Embed Size (px)
Citation preview

2386 East Heritage Way, Suite B, Salt Lake City, Utah 84109 USA Phone +1-877-628-7300 • Email—[email protected]
www.pachyonychia.org
15 March 2005
Use of Articles in the Pachyonychia Congenita Bibliography
The articles in the PC Bibliography may be restricted by copyright laws. These have been made available to you by PC Project for the exclusive use in teaching, scholar-ship or research regarding Pachyonychia Congenita. To the best of our understanding, in supplying this material to you we have followed the guidelines of Sec 107 regarding fair use of copyright materials. That section reads as follows:
Sec. 107. - Limitations on exclusive rights: Fair use Notwithstanding the provisions of sections 106 and 106A, the fair use of a copyrighted work, including such use by reproduction in copies or phonorecords or by any other means specified by that section, for purposes such as criticism, comment, news reporting, teaching (including multiple copies for classroom use), scholarship, or research, is not an infringement of copyright. In determining whether the use made of a work in any particular case is a fair use the factors to be considered shall include - (1) the purpose and character of the use, including whether such use is of a commercial nature or is for nonprofit educational purposes; (2) the nature of the copyrighted work; (3) the amount and substantiality of the portion used in relation to the copyrighted work as a whole; and (4) the effect of the use upon the potential market for or value of the copyrighted work. The fact that a work is unpublished shall not itself bar a finding of fair use if such finding is made upon consideration of all the above factors.
We hope that making available the relevant information on Pachyonychia Congenita will be a means of furthering research to find effective therapies and a cure for PC.

Statins Downregulate K6a Promoter Activity:A Possible Therapeutic Avenue forPachyonychia CongenitaYiwei Zhao1, Ulrike Gartner1, Frances J.D. Smith1 and W.H. Irwin McLean1
Pachyonychia congenita (PC) is a keratinizing disorder predominantly caused by mutations in keratin 6a (K6a)(B50% of cases) or K6b, K16, or K17. One means of treating PC is identification of small-molecule inhibitors ofPC-related keratins. Here, we cloned the human K6a promoter, and using a cell-based reporter gene assay, achemical library was screened for K6a inhibitors. One compound, compactin, the precursor of all cholesterol-lowering statins, was of particular interest. We found that, surprisingly, simvastatin and other statins inhibit K6apromoter activity and K6a protein expression. Further investigation showed that this effect works throughcholesterol/mevalonate pathway inhibition rather than an off-target effect. Inhibition of both basal and IFN-g-inducible K6a expression by statins was demonstrated. Both these K6a inhibitory effects were found to bemediated by Stat1 transcription factor, but only the IFN-g-inducible promoter activity was controlled via the Stat/JAK pathway. The repressive effect of statins was found to be mediated by the isoprenoid pathway downstreamof mevalonate (the intermediate following 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase) but upstream ofcholesterol, specifically the geranylgeranylation pathway. These data set the scene for further unravelingsignaling pathways that control the K6a promoter, as well as facilitating clinical trials for statins in PC patients.
Journal of Investigative Dermatology advance online publication, 10 March 2011; doi:10.1038/jid.2011.41
INTRODUCTIONPachyonychia congenita (PC) is a highly debilitatingkeratinizing disorder caused by mutations in any one of fourkeratin genes KRT6A, KRT6B, KRT16, or KRT17 (Bowdenet al., 1995; McLean et al., 1995; Smith et al., 1998). Thephenotypic and mutational spectrum found in PC is reportedby Wilson et al., 2011. For PC patients, the most painful,disabling feature is focal plantar keratoderma (Leachmanet al., 2005; Liao et al., 2007).
Currently, there are no specific therapies for PC but severalresearch groups are actively working on the development oftherapeutics (Kaspar et al., 2011), in particular using smallinterfering RNA (siRNA) to inhibit production of the mutantkeratin (Hickerson et al., 2006, 2008; Leachman et al., 2008;Smith et al., 2008). This preclinical work recently led to thefirst proof-of-concept siRNA clinical trial in the field ofdermatology, where a PC patient was treated by intradermalinjection of a mutation-specific siRNA (Leachman et al., 2010).
However, intradermal injection proved to be very painful andunsuitable for routine use. Hickerson et al., 2011, report apromising chemical approach to cutaneous siRNA delivery;however, this is still a long way from clinical application.
An alternative approach is to try to develop pharmaco-logical therapy for PC. Here, the problem of delivery is moretractable, as small-molecule drugs (typically o550 Da) maybe used systemically, or with suitable formulation and/orchemical modification, applied topically. However, thebiggest hurdle for development of small-molecule therapyfor an uncommon inherited disorder such as PC is theextremely high cost of drug development, typically hundredsof millions of dollars (Dickson and Gagnon, 2004; Kaspar,2005). Currently, the only drugs available to treat hyperker-atotic disorders such as PC are retinoids, which act viaretinoic acid response elements (RAREs) present in the genepromoters of many keratins and other structural molecules ofthe epidermis to repress gene expression (de The et al., 1990).These molecules are therefore not very specific for a givenkeratin or keratin-related gene, and their overall effect is tonon-specifically inhibit epidermal differentiation. For thisreason, retinoids are not well tolerated in keratinizingdisorders because, although they reduce hyperkeratosis, theythin the epidermis and lead to blistering, as well as a range ofother side-effects (DiGiovanna, 2010; Ormerod et al., 2010).
