Embed Size (px)
Citation preview

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Review
Silane coupling agents used for natural fiber/polymer composites: A review
Yanjun Xie a,b,*, Callum A.S. Hill b, Zefang Xiao a, Holger Militz a, Carsten Mai a
a Wood Biology and Wood Products, Burckhardt-Institute, Georg August University of Göttingen, Büsgenweg 4, D37077 Göttingen, Germanyb Centre for Timber Engineering, School of Engineering and the Built Environment, Edinburgh Napier University, 10 Colinton Road, EH10 5DT Edinburgh, United Kingdom
a r t i c l e i n f o
Article history:Received 9 November 2009Received in revised form 4 March 2010Accepted 4 March 2010
Keywords:A. Silane coupling agentsA. FibresB. Fiber/matrix bondB. Environmental degradation
a b s t r a c t
Natural fiber reinforced polymer composites (NFPCs) provide the customers with more alternatives in thematerial market due to their unique advantages. Poor fiber–matrix interfacial adhesion may, however,negatively affect the physical and mechanical properties of the resulting composites due to the surfaceincompatibility between hydrophilic natural fibers and non-polar polymers (thermoplastics and thermo-sets). A variety of silanes (mostly trialkoxysilanes) have been applied as coupling agents in the NFPCs topromote interfacial adhesion and improve the properties of composites. This paper reviews the recentprogress in using silane coupling agents for NFPCs, summarizes the effective silane structures from thesilane family, clarifies the interaction mechanisms between natural fibers and polymer matrices, and pre-sents the effects of silane treatments on the mechanical and outdoor performance of the resultingcomposites.
Crown Copyright � 2010 Published by Elsevier Ltd. All rights reserved.
1. Introduction and background
Natural fiber reinforced polymer composites (NFPCs), as animportant branch in the field of composite materials, have beenstudied for decades [1–11]. Natural fibers have different originssuch as wood, pulp, cotton, bark, nut shells, bagasse, corncobs,bamboo, cereal straw, and vegetable (e.g., flax, jute, hemp, sisal,and ramie) [10–13]. These fibers are mainly made of cellulose,hemicelluloses, lignin and pectins, with a small quantity ofextractives. The fiber constituents vary depending on their origi-
nation. Compared with conventional inorganic fillers such as glassfiber and carbon fibers, natural fibers provide many advantages:(1) abundance and therefore low cost, (2) biodegradability, (3)flexibility during processing and less resulting machine wear,(4) minimal health hazards, (5) low density, (6) desirable fiber as-pect ratio, and (7) relatively high tensile and flexural modulus.Incorporating the tough and light-weight natural fibers into poly-mer (thermoplastic and thermoset) matrices produces compositeswith a high specific stiffness and strength [14]. The renewableand biodegradable characteristics of natural fibers facilitate theirultimate disposal by composting or incineration, options not pos-sible with most industrial fibers. The fibers also contain seques-tered atmospheric carbon dioxide in their structure and areinvariably of lower embodied energy compared to industriallyproduced glass fibers.
Although natural fibers can offer the resulting composites manyadvantages, the usually polar fibers have inherently low compati-bility with non-polar polymer matrices, especially hydrocarbonmatrices such as polypropylene (PP) and polyethylene (PE)[15,16]. The incompatibility may cause problems in the compositeprocessing and material properties. Hydrogen bonds may form be-tween the hydrophilic fibers, and thus the fibers tend to agglomer-ate into bundles and unevenly distribute throughout the non-polarpolymer matrix during compounding processing [17,18]. There isalso insufficient wetting of fibers by the non-polar polymer matri-ces, resulting in weak interfacial adhesion. As a result, the stresstransfer efficiency from the matrix to the reinforcing fibers is re-duced. The incompatibility may not be an issue when using polarpolymers such as unsaturated polyester (UP) and epoxy resin as
1359-835X/$ - see front matter Crown Copyright � 2010 Published by Elsevier Ltd. All rights reserved.doi:10.1016/j.compositesa.2010.03.005
Abbreviations: ABI, acid–base interaction; APS, c-aminopropyl triethoxy silane;ASE, anti-swelling efficiency; BDMA, benzyl dimethylamine; BPO, benzoyl perox-ide; CTMP, chemithermomechanical pulp; DCUP, dicumyl peroxide; DCS, dichlo-rodiethylsilane; DGEBA, diglycidyl ethers of bisphenol A; DMA, dynamicmechanical analysis; DMVS, dichloro methyl vinyl silane; DSC, differential scanningcalorimetry; EMC, equilibrium moisture content; FTIR, Fourier transform infraredspectroscopy; GPS, c-glycidoxypropyltrimethoxy silane; HDS, hexadecyltrimethoxysilane; IPN, interpenetrating polymer network; ISS, interfacial shear strength; LDPE,low density polyethylene; MAPP, maleated polypropylene; MEKP, methyl ethylketone peroxide; MMS, methacryloxymethyltrimethoxy silane; MPS, c-methacryl-oxypropyl trimethoxy silane; MRPS, c-mercaptopropyltrimethoxy silane; NFPCs,natural fiber reinforced polymer composites; PALF, pineapple leaf fiber; PAPS, c-phenyl-aminopropyltrimethoxy silane; PE, polyethylene; PP, polypropylene; PS,polystyrene; PVC, polyvinyl chloride; SEC, size exclusion chromatography; TAS, c-diethylenetriaminopropyl trimethoxy silane; TGA, thermogravimetric analysis; UP,unsaturated polyester; VSPP, vinyltrimethoxysilane grafted polypropylene; VTS,vinyltrimethoxy silane; XPS, X-ray photoelectron spectroscopy.
* Corresponding author at: Centre for Timber Engineering, School of Engineeringand the Built Environment, Edinburgh Napier University, 10 Colinton Road, EH105DT Edinburgh, United Kingdom. Tel.: +44 131 4552494; fax: +44 131 4552239.
E-mail address: [email protected] (Y. Xie).
Composites: Part A 41 (2010) 806–819
Contents lists available at ScienceDirect
Composites: Part A
journal homepage: www.elsevier .com/locate /composi tesa

Author's personal copy
matrices; however, the resulting composites, similar to the com-posites with non-polar matrices, will be susceptible to moisturedeterioration and fungal damage during outdoor service. The mois-ture absorption of the natural fibers may cause dimensionalchanges of the resulting composites and weaken the interfacialadhesion [19,20]. Mould and decay fungi may also grow on/inthe composites, although more slowly than in the fibers alone,when they are utilized in the long-term under wet conditions[21]. In addition, natural fibers are of limited thermal stabilityand, therefore, thermal degradation may take place during com-posite processing at a high temperature, especially in the cases ofthermal extrusion and hot compression processes.
By this token, treatment of natural fibers is beneficial in order toimprove the water resistance of fibers, enhance the wettability ofthe natural fiber surface by polymers (mainly non-polar polymers)and promote interfacial adhesion. The performance of fibers is crit-ical to obtain the improved physical and mechanical properties ofthe resulting composites. Physical treatments (e.g. electronic dis-charge in the different media such as plasma and corona technol-ogies [22–25]) may create a hydrophilic or hydrophobic fibersurface by changing the surface energy to consequently increasethe compatibility of the treated fiber with the polymer matrices.These surface treatments only modify a very shallow surface of cellwalls and thus do not change the hygroscopic characteristics of fi-bers. Chemical modification provides the means of permanentlyaltering the nature of fiber cell walls [26]: by grafting polymersonto the fibers [27,28], crosslinking of the fiber cell walls [29], orby using coupling agents [4]. These modifying strategies have beengenerally reviewed recently [26,30]. The chemical modificationmay make the fiber cell walls more dimensionally stable, reducewater sorption, or increase resistance against fungal decay, butthere may be an associated reduced dynamic strength such as im-pact strength due to embrittlement. A coupling agent is a chemicalthat functions at the interface to create a chemical bridge betweenthe reinforcement and matrix. It improves the interfacial adhesionwhen one end of the molecule is tethered to the reinforcement sur-face and the functionality at the other end reacts with the polymerphase. Extensively used coupling agents for NFPCs are copolymerscontaining maleic anhydride such as maleated polypropylene(MAPP) or maleated polyethylene (MAPE) [18,31–33]. The anhy-dride groups of the copolymers may react with the surface hydro-xyl groups of natural fibers forming ester bonds whilst the otherend of copolymer entangles with the polymer matrix due to theirsimilar polarities [34]. Isocyanates have also been reported as thecoupling agents used in NFPCs. Urethane links can be formed be-tween the isocyanate functionality and the hydroxyl group of nat-ural fibers [35,36], consequently blocking these hygroscopichydroxyl sites [37].
Silanes are recognized as efficient coupling agents extensivelyused in composites and adhesive formulations [38]. They havebeen successfully applied in inorganic filler reinforced polymercomposites such as glass fiber reinforced polymer composites[e.g. 39,40] and mineral filled polymer composites [e.g. 41,42]. Si-lanes are also adhesion promoters in many adhesive formulationsor are used as substrate primers, giving stronger adhesion [43]. Thebifunctional structures of silanes have also been of interest inapplying them for natural fiber/polymer composites, since bothglass fibers and natural fibers bear reactive hydroxyl groups, andextensive researches have accordingly been carried out to screenthe varied silane structures for NFPC production.
The aim of this paper is to review the recent progress in usingsilane coupling agents for the production of natural fiber reinforcedpolymer composites. The effects of treatments of natural fiberswith silanes on the properties of fibers and the resulting compos-ites are discussed and the interaction mechanisms between phasesclarified.
2. Silane structures and hydrolysis processes
2.1. Silane structures
To effectively couple the natural fibers and polymer matrices,the silane molecule should have bifunctional groups which mayrespectively react with the two phases thereby forming a bridgein between them. Silane coupling agents have a generic chemicalstructure R(4�n)ASiA(R0X)n (n = 1,2) where R is alkoxy, X repre-sents an organofunctionality, and R0 is an alkyl bridge (or alkylspacer) connecting the silicon atom and the organofunctionality.In the past decades, various silane structures have been testedfor coupling of inorganic reinforcements such as glass fiber andorganic polymer matrices. The structures used to couple the nat-ural fibers and polymer matrices are relatively limited. Most ofthe established silanes used for NFPCs are trialkoxysilanes. Theorganofunctionality of the silane interacts with the polymermatrices with their interaction modes depending on the function-ality’s reactivity or compatibility towards the polymer. A non-reactive alkyl group of the silane may increase the compatibilitywith non-polar matrix due to their similar polarities; however,the reactive organofunctionality may covalently bond with aswell as being physically compatible with the polymer matrices.These organofunctionalities of silanes are typically amino, mer-capto, glycidoxy, vinyl, or methacryloxy groups. The most re-ported silanes and their applied target polymer matrices arelisted in Table 1.
With regard to these silanes shown in Table 1, aminosilanes,especially c-aminopropyltriethoxysilane (APS), are most exten-sively reported in the literature as coupling agents between naturalfibers and thermoplastics or thermosets. Vinyl- and acryl-silanesare coupling agents that are able to establish covalent bonds withpolymeric matrices in the presence of peroxide initiators. Methac-rylate–functional silanes can display high levels of reactivity withunsaturated polyester matrices [44], whilst azidosilanes can effi-ciently couple inorganic fillers with thermoplastic matrices[62,63]; however, there have been few reports of their use in nat-ural fiber reinforced thermoplastic composites. The application andeffects of these typical silanes on the NFPC’s properties will be pre-sented below.
