence to Dr Stephanie Lehoux, Lady Davis Institute for Medical Research, McGill University, 3755 Cote Ste Catherine, Montreal, QuebecH3T 1E2, Canada. E-mail [email protected]
© 2009 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.109.204040
869
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 4, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 4, 2016http://circres.ahajournals.org/
Dow
nloaded from

In spite of the fact that the roles of SS and Ang II in theblood vessel wall are well established, the interaction be-tween these 2 factors remains poorly investigated. However,indirect evidence exists showing that NO, which is producedby SS, modulates the biological functions of Ang II such asvascular smooth muscle cell migration15 and vascular reac-tivity.16,17 Moreover, the expression of AT1R is reduced invascular smooth muscle cells exposed to an NO donor,18,19
suggesting that SS may not only modulate the vascularresponse to Ang II through the opposing effects of NO butmay also directly influence AT1R expression in vascularcells. In the present study, we hypothesized that SS mayregulate the expression of AT1R in endothelial cells (ECs),which could account at least in part for the protective,antiatherogenic effects of SS.
MethodsImmunohistochemical AnalysisExperiments were performed in accordance with the EuropeanCommunity Standards on the Care and Use of Laboratory Animalsand were approved by the local ethics committee. Eight-week-oldC57BL/6 mice, eNOS-overexpressing transgenic mice (eNOS tg),and eNOS KO mice (eNOS KO) were euthanized by a lethal dose ofsodium pentobarbital (100 mg/kg). A separate group of C57BL/6mice was administered a 100 �L bolus of Ang II (10�6 mol/L; IV)6 hours before euthanasia for VCAM-1 expression analysis. Thevascular system was rinsed with PBS and fixed at 100 mm Hg with4% paraformaldehyde via a cannula inserted in the thoracic aorta.The aortic arch was removed and incubated in PBS/30% sucroseduring 24 hours. Arterial sections were included in OCT compound(Sakura). Successive longitudinal 7-�m sections were cut using acryostat, and transferred onto gelatin-coated slides. Nonspecificbinding sites on the tissue sections were blocked with 10% goatserum for 30 minutes at room temperature. Thereafter, anti-AT1Rantibody (Abcam) and anti-CD31 or anti–VCAM-1 (Santa CruzBiotechnology) were successively applied, followed by appropriatesecondary antibodies (Molecular Probes). The segments weremounted with medium containing DAPI (Vector).
Cell CultureHuman umbilical vein endothelial cells (HUVECs) (Promocell) weregrown to confluence on 0.2% gelatin-coated culture slides at 37°C ina humidified 5% CO2 incubator. Cells were cultured in endothelium
cell basal medium containing growth factors (human epidermalgrowth factor, human basic fibroblast growth factor, and human ECgrowth supplement; Promocell), 5% FCS (Boehringer–Mannheim)supplemented with streptomycin (100u/mL), penicillin (100 U/mL),and 10 �g/L Fungizone. In some experiments, cells were treated withAng II (10�6 mol/L), the NO synthase inhibitor NG-nitro-L-argininemethyl ester (L-NAME) (10�4 mol/L), the NO donor S-nitroso-L-acetyl penicillamine (SNAP) (10�6 mol/L), a protein kinase (PK)Ainhibitor (KT5720, 10�6 mol/L), a PKG inhibitor (KT5823, 10�6
mol/L), an AT1R antagonist (Losartan, 10�6 mol/L), an AT2Rantagonist (PD123319, 10�6 mol/L), or actinomycin D (5 mg/L). Foreach figure, each individual n corresponds to data obtained from anindividual batch of HUVECs processed separately from the next.
Application of SSA controlled level of laminar SS was applied to confluent HUVECsusing a parallel plate chamber connected to a perfusion circuit drivenby a peristaltic pump (Gilson). The circuit was placed in a sterile 5%CO2 incubator set at 37°C. HUVECs were placed in the perfusionsystem for 1, 3, 6, 12, or 24 hours and exposed to 0 or 15 dyn/cm2.Oscillatory flow was applied using the same circuit attached tocylinders producing 0�6 dyn/cm2 at 1 Hz.
Western Blot AnalysisHUVECs were washed twice with cold PBS and scraped off in 200�L of RIPA buffer (50 mmol/L Tris-HCL [pH 7.4], 1 mmol/LEDTA, 150 mmol/L NaCl, 20 mmol/L glycerophosphate, 0.1% SDS,0.1% deoxycholate, 1% Triton, complete protease inhibitor cocktailtablet [Roche]). Protein content was quantified using the Bradford(Bio-Rad) protein assay. Thirty micrograms of lysate were mixedwith reducing sample buffer for electrophoresis and subsequenttransfer onto nitrocellulose membranes (Amersham), and equalloading was verified using Ponceau red solution. Membranes wereincubated with anti-AT1R or anti-AT2R antibodies (Abcam) oranti–VCAM-1 (Santa Cruz). After secondary antibody incubation(Amersham), immunodetection proceeded using an enhanced chemi-luminescence kit (ECL Plus, Amersham), and bands were revealedusing the Las1000 imaging system and Image Gauge software (Fuji).After initial immunodetection, all membranes were stripped ofantibodies and reprobed with an anti-GAPDH antibody (Abcys).
Fluorescent–Angiotensin II Binding AssayHUVECs were either maintained in static conditions alone,static�AT1R blocker (ARB) (Losartan, 10�6 mol/L) or exposed toSS for 1 or 24 hours followed by an incubation of 30 minutes withfluorescent Ang II (10�8 mol/L, Sigma), diluted in a binding buffer(50mmol/L Tris (pH 7.4), 100 mmol/L NaCl, 5 mmol/L KCl,5 mmol/L mgCl2, 0.25% BSA). All cells were treated with AT2Rinhibitor (PD123319, 10�6 mol/L) added 20 minutes before fluores-cent Ang II incubation. Afterward, excess unbound Ang II wasremoved by 2 successive PBS washes and cells were fixed with 4%paraformaldehyde and mounted with Fluoprep containing DAPI(Vector). Green fluorescence was visualized and quantified usingHistolab software (Archimed), averaging threshold fluorescence areadata from a minimum of 5 fields per slide, for 4 slides perexperimental group.