From the International Pachyonychia Congenita ResearchRegistry data, the major PC gene is KRT6A, where about50% of patients in the registry carry mutations (Smith et al.,this issue). Interestingly, the four keratin genes involved
& 2011 The Society for Investigative Dermatology www.jidonline.org 1
ORIGINAL ARTICLE
Received 26 August 2010; revised 28 December 2010; accepted 5 January2011
1Division of Molecular Medicine, Medical Sciences Institute, University ofDundee, Dundee, UK
Correspondence: W.H. Irwin McLean, Division of Molecular Medicine,Medical Sciences Institute, University of Dundee, Dundee DD1 5EH, UK.E-mail: [email protected]
Abbreviations: CMV, cytomegalovirus; FPP, farnesyl pyrophosphate; GGPP,geranylgeranyl pyrophosphate; K, keratin; KRT, keratin gene; K6a-luciferase,KRT6A-promoter-driven firefly luciferase; PC, pachyonychia congenita;RARE, retinoic acid response element; siRNA, small interfering RNA

in PC differ from other keratins associated with humangenetic disorders because they are regulated in both aninducible and constitutive manner. The inducible componentincludes induction during UV light exposure, wound healing,viral infections, psoriasis, cancer, and other challengesto epithelial tissues (Bernerd et al., 1993; Moll et al., 1994;Komine et al., 2001; Hattori et al., 2002; Wakabayashi et al.,2003; Endo et al., 2008). Such naturally occurring inductionand repression mechanisms led us to hypothesize that certainsmall molecules may exist that can interfere with this naturalinduction/repression machinery and thereby modulate theexpression of these inducible keratins. The proximal promo-ter of human keratin 6 (K6) has been studied previously usinga clone originally derived from the KRT6B promoter(Ma et al., 1997). It has been shown that the KRT6Bexpression is strongly upregulated by IFN-g and down-regulated by retinoic acid through IFN-g activation sites andRAREs in the promoter (Tomic-Canic et al., 1996; Freedberget al., 2001; Hattori et al., 2002). Through studies of theKeap1/Nrf2 pathway involved in activation of detoxificationenzymes such as ketoreductase (Nioi et al., 2003), it hasemerged that the stress-inducible keratins such as K6 can beactivated by chemicals, such as sulforaphane (Wakabayashiet al., 2003; Kerns et al., 2007), that induce gene expressionvia antioxidant response elements. Bioinformatics analysisof the human KRT6A promoter sequence revealed that thisindeed contains RAREs, IFN-g activation site elements,and antioxidant response elements (Table 1; Figure 1a); andhence, one would predict that activity of this promotershould be induced by IFN-g and sulforaphane, and inhibitedby retinoic acid. Thus, these compounds represent a goodstarting point to validate a high-throughput chemical library-screening assay based on the KRT6A promoter.
One approach to development of new drugs for use in PCor other rare disorders is to screen molecules alreadyapproved for human use, in the hope that an existing drugmay have a beneficial effect for a new, undiscovered diseaseindication. This is a high-risk but low-cost approach tofinding new drugs to treat rare disorders. Here, we describesuch a small-molecule-screening campaign aimed at identi-fying compounds to inhibit K6a gene/protein expression, aspossible means of increasing the pharmacological repertoirefor treatment of PC. Unexpectedly, we found that members ofthe statin family of cholesterol-lowering drugs are able toinhibit human KRT6A promoter activity.
RESULTSThe KRT6A promoter responds as predicted to knowninducers/inhibitors
A 6,058-bp fragment comprising the promoter region of thehuman KRT6A gene, extending to the transcriptional start siteof the gene, was generated by PCR cloning (Figure 1a).Bioinformatics analysis showed that this promoter contains asingle RARE site close to the cap site, as well as threeantioxidant response and seven IFN-g activation site elementsscattered throughout the 6 kb promoter fragment (Figure 1a;Table 1). By transient transfection, the KRT6A-promoter-driven firefly luciferase construct (hereafter described as
‘‘K6a-luciferase’’ for brevity) was expressed in the strain ofHaCaT cells in use in our laboratory, which we hadpreviously found by RT-PCR, DNA sequencing, and quanti-tative real-time RT-PCR to express only K6a but not K6b orK6c (data not shown). In all transfections, a standard traceamount of cytomegalovirus (CMV) promoter-driven Renillaluciferase was cotransfected to act as an internal control forcell viability and transfection efficiency. Preliminary transienttransfection experiments showed that K6a-luciferase is activein HaCaT cells and primary keratinocytes but showsnegligible expression in fibroblasts (data not shown).
Figure 1b shows the K6-luciferase activity in HaCaT cells,after normalization against the Renilla luciferase signal inuntreated cells and cells treated with IFN-g, sulforaphane,and retinoic acid, at concentrations commonly used in theliterature. As expected from studies of the K6b promoter andpresence of seven IFN-g activation site elements in thepromoter, treatment with 100 U ml�1 IFN-g strongly inducedK6a-luciferase expression by approximately 3-fold. Similarly,10 mM sulforaphane induced K6a-luciferase expressionby approximately 2-fold, consistent with the antioxidantresponse elements present in the promoter. In contrast, 1 mM
retinoic acid inhibited K6a-luciferase expression by B50%,consistent with the RAREs present in the promoter. All threeagents showed dose-dependent responses (data not shown).
Table 1. Regulatory elements in the human K6apromoter
Site Sequence (50–30)
ARE consensus RTKAYNNNGCR
K6a ARE1 (�1473) CTGAGTTAGCA
K6a ARE2 (�4420) TTGAAGTGGCA
K6a ARE3 (�4900) ATGAACATGCG
GAS consensus TTCNNNKAA
K6a GAS1 (�309) TTCAGTGAA
K6a GAS2 (�1582) TTCCCAGAA
K6a GAS3 (�2038) TTCAATTAA
K6a GAS4 (�4904) TTATATGAA
K6a GAS5 (�5711) TTCCCTGAA
K6a GAS6 (�5864) TTCCAGGAA
K6a GAS7 (�6054) TTCCCTGAA
RARE consensus (half site) AGGTCA
K6a RARE (�139) AGCTCACCTTAGGACTGGG
Abbreviations: ARE, antioxidant response element; GAS, IFN-g activationsite; RARE, retinoic acid response element.Note: numbering refers to base pairs upstream of the K6a transcriptionalstart site (http://genome.ucc.edu; gene symbol KRT6A).R=A/G; K=G/T; Y=C/T; N=A/G/C/T.ARE consensus derived from Erickson et al, 2002; GAS consensussequence from Kanno et al, 1993; Decker et al, 1997; and Contursi et al,2000; and RARE consensus sequence from Perlmann et al, 1993;Radoja, 1997; and Umesono et al, 1991.
2 Journal of Investigative Dermatology
Y Zhao et al.Statins Downregulate K6a Promoter Activity

Thus, the K6a-luciferase construct appeared to behave asexpected based on predicted inducers/repressors. For con-venience and reproducibility in a high-throughput-screeningcontext, a stable HaCaT clone was generated via puromycinselection. A clone showing good levels of constitutiveK6a-luciferase expression that responded in an identicalmanner to these agents, as described above (Figure 1b), waschosen for chemical library screening (data not shown).