2.2. Hydrolysis processes of silanes
The alkoxysilanes have been demonstrated to be able to directlyreact with ASiAOH groups of silica thereby forming ASiAOASiAbonds [e.g. 43,67,68] without any requirement of prehydrolysis.However, silanes do not undergo the same reaction with the hy-droxyl groups of cellulosic fibers even at high temperature [69].This has been attributed to lower acidity of cellulosic hydroxylgroups compared with silanol [69]. In addition, cellulose is gener-ally unreactive to many chemicals and the OH groups of the micro-fibrils have very low accessibility. Based on the fact, an optionalstrategy is to activate the alkoxysilane by hydrolyzing the alkoxygroups off thereby forming the more reactive silanol groups. As aresult, the silanol may react with the hydroxyl groups of fibers orcondense themselves on the surfaces of fibers and/or in the cellwalls forming macromolecular network. Although the formedASiAOACA bonds are eventually not stable towards hydrolysis,blocking the hydroxyl groups (reversible to hydrolysis) and the for-mation of macromolecular network (permanent) under heatingcondition facilitate an enhancement of interfacial adhesion of trea-ted fibers and polymer matrices and of the properties of the result-ing composites. To hydrolyze the alkoxy groups off, participation ofwater is essential. Even though the natural fibers under roomcondition contain bound water which may act to hydrolyze the
Y. Xie et al. / Composites: Part A 41 (2010) 806–819 807
Author's personal copy
silanes; however, additional water is required to achieve a com-plete hydrolysis of silanes [70–72].
2.2.1. Effect of silane structures on the hydrolysisAlkoxy groups associated with silane coupling agents prior to
utilization can be hydrolyzed off thereby liberating the corre-sponding alcohols in the presence of water and generating reactivesilanol groups. The alkoxy groups are usually ethoxy or methoxy.Under the same hydrolytic conditions the methoxy groups of tri-methoxysilane hydrolyzes more rapidly than the ethoxy groupsof triethoxysilane [73]. The hydrolysis of trimethoxysilane produc-ing methanol may be more environmentally problematic than thetriethoxysilane releasing ethanol because methanol is more toxicthan ethanol. The number of alkoxy groups will determine theamount of water used to fully hydrolyze them and influence theadhesion between silanes and filler. Di- and tri-alkoxy silanes pro-duce stronger adhesion strength than mono-alkoxy silanes sincethey form more binding sites after they are hydrolyzed [74]. If itis desired to deliver an alkoxy functional silane to the interior ofthe fiber this can be accomplished by dissolving the silane in theappropriate alcohol (e.g. ethanol for an ethoxy silane). Hydrolysiscan then be achieved by subsequent water treatment.
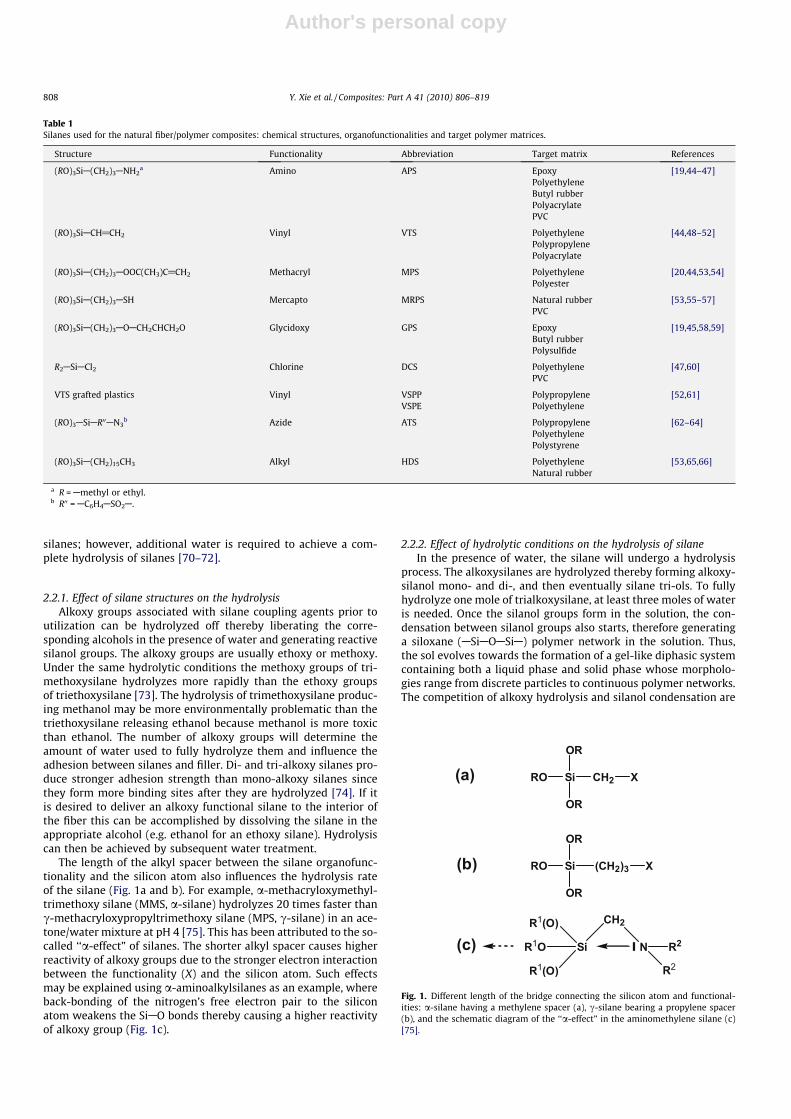
The length of the alkyl spacer between the silane organofunc-tionality and the silicon atom also influences the hydrolysis rateof the silane (Fig. 1a and b). For example, a-methacryloxymethyl-trimethoxy silane (MMS, a-silane) hydrolyzes 20 times faster thanc-methacryloxypropyltrimethoxy silane (MPS, c-silane) in an ace-tone/water mixture at pH 4 [75]. This has been attributed to the so-called ‘‘a-effect” of silanes. The shorter alkyl spacer causes higherreactivity of alkoxy groups due to the stronger electron interactionbetween the functionality (X) and the silicon atom. Such effectsmay be explained using a-aminoalkylsilanes as an example, whereback-bonding of the nitrogen’s free electron pair to the siliconatom weakens the SiAO bonds thereby causing a higher reactivityof alkoxy group (Fig. 1c).
2.2.2. Effect of hydrolytic conditions on the hydrolysis of silaneIn the presence of water, the silane will undergo a hydrolysis
process. The alkoxysilanes are hydrolyzed thereby forming alkoxy-silanol mono- and di-, and then eventually silane tri-ols. To fullyhydrolyze one mole of trialkoxysilane, at least three moles of wateris needed. Once the silanol groups form in the solution, the con-densation between silanol groups also starts, therefore generatinga siloxane (ASiAOASiA) polymer network in the solution. Thus,the sol evolves towards the formation of a gel-like diphasic systemcontaining both a liquid phase and solid phase whose morpholo-gies range from discrete particles to continuous polymer networks.The competition of alkoxy hydrolysis and silanol condensation are
Table 1Silanes used for the natural fiber/polymer composites: chemical structures, organofunctionalities and target polymer matrices.
Structure Functionality Abbreviation Target matrix References
(RO)3SiA(CH2)3ANH2a Amino APS Epoxy [19,44–47]
PolyethyleneButyl rubberPolyacrylatePVC
(RO)3SiACH@CH2 Vinyl VTS Polyethylene [44,48–52]PolypropylenePolyacrylate
(RO)3SiA(CH2)3AOOC(CH3)C@CH2 Methacryl MPS Polyethylene [20,44,53,54]Polyester
(RO)3SiA(CH2)3ASH Mercapto MRPS Natural rubber [53,55–57]PVC
(RO)3SiA(CH2)3AOACH2CHCH2O Glycidoxy GPS Epoxy [19,45,58,59]Butyl rubberPolysulfide
R2ASiACl2 Chlorine DCS Polyethylene [47,60]PVC
VTS grafted plastics Vinyl VSPP Polypropylene [52,61]VSPE Polyethylene
(RO)3ASiAR00AN3b Azide ATS Polypropylene [62–64]
PolyethylenePolystyrene
(RO)3SiA(CH2)15CH3 Alkyl HDS Polyethylene [53,65,66]Natural rubber
a R = Amethyl or ethyl.b R00 = AC6H4ASO2A.
RO Si
OR
OR
(CH2)3 X
RO Si
OR
OR
CH2 X
R1O Si
R1(O)
R1(O) CH2
N R2
R2
(a)
(b)
(c)
Fig. 1. Different length of the bridge connecting the silicon atom and functional-ities: a-silane having a methylene spacer (a), c-silane bearing a propylene spacer(b), and the schematic diagram of the ‘‘a-effect” in the aminomethylene silane (c)[75].
808 Y. Xie et al. / Composites: Part A 41 (2010) 806–819

Author's personal copy
affected by the hydrolysis condition used such as the solvent, tem-perature, pH, concentration of silanes [76–78]. Under optimumconditions hydrolysis may be accelerated, but condensation of sil-anols is inhibited to maintain a stable intermediary structures suchas silanol monomer or dimers [76,79,80].
The dynamics of the hydrolysis and ensuing condensation ofseveral trimethoxysilanes bearing different chain length of alkylfunctionalities (methyl, ethyl, propyl, or butyl) have been investi-gated in water–acetone mixtures by quantifying the producedmethanol using internal reflection Fourier transform infrared spec-troscopy (FTIR) combined with statistical regression. The resultsshow that the relative rates of the consecutive hydrolysis reactionsand the rate of condensation to form siloxane bonds depend on thesilane organofunctionalities, and this FTIR technique permits thetailoring of the composition of the alkoxysilane compounds insolution to obtain the desired rate and degree of hydrolysis of alk-oxy groups, and the extent of siloxane bond formation [82]. Thehydrolysis rates of APS, c-diethylenetriaminopropyl trimethoxy si-lane (TAS) and MPS were established using 1H NMR by measuringthe evolution of liberated free alcohol, and it showed that the rateincreased in the order MPS < APS < TAS. The formation of the sila-nol groups was followed by their condensation to generate oligo-meric structures. Whereas APS and MPS only gave solubleproducts, colloidal particles by contrast precipitated in the med-ium when the TAS was hydrolyzed [83,84]. At alkaline pH, the alk-oxy groups of APS were hydrolyzed almost immediately in waterand the resulting aminosilanols formed a cyclic structure with aninternal hydrogen-bonded ring (Fig. 2). Bonding of the silanolhydrogen makes the silanol stable against condensation in the con-centrated aqueous solutions. As a result, there was no insoluble gelin an alkaline aqueous solution of the aminoalkylsilanes after sev-eral months of storage [43]. In the pH range of 3–6, aminosilanolgroups in the solution were relatively more stable, existing as zwit-terionic structures [43]. In contrast, MPS hydrolyzes much slowerand requires the use of an amine as catalyst in the water/solventsolution [83].
Generally, under acid-catalyzed conditions, the hydrolysis rateof silanes forming silanol groups is greater than the condensationrate of the ensuing silanols forming siloxane bonds [72,79]. Thesecompeting reactions of hydrolysis and condensation in an etha-nol/water (80/20, w/w) solution at different pH values were ascer-tained in situ using 1H, 13C, and 29Si NMR spectroscopy [72]. Afterhydrolysis of the silane, the ensuing silanol groups gradually self-condensed into the structures of dimer (T1) (Fig. 3a), linear siloxane(T2) (Fig. 3b), and ultimately rigidly three-dimensional polysilox-ane cage structures (T3) (Fig. 3c). In an acidic solution, the hydroly-sis rate of MPS was accelerated and the self-condensation rate wasreduced compared to those in the alkaline solution. The entitieswith T3 units appeared after more than 1 month under acidic con-ditions; however, they appeared after only 6 h in the alkaline solu-tion. The formation of a T3 structure in the treating solution mustbe inhibited since this may reduce the number of silanol groupsadsorbed to fibers. The silanol condensation will also make the cellwall penetration by silane impossible due to the increased molec-ular size.