Quantitative RT-PCRRT-PCR was performed to quantify AT1R mRNA levels using theRNeasy micro protocol (Qiagen) to isolate total RNA from cells.One microgram of RNA was mixed with random primers and reversetranscribed according to the first-strand method (Supershift, Invitro-gen). cDNA thus obtained was amplified by PCR under the follow-ing conditions: 30 seconds at 94°C, 30 seconds at 57.6°C, 30 secondsat 72°C, for 40 cycles. PCR primers used were as follows: AT1Rsense, 5�-GGC GCG GGT TTG ATA TTT GAC A-3�; AT1Rantisense, 3�-CAA AGG GCC AGC GGT ATT CCA TAG-5�;GAPDH sense, 5�-GAA GGT GAA GGT CGG AGT C-3�; GAPDHantisense, 3�-GAA GAT GGT GAT GGG ATT TC-5�.
Non-standard Abbreviations and Acronyms
Ang II angiotensin II
ApoE apolipoprotein E
ARB AT1R blocker
AT1R angiotensin type 1 receptor
AT2R angiotensin type 2 receptor
eNOS endothelial NO synthase
HUVEC human umbilical vein endothelial cell
KO knockout
L-NAME NG-nitro-L-arginine methyl ester
PK protein kinase
SNAP S-nitroso-L-acetyl penicillamine
SS shear stress
tg transgenic
VCAM vascular cell adhesion molecule
870 Circulation Research October 23, 2009
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from

The same cDNA samples were used for GAPDH and AT1Ramplification. PCR amplification resulted in 485 bp fragmentsoriginated from AT1R mRNA and 226 bp from GAPDH mRNA. Forquantification, the number of PCR cycles was chosen within thelinear exponential phase with respect to the amount of cDNAtemplate and the PCR performed.
Statistical AnalysisData presented as means�SEM were obtained in at least 3 indepen-dent experiments, obtained with different sets of cells. Valuesreported from western blots are expressed with respect to staticcontrol experiments. Statistical significance was determined by thenonparametric Mann–Whitney test, with P�0.05 considered asstatistically significant.
ResultsDifferential SS–Dependent Expression of AT1R inthe Vascular WallTo assess the expression of AT1R in arteries, we comparedthe inner curvature of the aortic arch, where atheroscleroticplaques tend to form, to the neighboring outer curvature ofthe arch that is comparatively protected. Immunostaining ofC57BL/6 mouse aortic arches showed a distinctive endothe-lial AT1R staining at the inner curvature of the aortic arch butnot in the adjoining outer curvature (Figure 1). AT1R expres-sion was similarly absent from the outer curvature of eNOS tgmice, but in these animals, even the inner curvature failed toshow endothelial AT1R staining. On the contrary, in vesselsof eNOS KO mice, both the inner and outer curvaturedisplayed positive AT1R staining in the endothelium (Figure1). Hence, endothelial AT1R expression is limited to plaque-prone areas having low SS in wild-type animals and ismodulated by endothelial eNOS expression.
Laminar Flow Decreases AT1R Protein Levelsin HUVECsTo explain the selective regional distribution of AT1R ob-served in immunostaining experiments, we examined theeffects of SS on AT1R protein expression. HUVECs wereexposed to laminar flow with a SS of 0 or 15 dyn/cm2 for 1,3, 6, 12, or 24 hours. Western blotting revealed that AT1Rprotein levels followed a biphasic regulation, characterized
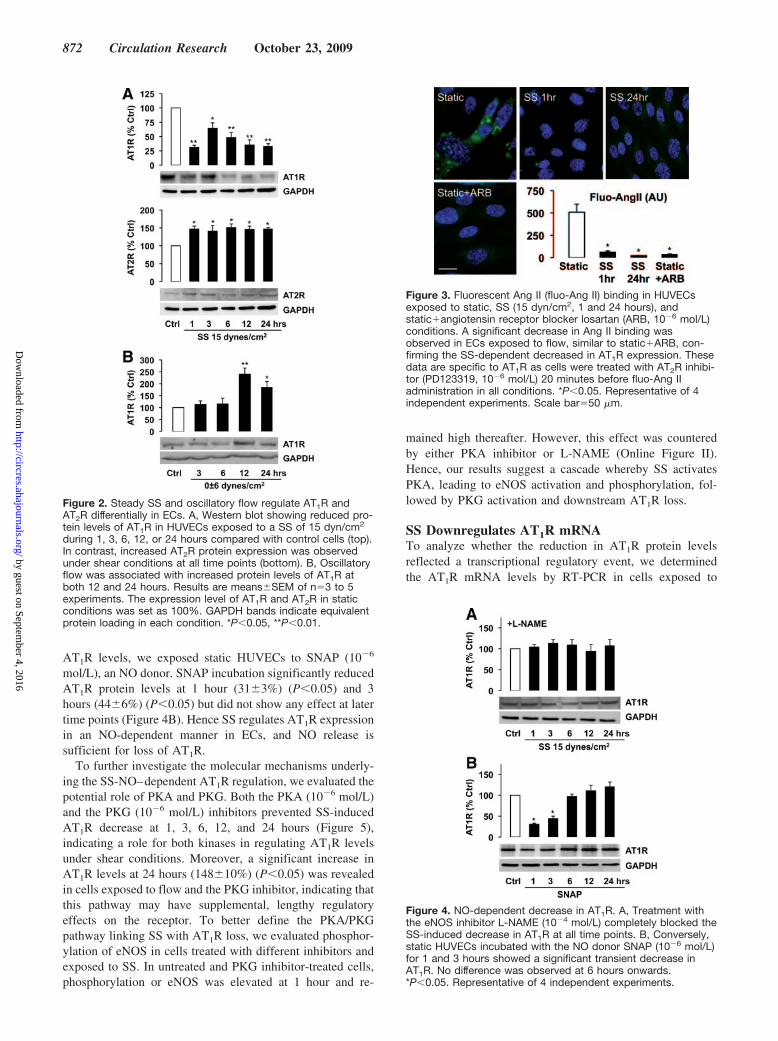
by an initial decrease at 1 hour (31�4% of static control,P�0.001), partial recovery at 3 hours (65�9%, P�0.05), anda second more prolonged decrease at 6, 12, and 24 hours(48�9%, 36�9%, 33�5% of static control, respectively;P�0.01) (Figure 2A). Interestingly, we found that oscillatoryflow (0�6 dyn/cm2) had the opposite effect, increasing AT1Rlevels at 12 hours (240�26%, P�0.01) and 24 hours(185�25, P�0.05) (Figure 2B). On the other hand, AT2Rlevels increased significantly at 1 hour of SS and remainedelevated thereafter (P�0.05) (Figure 2A), suggesting a dif-ferential modulation of the Ang II receptors by SS.