Small-molecule library screening identified nine confirmedinhibitors of KRT6A
The chemical library chosen for screening was the NationalCancer Institute (NCI) 2,522 diversity set, which consists of2,522 small drug-like chemical compounds. After initialscreening of all compounds at a concentration of 20 mM,110 compounds were found that decreased K6a-luciferaseexpression by 40% compared with untreated controls. Ofthese, 59 compounds decreased K6a-luciferase activity by450% after normalization to Renilla luciferase expression. Inaddition, 61 compounds increased K6a promoter activityby B50% after normalization, which were not considered
further here. An additional 146 compounds up- or down-regulated K6a promoter activity but were found to havecytotoxic effects, and therefore were discarded from subse-quent analysis. All hit compounds were retested in triplicateusing the K6a-luciferase assay and were also counterscreenedagainst a CMV-driven luciferase construct to eliminate falsepositives, such as inhibitors of luciferase enzymatic activity,rather than K6a promoter activity or general inhibitors oftranscription/translation. Following counterscreening and hitvalidation, nine confirmed hits remained that inhibited K6apromoter activity by 450%, as exemplified in Figure 2a. Anindependent sample of all these compounds was sourced andin all cases, the activity was retained, further validating thesehit compounds (data not shown). Of these, eight werechemical structures that were not closely related to knowndrugs and are not discussed further here; however, one ofthe validated hits, compactin (Brown et al., 1978), is theprecursor of the cholesterol-lowering statin class of drugscurrently in widespread clinical use (Miller, 1999).
Statins inhibit KRT6A basal and inducible promoter activity
As shown in Figure 2b, simvastatin was able to inhibit K6a-luciferase activity to a similar degree to the initial validatedhit compound, compactin. The level of inhibition of the K6a-luciferase reporter was also similar to that seen with retinoicacid (Figure 1b). Figure 2c shows the effect of simvastatin onactual K6a protein expression in HaCat cells by westernblotting. K6a protein expression was reduced compared withDMSO (vehicle) control, but the effect was less marked thanwith 1 mM retinoic acid (Figure 2c). In a separate experiment,quantification of western blots was performed using the Li-CorOdyssey infrared imaging system (Li-Cor, Cambridge, UK).Specifically, K6a protein expression, normalized against aninternal actin standard, was substantially inhibited in a dose-dependent and time-dependent manner (Figure 2c). Importantly,we did not observe inhibition of K5 or K14 by western blotting(data not shown), indicating that the inhibitory effect hassome degree of specificity. Other statins tested includedpravastatin, fluvastatin, lovastatin, and mevastatin, all ofwhich inhibited K6a promoter activity in a similar manner(data not shown). Thus, it appeared that, unexpectedly,statins are somehow able to inhibit expression of the K6apromoter (Figure 2a and b), which was confirmed at the levelof protein expression (Figure 2c and d). For simplicity,simvastatin was employed in all subsequent experimentsdescribed here, although very similar effects were seen withother statins (data not shown).
To investigate whether the inhibitory effect of statins waslimited to the basal K6a promoter activity or whether statinsare also able inhibit inducible expression, simvastatin wasused in combination with IFN-g. Two concentrations of IFN-g(25 and 50 U ml�1) were shown to upregulate K6a-luciferasein a dose-dependent manner (Figure 3); however, in thepresence of simvastatin, not only was this inductioncompletely abolished but also, in addition, K6a-luciferasewas reduced to well below the basal expression level. Thus,statins appear to fully block the inducible expression of theK6a promoter as well as its basal activity.
Human KRT6A promoter (6,058 bp)
RARE site:
ARE sites:
GAS sites:
–6,000
350%
300%
250%
200%
150%
100%
50%
Nor
mal
ized
K6a
-Iuc
ifera
se a
ctiv
ity (
%)
0%K6a-luc IFN-γ SuI RA
–5,000 –4,000 –3,000 –2,000 –1,000KRT6A exon 1
Figure 1. K6a promoter elements and activities. (a) The human K6a promoter.
Schematic of the human KRT6A promoter (6,058 bp) with recognized elements
annotated (Table 1). (b) The K6a promoter responds to predicted inducers or
inhibitors. HaCaT cells were transiently transfected with the K6a-luciferase
construct and cytomegalovirus-Renilla luciferase. The resultant luciferase data
obtained at 24hours posttransfection were normalized against Renilla and the
untreated cells set to 100% (K6a-luc bar). In all, 100U ml�1 IFN-g resulted in an
approximately 3-fold increase in K6a-luciferase, and 10mM sulforaphane resulted
in an approximately 2-fold increase in K6a expression, whereas 1 mM retinoic
acid treatment reduced K6a expression by B50%. Error bars represent
standard error of the mean derived from assay replicates. ARE, antioxidant
response element; GAS, IFN-g activation site; K, keratin; KRT, keratin gene;
K6a-luciferase, KRT6A-promoter-driven firefly luciferase; luc, luciferase;
RA, retinoic acid; RARE, retinoic acid response element; Sul, sulforaphane.
www.jidonline.org 3
Y Zhao et al.Statins Downregulate K6a Promoter Activity

Inhibition of KRT6A by statins is mediated by the cholesterolpathway
Statins act by competitively inhibiting the enzyme HMGCoA reductase (3-hydroxy-3-methyl-glutaryl-CoA reductase),
which is the rate-limiting step in cholesterol biosynthesis, andtherefore the prime target for anti-cholesterol therapy(Russell, 1992). A schematic illustrating key features of thecholesterol biosynthesis pathway is shown in SupplementaryFigure 1. To determine whether inhibition of K6a promoteractivity was due to the effect of statins on inhibiting thecholesterol biosynthetic pathway rather than an off-targeteffect, e.g., binding directly to a transcription factor involvedin control of the K6a promoter, the intermediate in thecholesterol pathway downstream of 3-hydroxy-3-methyl-glutaryl-CoA reductase, mevalonate (Russell, 1992), wasused to see whether this could rescue K6a promoter activity.Figure 4 shows that addition of mevalonate alone has noappreciable effect on the constitutive expression level of theK6a-luciferase construct. In contrast, mevalonate is able torescue the inhibition of K6a-luciferase by simvastatin(Figure 4). Furthermore, mevalonate is able to rescuesimvastatin’s inhibition of K6a induction by IFN-g (Figure 4).In contrast, addition of cholesterol had no effect on basal K6apromoter activity in the presence or absence of simvastatin,plus or minus IFN-g (data not shown). Overall, these datasuggest that the effect of statins on K6a promoter activity is infact due to inhibition of the mevalonate/cholesterol biosyn-thetic pathway rather than via an off-target mechanism.The fact that mevalonate, but not cholesterol, can rescue theeffects of simvastatin implies that the key determinant in thisK6a inhibitory mechanism is a molecule downstream ofmevalonate but upstream of cholesterol in this biosynthetic
120
100
80
60
40
20
K6a
GAPDH
0DMSO
DM
SO
Compactin
RA
1µM
SS
8µM
DMSO
120,000
100,000
80,000
40,000
20,000
0Nor
mal
ized
K6a
-luci
fera
se (
%)
Untreated 1 µM SS 8 µM SS
24 Hours
48 Hours72 Hours
60,000
120
100
80
60
40
20
0SS
Nor
mal
ized
K6a
-luci
fera
se(%
)
Nor
mal
ized
K6a
-luci
fera
se(%
)
Figure 2. Small-molecule library screening identified statins as K6a inhibitors. (a) K6a-luciferase expression data were normalized as described in Figure 2.