3. Interaction mechanisms between silanes and natural fibers
3.1. Fiber surface coating and cell wall modification
In the present literature, there are several different means re-ported to apply silanes to natural fibers. These can be divided intofiber surface treatment and cell wall modification. Spraying is a rel-atively easy way to treat the fiber surface with a silane solution.The silanes are dissolved into certain organic solvents or solvent/water mixtures and the prepared solution is directly sprayed ontothe fibers. If the solution is water-free, the sprayed silanes can bepartly hydrolyzed via the reaction with water from fibers and air.Due to the fast evaporation of solvent in air, the nano-pores of cellwalls cannot be opened up and thereby the silane molecules willhardly penetrate into fiber cell walls, because it will take manyhours for the chemical to diffuse into dense fiber cell walls fromthe solution [29]. As a result, spraying only results in a surfacecoating with silanes and the inside of cell walls is left untreated.
In one method, silane solution and initiator were directlypumped into an extruder during an extrusion process of natural fi-bers and thermoplastic matrices. The extruded composites weresubsequently exposed to an environment with high humidity andtemperature (ca. 100% relative humidity and 90 �C) in order tocomplete the hydrolysis and condensation processes of the silane[50]. This technique is simple at the initial stage but it will take along time at the hydrolysis and condensation stage. In addition,the silanes added will not only distribute on the interface of fiberand matrices, but some also in the matrices. This causes inefficientutilization of the silanes added. This process technique is also to beseen as a surface treatment.
In another approach, thermoplastic matrices were grafted withsilanes and the grafted pre-polymers were used as a new couplingagent directly blending with natural fibers and thermoplastic
OH NH2
Si
CH2CH2O
O
CH2
Fig. 2. A proposed cyclic hydrogen-bonded amine structure in the hydrolytic APSsolution [81].
SiRO
RO
R'
OSi
OROR
R'
SiRO
RO
R'
OSi
ORR'
OSi
R'OR
OR
SiRO
RO
R'
OSi
R'
OSi
R'OR
ORO
SiORRO
R'
T1 structuredimer or chain end
T2 structurelinear link
T3 structurethree dimensional
R' = functional group; R = alkyl group
(a)
(b)
(c)
Fig. 3. Schematic presentation of Ti silane structures [72].
Y. Xie et al. / Composites: Part A 41 (2010) 806–819 809

Author's personal copy
matrices [52]. Through this method of application, the alkoxysil-anes do not undergo any hydrolysis.
Impregnation processes where natural fibers were treated withprehydrolyzed silane solution to allow the silane penetrate into thefiber lumina and further diffuse into the cell walls have been re-ported [85,86]. Consequently, both fiber surfaces and cell wallsare modified with silanes (bulking treatment). The penetration ofsilanes into cell walls is influenced by the molecular size of silanewhich is influenced by the aging of the hydrolyzed silane solution.Improper hydrolysis processes may result in a fast condensation ofsilanols thereby prematurely increasing the molecular size of thesilanes. In this case diffusion of silanes into the cell walls will belimited or prevented entirely. The bulking treatment of fiber cellwalls can change the properties of cell walls, and as a result pro-mote the performance improvement of the ensuing composites.Compared to the surface treatment such as spraying, the impreg-nation process may cause problems for some type of fibers. Forexample, fine short fibers may aggregate and cannot thus evenlydisperse in the solution; the drying process may also consumeenergy.
3.2. Interaction mechanisms between silanes and natural fibers
3.2.1. Adsorption between silanols and hydroxyl groups of fibersWhen hydrolyzed silane solutions are mixed with natural fibers,
the reactive silanol groups have a high affinity for each other, form-ing ASiAOASiA bonds and also for the hydroxyl sites of fibers viahydrogen bonds. The adsorption isotherms of silanol groups ontocellulose fiber surfaces have been established using the techniquesof FTIR and UV spectroscopy [87]. The silanols of APS, TAS, or MPSfirstly form a monolayer on the fiber, and then are further adsorbeddue to the temperature-driven condensation reaction, which re-sults in the formation of a rigid polysiloxane layer on the fiber sur-face. The polysiloxane layer on the fiber surfaces may hinder anydiffusion of the silane molecules into the cell walls, or result in aconcentration gradient in the cell walls. Consequently, the hydro-lysis conditions such as pH of solution have to be adjusted to slowdown the condensation thereby allowing the reactive silanol ac-cess to hydroxyl sites in the fiber cell walls. Both, APS and TAS, dis-played higher adsorption towards the surfaces of cellulose fibersthan did MPS, since the former two may establish strong hydrogenbonds between the amino groups and the fiber hydroxyl groups.Owing to steric hindrance by the aromatic ring, c-phenyl-amino-propyltrimethoxy silane (PAPS) shows the lowest adsorption affin-ity for cellulose fibers compared to APS, TAS, and MPS. Theadsorption of silanols in the fiber cell walls is not clear in the caseof bulking treatments (impregnation treatment) due to the difficul-ties of in situ determination; however, the affinity of silanols forthe microfibrils of the cell walls may be similar to that displayedwith the fiber surface. To fully modify the cell walls, the silanolmonomers or oligomers have to be small enough to get access tothe hydroxyl groups of the interior of the cell walls.
3.2.2. Bonding of silanols and the hydroxyl groups of fibersThe free silanol groups may undergo further condensation
forming ASiAOASiA network linkages on/in the fibers during sol-vent evaporation as evidenced by 29Si NMR study [72]. Unlike theSiAOAC bonds, the ASiAOASiA bonds formed are very stable to-wards hydrolysis [43]. The hydrogen bonds formed between theadsorbed silanols and hydroxyl groups of natural fibers at theadsorption sites do not, however, convert into the covalent bondsof ASiAOACA linkages at room temperature [88–90], and thusthe adsorbed silanes may be leached off the fibers by Soxhletextraction with ethanol [87]. Heating (e.g. 110 �C for 2 h) may re-move the water or solvents in the fibers and drive the dehydrationreaction at the adsorption sites between silanols and fiber hydroxyl
groups thereby forming ASiAOACA bonds [69,87,91]. Further evi-dence on the interfacial bonds of silane-modified cellulose fibersmeasured using X-ray photoelectron spectroscopy (XPS) and FTIRspectroscopy confirm the occurrence of ASiAOACA bonds[87,92–94]. Heating also promotes the condensation of free silanolgroups resultantly forming the solid polysiloxane layers on the fi-ber surface and the entangled polysiloxane networks in the cellwalls. Hydrogen bonding is also possible between the ASiAOASiAbackbone and the hydroxyl groups of fibers. In the case of a bulkingtreatment, the polysiloxane networks in the cell walls may reducethe size of the cell wall nano-pores where the water is normallyable to gain access. As a result, the water sorption of the cell wallsmay be reduced. In addition, the aminosilanes such as APS mayform the strong intra- and inter-molecular hydrogen bonds withits amino group also possessing a strong affinity towards the hy-droxyl groups of fibers. These interactions in the aminosilane-trea-ted fibers may result in the formation of a cage-like structure in thefibers [44,95]. However, it must be emphasized that the bonds ofASiAOACA are not stable towards hydrolysis under a moist envi-ronment. Hydrolysis of ASiAOACA bonds will produce the free sil-anol groups again. The reproduced silanols may condensethemselves or form ASiAOACA bonds again under heatingconditions.
In general, interaction of silane coupling agents with natural fi-bers may mainly proceed through following steps [72,96]:
(1) Hydrolysis (Fig. 4a): The silane monomers are hydrolyzed inthe presence of water and catalyst (normally acid or base)liberating alcohol and yielding reactive silanol groups.
(2) Self-condensation (Fig. 4b): During the hydrolysis process,the concomitant condensation of silanols (aging) also takesplace. The condensation should be minimized at this stageto leave the silanols free for being adsorbed to the hydroxylgroups in the natural fibers. For the bulking treatment offibers, the condensation should also be controlled in orderto retain a small molecular size of monomers or oligomersto diffuse into the cell walls. The condensation rate of sila-nols is controllable by adjusting the pH of the hydrolysis sys-tem. An acidic pH environment is usually preferable toaccelerate the hydrolysis rate of silanes but slow down thecondensation rate of silanols.
(3) Adsorption (Fig. 4c): The reactive silanol monomers or olig-omers are physically adsorbed to hydroxyl groups of naturalfibers by hydrogen bonds on the fiber surfaces (surface coat-ing) and/or in the cell walls (cell wall bulking), whichdepends on the molecular size of silanol monomers/oligo-mers formed. The free silanols also adsorb and react witheach other thereby forming a rigid polysiloxane structureslinked with a stable ASiAOASiA bond.
(4) Grafting (Fig. 4d): Under heating conditions, the hydrogenbonds between the silanols and the hydroxyl groups of fiberscan be converted into the covalent ASiAOACA bonds andliberating water. The residual silanol groups in the fibers willfurther condense with each other. The bonds of ASiAOACAmay not be stable towards hydrolysis; however, this bond isreversible when the water is removed at a raisedtemperature.
3.3. Effect of silane treatments on the fiber properties
3.3.1. Stabilization against hydrolysisAlthough the SiAOAC bond is not stable towards hydrolysis
[43], several authors have found that the silanes in the treated fi-bers or wood are nonetheless resistant against water leaching[85,87,97]. Commercial microcrystalline cellulosic fibers treatedwith MPS, APS, and TAS silanes only had slight weight loss after
810 Y. Xie et al. / Composites: Part A 41 (2010) 806–819

Author's personal copy
15 h Soxhlet extraction with ethanol [87]. Donath et al. [97] deter-mined the anti-leaching ability of wood treated with the oligomersof methyl- and propyl-triethoxysilane and a multifunctional aque-ous siloxane HS 2909 (with the functionalities of alkyl and aminogroups). Both silane oligomers, especially the latter, cannot be lea-ched out of pine wood after treatment. Both silanes MPS and vinyl-trimethoxy silane (VTS) can penetrate into and react with the cellwalls of Corsican pine (Pinus nigra) sapwood via radical grafting orcondense in the cell walls [85]. After five leaching cycles involvingwater saturation followed by oven drying, only small amounts ofsilane were leached out and the treated wood still maintained ahigh anti-swelling efficiency (ASE), indicating the silanes still bondto cell walls after leaching. The high efficiency in resisting waterleaching may be apparently attributed to the highly hydrophobicalkyl groups and/or high affinity of silanols for cellulosic hydroxylgroups. In case of aminosilane, the strong affinity of amino groupsfor the hydroxyl groups of fibers should be also considered as a rea-son. In addition, X-ray mapping (SEM-EDX) of silane treated woodreveals an accumulation of silicon in the cell lumina and borderedpits thereby plugging of these typical penetration pathways forwater [97]. Where polymerization occurs within the cell wall, thesilane polymers become entangled with the cell wall polymericnetwork and are leach-resistant.
3.3.2. Hydrophobation of fibersBy a proper surface and/or bulking treatment with silanes, the
normally hygroscopic natural fibers can be converted into a hydro-phobic reinforcement for non-polar polymer matrices. A surfacecoating only decreases the water sorption rate and does not sub-
stantially reduce the amount of water to be absorbed becausethe cell walls are not filled with polymeric silane. In contrast, a bul-king treatment may reduce the cell wall nano-pore size and deac-tivate or mask the hydroxyl functionalities thereby decreasingwater sorption [86]. Hydrophobation treatments can weaken thehydrogen bonds between natural fibers and the agglomeration offibers is thus reduced during compounding with non-polar poly-mer matrices.