To confirm the decreased protein expression of AT1Robserved under flow conditions, we conducted binding assayswith fluorescent Ang II in cells exposed to static conditions or1 or 24 hours of SS. As demonstrated in Figure 3, cellsexposed to flow showed a significant decrease in Ang IIbinding (62�13 at 1 hour, 23�2 at 24 hours, P�0.05),similar to losartan-treated static cells (33�9, P�0.05), com-pared with untreated static cells (509�88). These results areAT1R-specific because all cells were treated with an AT2Rinhibitor just before incubation with the fluorescent Ang II.The punctate Ang II binding pattern corresponded to thesurface distribution of the AT1R receptor observed by immuno-cytochemistry in HUVECs (data not shown). Hence, the de-crease in AT1R protein expression observed in cells exposed toSS is associated with a reduction in Ang II binding.
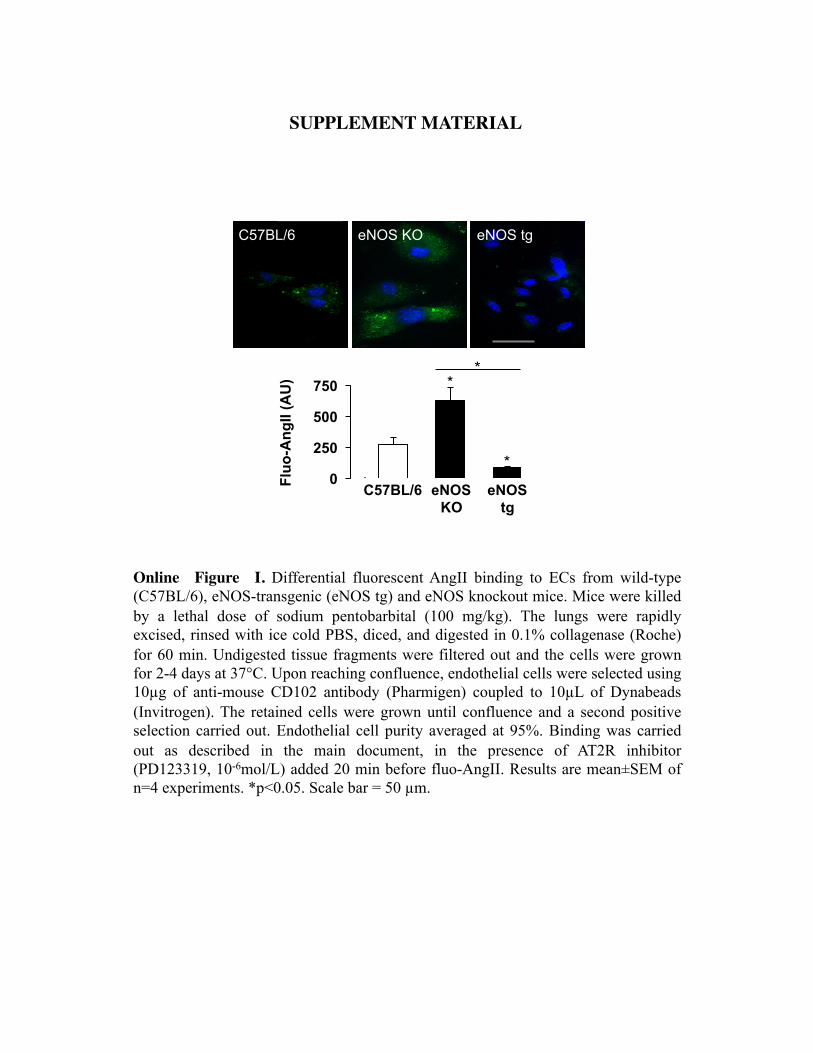
Pathways of SS-Induced AT1R RegulationIt is well known that SS triggers NO formation through thephosphorylation and activation of eNOS. To assess whetherthis pathway participates in the regulation of AT1R expres-sion, we treated HUVECs with the eNOS inhibitor L-NAME(10�4 mol/L). Figure 4A demonstrates that the SS-induceddecrease of AT1R was completely abolished by this treat-ment. These data are upheld by the finding that comparedwith ECs from wild-type mice, binding of Ang II to ECs fromeNOS KO mice was enhanced, whereas binding to ECs ofeNOS overexpressing mice was diminished (Online Figure I,available at http://circres.ahajournals.org). Furthermore, tofurther assess whether NO is sufficient for regulation of
Figure 1. Longitudinal sectionsshowing colocalization of AT1Rexpression and endothelial(CD31) staining in the plaque-prone inner curvature of the aorticarch but not the protected outercurvature of the aortic arch fromC57BL/6 mice (left images). AT1Rstaining is absent from ECs liningthe inner curvature of aortas frommice overexpressing eNOS(eNOS tg; middle images),whereas endothelial AT1R stain-ing is enhanced in the outer cur-vature of eNOS-deficient mice(right images). Results are repre-sentative of 5 (C57BL/6) and 4(eNOS tg, eNOS KO) indepen-dent experiments. All vessels aredisplayed with the luminal aspectfacing down. Scale bar�50 �m.
Ramkhelawon et al Shear Stress Regulates AT1R Expression 871
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from
AT1R levels, we exposed static HUVECs to SNAP (10�6
mol/L), an NO donor. SNAP incubation significantly reducedAT1R protein levels at 1 hour (31�3%) (P�0.05) and 3hours (44�6%) (P�0.05) but did not show any effect at latertime points (Figure 4B). Hence SS regulates AT1R expressionin an NO-dependent manner in ECs, and NO release issufficient for loss of AT1R.