Compared with untreated cells (K6a-luc), a significant library hit that reproducibly inhibited K6a-luciferase expression by B50% was compactin. (b) As
compactin is the precursor from which all cholesterol-lowering statins are synthesized, we tested a number of statins for K6a inhibitory effects. As shown here,
SS reduced K6a-luciferase expression by 450% compared with untreated cells (K6a-luc). Error bars represent standard error of the mean derived from assay
replicates. (c) Western blotting showed that 24 hours incubation with SS (8 mM) reduces K6a protein expression in HaCat cells compared with mock (DMSO)-
treated cells. In all, 1mM RA was used as a positive control. The blot was reprobed with GAPDH as a loading control. No appreciable effect of SS on K5 or K14
expression was observed (data not shown). (d) In an experiment separate from that of c, above, infrared imaging (Li-Cor Odyssey system) was used to quantify
K6a protein expression on western blots, normalizing against an internal actin-loading control. Over a time course of 24–72 hours, SS was seen to inhibit
K6a protein expression at both 1 and 8 mM concentrations. The equivalent quantity of DMSO (used to deliver SS) was used as a negative control. Note that
the 1 and 8 mM SS samples were run on the same gel but in non-consecutive lanes (as denoted by the bar). GAPDH, glyceraldehyde-3-phosphate dehydrogenase;
K, keratin; KRT, keratin gene; K6a-luciferase, KRT6A-promoter-driven firefly luciferase; RA, retinoic acid; SS, simvastatin.
400
350
300
250
200
150
100
50
0
Nor
mal
ized
K6a
-luci
fera
se a
ctiv
ity (
%)
K6a-luc SS IFN 25U IFN 50U SS+IFN 25U SS+IFN 50U
Figure 3. Statins inhibit both constitutive and IFN-inducible K6a expression.
K6a-luciferase expression data were normalized as described in Figure 2.
Again, SS (10 mM) reduced K6a-luciferase activity by 450% compared with
untreated cells (K6a-luc). Two different doses of IFN-g (25 and 50 U ml�1)
strongly induced K6a expression in a dose-dependent manner; however,
addition of 10 mM SS in addition to IFN-g (SSþ IFN 25U; SSþ IFN 50U) not
only blocked the 2- to 3-fold induction observed but also reduced K6a-
luciferase expression below the basal level (K6a-luc). Thus, statins inhibit both
the constitutive and inducible expression of the K6a promoter. Error bars
represent standard error of the mean derived from assay replicates. K, keratin;
KRT, keratin gene; K6a-luciferase, KRT6A-promoter-driven firefly luciferase;
luc, luciferase; SS, simvastatin.
4 Journal of Investigative Dermatology
Y Zhao et al.Statins Downregulate K6a Promoter Activity
pathway. The most obvious candidates are components of theisoprenylation pathways, which are important posttransla-tional modifications of G-proteins, which in turn are keysignaling molecules.
Effect of statins on KRT6A activity is mediated by isoprenylation
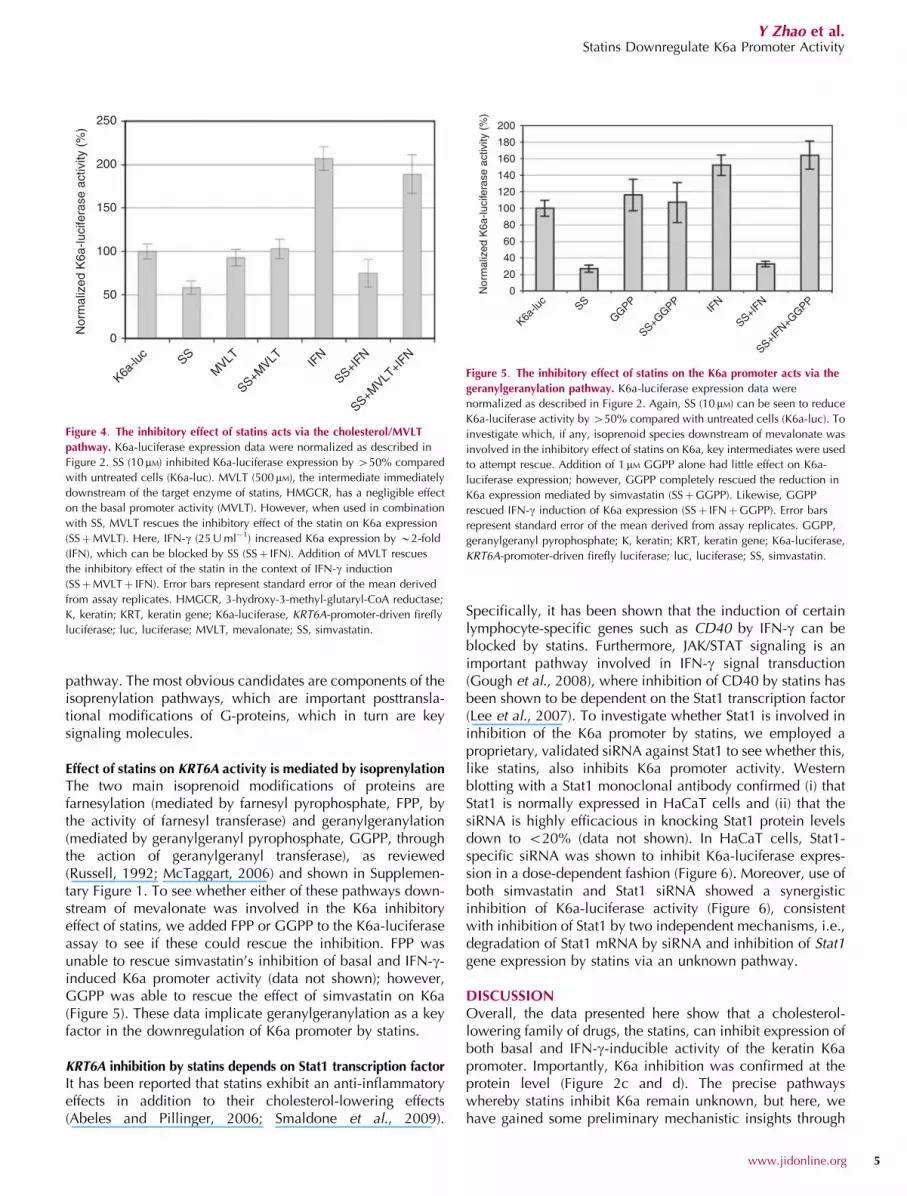
The two main isoprenoid modifications of proteins arefarnesylation (mediated by farnesyl pyrophosphate, FPP, bythe activity of farnesyl transferase) and geranylgeranylation(mediated by geranylgeranyl pyrophosphate, GGPP, throughthe action of geranylgeranyl transferase), as reviewed(Russell, 1992; McTaggart, 2006) and shown in Supplemen-tary Figure 1. To see whether either of these pathways down-stream of mevalonate was involved in the K6a inhibitoryeffect of statins, we added FPP or GGPP to the K6a-luciferaseassay to see if these could rescue the inhibition. FPP wasunable to rescue simvastatin’s inhibition of basal and IFN-g-induced K6a promoter activity (data not shown); however,GGPP was able to rescue the effect of simvastatin on K6a(Figure 5). These data implicate geranylgeranylation as a keyfactor in the downregulation of K6a promoter by statins.