Treatments of a commercial microcrystalline cellulose fiberwith a silane solution of APS, MPS, hexadecyltrimethoxy silane(HDS), or c-mercaptopropyltrimethoxy silane (MRPS) in an etha-nol/water (80/20 v/v) mixture caused a significant increase in thewater contact angle of treated fibers from an original value of20� (untreated control) up to 110� determined by a dynamic con-tact angle test [65,66]. The polar component of the fiber surface en-ergy is reduced and the non-polar component increases due to thetreatments with alkylsilane such as propyltrimethoxysilane andHDS [59,66,98–100]. Optical microscopy showed that treatmentof flax fibers with VTS reduces the heterogeneous nucleation ofPP crystals on flax fibers and the treated fiber surfaces weresmoother than the untreated fibers [99]. Treatment of sisal fiberswith 1% MPS in benzene has been reported to reduce the equilib-rium moisture content (EMC) of fibers from 12.8% to 1.7% andthe water absorption from 123.8% to 64.4% [20]. However, sincebenzene does not swell the fiber cell wall the penetration of MPSin the cell walls may consequently be limited and the reductionin moisture/water sorption may mainly be due to the hydrophobiceffect of the surface coating. Compared to the silanes bearing non-polar alkyl groups, treatments with APS or PAPS which bear polar
Si OR
OR
OR
R' Si OH
OH
OH
R'+ 3 H2OH or OH
+ 3 ROH
Si OH
OH
OH
R' SiHO
OH
OH
R'+ Si O
OH
OH
R' Si
OH
OH
R' + H2O
R' Si
O
O
O Si
Si
O
R' O
R'
O
O
Si R'
O
+ x H2O
HO Si R'
OH
OH
HO Si O
R'
OH
Si
OH
R'
OH
O
O HO Si R'
OH
OHH
HHO Si O
R'
OH
Si
OH
R'
OH
O
OH
H
O
O HO Si R'
OH
OHH
HHO Si O
R'
OH
Si
OH
R'
OH
O Si R'
O
O
O Si O
R'
Si
O
R'
O
Δ
+
(a) Hydrolysis:
(b) Self-condensation:
(c) Adsorption:
(d) Chemically grafting:+ x H2O
Fig. 4. Interaction of silane with natural fibers by hydrolysis process.
Y. Xie et al. / Composites: Part A 41 (2010) 806–819 811

Author's personal copy
amino groups only resulted in a minor reduction of the moisturecontent of jute fibers [59]. This may be due to the highly hydro-philic characteristics of the amino end group.
3.3.3. Effect on the thermostabilizationSilane treatment of fibers could affect the thermal stability of
natural fibers. During the processing of composites, the natural fi-bers are subject to intense heat in the compounding machine orextruder. Therefore, a positive effect on the thermal properties isdesired. The thermal stability of silane-treated flax fibers was stud-ied using thermogravimetric analysis (TGA) at a temperature rangeof 30–800 �C in a helium atmosphere [101]. The fibers were treatedby immersing them into the VTS aqueous solution for 1 h to obtaina weight percent gain of 2.5%. The treated fibers exhibited an im-proved thermal stability of hemicellulose and pectin. Kinetic anal-ysis of the degradation process using Kissinger equation showed anactivation energy which proved to be 22% higher for VTS-treatedfibers than the untreated controls.
3.3.4. Effect on the fiber strengthThe treating system should not contain any fiber-damaging ele-
ment such as an acidic catalyst (for hydrolysis) or include hightemperature. The acidic conditions (ca. pH 4.0), in some cases, forthe acid-catalyzed hydrolysis of silanes were moderate and thepH was close to that of many natural fibers. As a result, silane treat-ment caused little effect on the fiber tensile strength [47,59,102].Varma et al. [103] reported, however, a 35% and 19% tenacity lossof coir fibers after refluxing in a benzene solution of 0.5% dichloromethyl vinyl silane (DMVS) or of 2% MPS, respectively (Table 2);however, these tenacity losses are mostly thought to have beencaused by the benzene (24%), rather than the silane itself (Table 2).
4. Interaction with polymer matrices
4.1. Coupling with thermoplastic matrices
4.1.1. Inter-phase compatibilityThe interaction mode between the silane-treated fiber and the
polymer matrix is a crucial factor for the mechanical propertiesof the resulting NFPCs. Physical blending of the silane-treated fi-bers and the thermoplastic matrices enhances their mutual adher-ence via inter-molecular entanglement, or acid–base interactions(ABI) [43]. The interfacial shear strength (ISS) between jute fibersand PP, determined by a microdroplet micromechanical test, wasimproved by treating jute fibers with a 0.5% APS aqueous solution[104]. Treatment of aspen chemithermomechanical pulp (CTMP,mesh 60 lm) with a 4 wt.% ethanol solution of silane VTS or MPSdid not substantially enhance the tensile properties of the resultingpolyethylene composites compared to the untreated composites[105]. The composites made from the fibers treated with silane,such as HDS or dichlorodiethylsilane (DCS) merely bearing a non-reactive aliphatic chain, displayed only a moderate improvementin the mechanical properties of the composite [47,53,106,107].
The limited coupling effects on the mechanical strength of theNFPCs due to the absence of covalent bonds between the silanefunctionality and thermoplastic matrices are exemplified in Ta-ble 3. The marginal improvements in the mechanical propertiesof the composites were mainly attributed to the increased compat-ibility resulting from the uniform dispersion of silane-treated fi-bers into the thermoplastic matrices [47]. The thermoplasticmolecular chains may also diffuse into the rigid polysiloxane struc-tures on the fiber surface forming an entangled ‘‘interpenetratingpolymer network (IPN)” [43,94,108,109].
In the case of aminosilanes, the amino groups cannot react withthe hydrocarbon backbone of PP or PE, but the natural fibers andthermoplastic composites coupled with APS was reported to pro-vide somewhat better mechanical properties than the uncoupledones [47,110]. Such improvements have been proposed to be dueto the strong affinity of the amino group towards the hydroxylgroups of fibers and to the formation of a cage-like IPN composedof the polysiloxane structures [43,44]. The IPN network can entrapthe thermoplastic molecules thereby anchoring the thermoplasticmatrices to the treated fiber surface. In addition, acid–base interac-tions (ABI) may also play a role in determining the interfacial adhe-sion of composites composed of the aminosilane-treated fibers andspecific thermoplastic matrices bearing acidic or basic characteris-tics, e.g. polystyrene (PS) and polyvinyl chloride (PVC) [47,111].The calculation according to electron donor and acceptor numbersof organic molecules indicates that the cellulosic fibers treatedwith aminosilane have both the acidic (KA) and basic (KB) charac-teristics, and the basicity is more prominent than the acidity(KA = 0.33, KB = 0.52) [111]. This ‘bipolar’ characteristic enhancesthe interactions of aminosilane-treated fibers with the ‘bipolar’polystyrene (KA = 0.28, KB = 0.46) [111] or PVC (KA = 1.43,KB = 0.65) [47] since the acidic sites from the aminosilane-treatedfibers can interact with the basic sites of thermoplastic matricesand vice versa. Reinforced polystyrene composites with the cellu-lose fibers treated with different structures of silanes show a linearincrease in the maximum interfacial shear stress (ISS) with theacid–base interaction value (Ia–b) between the treated cellulose fi-bers and polystyrene matrix [111]. For the studied silanes with afunctionality of phenyl (C6H5), phenylamino ((CH2)3NHC6H5), ami-no ((CH2)3NH2) or octadecyl ((CH2)17CH3), the aminosilane-treatedfibers display a stronger acid–base interaction with polystyrenethan the other silanes, and hence obtain stronger interfacial adhe-sion [111]. Similar to a polystyrene matrix, interaction of the acidicPVC and basic APS also showed acid–base characteristics (Fig. 5)[47,57]. Compared to the untreated fiber/PVC composites, thePVC composites reinforced with the APS-modified fibers displayedan increase up to 36% in the tensile strength due to the high Ia–b va-lue between treated fiber and PVC; however, treatment with DCSdid not change the Ia–b of two components of the composite andthus the tensile strength of resulting PVC composites did notchange [47].
4.1.2. Radical graftingAs presented above, physically blending of the silane-modified
natural fibers and thermoplastic matrices only produces a limitedimprovement in the mechanical properties of the resulting com-posites (Table 3). To substantially improve the mechanical prop-erties of natural fiber and thermoplastic composites, theformation of a covalent bond between the silane and non-polarthermoplastic matrix seems to be necessary [113]. It should, how-ever, be noted that a strong coupling between a reinforcing fiberand the matrix can result in a brittle composite especially withthermoset matrices [114]. However, most thermoplastic resinsdo not bear any reactive functionality. As a result, the silane-trea-ted natural fibers cannot react with the target thermoplastic toform any covalent bond during the compounding process, either
Table 2Effect of silane treatment on mechanical properties of coir fibers [103].
Treatment Time(h)
Tenacity (g/denier)
Elongation atbreak (%)
Initial modulus (g/denier)
Untreated – 2.26 28.80 38.302% MPS 1.00 1.82 23.52 36.090.5%
DMVS0.50 1.47 22.44 30.13
0.5%DMVS
0.25 1.80 26.12 34.20
Benzene 1.00 1.71 23.72 35.94
812 Y. Xie et al. / Composites: Part A 41 (2010) 806–819

Author's personal copy
in the mixers or in the extruders. An effective means to covalentlybond the silane to a thermoplastic matrix is by free radical graft-ing [113,115] or by plasma discharge [116]. The mostly reportedapplications are to graft VTS or MPS onto a thermoplastic matrixin the presence of peroxide initiators such as benzoyl peroxide(BPO) or dicumyl peroxide (DCUP) [e.g. 44,49]. At an elevatedtemperature the peroxide first decomposes, generating oxy radi-cals. The oxy radicals not only have the potential to abstracthydrogen from the backbone of thermoplastic molecules or natu-ral fibers, but can also add to vinyl double bonds of vinylsilane,producing vinyl radicals. The vinyl free electron may either com-bine with each other (homo-polymerization) or attack the other
molecules in a similar fashion to propagate the free radical reac-tion. A previous study showed, however, that VTS did not homo-polymerize during reaction due to its bulky silane side groups[117]. Accordingly, the radical reaction would ultimately resultin a grafting of vinylsilane onto thermoplastics as shown inFig. 6 [44,118]. The grafting of vinylsilanes onto thermoplasticmatrices has two options: one approach is to graft vinylsilanesonto matrices with the resulting copolymer being used as a cou-pling agent to bond the natural fiber and matrices; the other wayis to treat the fiber with the vinylsilane solution and the graftingreaction appears in the thermal compounding process of vinylsi-lane treated fibers and thermoplastic matrices.
Table 3Improvement (%) in tensile properties of natural fiber/thermoplastic composites coupled with different functionalities of silanes in the absence of initiators.
Silane (%) Fiber treatment with silane Fiber (%) Polymer Improvement (%) References
Tensilestrength
Tensilemodulus
Elongation at break Energy at yield
VTS (4) Immersion in ethanol/water CTMP (40) LDPE �27 �65 107 16 [105]MPS (4) Immersion in ethanol/water CTMP (40) LDPE �9 �74 245 25 [105]APS (4) Immersion in p-xylene CTMP (30) PS 6 25 �18 19 [44]MPS (2) Spray with aqueous solution Wheat straw fiber PP 2 �10 �6 – [112]HDS (3) Immersion in ethanol/water Cellulosic fibers LDPE 6 7 – – [53]MRPS (3) Immersion in ethanol/water Cellulosic fibers LDPE 12 6 – – [53]APS (0.01) Immersion in acetone Pine TMP (10)a PE 1 – – – [47]DCS (0.1) Dry blending Newsprint fiber (45) PVC 5 �15 30 – [47]
a TMP: thermomechanical pulp.
+ Cl
Cl
PVC
O (CH2)3SiO
ONHO (CH2)3Si
O
ONH2
APS grafted fiber
Δ
Fig. 5. Acid–base reaction between APS-modified fibers and PVC matrix [47].