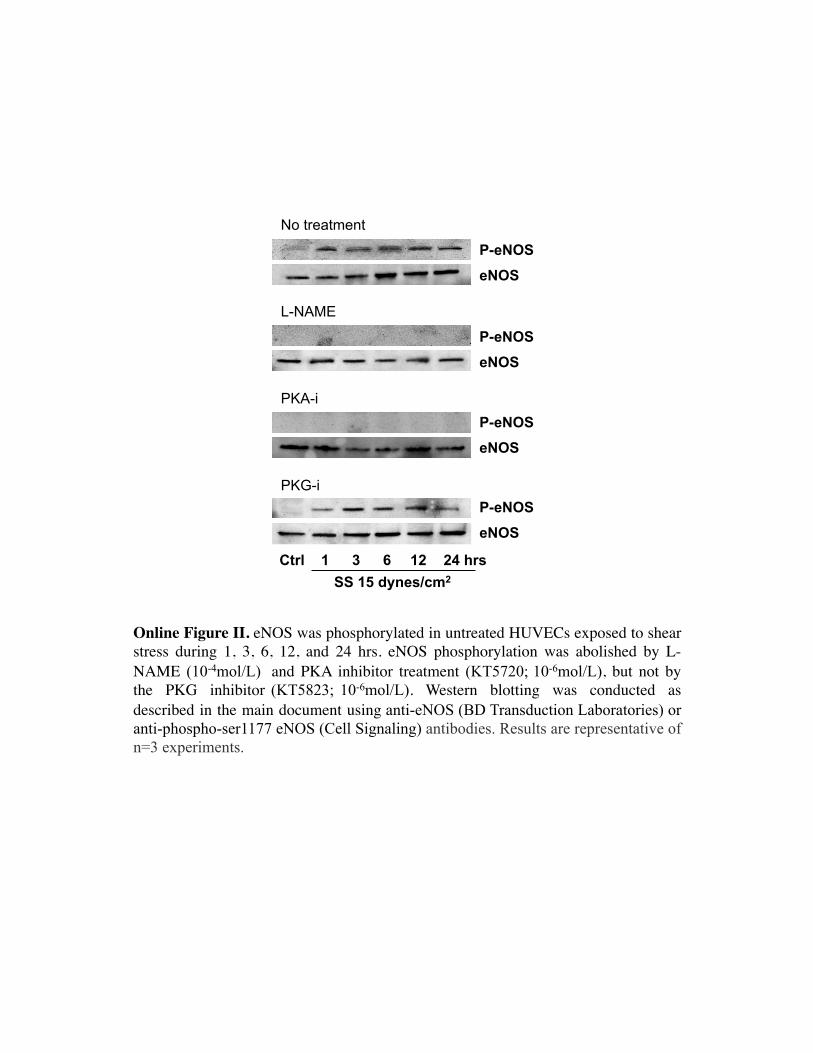
To further investigate the molecular mechanisms underly-ing the SS-NO–dependent AT1R regulation, we evaluated thepotential role of PKA and PKG. Both the PKA (10�6 mol/L)and the PKG (10�6 mol/L) inhibitors prevented SS-inducedAT1R decrease at 1, 3, 6, 12, and 24 hours (Figure 5),indicating a role for both kinases in regulating AT1R levelsunder shear conditions. Moreover, a significant increase inAT1R levels at 24 hours (148�10%) (P�0.05) was revealedin cells exposed to flow and the PKG inhibitor, indicating thatthis pathway may have supplemental, lengthy regulatoryeffects on the receptor. To better define the PKA/PKGpathway linking SS with AT1R loss, we evaluated phosphor-ylation of eNOS in cells treated with different inhibitors andexposed to SS. In untreated and PKG inhibitor-treated cells,phosphorylation or eNOS was elevated at 1 hour and re-
mained high thereafter. However, this effect was counteredby either PKA inhibitor or L-NAME (Online Figure II).Hence, our results suggest a cascade whereby SS activatesPKA, leading to eNOS activation and phosphorylation, fol-lowed by PKG activation and downstream AT1R loss.
SS Downregulates AT1R mRNATo analyze whether the reduction in AT1R protein levelsreflected a transcriptional regulatory event, we determinedthe AT1R mRNA levels by RT-PCR in cells exposed to
Figure 2. Steady SS and oscillatory flow regulate AT1R andAT2R differentially in ECs. A, Western blot showing reduced pro-tein levels of AT1R in HUVECs exposed to a SS of 15 dyn/cm2
during 1, 3, 6, 12, or 24 hours compared with control cells (top).In contrast, increased AT2R protein expression was observedunder shear conditions at all time points (bottom). B, Oscillatoryflow was associated with increased protein levels of AT1R atboth 12 and 24 hours. Results are means�SEM of n�3 to 5experiments. The expression level of AT1R and AT2R in staticconditions was set as 100%. GAPDH bands indicate equivalentprotein loading in each condition. *P�0.05, **P�0.01.
Figure 3. Fluorescent Ang II (fluo-Ang II) binding in HUVECsexposed to static, SS (15 dyn/cm2, 1 and 24 hours), andstatic�angiotensin receptor blocker losartan (ARB, 10�6 mol/L)conditions. A significant decrease in Ang II binding wasobserved in ECs exposed to flow, similar to static�ARB, con-firming the SS-dependent decreased in AT1R expression. Thesedata are specific to AT1R as cells were treated with AT2R inhibi-tor (PD123319, 10�6 mol/L) 20 minutes before fluo-Ang IIadministration in all conditions. *P�0.05. Representative of 4independent experiments. Scale bar�50 �m.
Figure 4. NO-dependent decrease in AT1R. A, Treatment withthe eNOS inhibitor L-NAME (10�4 mol/L) completely blocked theSS-induced decrease in AT1R at all time points. B, Conversely,static HUVECs incubated with the NO donor SNAP (10�6 mol/L)for 1 and 3 hours showed a significant transient decrease inAT1R. No difference was observed at 6 hours onwards.*P�0.05. Representative of 4 independent experiments.