KRT6A inhibition by statins depends on Stat1 transcription factor
It has been reported that statins exhibit an anti-inflammatoryeffects in addition to their cholesterol-lowering effects(Abeles and Pillinger, 2006; Smaldone et al., 2009).
Specifically, it has been shown that the induction of certainlymphocyte-specific genes such as CD40 by IFN-g can beblocked by statins. Furthermore, JAK/STAT signaling is animportant pathway involved in IFN-g signal transduction(Gough et al., 2008), where inhibition of CD40 by statins hasbeen shown to be dependent on the Stat1 transcription factor(Lee et al., 2007). To investigate whether Stat1 is involved ininhibition of the K6a promoter by statins, we employed aproprietary, validated siRNA against Stat1 to see whether this,like statins, also inhibits K6a promoter activity. Westernblotting with a Stat1 monoclonal antibody confirmed (i) thatStat1 is normally expressed in HaCaT cells and (ii) that thesiRNA is highly efficacious in knocking Stat1 protein levelsdown to o20% (data not shown). In HaCaT cells, Stat1-specific siRNA was shown to inhibit K6a-luciferase expres-sion in a dose-dependent fashion (Figure 6). Moreover, use ofboth simvastatin and Stat1 siRNA showed a synergisticinhibition of K6a-luciferase activity (Figure 6), consistentwith inhibition of Stat1 by two independent mechanisms, i.e.,degradation of Stat1 mRNA by siRNA and inhibition of Stat1gene expression by statins via an unknown pathway.
DISCUSSIONOverall, the data presented here show that a cholesterol-lowering family of drugs, the statins, can inhibit expression ofboth basal and IFN-g-inducible activity of the keratin K6apromoter. Importantly, K6a inhibition was confirmed at theprotein level (Figure 2c and d). The precise pathwayswhereby statins inhibit K6a remain unknown, but here, wehave gained some preliminary mechanistic insights through
250
200
150
100
50
0
Nor
mal
ized
K6a
-luci
fera
se a
ctiv
ity (
%)
K6a-lu
c SSM
VLT
SS+MVLT IF
N
SS+IFN
SS+MVLT
+IFN
Figure 4. The inhibitory effect of statins acts via the cholesterol/MVLT
pathway. K6a-luciferase expression data were normalized as described in
Figure 2. SS (10 mM) inhibited K6a-luciferase expression by 450% compared
with untreated cells (K6a-luc). MVLT (500 mM), the intermediate immediately
downstream of the target enzyme of statins, HMGCR, has a negligible effect
on the basal promoter activity (MVLT). However, when used in combination
with SS, MVLT rescues the inhibitory effect of the statin on K6a expression
(SSþMVLT). Here, IFN-g (25 U ml�1) increased K6a expression by B2-fold
(IFN), which can be blocked by SS (SSþ IFN). Addition of MVLT rescues
the inhibitory effect of the statin in the context of IFN-g induction
(SSþMVLTþ IFN). Error bars represent standard error of the mean derived
from assay replicates. HMGCR, 3-hydroxy-3-methyl-glutaryl-CoA reductase;
K, keratin; KRT, keratin gene; K6a-luciferase, KRT6A-promoter-driven firefly
luciferase; luc, luciferase; MVLT, mevalonate; SS, simvastatin.
200
180
160
140
120
100
80
60
40
0Nor
mal
ized
K6a
-luci
fera
se a
ctiv
ity (
%)
K6a-lu
cSS
GGPP
SS+GGPP
IFN
SS+IFN
SS+IFN+G
GPP
20
Figure 5. The inhibitory effect of statins on the K6a promoter acts via the
geranylgeranylation pathway. K6a-luciferase expression data were
normalized as described in Figure 2. Again, SS (10 mM) can be seen to reduce
K6a-luciferase activity by 450% compared with untreated cells (K6a-luc). To
investigate which, if any, isoprenoid species downstream of mevalonate was
involved in the inhibitory effect of statins on K6a, key intermediates were used
to attempt rescue. Addition of 1 mM GGPP alone had little effect on K6a-
luciferase expression; however, GGPP completely rescued the reduction in
K6a expression mediated by simvastatin (SSþGGPP). Likewise, GGPP
rescued IFN-g induction of K6a expression (SSþ IFNþGGPP). Error bars
represent standard error of the mean derived from assay replicates. GGPP,
geranylgeranyl pyrophosphate; K, keratin; KRT, keratin gene; K6a-luciferase,
KRT6A-promoter-driven firefly luciferase; luc, luciferase; SS, simvastatin.
www.jidonline.org 5
Y Zhao et al.Statins Downregulate K6a Promoter Activity

the use of siRNA against candidate genes, rescue ofexpression using pathway intermediates and specific kinaseinhibitors. Our data clearly show that the inhibitory effectsof statins on K6a are dependent on Stat1 transcription factor.Interestingly, a similar inhibitory effect has been shownin lymphocytes for another IFN-g-inducible gene, CD40(Lee et al., 2007).