CH2
CH2
CH2
R-O-O-R
Δ
CH2
HC
CH2
SiCH
OC2H5
OC2H5
OC2H5H2CR-O-O-R
ΔΔ
.
CH2
HC
CH2
. . GraftingCH2
HC
CH2
SiCH
OC2H5
OC2H5
OC2H5
CH2
OR
SiCH
OC2H5
OC2H5
OC2H5
CH2
OR
+
. SiCH
OC2H5
OC2H5
OC2H5
CH2
OR
PE radicalPE chain
Fig. 6. Radical grafting of vinylsilane onto polyethylene matrix.
Y. Xie et al. / Composites: Part A 41 (2010) 806–819 813

Author's personal copy
4.1.2.1. Copolymer as new coupling agent. Grafting of VTS alone ontolow density polyethylene (LDPE) with different concentrations ofVTS (0–27.33 part per hundred (phr)) and BPO (0–1.25 phr) has re-vealed that the VTS grafted polyethylene produced a maximumtensile strength at the concentrations of 5.0 phr VTS and 0.25 phrBPO [118]. The grafting efficacy of VTS and maleic anhydride ontoPP in the presence of DCUP were studied and the results demon-strated that VTS has a higher reactivity towards PP than maleicanhydride [61,119]. When the copolymers of VTS grafted polypro-pylene (VSPP) and MAPP were respectively used as a new couplingagent to couple wood flour and PP matrix, VSPP coupled compos-ites were 47% stronger in tensile strength and 35% lower in wateruptake than the composites coupled with the copolymer MAPP[52]. The study from Arbelaiz et al. [120] also reveals a superiorityof VSPP to MAPP used as the coupling agents. MAPP has beenextensively studied and is well known to be an excellent couplingagent in the bonding of natural fibers and thermoplastic matrices.This provides a means to further improve the interfacial propertiesof the NFPCs by using the VSPP copolymer.
4.1.2.2. Grafting reaction between the silane-treated fiber and matri-ces. A study of the melt rheological behavior showed that treat-ment of pineapple fibers with VTS (4.0 wt.% of fiber) andperoxide DCUP (2.0 wt.% of fiber) caused an increase in the meltviscosity of the resulting LDPE composites. This was attributed tothe increased fiber–matrix interfacial interaction due to a graftingreaction. Treatment of pineapple fibers with the peroxides DCUP(0.5 wt.% of LDPE) or BOP (1.0 wt.% of LDPE) alone also increasedthe melt viscosity of resulting LDPE composites due to the free rad-ical grafting reaction between fibers and LDPE at a high tempera-ture (125–145 �C) [49]. The grafting reaction of VTS-treated woodflour with PE in the presence of DCUP initiator caused a significantincrease in the flexural strength (up to 87%) and impact strength(more than 100%) of the resulting extruded composites[50,121,122]. The reported effects of grafting vinylsilanes with dif-ferent thermoplastic matrices on the mechanical properties ofresulting NFPCs are exemplified in Table 4.
Grafting effects of vinylsilane on the mechanical properties ofthe resulting NFPCs depended on the types of thermoplastic matri-ces [123,124]. Treatment of fibers with VTS or MPS in the presenceof DCUP initiator resulted in an increase of up to 62% in the tensilestrength of the PE matrix composites [51]; however, incorporationof fibers with the same treatment into a polystyrene matrix did notsubstantially improve the mechanical properties of the ensuingcomposites compared to the untreated fiber reinforced composites[44]. It may be concluded that the proper match of silane organo-functionality with the target polymer matrices is important to at-tain a desired coupling effect (Table 1).
Azidosilanes with a general formula (CH3O)3SiAR0ASO2N3
(Fig. 7) have been reported to be highly efficient in coupling min-eral fillers with polyolefin matrices [62–64,125]. Azidosilanes canalso be hydrolyzed thereby forming the reactive silanols whichare able to react with or condense in the fibers. The organofunc-
tional azido group can react with thermoplastic matrices formingcovalent bonds (Fig. 8). Upon heating, the azide may decomposeforming a highly reactive intermediate nitrene and simultaneouslyreleasing a nitrogen molecule. The nitrene can react with any of theCAH bonds in the thermoplastic polymer by inserting itself intothe carbon–hydrogen single bond or by adding to a carbon–carbondouble bond or aromatic system [126]. The required temperaturefor the covalent reaction is below 200 �C (in the temperature rangeof thermal process of NFPCs) and the reaction does not need anyperoxide initiator. Accordingly, azidosilanes would also be of greatinterest to couple natural fibers and polymer matrices. However,the application of azidosilanes on the NFPCs is little reported.
4.2. Coupling with thermosetting resins
In contrast with thermoplastic resins, the thermosetting resins(thermosets) are usually liquids prior to curing, bear reactiveorganofunctionalities and thereby are able to form three-dimen-sional networks after a curing reaction. Compared to thermoplas-tics, thermoset resins are more reactive towards theorganofunctionalities of silanes. Unlike the extrusion process ofnatural fiber/thermoplastic composites where there is a shear fail-ure of long fibers, the long natural fibers can be easily incorporatedinto the thermosetting resins without any damage from the pro-
Table 4Mechanical properties of natural fiber/thermoplastic composites coupled with the representative vinylsilanes in the presence of peroxide initiator.
Components of composite Improvement in mechanical properties (%) References
Fiber (%) Polymer Silane (%) Initiator (%)
Henequen (20) HDPE VTS (1) DCUP (0.5) ca. 30% in tensile strength, 50% in Iosipescu shear strength [91,94,124]CTMP of aspen (30) LDPE MPS (4) DCUP (unclear) 38% in tensile strength, 13% in tensile modulus [51]Wood flour (40) HDPE VTS (2) DCUP (0.17) 87% in flexural strength, 18.8% in flexural modulus,
more than 100% in impact strength[50,121,122]
Flax (30) PP MPS (3) DCUP (0.3) More than 60% in tensile strength [106]CTMP of aspen (30) LDPE VTS (4) DCUP (unclear) 66% in tensile strength, 44% in tensile modulus [51]CTMP of spruce and balsam fir (20) PVC MRPS (2) BPO (0.8) 98% in tensile strength [57]
RO Si
OR
R' S
O
O
N3
OR
Fig. 7. A general chemical formula of azidosilane. Representatively, R0 = aryl-containing group.
OH RO Si R"
OR
OR
N3 O Si R"
OR
OR
N3+
O Si R"
OR
OR
N3 O Si R"
OR
OR
N:Δ
+ N2
O Si R"
OR
OR
N: +CH2
CH2
n
O Si R"
OR
OR
NH CH
CH2
n
Fig. 8. A proposed coupling reaction between azidosilane-treated fibers andthermoplastic matrices [63].
814 Y. Xie et al. / Composites: Part A 41 (2010) 806–819
Author's personal copy
cessing. In the published literature, the most reported thermosetsfor NFPCs are epoxy compounds and unsaturated polyesters[1,114,127–130].
4.2.1. Reaction with epoxy resinBleached soda pulp fibers were modified with several silanes
(3% of APS, MPS, HDS, or MRPS) and then used as a reinforcement(40 vol.%) for epoxy resin. The results show that APS was the mostefficient in improving the mechanical properties and reducing thewater absorption of the resulting composites [45]. The reactionkinetics of epoxy pre-polymer diglycidyl ethers of bisphenol A(DGEBA, as shown in Fig. 9) with APS was systematically studiedusing differential scanning calorimetry (DSC), FTIR, and size exclu-sion chromatography (SEC), indicating that the epoxy is highlyreactive towards the amine group producing an insoluble fractionafter reaction [131]. The insoluble fraction is formed via the gela-tion of silane ethoxy and methylol groups which are produced by
the amine-epoxy reaction [46]. The interfacial interactions of DGE-BA and silanes were studied by incorporating the polysiloxanepowders formed from condensation of hydrolyzed APS into DGEBAmatrix [108,132]. FTIR evidence showed that the epoxy resin re-acted with polyaminosiloxane forming covalent bonds at the inter-face [132]. Use of a polymerizing accelerator benzyldimethylamine (BDMA) increased the reaction rate of the epoxyresin and polyaminosiloxane. The coupling mechanism of amino-silanes in the fiber/epoxy composites have been proposed by sev-eral authors as summarized in Fig. 10 [e.g. 45,46,108,132].Treatments of flax fibers with APS resulted in an enhancement oftensile strength up to 17%, and of the modulus up to 25%, but didnot change the impact toughness of the resulting epoxy compos-ites [133]. Sisal fiber/epoxy composites (40 vol.% of fiber) coupledwith 5% APS showed a considerable improvement in the compres-sion strength and wet tensile strength [19].
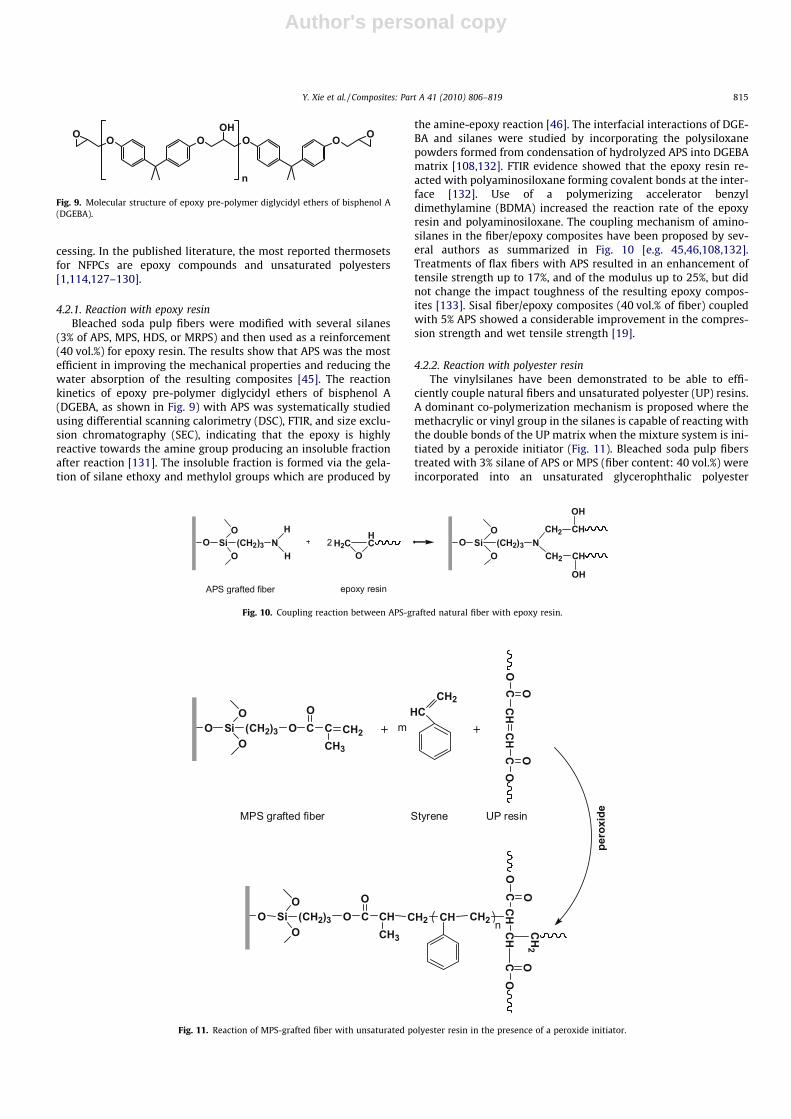
4.2.2. Reaction with polyester resinThe vinylsilanes have been demonstrated to be able to effi-
ciently couple natural fibers and unsaturated polyester (UP) resins.A dominant co-polymerization mechanism is proposed where themethacrylic or vinyl group in the silanes is capable of reacting withthe double bonds of the UP matrix when the mixture system is ini-tiated by a peroxide initiator (Fig. 11). Bleached soda pulp fiberstreated with 3% silane of APS or MPS (fiber content: 40 vol.%) wereincorporated into an unsaturated glycerophthalic polyester
OO O O O OOH
n
Fig. 9. Molecular structure of epoxy pre-polymer diglycidyl ethers of bisphenol A(DGEBA).