872 Circulation Research October 23, 2009
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from

different periods of SS. AT1R mRNA levels were equivalentin unstimulated controls and HUVECs exposed to 1 hour(115�8%) of SS. However, a significant (P�0.05) decreaseof AT1R mRNA was observed in HUVECs exposed to SS for3 hours (6�2% of static control), 6 hours (4�1%), 12 hours(4�1%), and 24 hours (15�4%) (Figure 6A). To determinewhether this reduction in AT1R by SS was dependent onchanges in AT1R mRNA stability, we treated cells withactinomycin D, an inhibitor of gene transcription. Actinomy-cin D was added to cell medium in either static or SSconditions, and AT1R mRNA was examined 1, 3, 6, 12, and24 hours thereafter. As shown in Figure 6B, no significantdifference was observed in the time-dependent decrease ofAT1R mRNA between the static group and SS-inducedgroup. These results support the notion that SS primarilydecreases the transcription rate of the AT1R gene, rather thanaltering mRNA stability.
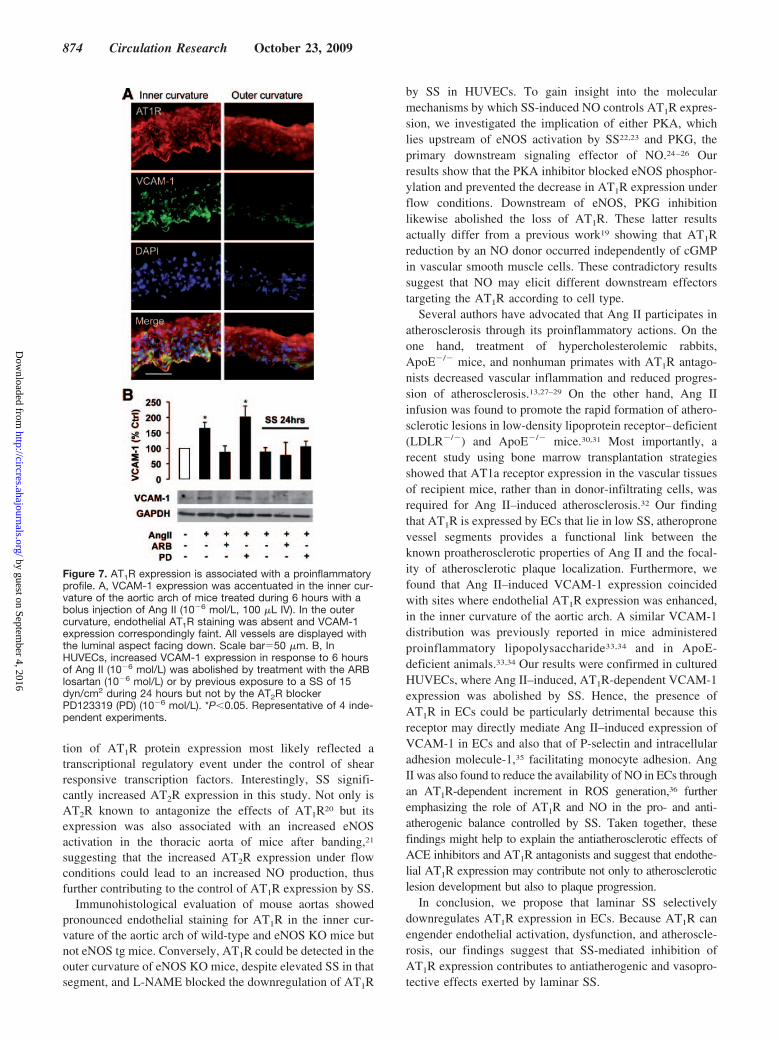
Lower AT1R Expression Is Associated With aReduced Proinflammatory ResponseTo verify that the loss of AT1R expression in ECs couldtranslate into reduced atherosclerotic potential, wild-typemice were injected with Ang II and the vascular expression ofVCAM-1 was evaluated after 6 hours. As demonstrated inFigure 7A, both AT1R and VCAM-1 expression could bereadily detected in ECs in the inner curvature of the aorticarch, whereas the outer curvature displayed little VCAM-1straining and no endothelial AT1R. VCAM-1 could not bedetected in vessels from control mice not injected with Ang II(data not shown). These results were confirmed in HUVECs;cells were maintained in static conditions or exposed to SSduring 24 hours, then stimulated with Ang II (10�6 mol/L)during 6 hours, with or without concomitant treatment withlosartan or PD123319. VCAM-1 was upregulated signifi-
cantly (P�0.05) in static HUVECs exposed to Ang II alone(164�20% of unstimulated control) or in combination withPD123319 (201�36%) but not in cells treated with the ARB(Figure 7B). In comparison, Ang II failed to induce VCAM-1expression in all cells exposed to SS.
DiscussionLaminar SS plays a fundamental role in the regulation of ECfunctions. The results presented herein identify for the firsttime a new mechanism whereby SS may convey some of itsatheroprotective effects. Firstly, we demonstrated that endo-thelial AT1R expression in whole vessels was only localizedin the inner aortic arch, characterized by disturbed or oscil-latory SS, but not in the outer aortic arch exposed to high SS.Secondly, in vitro assays revealed that SS was associated withthe downregulation of AT1R both at the protein and at themRNA levels in ECs, in an NO-dependent manner. PKA andPKG were implicated in this regulatory mechanism as well.Finally, absence of endothelial AT1R expression was associ-ated with lower VCAM-1 induction by Ang II, both in vivoand in vitro.