We have shown that the K6a inhibitory effect of statins isdue bona fide inhibition of the cholesterol/mevalonatebiosynthesis pathway rather than an off-target effect (Figure 4),and interestingly, that this effect can be rescued bymevalonate but not cholesterol itself. Mevalonate is a keymolecule in biosynthesis of the isoprenoids, the farnesyl andgeranylgeranyl groups added to many G-proteins posttransla-tionally, such as members of the Rho and Ras families ofsmall guanosine triphosphate-binding proteins, which are keysignaling molecules and prime suspects in the immuno-modulatory effects of statins (Smaldone et al., 2009). Our datastrongly suggest that it is the geranylgeranylation pathwayrather than the farnesylation pathway that is responsiblebecause GGPP (Figure 5) can rescue K6a promoter inhibitionby statins, but not FPP (McTaggart, 2006).
Only in recent years, through the availability of bothcommercial and non-profit chemical compound libraries(such as the free NCI library used here), coupled with theincreased availability of robotics suitable for high-throughoutscreening, has the advent of drug discovery been seen in anacademic context, outside of the biopharmaceutical industry.Our study illustrates two key outcomes of academic drugdiscovery. First, fresh insights have been gained into thesignaling pathways that control keratin expression. As therewas no previous obvious clue that keratin gene expressionwas in any way dependent upon the cholesterol biosyntheticpathway, this somewhat surprising result opens up anew avenue for research into the control of keratin geneexpression. Second, and importantly, we have identified analready approved, widely used, and inexpensive drug that
might prove to be of use in a neglected inherited skin disorderor group of disorders, immediately opening up the possibilityof clinical trials. This circumvents the hundreds of millions ofdollars required to develop a drug from scratch, which isunfeasible in an ultra-rare disorder. Because of the rarity of PCand related keratinizing disorders, a chemical library screenbased on a simple reporter gene promoter assay is somethingwhich the profit-driven pharmaceutical industry is likely tohave considered to be too high risk in relation to potentialreturn on the investment; however, this makes an idealacademic drug discovery project. In the future, it is likely thatthere will be closer collaboration between academia andindustry in drug development (Vallance et al., 2010).
We are continuing our efforts to elucidate the precisemechanism whereby statins inhibit K6a gene expression.However, given the excellent safety profile of statins, we arealready involved in open-label clinical trials, with recruitment ofa small case series of mutation-defined PC patients identifiedthrough the International Pachyonychia Congenita ResearchRegistry. These trials will make use of both systemic andtopically formulated statins. While it is highly unlikely thatstatins will show complete specificity for keratins, their generalanti-inflammatory effects might also be useful in inhibiting theprocess of hyperkeratosis, whose underlying pathomechanismsremain poorly understood, but is thought to involve inflamma-tory factors such as tumor necrosis factor-a. Even if onlypartially effective, the use of statins in the treatment-keratinizingdisorders such as PC might complement other broad-spectrumdrugs such as retinoids. It is also possible that a combinationtherapy of low-dose systemic or topical retinoids, supplementedby statins, may prove to be more effective.
In conclusion, we have identified statins as potential K6ainhibitory drugs, facilitating a range of low-cost, safe clinicaltrials in the orphan disease PC.
MATERIALS AND METHODSPromoter constructs
A 6,058 bp DNA fragment comprising the KRT6A promoter
sequence upstream of, and including the transcription start site,
was generated from genomic DNA of a normal, healthy control
individual using primers PRM3.L (50-TTCCTAGCCATGTTGTGTGT
TC-30) and PRM3.R (50-GAGGGAAGAGAAGCAGGACTAG-30). This
fragment was cloned into plasmid pCR2.1 (Invitrogen, Paisley, UK)
and fully sequenced. The full-length insert from a sequence-verified
clone was subcloned into the pGL4.21 promoterless vector contain-
ing a humanized, high-turnover firefly luciferase gene (luc2P ;
Promega, Southampton, UK) using KpnI and XhoI. A control plasmid
was generated by subcloning the CMV promoter (BamHI/BglII
fragment) from pcDNA3 (Invitrogen) into pGL4.21 (BamHI site). The
pRL-CMV plasmid (Renilla luciferase driven by the CMV promoter)
was obtained from Promega.
Cell lines
The HaCaT cell line was obtained from Professor Birgit Lane’s Laboratory,
School of Life Sciences, University of Dundee, UK, originally derived
from human adult skin keratinocytes (Boukamp et al., 1988). HaCaT cells
were maintained in Gibco DMEM (Invitrogen, Paisley, UK) supplemented
with 10% keratinocytes, Gibco) at 37 1C with a 5% CO2 atmosphere.
120
100
80
60
40
20
0Nor
mal
ized
K6a
-luci
fera
se a
ctiv
ity (
%)
K6a-luc SS siRNA 7nM
siRNA 20nM
siRNA 67nM
siRNA 7 nM+SS
siRNA 20 nM+SS
siRNA 67 nM+SS
Figure 6. Stat1 transcription factor, known to be involved in IFN-c signaling,
is linked to K6a expression. Stat1 has been linked to expression of IFN-g-
inducible genes in lymphocytes, e.g., CD40. To see whether Stat1 was linked
to K6a expression, we used a specific siRNA for Stat1. K6a expression was
inhibited by Stat1 siRNA in a dose-dependent manner (siRNA 7–67 nM). When
SS was used in combination with the Stat1 siRNA, the dose-dependent
inhibitory effect increased. It has been suggested that statins inhibit expression
of Stat1 via an unknown pathway (Lee et al., 2007). Our data are consistent
with this, as action of statins at the transcriptional level synergizes with
degradation of the Stat1 mRNA. Error bars represent standard error of the
mean. K, keratin; siRNA, small interfering RNA; SS, simvastatin.