O SiO
ON
APS grafted fiber
H
HH2C
HC
O2 O (CH2)3Si
O
ON
CH2
CH2
CH
CH
OH
OH
(CH2)3
epoxy resin
Fig. 10. Coupling reaction between APS-grafted natural fiber with epoxy resin.
O SiO
OO(CH2)3 C
OC CH2CH3
HCCH2
O SiO
OO(CH2)3 C
OCHCH3
OC O
CH
CH
CO
O
CH2 CH CH2
OC O
CH
CH
CO
OC
H2
n
+ +
MPS grafted fiber Styrene UP resin
pero
xide
m
Fig. 11. Reaction of MPS-grafted fiber with unsaturated polyester resin in the presence of a peroxide initiator.
Y. Xie et al. / Composites: Part A 41 (2010) 806–819 815

Author's personal copy
(60 wt.% in styrene) with benzoyl peroxide used as a free radicalinitiator [45]. Treatment with APS caused a slight increase in thetensile Young’s modulus of the composite, but did not change theother mechanical properties. The results are consistent with a pre-vious study [134]. This is due to the lack of reactivity between theamino groups of APS and the polyester matrix. In contrast to APS,treatment of fibers with MPS improved the flexural strength ofthe resulting composites and slightly reduced the water uptakein a 4-week water immersion test [45].
Compared to bleached soda pulp fibers [45], the application ofMPS (1–5%) to the other types of natural fibers such as fique andsisal fibers resulted in an enhancement in the mechanical proper-ties of the resulting UP matrix composites. In the presence of theradical initiator methyl ethyl ketone peroxide (MEKP), the tensilestrength and modulus of the fique fiber/UP composite increaseup to 60% and 80%, respectively, due to the MPS treatment of fibers[135]. The flexural strength of sisal/UP composites can be im-proved up to 63% [20]. Comparably, the tensile strength of pineap-ple leaf fiber (PALF)/UP composites coupled with VTS increased40% as compared to the uncoupled composites [134]. Dynamicmechanical analysis (DMA) showed that treatment of banana fi-bers with MPS or VTS caused an increase in the dynamic storagemodulus and a reduction in the damping value of the resultingpolyester composites, suggesting the interfacial adhesion betweenbanana fiber and the polyester is improved [54,136]. The varyingmechanical improvement due to the different fiber types aftertreatment with MPS may be due to the different fiber constituentsand preparation methods of composites in the studies.
5. Outdoor performance
The moisture sorption of NFPCs measured according to standardASTM D-1037 [137] generally indicates very low moisture contents(in a range of ca. 2%), although this depends upon the fiber con-tents. This has been attributed to the effect of the encapsulationof fibers by polymers [138]; however, with the NFPCs in servicethere often appears deleterious effects such as surface colour fad-ing and erosion, warpage, mold growth, fungal decay, and strengthloss after a long-term exposure to an external environment[21,139–144]. These issues are mostly associated with the mois-ture sorption of the natural fibers. The moisture sorption of NFPCsin service is slow and seldom reaches an equilibrium condition in amoist environment. The core of the composites may have very lowmoisture contents whilst the surface layers may be significantlysaturated with water [145]. However, even though it may taketime for the water to penetrate the core of such composites, thewater susceptibility of natural fibers remains a cause of concern,which is particularly important if the fibers are exposed to mois-ture either through damage or cutting of the composite. Conse-quently, hydrophobation treatments of fibers thereby reducingthe moisture sorption are of importance for the NFPCs during envi-ronmental exposure in order to reduce potential damage associ-ated with water.
Oil palm fibers modified with VTS in an alcohol–wax mixture(60:40) were soaked in water at different temperatures (30, 50,70, and 90 �C) to determine their water uptake behavior and the re-sults showed that VTS-treatment reduced the water uptake of fi-bers at all temperatures used [146]. This makes the resultingpolymer composites drier under a moist environment therebyreducing the risk of environmental damage such as deformationand fungal decay [147]. Epoxy composites reinforced with sisalwhich had been treated with APS or with jute fiber treated withc-glycidoxypropyltrimethoxy silane (GPS) exhibited a reduced sus-ceptibility of moisture on the flexural strength [19,58]. Coir or oilpalm fibers modified with MPS were incorporated into polyester
resin and the modifying effects on the resulting composite’s prop-erties of water resistance and fungal decay were respectivelyexamined during one year weathering [148]. The mass loss dueto fungal decay reached over 15% for unmodified composites. Incomparison, MPS modified composites only lost 6% of the mass.The modified composites after fungal test obtained a moisture con-tent of 6%, less than the 12% observed with the unmodified com-posites. The low moisture content may be related to the reducedmass loss due to fungal decay [147]. The tensile and flexuralstrength of the modified composites were slightly reduced (up to8%), which is distinctly less than over 30% for the unmodifiedcontrols.
6. Summary
Most established silanes used for natural fiber/polymer com-posites are trialkoxysilanes bearing a non-reactive alkyl or reactiveorganofunctionality. Silane is hydrolyzed forming reactive silanolsand is then adsorbed and condensed on the fiber surface (sol–gelprocess) at a specific pH and temperature. The hydrogen bondsformed between the adsorbed silanols and hydroxyl groups of nat-ural fibers may be further converted into covalent bonds by heat-ing the treated fibers at a high temperature, although such bondsare susceptible to hydrolysis.
The interaction modes of the silane and matrix are dominatedby the organofunctionality of silane and the matrix characteristics.Physical compatibility (such as molecular entanglement, or acid–base interactions) between silane-grafted fiber and thermoplasticmatrices only provides a limited improvement in the mechanicalproperties of the resulting composites. To substantially improvethe interfacial adhesion, a chemical bonding between the organo-functionalities of silanes and the matrices is required. For the ‘‘in-ert” thermoplastic matrices, a free radical process is an effectivemeans to couple the vinylsilane treated fiber and matrices. In thecases of thermoset matrices, the organofunctionalities of silanescan react with the functional groups of thermoset matrices in thepresence of catalysts or radical initiators. Proper treatment of fi-bers with silane can increase the interfacial adhesion to the targetpolymer matrices and improve the mechanical and outdoor perfor-mance of the resulting fiber/polymer composites.
Acknowledgement
The author Dr. Yanjun Xie would like to thank the German Aca-demic Exchange Service (DAAD) for a research grant support.
References
[1] Hill CAS, Abdul Khalil HPS. Effect of fiber treatments on mechanical propertiesof coir or oil palm fiber reinforced polyester composites. J Appl Polym Sci2000;78:1685–97.
[2] Zadorecki P, Michell AJ. Future prospects for wood cellulose as reinforcementin organic polymer composites. Polym Compos 1989;10:69–77.
[3] Bledzki AK, Gassan J. Composites reinforced with cellulose based fibres. ProgPolym Sci 1999;24:221–74.
[4] Lu JZ, Wu Q, McNaabb HS. Chemical coupling in wood fiber and polymercomposites: a review of coupling agents and treatments. Wood Fiber Sci2000;32:88–104.
[5] Mukhopadhyay S, Deopura BL, Alagiruswamy R. Interface behavior inpolypropylene composites. J Thermoplast Compos 2003;16:479–95.
[6] George J, Sreekala MS, Thomas S. A review on interface modification andcharacterization of natural fiber reinforced plastic composites. Polym Eng Sci2001;41:1471–85.
[7] Narkis M, Chen JH. Review of methods for characterization of interfacialfiber–matrix interactions. Polym Compos 1988;9:245–51.
[8] Ishida H. A review of recent progress in the studies of molecular andmicrostructure of coupling agents and their functions in composites, coatingsand adhesive joints. Polym Compos 1984;5:101–23.
[9] Jiang H, Kamdem DP. Development of poly(vinyl chloride)/wood composites.A literature review. J Vinyl Addit Technol 2004;10:59–69.
816 Y. Xie et al. / Composites: Part A 41 (2010) 806–819

Author's personal copy
[10] Li Y, Mai YW, Ye L. Sisal fibre and its composites: a review of recentdevelopments. Compos Sci Technol 2000;60:2037–55.
[11] Sabu T, Pothan L. Cellulose fibre reinforced polymercomposites. Philadelphie: Old City Publishing; 2009.
[12] Xiao Z, Zhao LB, Xie Y, Wang QW. Review for development of wood plasticcomposites. J Northeast Forest Univ 2003;31:89–93.
[13] Seymour RB. Cellulose-filled polymer composites. Pop Plast 1978;23:27–30.[14] Wambua P, Ivens J, Verpoest I. Natural fibres: can they replace glass in fibre
reinforced plastics? Compos Sci Technol 2003;63:1259–64.[15] Bledzki AK, Gassan J, Theis S. Wood-filled thermoplastic composites. Mech
Compos Mater 1998;34:563–8.[16] Cantero G, Arbeliaz A, Liano-Ponte R, Mondargon I. Effects of fibre treatment
on wettability and mechanical behavior of flax/polypropylene composites.Compos Sci Technol 2003;63:1247–54.
[17] Raj RG, Kokta BV. Compounding of cellulose fibers with polypropylene: effectof fiber treatment on dispersion in the polymer matrix. J Appl Polym Sci1989;38:1987–96.
[18] Kazayawoko M, Balatinecz JJ. Matuana LM surface modification and adhesionmechanisms in woodfiber–polypropylene composites. J Mater Sci1999;34:6189–99.
[19] Bisanda ETN, Ansell MP. The effect of silane treatment on the mechanical andphysical properties of sisal–epoxy composites. Compos Sci Technol1991;41:165–78.
[20] Singh B, Gupta M, Verma A. Influence of fiber surface treatment on theproperties of sisal–polyester composites. Polym Compos 1996;17:910–8.
[21] Schirp A, Wolcott M. Influence of fungal decay and moisture absorption onmechanical properties of extruded wood–plastic composites. Wood Fiber Sci2005;37:643–52.
[22] Li R, Ye L, Mai YW. Application of plasma technologies in fibre-reinforcedpolymer composites: a review of recent developments. Compos Part A – ApplSci 1997;28:73–86.
[23] Sun D. Investigating the plasma modification of natural fiber fabrics-theeffect on fabric surface and mechanical properties. Text Res J 2005;75:639–44.
[24] Belgacem MN, Bataille P, Sapieha S. Effect of corona modification on themechanical properties of polypropylene/cellulose composites. J Appl PolymSci 1994;53:379–85.
[25] Sakata I, Morita M, Tsuruta N, Morita K. Activation of wood surface by coronatreatment to improve adhesive bonding. J Appl Polym Sci 1993;49:1251–8.
[26] Hill CAS. Wood–plastic composites: strategies for compatibilising the phases.J Inst Wood Sci 2000;15:140–6.
[27] Daneault C, Kokta BV, Maldas D. Grafting of vinyl monomers onto wood fibersinitiated by peroxidation. Polym Bull 1988;20:137–41.
[28] Hong CK, Kim N, Kang SL, Nah C, Lee YS, Cho BH, et al. Mechanical propertiesof maleic anhydride treated jute fibre/polypropylene composites. PlastRubber Compos 2008;37:325–30.
[29] Hill CAS. Wood modification: chemical, thermal and other processes. JohnWiley & Sons, Ltd.; 2006.
[30] Belgacem MN, Gandini A. The surface modification of cellulose fibres for useas reinforcing elements in composite materials. Compos Interf2005;24:41–75.