We found that exposure to SS was associated with abiphasic reduction in AT1R protein expression, characterizedby a transient decrease at 1 hour and a more prolonged loss at24 hours. Reduced binding of fluorescent Ang II to the AT1Robserved in HUVECs exposed to SS uphold these findings.The rapid decrease in receptor number after SS exposuresuggests that the protein half-life is short and could beexplained by an intracellular degradation of the receptorfollowing its activation by SS conditions. The rise in AT1Rexpression observed at 3 hours could result from a lower rateof endocytosis, reduced AT1R vesicle lysis in the favor ofenhanced recycling, or posttranscriptional regulation. At latertime points, from 3 hours of SS onwards, RT-PCR revealedthat AT1R mRNA expression was downregulated. Experi-ments with actinomycin D suggest that the long-term reduc-
Figure 6. AT1R mRNA quantification by quantitative PCR incells normalized to GAPDH levels. A, AT1R mRNA significantlydecreased in HUVECS exposed to SS from 3 hours onwardscompared with static controls. B, Analysis of AT1R mRNA stabil-ity by treatment with the transcription inhibitor actinomycin D (5�g/mL) revealed no significant difference in the time-dependentdecrease in AT1R mRNA between static and shear conditions.Results are means�SEM of n�3 experiments. *P�0.05.
Figure 5. PKA- and PKG-dependent AT1R regulation. Top,HUVECs incubated with PKA inhibitor (KT5720) (10�6 mol/L) nolonger demonstrated the SS-induced decrease in AT1R expres-sion. Bottom, Similarly, PKG inhibitor treatment (KT5823) (10�6
mol/L) prevented the loss in AT1R protein in HUVECs exposedto flow. A significant increase in AT1R was observed in treatedHUVECs at 24 hours. *P�0.05. Representative of 3 separateexperiments.
Ramkhelawon et al Shear Stress Regulates AT1R Expression 873
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from
tion of AT1R protein expression most likely reflected atranscriptional regulatory event under the control of shearresponsive transcription factors. Interestingly, SS signifi-cantly increased AT2R expression in this study. Not only isAT2R known to antagonize the effects of AT1R20 but itsexpression was also associated with an increased eNOSactivation in the thoracic aorta of mice after banding,21
suggesting that the increased AT2R expression under flowconditions could lead to an increased NO production, thusfurther contributing to the control of AT1R expression by SS.
Immunohistological evaluation of mouse aortas showedpronounced endothelial staining for AT1R in the inner cur-vature of the aortic arch of wild-type and eNOS KO mice butnot eNOS tg mice. Conversely, AT1R could be detected in theouter curvature of eNOS KO mice, despite elevated SS in thatsegment, and L-NAME blocked the downregulation of AT1R
by SS in HUVECs. To gain insight into the molecularmechanisms by which SS-induced NO controls AT1R expres-sion, we investigated the implication of either PKA, whichlies upstream of eNOS activation by SS22,23 and PKG, theprimary downstream signaling effector of NO.24–26 Ourresults show that the PKA inhibitor blocked eNOS phosphor-ylation and prevented the decrease in AT1R expression underflow conditions. Downstream of eNOS, PKG inhibitionlikewise abolished the loss of AT1R. These latter resultsactually differ from a previous work19 showing that AT1Rreduction by an NO donor occurred independently of cGMPin vascular smooth muscle cells. These contradictory resultssuggest that NO may elicit different downstream effectorstargeting the AT1R according to cell type.
Several authors have advocated that Ang II participates inatherosclerosis through its proinflammatory actions. On theone hand, treatment of hypercholesterolemic rabbits,ApoE�/� mice, and nonhuman primates with AT1R antago-nists decreased vascular inflammation and reduced progres-sion of atherosclerosis.13,27–29 On the other hand, Ang IIinfusion was found to promote the rapid formation of athero-sclerotic lesions in low-density lipoprotein receptor–deficient(LDLR�/�) and ApoE�/� mice.30,31 Most importantly, arecent study using bone marrow transplantation strategiesshowed that AT1a receptor expression in the vascular tissuesof recipient mice, rather than in donor-infiltrating cells, wasrequired for Ang II–induced atherosclerosis.32 Our findingthat AT1R is expressed by ECs that lie in low SS, atheropronevessel segments provides a functional link between theknown proatherosclerotic properties of Ang II and the focal-ity of atherosclerotic plaque localization. Furthermore, wefound that Ang II–induced VCAM-1 expression coincidedwith sites where endothelial AT1R expression was enhanced,in the inner curvature of the aortic arch. A similar VCAM-1distribution was previously reported in mice administeredproinflammatory lipopolysaccharide33,34 and in ApoE-deficient animals.33,34 Our results were confirmed in culturedHUVECs, where Ang II–induced, AT1R-dependent VCAM-1expression was abolished by SS. Hence, the presence ofAT1R in ECs could be particularly detrimental because thisreceptor may directly mediate Ang II–induced expression ofVCAM-1 in ECs and also that of P-selectin and intracellularadhesion molecule-1,35 facilitating monocyte adhesion. AngII was also found to reduce the availability of NO in ECs throughan AT1R-dependent increment in ROS generation,36 furtheremphasizing the role of AT1R and NO in the pro- and anti-atherogenic balance controlled by SS. Taken together, thesefindings might help to explain the antiatherosclerotic effects ofACE inhibitors and AT1R antagonists and suggest that endothe-lial AT1R expression may contribute not only to atheroscleroticlesion development but also to plaque progression.
In conclusion, we propose that laminar SS selectivelydownregulates AT1R expression in ECs. Because AT1R canengender endothelial activation, dysfunction, and atheroscle-rosis, our findings suggest that SS-mediated inhibition ofAT1R expression contributes to antiatherogenic and vasopro-tective effects exerted by laminar SS.
Figure 7. AT1R expression is associated with a proinflammatoryprofile. A, VCAM-1 expression was accentuated in the inner cur-vature of the aortic arch of mice treated during 6 hours with abolus injection of Ang II (10�6 mol/L, 100 �L IV). In the outercurvature, endothelial AT1R staining was absent and VCAM-1expression correspondingly faint. All vessels are displayed withthe luminal aspect facing down. Scale bar�50 �m. B, InHUVECs, increased VCAM-1 expression in response to 6 hoursof Ang II (10�6 mol/L) was abolished by treatment with the ARBlosartan (10�6 mol/L) or by previous exposure to a SS of 15dyn/cm2 during 24 hours but not by the AT2R blockerPD123319 (PD) (10�6 mol/L). *P�0.05. Representative of 4 inde-pendent experiments.