6 Journal of Investigative Dermatology
Y Zhao et al.Statins Downregulate K6a Promoter Activity

TransfectionFor transient transfection of HaCaT or fibroblasts, cells were plated
in 96-well plates at a density of B1� 104 cells per well in 100ml of
DMEM with 10% fetal calf serum 24 hours before transfection. These
conditions routinely gave 70% confluency at the time of transfec-
tion. In all, 0.05 mg of plasmid DNA, with or without 0.005 mg pRL-
CMV Renilla plasmid, was complexed with 0.15 ml Fugene 6 (Roche,
West Sussex, UK) in 4.85 ml of DMEM with no fetal calf serum or
antibiotics. DNA–Fugene 6 complexes were mixed gently and incubated
at room temperature for 20 minutes before adding to cells. For stable
transfection, HaCaT cells were transfected with linearized plasmid
(KRT6A promoter in pGL4.21, above) in 10 cm dishes using a scaled-
up version of the transfection protocol and clones selected using
1mg ml�1 puromycin (Sigma-Aldrich, Poole, UK).
Dual-luciferase reporter assay system
At 24 hours after transfection, medium was removed and cells in
96-well plates were rinsed in phosphate-buffered saline solution
(Invitrogen). Cells were lysed in 20 ml 1� luciferase lysis buffer
(Dual-Luciferase Reporter Assay System, Promega) at room tem-
perature for 15 min. Both firefly and Renilla luciferase activities
were measured by LUMI Star OPTIMA luminometer (BMG Labtech,
Aylesbury, UK) according to manufacturer’s protocol.
Western blottingTotal protein extracts of HaCat cells were subjected to Coomassie
blue staining and western blotting, as described previously (Smith
et al., 2008). Keratin expression was detected by immunoblotting
using 1 hour incubations of the following primary antibodies: 1:200
dilution anti-K6 (Ks6.KA12; Progen Biotechnik, GmbH, Heidelberg,
Germany); 1:1,000 anti-K14 (LL001); and 1:10,000 dilution anti-K5
(BL18). All antibodies were kindly donated by EB Lane, College of
Life Sciences, University of Dundee, Dundee, UK. On western blots,
protein bands were either subjected to enhanced chemilumin-
secence staining or, alternatively, were detected and quantified
using a Li-Cor Odyssey infrared protein imager according to the
manufacturer’s recommended protocols (Li-Cor, Cambridge, UK),
and data normalized against an internal actin standard.
Reagents
The NCI 2,522 chemical library was obtained from the NCI,
National Institutes of Health, Bethesda, MD. IFN-g, simvastatin,
pravastatin, fluvastatin, lovastatin, and mevastatin were purchased
from Calbiochem (San Diego, CA). Retinoic acid, DL-mevalonate,
cholesterol, GGPP, and FPP were obtained from Sigma-Aldrich.
Sulforaphane was kindly provided by Professor John Hayes (Nine-
wells Medical School, Dundee, UK). Stat1 siRNA and antibody were
purchased from Cell Signaling Technology (Beverly, MA).
CONFLICT OF INTERESTYZ, FJDS, and WHIM have filed a patent on the use of statins to treatkeratinizing disorders.
ACKNOWLEDGMENTSWe are very grateful to Professor Sir David Lane, Division of MolecularMedicine, University of Dundee, UK, and Dr Sonia Lain, Karolinska Institute,Stockholm, Sweden, for help and advice on assay development and small-molecule screening, and Dr Nick Westwood, Department of Chemistry,St Andrews University, UK, for his enthusiastic encouragement and expertadvice on chemical structures. We thank Professor Inke Nathke, College of
Life Sciences, University of Dundee, for use of the Li-Cor Odyssey system.This work was funded by Pachyonychia Congenita Project (FJDS and WHIM),and therapy development in the McLean/Smith lab is also funded by grantsfrom DEBRA UK (WHIM) and the Medical Research Council (WHIM).
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at http://www.nature.com/jid
REFERENCES
Abeles AM, Pillinger MH (2006) Statins as antiinflammatory and immuno-modulatory agents: a future in rheumatologic therapy? Arthritis Rheum54:393–407
Bernerd F, Magnaldo T, Freedberg IM et al. (1993) Expression ofthe carcinoma-associated keratin K6 and the role of AP-1 proto-oncoproteins. Gene Expr 3:187–99
Boukamp P, Petrussevska RT, Breitkreutz D et al. (1988) Normal keratiniza-tion in a spontaneously immortalized aneuploid human keratinocyte cellline. J Cell Biol 106:761–71
Bowden PE, Haley JL, Kansky A et al. (1995) Mutation of a type II keratin gene(K6a) in pachyonychia congenita. Nat Genet 10:363–5
Brown MS, Faust JR, Goldstein JL et al. (1978) Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblastsincubated with compactin (ML-236B), a competitive inhibitor of thereductase. J Biol Chem 253:1121–8
Contursi C, Wang IM, Gabriele L et al. (2000) IFN consensus sequencebinding protein potentiates STAT1-dependent activation of IFNgamma-responsive promoters in macrophages. Proc Natl Acad Sci USA 97:91–6
de The H, Vivanco-Ruiz MM, Tiollais P et al. (1990) Identification of aretinoic acid responsive element in the retinoic acid receptor beta gene.Nature 343:177–80
Decker T, Kovarik P, Meinke A (1997) GAS elements: a few nucleotides witha major impact on cytokine-induced gene expression. J InterferonCytokine Res 17:121–34
Dickson M, Gagnon JP (2004) Key factors in the rising cost of new drugdiscovery and development. Nat Rev Drug Discov 3:417–29
DiGiovanna JJ (2010) Fracturing support for the role of systemic retinoidtherapy as a cause of bone demineralization. Arch Dermatol 146:551–3
Endo H, Sugioka Y, Nakagi Y et al. (2008) A novel role of the NRF2transcription factor in the regulation of arsenite-mediated keratin 16 geneexpression in human keratinocytes. Environ Health Perspect 116:873–9
Erickson AM, Nevarea Z, Gipp JJ et al. (2002) Identification of a variantantioxidant response element in the promoter of the human glutamate-cysteine ligase modifier subunit gene. Revision of the ARE consensussequence. J Biol Chem 277:30730–7
Freedberg IM, Tomic-Canic M, Komine M et al. (2001) Keratins and thekeratinocyte activation cycle. J Invest Dermatol 116:633–40
Gough DJ, Levy DE, Johnstone RW et al. (2008) IFNgamma signaling-doesit mean JAK-STAT? Cytokine Growth Factor Rev 19:383–94
Hattori N, Komine M, Yano S et al. (2002) Interferon-gamma, a strongsuppressor of cell proliferation, induces upregulation of keratin K6, oneof the inflammatory- and proliferation-associated keratins. J InvestDermatol 119:403–10
Hickerson RP, Flores MA, Leake D et al. (2011) Use of self-delivery siRNAsto inhibit gene expression in an organotypic pachyonychia congenitamodel. J Invest Dermatol; e-pub ahead of print 20 Jan 2011
Hickerson RP, Smith FJD, McLean WHI et al. (2006) SiRNA-mediatedselective inhibition of mutant keratin mRNAs responsible for the skindisorder pachyonychia congenita. Ann N Y Acad Sci 1082:56–61.