[31] Mohanty AK, Dryal LT, Misra M. Novel hybrid coupling agent as an adhesionpromoter in natural fiber reinforced powder polypropylene composites. JMater Sci Lett 2002;21:1885–8.
[32] Sun ZY, Han HS, Dai GC. Mechanical properties of injection-molded naturalfiber-reinforced polypropylene composites: formulation and compoundingprocesses. J Reinf Plast Compos 2009. doi:10.1177/0731684408100264, 2009.
[33] Gassan J, Bledzki AK. The influence of fiber-surface treatment on themechanical properties of jute–polypropylene composites. Compos Part A –Appl Sci 1997;28:1001–5.
[34] Felix JM, Gatenholm P. The nature of adhesion in composites of modifiedcellulose fibers and polypropylene. J Appl Polym Sci 1991;42:609–20.
[35] Botaro VR, Gandini A, Belgacem Naceur. Heterogeneous chemicalmodification of cellulose for composite materials. J Thermoplast Compos2005;18:107–17.
[36] Maldas D, Kokta BV. Improving adhesion of wood fiber with polystyrene bychemical treatment of fiber with coupling agent and the influence on themechanical properties of the composites. J Adhes Sci Technol 1989;3:529–39.
[37] Richelt L, Poller S. Uber die umsetzung von cellulose und lignin mitisocyanates bzw. Isocyanatgruppenhaltigen prapolymeren. Acta Polym1981;32:172–6.
[38] Rider AN, Arnott DR. Boiling water and silane pre-treatment of aluminiumalloys for durable adhesive bonding. Int J Adhes Adhes 2000;20:209–20.
[39] Wu HF, Dwight DW, Huff NT. Effects of silane coupling agents on theinterphase and performance of glass–fiber-reinforced polymer composites.Compos Sci Technol 1997;57:975–83.
[40] Clark HA, Plueddemann EP. Bonding of silane coupling agents in glass-reinforced plastics. Mod Plast 1963;40. p. 133–5, 137–8, 195–6.
[41] Park JM, Subramanian RV, Bayoumi AE. Interfacial shear strength anddurability improvement by silanes in single-filament composite specimensof basalt fiber in brittle phenolic and isocyanate resins. J Adhes Sci Technol1994;8:133–50.
[42] Favis BD, Blanchard LP, Leonard J, Prud’Homme RE. The interaction of acationic silane coupling agent with mica. J Appl Polym Sci 2003;28:1235–44.
[43] Plueddemann EP. Silane coupling agents. 2nd ed. New York andLondon: Plenum Press; 1991.
[44] Maldas D, Kokta BV, Daneault C. Influence of coupling agents and treatmentson the mechanical properties of cellulose fiber–polystyrene composites. JAppl Polym Sci 1989;37:751–75.
[45] Abdelmouleh M, Boufi S, Belgacem MN, Dufresne A, Gandini A. Modificationof cellulose fibers with functionalized silanes: effect of the fiber treatment onthe mechanical performances of cellulose–thermoset composites. J ApplPolym Sci 2005;98:974–84.
[46] Serier A, Pascault JP, Lam TM. Reaction in aminosilane–epoxy prepolymersystems. II. Reactions of alkoxysilane groups with or without the presence ofwater. J Polym Sci Polym Chem 1991;29:1125–31.
[47] Matuana LM, Woodhams RT, Balatinecz JJ, Park CB. Influence of interfacialinteractions on the properties of PVC/cellulosic fiber composites. PolymCompos 1998;19:446–55.
[48] George J, Bhagawan SS, Thomas S. Thermogravimetric and dynamicmechanical thermal analysis of pineapple fibre reinforced polyethylenecomposites. J Therm Anal 1996;47:1121–40.
[49] George J, Janardhan R, Anand JS, Bhagawan SS, Thomas S. Melt rheologicalbehaviour of short pineapple fibre reinforced low density polyethylenecomposites. Polymer 1996;37:5421–31.
[50] Bengtsson M, Oksman K. Silane crosslinked wood plastic composites:processing and properties. Compos Sci Technol 2006;66:2177–86.
[51] Raj RG, Kokta BV, Maldas D, Daneault C. Use of wood fibers in thermoplastics.VII. The effect of coupling agents in polyethylene–wood fiber composites. JAppl Polym Sci 1989;37:1089–103.
[52] Nachtigall SMB, Cerveira GS, Rosa SML. New polymeric-coupling agent forpolypropylene/wood–flour composites. Polym Test 2007;26:619–28.
[53] Abdelmouleh M, Boufi S, Belgacem MN, Dufresne A. Short natural-fibrereinforced polyethylene and natural rubber composites: effect of silanecoupling agents and fibres loading. Compos Sci Technol 2007;67:1627–39.
[54] Pothan LA, Thomas S, Groeninckx G. The role of fibre/matrix interactions onthe dynamic mechanical properties of chemically modified banana fibre/polyester composites. Compos Part A – Appl Sci 2006;37:1260–9.
[55] Ismail H. The effect of filler loading and a silane coupling agent on thedynamic properties and swelling behaviour of bamboo filled natural rubbercompounds. J Elastom Plast 2003;35:149–59.
[56] Ismail H, Shuhelmy S, Edyham MR. The effects of a silane coupling agent oncuring characteristics and mechanical properties of bamboo fibre fillednatural rubber composites. Eur Polym J 2002;38:39–47.
[57] Beshay A, Hoa SV. Reinforcement of polyvinyl chloride (PVC) and polystyrene(PS) with cellulose fibers treated with silane. J Thermoplast Compos1990;3:264–74.
[58] Gassan J, Bledzki AK. Effect of moisture content on the properties of silanizedjute–epoxy composites. Polym Compos 1997;18:179–84.
[59] Doan TTL. Investigation on jute fibres and their composites based onpolypropylene and epoxy matrices. PhD thesis, Technischen UniversitätDresden; 2006.
[60] Pickering KL, Abdalla A, Ji C, McDonald AG, Franich RA. The effect of silanecoupling agents on radiata pine fibre for use in thermoplastic matrixcomposites. Compos Part A – Appl Sci 2003;34:915–26.
[61] Nachtigall SMB, Stedile FC, Felix AHO, Mauler RS. Polypropylenefunctionalization with vinyltriethoxysilane. J Appl Polym Sci 1999;72:1313–9.
[62] Miller JD, Ishida H. Controlling and monitoring interfacial reactions incomposites of azidosilane modified glass filled polyethylene. Polym Compos1988;9:12–9.
[63] Miller JD, Ishida H, Maurer FHJ. Dynamic-mechanical properties of interfaciallymodified glass sphere filled polyethylene. Rheol Acta 1988;27:397–404.
[64] McFarren GA, Sanderson TF, Schappell FG. Azidosilane polymer–fillercoupling agent. Polym Eng Sci 1977;17:46–9.
[65] Gliesche K, Mäder E. Langfaserverstärkte Kunststoffe auf der Basis vonNaturfasern. In: Proceedings of 7th international techtextil symposium,Frankfurt, Germany; 1995.
[66] Abdelmouleh M, Boufi S, Belgacem MN, Duarte AP, Ben Salah A, Gandini A.Modification of cellulosic fibres with functionalised silanes: development ofsurface properties. Int J Adhes Adhes 2004;24:43–54.
[67] Hertl W. Mechanism of gaseous siloxane reaction with silica. I. J Phys Chem1968;72:1248–53.
[68] Krasnoslobodtsev AV, Smirnov SN. Effect of water on silanization of silica bytrimethoxysilanes. Langmuir 2002;18:3181–4.
[69] Castellano M, Gandini A, Fabbri P, Belgacem MN. Modification of cellulosefibres with organosilanes: under what conditions does coupling occur? JColloid Interf Sci 2004;273:505–11.
[70] Schneider MA, Brebner KI. Wood–polymer combinations: the chemicalmodification of wood by alkoxysilane coupling agents. Wood Sci Technol1985;19:67–73.
[71] Matuana LM, Balatinecz JJ, Park CB, Sodhi RNS. X-ray photoelectronspectroscopy study of silane-treated newsprint-fibers. Wood Sci Technol1999;33:259–70.
[72] Salon MCB, Gerbaud G, Abdelmouleh M, Bruzzese C, Boufi S, Belgacem MN.Studies of interactions between silane coupling agents and cellulose fiberswith liquid and solid-state NMR. Magnet Reson Chem 2007;45:473–83.
[73] Kang HJ, Meesiri W, Blum FD. NMR studies of the hydrolysis and molecularmotion of aminopropylsilane. Mater Sci Eng A – Struct 1990;126:265–70.
[74] Miller AC, Berg JC. Effect of silane coupling agent adsorbate structure onadhesion performance with a polymeric matrix. Compos Part A – Appl Sci2003;34:327–32.
Y. Xie et al. / Composites: Part A 41 (2010) 806–819 817

Author's personal copy
[75] Wacker Chemical AG publication. Products information brochure:organofunctional silanes from Wacker. <http://www.wacker.com/>.
[76] Tesoro G, Wu Y. Silane coupling agents: the role of the organofunctionalgroup. J Adhes Sci Technol 1991;5:771–84.
[77] Navoroj S, Culler R, Koenig JL, Ishida H. Structure and adsorptioncharacteristics of silane coupling agents on silica and E-glass fiber;dependence on pH. J Colloid Interf Sci 1984;97:309–17.
[78] Riegel B, Blittersdorf S, Kiefer W, Hofacker S, Mueller M, Schottner G. Kineticinvestigations of hydrolysis and condensation of theglycidoxypropyltrimethoxysilane/aminopropyltriethoxy-silane system bymeans of FT-Raman spectroscopy I. J Non-Cryst Solids 1998;226:76–84.
[79] Pohl ER, Osterholtz FD. Kinetics and mechanism of aqueous hydrolysis andcondensation of alkyltrialkoxysilanes. In: Ishida H, editor. Molecularcharacterization of composite interfaces. New York: Plenum Press; 1983.
[80] Pantoja M, Díaz-Benito B, Velasco F, Abenojar J, del Real JC. Analysis ofhydrolysis process of c-methacryloxypropyltrimethoxysilane and itsinfluence on the formation of silane coatings on 6063 aluminum alloy. ApplSurf Sci 2009;255:6386–90.
[81] Chiang CH, Ishida H, Koenig J. The structure of c-aminopropyltriethoxysilaneon glass surfaces. J Colloid Interf Sci 1980;74:396–404.
[82] Leyden DE, Atwater JB. Hydrolysis and condensation of alkoxysilanesinvestigated by internal reflection FTIR spectroscopy. J Adhes Sci Technol1991;5:815–29.
[83] Salon MCB, Abdelmouleh M, Boufi S, Belgacem MN, Gandini A. Silaneadsorption onto cellulose fibers: hydrolysis and condensation reactions. JColloid Interf Sci 2005;289:249–61.
[84] Salon MCB, Bayle PA, Abdelmouleh M, Boufi S, Belgacem MN. Kinetics ofhydrolysis and self-condensation reaction of silanes by NMR spectroscopy.Colloid Surf A 2008;312:83–91.
[85] Hill CAS, Mastery Farahani MR, Hale MDC. The use of organo alkoxysilanecoupling agents for wood preservation. Holzforschung 2004;58:316–25.
[86] Donath S, Militz H, Mai C. Creating water-repellent effects on wood bytreatment with silanes. Holzforschung 2006;60:40–6.
[87] Abdelmouleh M, Boufi S, ben Salah A, Belgacem MN, Gandini A. Interaction ofsilane coupling agents with cellulose. Langmuir 2002;18:3203–8.
[88] Daniels MW, Francis LF. Silane adsorption behavior, microstructure, andproperties of glycidoxypropyltrimethoxysilane-modified colloidal silicacoatings. J Colloid Interf Sci 1998;205:191–200.