874 Circulation Research October 23, 2009
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from

Sources of FundingThis work is supported by the European Vascular Genomics Net-work (http://www.evgn.org), a Network of Excellence supported bythe European Community’s Sixth Framework Programme for Re-search Priority 1 “Life sciences, genomics and biotechnology forhealth” (contract no. LSHM-CT-2003-503254) and by the CanadianInstitutes of Health Research Institute of Circulatory and RespiratoryHealth (contract no. CRH-89063).
DisclosuresNone.
References1. Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in
atherosclerosis. JAMA. 1999;282:2035–2042.2. Caro CG, Fitz-Gerald JM, Schroter RC. Arterial wall shear and distri-
bution of early atheroma in man. Nature. 1969;223:1159–1160.3. DiChiara mR, Kiely JM, Gimbrone mA Jr, Lee ME, Perrella mA, Topper
JN. Inhibition of E-selectin gene expression by transforming growthfactor � in endothelial cells involves coactivator integration of Smad andnuclear factor �B-mediated signals. J Exp Med. 2000;192:695–704.
4. Ando J, Tsuboi H, Korenaga R, Takada Y, Toyamasorimachi N,Miyasaka M, Kamiya A. Shear stress inhibits adhesion of cultured mouseendothelial cells to lymphocytes by downregulating VCAM-1 expression.Am J Physiol. 1994;267:C679–C687.
5. Dimmeler S, Haendeler J, Rippmann V, Nehls M, Zeiher AM. Shearstress inhibits apoptosis of human endothelial cells. FEBS Lett. 1996;399:71–74.
6. Topper JN, Cai JX, Falb D, Gimbrone MA. Identification of vascularendothelial genes differentially responsive to fluid mechanical stimuli:cyclooxygenase-2, manganese superoxide dismutase, and endothelial cellnitric oxide synthase are selectively up-regulated by steady laminar shearstress. Proc Natl Acad Sci U S A. 1996;93:10417–10422.
7. Walpola PL, Gotlieb AI, Cybulsky MI, Langille BL. Expression ofICAM-1 and VCAM-1 and monocyte adherence in arteries exposed toaltered shear stress. Arterioscler Thromb Vasc Biol. 1995;15:2–10.
8. Bao X, Lu C, Frangos JA. Temporal gradient in shear but not steady shearstress induces PDGF-A and MCP-1 expression in endothelial cells: roleof NO, NF kappa B, and egr-1. Arterioscler Thromb Vasc Biol. 1999;19:996–1003.
9. Kuhlencordt PJ, Chen J, Han F, Astern J, Huang PL. Genetic deficiencyof inducible nitric oxide synthase reduces atherosclerosis and lowersplasma lipid peroxides in apolipoprotein E-knockout mice. Circulation.2001;103:3099–3104.
10. Hayashi T, Sumi D, Juliet PA, Matsui-Hirai H, Asai-Tanaka Y, Kano H,Fukatsu A, Tsunekawa T, Miyazaki A, Iguchi A, Ignarro LJ. Genetransfer of endothelial NO synthase, but not eNOS plus inducible NOS,regressed atherosclerosis in rabbits. Cardiovasc Res. 2004;61:339–351.
11. Hollenberg NK, Fisher ND, Price DA. Pathways for angiotensin II gen-eration in intact human tissue: evidence from comparative pharmaco-logical interruption of the renin system. Hypertension. 1998;32:387–392.
12. Chobanian AV, Alexander RW. Exacerbation of atherosclerosis byhypertension. Potential mechanisms and clinical implications. Arch InternMed. 1996;156:1952–1956.
13. Dol F, martin G, Staels B, mares AM, Cazaubon C, Nisato D, BidouardJP, Janiak P, Schaeffer P, Herbert JM. Angiotensin AT1 receptor antag-onist irbesartan decreases lesion size, chemokine expression, and macro-phage accumulation in apolipoprotein E-deficient mice. J CardiovascPharmacol. 2001;38:395–405.
14. Schmidt-Ott KM, Kagiyama S, Phillips MI. The multiple actions ofangiotensin II in atherosclerosis. Regul Pept. 2000;93:65–77.
15. Dubey RK, Jackson EK, Luscher TF. Nitric oxide inhibits angiotensinII-induced migration of rat aortic smooth muscle cell - role of cyclic-nu-cleotides and angiotensin(1) receptors. J Clin Invest. 1995;96:141–149.
16. Akimoto H, Ito H, Tanaka M, Adachi S, Hata M, Lin MH, Fujisaki H,Marumo F, Hiroe M. Heparin and heparan sulfate block angiotensinII-induced hypertrophy in cultured neonatal rat cardiomyocytes: apossible role of intrinsic heparin-like molecules in regulation of cardio-myocyte hypertrophy. Circulation. 1996;93:810–816.
17. Madrid MI, Garcia-Salom M, Tornel J, de Gasparo M, Fenoy FJ. Inter-actions between nitric oxide and angiotensin II on renal cortical andpapillary blood flow. Hypertension. 1997;30:1175–1182.
18. Cahill PA, Redmond EM, Foster C, Sitzmann JV. Nitric oxide regulatesangiotensin II receptors in vascular smooth muscle cells. EurJ Pharmacol. 1995;288:219–229.
19. Ichiki T, Usui M, Kato M, Funakoshi Y, Ito K, Egashira K, Takeshita A.Downregulation of angiotensin II type 1 receptor gene transcription bynitric oxide. Hypertension. 1998;31:342–348.
20. Berk BC. Angiotensin type 2 receptor (AT2R): a challenging twin. SciSTKE. 2003;2003:PE16.
21. Yayama K, Hiyoshi H, Imazu D, Okamoto H. Angiotensin II stimulatesendothelial NO synthase phosphorylation in thoracic aorta of mice withabdominal aortic banding via type 2 receptor. Hypertension. 2006;48:958–964.
22. Boo YC. Shear stress stimulates phosphorylation of protein kinase Asubstrate proteins including endothelial nitric oxide synthase in endothe-lial cells. Exp Mol Med. 2006;38:63–71.
23. Dixit M, Loot AE, Mohamed A, Fisslthaler B, Boulanger CM,Ceacareanu B, Hassid A, Busse R, Fleming I. Gab1, SHP2, and proteinkinase A are crucial for the activation of the endothelial NO synthase byfluid shear stress. Circ Res. 2005;97:1236–1244.