Hickerson RP, Smith FJD, Reeves RE et al. (2008) Single-nucleotide-specificsiRNA targeting in a dominant-negative skin model. J Invest Dermatol128:594–605
Kanno Y, Kozak CA, Schindler C et al. (1993) The genomic structure of themurine ICSBP gene reveals the presence of the gamma interferon-responsive element, to which an ISGF3 alpha subunit (or similar)molecule binds. Mol Cell Biol 13:3951–63
www.jidonline.org 7
Y Zhao et al.Statins Downregulate K6a Promoter Activity

Kaspar RL (2005) Challenges in developing therapies for rare diseasesincluding pachyonychia congenita. J Investig Dermatol Symp Proc10:62–6
Kaspar RL, Leachman SA, McLean WHI et al. Toward a treatment for PC:report on the 7th annual International Pachyonychia CongenitaConsortium meeting. J Invest Dermatol (in press)
Kerns ML, DePianto D, Dinkova-Kostova AT et al. (2007) Reprogramming ofkeratin biosynthesis by sulforaphane restores skin integrity in epidermo-lysis bullosa simplex. Proc Natl Acad Sci USA 104:14460–5
Komine M, Rao LS, Freedberg IM et al. (2001) Interleukin-1 inducestranscription of keratin K6 in human epidermal keratinocytes.J Invest Dermatol 116:330–8
Leachman SA, Hickerson RP, Hull PR et al. (2008) Therapeutic siRNAs fordominant genetic skin disorders including pachyonychia congenita.J Dermatol Sci 51:151–7
Leachman SA, Hickerson RP, Schwartz ME et al. (2010) First-in-humanmutation-targeted siRNA phase Ib trial of an inherited skin disorder.Mol Ther 18:442–6
Leachman SA, Kaspar RL, Fleckman P et al. (2005) Clinical and pathologicalfeatures of pachyonychia congenita. J Investig Dermatol Symp Proc10:3–17
Lee SJ, Qin H, Benveniste EN (2007) Simvastatin inhibits IFN-gamma-inducedCD40 gene expression by suppressing STAT-1alpha. J Leukoc Biol82:436–47
Liao H, Sayers JM, Wilson NJ et al. (2007) A spectrum of mutations in keratinsK6a, K16 and K17 causing pachyonychia congenita. J Dermatol Sci48:199–205
Ma S, Rao L, Freedberg IM et al. (1997) Transcriptional control of K5, K6, K14,and K17 keratin genes by AP-1 and NF-kappaB family members.Gene Expr 6:361–70
McLean WHI, Rugg EL, Lunny DP et al. (1995) Keratin 16 and keratin 17mutations cause pachyonychia congenita. Nat Genet 9:273–8
McTaggart SJ (2006) Isoprenylated proteins. Cell Mol Life Sci 63:255–67
Miller M (1999) New developments in the treatment of low high-densitylipoprotein cholesterol. Curr Atheroscler Rep 1:24–30
Moll I, Bohnert E, Treib U et al. (1994) Effects of ultraviolet B radiationon cytoskeletal and adhesion molecules in human epidermis.Photodermatol Photoimmunol Photomed 10:26–32
Nioi P, McMahon M, Itoh K et al. (2003) Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensussequence. Biochem J 374:337–48
Ormerod AD, Campalani E, Goodfield MJ (2010) British Association ofDermatologists guidelines on the efficacy and use of acitretin indermatology. Br J Dermatol 162:952–63
Perlmann T, Rangarajan PN, Umesono K et al. (1993) Determinants forselective RAR and TR recognition of direct repeat HREs. Genes Dev7:1411–22
Radoja N, Diaz DV, Minars TJ et al. (1997) Specific organization of thenegative response elements for retinoic acid and thyroid hormonereceptors in keratin gene family. J Invest Dermatol 109:566–72
Russell DW (1992) Cholesterol biosynthesis and metabolism. CardiovascDrugs Ther 6:103–10
Smaldone C, Brugaletta S, Pazzano V et al. (2009) Immunomodulator activityof 3-hydroxy-3-methilglutaryl-CoA inhibitors. Cardiovasc HematolAgents Med Chem 7:279–94
Smith FJD, Hickerson RP, Sayers JM et al. (2008) Development of therapeuticsiRNAs for pachyonychia congenita. J Invest Dermatol 128:50–8
Smith FJD, Jonkman MF, van Goor H et al. (1998) A mutation in humankeratin K6b produces a phenocopy of the K17 disorder pachyonychiacongenita type 2. Hum Molec Genet 7:1143–8
Tomic-Canic M, Freedberg IM, Blumenberg M (1996) Codominant regulationof keratin gene expression by cell surface receptors and nuclearreceptors. Exp Cell Res 224:96–102
Umesono K, Murakami KK, Thompson CC et al. (1991) Direct repeats asselective response elements for the thyroid hormone, retinoic acid, andvitamin D3 receptors. Cell 65:1255–66
Vallance P, Williams P, Dollery C (2010) The future is much closercollaboration between the pharmaceutical industry and academicmedical centers. Clin Pharmacol Ther 87:525–7
Wakabayashi N, Itoh K, Wakabayashi J et al. (2003) Keap1-null mutationleads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet35:238–45
Wilson NJ, Leachman SA, Hansen CD et al. (2011) A large mutational studyin pachyonychia congenita. J Invest Dermatol; e-pub ahead of print17 Feb 2011
8 Journal of Investigative Dermatology
Y Zhao et al.Statins Downregulate K6a Promoter Activity

35
Zhao Y, Gartner U, Smith FJD and McLean WHI
Statins down-regulate K6a promoter activity – a possible therapeutic avenue for
pachyonychia congenita
Supplementary Figure 1
Summary of the cholesterol biosynthetic pathway. CoA = Coenzyme-A; PP =
pyrophosphate; and HMG = 3-hydroxy-3-methyl-glutaryl. Statins inhibit the enzyme
HMG-CoA redeuctase, which normally catalyzes the conversion of HMG-CoA to form
L-mevalonate.





