[89] Nishiyama N, Horie K, Asakura T. Adsorption behavior of a silane couplingagent onto a colloidal silica surface studied by 29Si NMR spectroscopy. JColloid Interf Sci 1989;129:113–9.
[90] Vrancken KC, Coster LD, Voort PVD, Grobet PJ, Vansant EJ. The role of silanolsin the modification of silica gel with aminosilanes. J Colloid Interf Sci1995;170:71–7.
[91] Valadez-Gonzalez A, Cervantes-Uc JM, Olayo R, Herrera-Franco PJ. Effect offiber surface treatment on the fiber-matrix bond strength of natural fiberreinforced composites. Compos Part B – Eng 1999;30:309–20.
[92] Matias MC, De La Orden MU, Gonzalez-Sanchenz C, Martinez-Urreaga J.Comparative spectroscopic study of the modification of cellulosic materialswith different coupling agents. J Appl Polym Sci 2000;75:256–66.
[93] Valadez-Gonzalez A, Cervantes-Uc JM, Olayo R, Herrera-Franco PJ. Chemicalmodification of henequen fibers with an organosilane coupling agent.Compos Part B – Eng 1999;30:321–31.
[94] Herrera-Franco PJ, Valadez-Gonzalez A. A study of the mechanical propertiesof short natural-fiber reinforced composites. Compos Part B – Eng2005;36:597–608.
[95] Kokta BV, Maldas D, Daneault C, Beland P. Composites of polyvinyl chloride-wood fibers. III: effect of silane as coupling agent. J Vinyl Technol 1990;12:146–53.
[96] Arkles B, Steinmetz JR, Zazyczny J, Mehta P. Factors contributing to thestability of alkoxysilanes in aqueous solution. J Adhes Sci Technol 1992;6:193–206.
[97] Donath S, Militz H, Mai C. Wood modification with alkoxysilanes. Wood SciTechnol 2004;38:555–66.
[98] Van de Velde K, Kiekens P. Influence of fiber surface characteristics on theflax/polypropylene interface. J Thermoplast Compos 2001;14:244–60.
[99] Biagiotti J, Puglia D, Torre L, Kenny JM. A systematic investigation on theinfluence of the chemical treatment of natural fibers on the properties of theirpolymer matrix composites. Polym Compos 2004;25:470–9.
[100] Mohammed-Ziegler I, Marosi G, Matko S, Horvölgyi Z, Toth A. Silylation ofwood for potential protection against biodegradation. An ATR-FTIR, ESCA andcontact angle study. Polym Adv Technol 2003;14:790–5.
[101] Arbelaiz A, Fernández B, Ramos JA, Mondragon I. Thermal and crystallizationstudies of short flax fibre reinforced polypropylene matrix composites: effectof treatments. Thermochim Acta 2006;440:111–21.
[102] Sreekala MS, Kumaran MG, Thomas S. Oil palm fibers: morphology, chemicalcomposition, surface modification, and mechanical properties. J Appl PolymSci 1997;66:821–35.
[103] Varma DS, Varma M, Varma IK. Coir fiber II: evaluation as a reinforcement inunsaturated polyester resin composites. J Reinf Plast Comp 1985;4:419–31.
[104] Park JM, Kim PG, Jang JH, Wang Z, Hwang BS, DeVries KL. Interfacialevaluation and durability of modified Jute fibers/polypropylene (PP)composites using micromechanical test and acoustic emission. Compos PartB – Eng 2008;39:1042–61.
[105] Bashay AD, Kokta BV. Use of wood fibers in thermoplastic composites II:polyethylene. Polym Compos 1985;6:261–71.
[106] Mieck KP, Nechwatal A, Knobelsdorf C. Faser-matrix-haftung inkunststoffverbunden aus thermoplastischer matrix und flachs. 1. Dieausrüstung mit silanen. Die Angew Makromol Chem 1995;224:73–88.
[107] Xie Y. Silane treated wood flour as reinforcement for thermoplasticcomposites. Project report of German academic exchange service, Gerog-August-University Goettingen, Germany; 2008.
[108] Jensen RE, Palmese GR, Mcknight SH. Viscoelastic properties of alkoxy silane-epoxy interpenetrating networks. Int J Adhes Adhes 2006;26:103–15.
[109] Laly AP, Sabu T. Polarity parameters and dynamic mechanical behaviours ofchemically modified banana fiber reinforced polyester composites. ComposSci Technol 2003;63:1231–40.
[110] Xie Y. Silane treated wood flour as reinforcement for thermoplasticcomposites. Project progressive report of German academic exchangeservice, Gerog-August-University Goettingen, Germany; 2007.
[111] Tze WTY, Gardner DJ, Tripp CP, O’Neill SC. Cellulosic fiber/polymer adhesion:effect of fiber/matrix interfacial chemistry on the micromechanics of theinterphase. J Adhes Sci Technol 2006;20:1649–68.
[112] Le Digabel F, Boquillon N, Dole P, Monties B, Averous L. Properties ofthermoplastic composites based on wheat–straw lignocellulosic fillers. J ApplPolym Sci 2004;93:428–36.
[113] Beshay A, Hoa SV. Improved interface bonding between cellulosic fibers andthermoplastics. Sci Eng Compos Mater 1992;2(2):85–97.
[114] Hughes M, Carpenter J, Hill CAS. Deformation and fracture behaviour of flaxfibre reinforced thermosetting polymer matrix composites. J Mater Sci2007;42:2499–511.
[115] Xanthos M. Processing conditions and coupling agent effects inpolypropylene/wood flour composites. Plast Rubber Proc Appl 1983;3:223–8.
[116] Gaiolas C, Costa AP, Nunes M, Santos Siva MJ, Belgacem MN. Grafting of paperby silane coupling agents using cold-plasma discharge. Plasma Process Polym2008;5:444–52.
[117] Kim H, Jang J. Synthesis and characterization of vinyl silane modifiedimidazole copolymer as a novel corrosion inhibitor. Polym Bull 1997;38:249–56.
[118] Shah GB, Fuzail M, Anwar J. Aspects of the crosslinking of polyethylene withvinyl silane. J Appl Polym Sci 2004;92:3796–803.
[119] Shieh YT, Tsai TH. Silane grafting reactions of low-density polyethylene. JAppl Polym Sci 1998;69:255–61.
[120] Arbelaiz A, Fernández B, Cantero G, Llano-Ponte R, Valea A, Mondragon I.Mechanical properties of flax fibre/polypropylene composites. Influence offibre/matrix modification and glass fibre hybridization. Compos Part A – ApplSci 2005;36:1637–44.
[121] Bengtsson M, Oksman K. Profile extrusion and mechanical properties ofcrosslinked wood–thermoplastic composites. Polym Compos 2006;27: 184–94.
[122] Bengtsson M, Oksman K. The use of silane technology in crosslinkingpolyethylene/wood flour composites. Compos Part A – Appl Sci2006;37:752–65.
[123] Wong WK, Varrall DC. Role of molecular structure on the silane crosslinkingof polyethylene: the importance of resin molecular structure change duringsilane grafting. Polymer 1994;35:5447–52.
[124] Herrera-Franco PJ, Valadez-González A. Mechanical properties of continuousnatural fibre-reinforced polymer composites. Compos Part A – Appl Sci2004;35:339–45.
[125] Marsden JG, Orenski PJ. Azido-silane compositions. United States patent3944574; 1976.
[126] Arenas JF, Marcos JI, Otero JC, Tocón IL, Soto J. Nitrenes as intermediates inthe thermal decomposition of aliphatic azides. Int J Quantum Chem 2001;84:241–8.
[127] Towo AN, Ansell MP. Fatigue evaluation and dynamic mechanical thermalanalysis of sisal fibre–thermosetting resin composites. Compos Sci Technol2008;68:925–32.
[128] Kim HJ, Seo DW. Effect of water absorption fatigue on mechanical propertiesof sisal textile-reinforced composites. Int J Fatigue 2006;28:1307–14.
[129] Baley C, Busnel F, Grohens Y, Sire O. Influence of chemical treatments onsurface properties and adhesion of flax fibre–polyester resin. Compos Part A –Appl Sci 2006;37:1626–37.
[130] Hughes M, Hill CAS, Hague JRB. The fracture toughness of bast fibrereinforced polyester composites Part 1: evaluation and analysis. J Mater Sci2002;37:4669–76.
[131] Serier A, Pascault JP, My LT. Reaction in aminosilane–epoxy prepolymersystems I. Kinetics of epoxy–amine reactions. J Polym Sci Polym Chem1991;29:209–18.
[132] Chiang CH, Koenig JL. Chemical reactions occurring at the interface of epoxymatrix and aminosilane coupling agents in fiber-reinforced composites.Polym Compos 1980;1:88–92.
[133] George J, Ivens J, Verpoest I. Mechanical properties of flax fibre reinforcedepoxy composites. Die Angew Makromol Chem 1999;272:41–5.
[134] Uma Devi L, Bhagawan SS, Thomas S. Mechanical properties of pineapple leaffiber-reinforced polyester composites. J Appl Polym Sci 1997;64: 1739–48.
[135] Ganan P, Mondragon I. Fique fiber-reinforced polyester composites: effects offiber surface treatments on mechanical behavior. J Mater Sci 2004;39:3121–8.
[136] Pothan LA, Thomas S. Polarity parameters and dynamic mechanicalbehaviour of chemically modified banana fiber reinforced polyestercomposites. Compos Sci Technol 2003;63:1231–40.
[137] ASTM D1037-99. Standard test methods for evaluating properties of wood-base fiber and particle panel materials.
818 Y. Xie et al. / Composites: Part A 41 (2010) 806–819

Author's personal copy
[138] Wolcott M. The role of thermoplastics in conventional wood composites. In:Proceedings 30th international particleboard/composite materialssymposium. Washington State University;1996.
[139] Lundin T, Falk RH, Felton C. Accelerated weathering of natural fiber–thermoplastic composites: effects of ultraviolet exposure on bendingstrength and stiffness. In: The sixth international conference on woodfiber–plastic composites, Madison, WI; 2001.
[140] Wallenberger FT, Weston N. Natural fibers, plastics and composites. KluwerAcademic Publishers; 2004.
[141] Mohanty AK, Misra M, Drzal LT. Natural fibers, biopolymers, andbiocomposites. Taylor & Francis; 2005.
[142] Schirp A, Wolcott MP. Fungal degradation of wood–plastic composites andevaluation using dynamic mechanical analysis. J Appl Polym Sci2006;99:3138–46.
[143] Morris PI, Cooper PA. Recycled plastic/wood composite lumber attacked byFungi. Forest Prod J 1997;48:86–8.
[144] Lomelí-Ramírez MG, Ochoa-Ruiz HG, Fuentes-Talavera FJ, García-Enriquez S,Cerpa-Gallegos MA, Silva-Guzmán JA. Evaluation of accelerated decay ofwood plastic composites by Xylophagus fungi. Int Biodeter Biodegr2009;63:1030–5.
[145] Gnatowski M. Water absorption by wood plastic composites: field andlaboratory challenges. In: The 10th international conference on progress inbiofibre plastic composites, Toronto, Ontario; 2008.
[146] Sreekala MS, Thomas S. Effect of fibre surface modification on water-sorptioncharacteristics of oil palm fibres. Compos Sci Technol 2003;63:861–9.
[147] Ibach RE, Clemons CM. Biological resistance of polyethylene compositesmade with chemically modified fiber or flour. In: Proceedings of the 6thpacific rim bio-based composites symposium, Portland, OR, USA, 2002. p.574–84.
[148] Hill CAS, Abdul Khalil HPS. The effect of environmental exposure upon themechanical properties of coir and oil palm fiber reinforced composites. J ApplPolym Sci 2000;77:1322–30.
Y. Xie et al. / Composites: Part A 41 (2010) 806–819 819





