24. Hood J, Granger HJ. Protein kinase G mediates vascular endothelialgrowth factor-induced Raf-1 activation and proliferation in human endo-thelial cells. J Biol Chem. 1998;273:23504–23508.
25. Chang YS, Yaccino JA, Lakshminarayanan S, Frangos JA, Tarbell JM.Shear-induced increase in hydraulic conductivity in endothelial cells ismediated by a nitric oxide-dependent mechanism. Arterioscler ThrombVasc Biol. 2000;20:35–42.
26. Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, SickmannA, Meyer HE, Lohmann SM, Schmidt HH. Endothelial nitric-oxidesynthase (type III) is activated and becomes calcium independent uponphosphorylation by cyclic nucleotide-dependent protein kinases. J BiolChem. 2000;275:5179–5187.
27. Hayek T, Attias J, Coleman R, Brodsky S, Smith J, Breslow JL, KeidarS. The angiotensin-converting enzyme inhibitor, fosinopril, and the an-giotensin II receptor antagonist, losartan, inhibit LDL oxidation andattenuate atherosclerosis independent of lowering blood pressure in apo-lipoprotein E deficient mice. Cardiovasc Res. 1999;44:579–587.
28. Strawn WB, Chappell MC, Dean RH, Kivlighn S, Ferrario CM. Inhibitionof early atherogenesis by losartan in monkeys with diet-induced hyper-cholesterolemia. Circulation. 2000;101:1586–1593.
29. Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH,Skatchkov M, Heitzer T, Stasch JP, Griendling KK, Harrison DG, BohmM, Meinertz T, Munzel T. Increased NADH-oxidase-mediatedsuperoxide production in the early stages of atherosclerosis: evidence forinvolvement of the renin-angiotensin system. Circulation. 1999;99:2027–2033.
30. Daugherty A, manning MW, Cassis LA. Angiotensin II promotes athero-sclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J ClinInvest. 2000;105:1605–1612.
31. Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertensionaccelerates the development of atherosclerosis in apoE-deficient mice.Circulation. 2001;103:448–454.
32. Cassis LA, Rateri DL, Lu H, Daugherty A. Bone marrow transplantationreveals that recipient AT1a receptors are required to initiate angiotensinII-induced atherosclerosis and aneurysms. Arterioscler Thromb VascBiol. 2007;27:380–386.
33. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. TheNF-�B signal transduction pathway in aortic endothelial cells is primedfor activation in regions predisposed to atherosclerotic lesion formation.Proc Natl Acad Sci U S A. 2000;97:9052–9057.
34. Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulationof VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothe-lium in the ApoE-deficient mouse. Arterioscler Thromb Vasc Biol. 1998;18:842–851.
35. Soehnlein O, Schmeisser A, Cicha I, Reiss C, Ulbrich H, Lindbom L,Daniel WG, Garlichs CD. ACE inhibition lowers angiotensin-II-inducedmonocyte adhesion to HUVEC by reduction of p65 translocation and AT1 expression. J Vasc Res. 2005;42:399–407.
36. Desideri G, Bravi MC, Tucci M, Croce G, Marinucci MC, Santucci A,Alesse E, Ferri C. Angiotensin II inhibits endothelial cell motility throughan AT1-dependent oxidant-sensitive decrement of nitric oxide avail-ability. Arterioscler Thromb Vasc Biol. 2003;23:1218–1223.
Ramkhelawon et al Shear Stress Regulates AT1R Expression 875
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from

Stéphanie LehouxBhama Ramkhelawon, Jose Vilar, Daniel Rivas, Barend Mees, Rini de Crom, Alain Tedgui and
Shear Stress Regulates Angiotensin Type 1 Receptor Expression in Endothelial Cells
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2009 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.109.2040402009;105:869-875; originally published online September 17, 2009;Circ Res.
http://circres.ahajournals.org/content/105/9/869World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2009/09/17/CIRCRESAHA.109.204040.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on September 4, 2016
http://circres.ahajournals.org/D
ownloaded from
0
250
500
750 *
*
*
C57BL/6
Fluo
-Ang
II (A
U)
eNOS KO
C57BL/6 eNOS tg
eNOS KO eNOS tg
SUPPLEMENT MATERIAL
Online Figure I. Differential fluorescent AngII binding to ECs from wild-type (C57BL/6), eNOS-transgenic (eNOS tg) and eNOS knockout mice. Mice were killed by a lethal dose of sodium pentobarbital (100 mg/kg). The lungs were rapidly excised, rinsed with ice cold PBS, diced, and digested in 0.1% collagenase (Roche) for 60 min. Undigested tissue fragments were filtered out and the cells were grown for 2-4 days at 37°C. Upon reaching confluence, endothelial cells were selected using 10µg of anti-mouse CD102 antibody (Pharmigen) coupled to 10µL of Dynabeads (Invitrogen). The retained cells were grown until confluence and a second positive selection carried out. Endothelial cell purity averaged at 95%. Binding was carried out as described in the main document, in the presence of AT2R inhibitor (PD123319, 10-6mol/L) added 20 min before fluo-AngII. Results are mean±SEM of n=4 experiments. *p<0.05. Scale bar = 50 µm.
eNOS
P-eNOS
eNOS
P-eNOS
eNOS
P-eNOS
eNOS
P-eNOS
No treatment
PKG-i
PKA-i
L-NAME
Ctrl 1 24 hrs 3 12 6 SS 15 dynes/cm2
Online Figure II. eNOS was phosphorylated in untreated HUVECs exposed to shear stress during 1, 3, 6, 12, and 24 hrs. eNOS phosphorylation was abolished by L-NAME (10-4mol/L) and PKA inhibitor treatment (KT5720; 10-6mol/L), but not by the PKG inhibitor (KT5823; 10-6mol/L). Western blotting was conducted as described in the main document using anti-eNOS (BD Transduction Laboratories) or anti-phospho-ser1177 eNOS (Cell Signaling) antibodies. Results are representative of n=3 experiments.





























