Embed Size (px)
Citation preview

3
1 INTRODUÇÃO
Os principais avanços científicos e tecnológicos são
as principais causas de mudanças na pratica da moderna
hematologia, influindo especialmente no diagnostico
laboratorial e nos procedimentos terapêuticos de algumas
patologias com destaque para anemias falciformes,
hemofilias e leucemias. O progresso obtido pela imunologia
tem alavancado o desenvolvimento do diagnostico
laboratorial de doenças hematológicas mielo e
linfoproliferativas, bem como determinando especificidades
da fisiologia leucocitária diante das toxicidades de
proveniências bacterianas e virais. Essas conquistas
ultrapassaram as fronteiras das aplicações laboratoriais e
forneceram conhecimentos (ANDRIOLO, 2000).
A anemia é considerada a doença mais prevalente em
todo o mundo sendo consequência da incapacidade do setor
hematopoiético em manter a concentração de hemoglobina
acima de 12 g/dL. Ocorre como problema de ocorrência de
saúde pública em diversos países (principalmente naqueles
em desenvolvimento como o Brasil), com importantes
consequências para a saúde humana e desenvolvimento das
nações (ADAMSON, 2006).
Estima-se que na América Latina aproximadamente 10-30%
das mulheres em idade fértil apresentem algum tipo de
anemia, principalmente associada à deficiência de ferro.
Reduções nos níveis de hemoglobina disponíveis promovem
alterações morfológicas, bioquímicas e estruturais, com

4
sérias conseqüências para o desenvolvimento do indivíduo
(CANÇADO; JESUS, 2007).
De acordo com estimativa do Ministério da Saúde, cerca
de 45% das crianças brasileiras de até 5 anos (10 milhões
de pessoas) tem anemia (ANVISA, 2006). O Brasil tem uma
alta incidência de anemia por deficiência de ferro: 50 por
cento em crianças (menores de 2 anos de idade) e 35 por
cento em gestantes. (ADAMSON, 2006).
O hemograma constitui um importante exame laboratorial
que permite avaliar o estado de saúde geral de um
indivíduo. As alterações observadas neste exame permitem ao
médico avaliar patologias relacionadas às séries vermelha
(anemias, policitemia, malária), branca (leucemias,
infecções diversas) e plaquetas (púrpuras,
trombocitopenias) e relacioná-las aos achados clínicos
observados no paciente (TEIXEIRA JUNIOR; SILVA; MAGALHAES,
2011).
Na interpretação do hemograma, atenção especial deve
ser dada ao número de eritrócitos, valores de hemoglobina,
hematócrito, assim como aos índices hematimétricos (VCM,
HCM, CHCM), possibilitando ao médico vislumbrar as
possibilidades diagnósticas (TERRA, 2000).
A partir dos dados do hemograma, as anemias podem ser
classificadas em três tipos fundamentais: normocítica
normocrômicas, microcítica hipocrômica e macrocítica.
Anemia normocítica pode ocorrer em diversas patologias como
nas anemias hemolíticas ou ser causada por perda sanguínea

5
aguda, doença crônica, problemas hepáticos e renais (MOURA;
WADA; PURCHIO, 2006).
A avaliação diagnóstica do paciente com anemia inclui
uma história e exame físico detalhados, e um mínimo de
exames laboratoriais. O primeiro exame a ser solicitado é o
hemograma, o qual deve ser sempre acompanhado da contagem
de reticulócitos, para definirmos a causa da anemia como
secundária; a diminuição de produção ou aumento de
destruição de dos glóbulos vermelhos (ADAMSON, 2006).
Segundo a Organização Mundial da Saúde, considera-se
anêmico um indivíduo com nível de hemoglobina menor que 12
g/dL. Laboratorialmente, a anemia caracteriza-se por
diminuição do hematócrito, queda na concentração de
hemoglobina no sangue ou variação dos índices
hematimétricos, que dependem de fatores como fase de
desenvolvimento individual, estimulação hormonal, à tensão
de oxigênio no ambiente, idade e sexo (LIMA; GROTTO, 2004).
DESENVOLVIMENTO
2.0 Leucócitos

6
Os leucócitos são as principais células sanguíneas
envolvidas na resposta inflamatória, embora plaquetas e
eritrócitos também participem. Os leucócitos são
classificados em neutrófilos (40%-75%), linfócitos (20%-
50%), monócitos (2%-10%), eosinófilos (1%-6%) e basófilos
(<1%). Destes, os neutrófilos são os mais importantes na
patogênese da inflamação. São as células predominantes nas
primeiras 6 a 24 horas nas inflamações agudas. Medem de 12
a 18 micrômetros, duram de 7-10 horas na circulação e
sofrem apoptose em 24 horas. (JUNQUEIRA; CARNEIRO, 2004).
Os leucócitos são produzidos por hematopoiese, na fase
fetal esse processo ocorre no timo, baço, fígado e medula
óssea, porém na fase pós fetal, somente a medula óssea é
que realiza o processo, ativando os outros órgãos somente
se a medula tiver alguma deficiência ou em caso de
urgência. Os leucócitos são são originados na hematopoiese
através de uma única célula, a célula tronco pluripotencial
e liberados na corrente sanguínea, são células grandes que
possuem núcleo e podem mudar de forma e atravessar os
capilares e também se direcionar aos tecidos para fazer
resposta imunológica. (FAILACE, 1992).
Os Monócitos possuem grânulos no citoplasma que tem
cor azulada, tem o núcleo muito denso que ocupa quase toda
a célula, é o leucócito de maior tamanho, aparecem no
sangue no caso de infecções, recuperação de infecções e
outras doenças como lupus e artrite reumatóide. Os
monócitos ficam cerca de 8 horas no sanguem e migram para o
tecido dando origem aos macrófagos, que são responsáveis

7
por fagocitar microorganismos, células mortas, células
tumorais, partículas estranhas e participa da imunidade
humoral como célula apresentadora de antígeno ao linfócito
T. (JUNQUEIRA; CARNEIRO, 2004).
Já os Linfócitos são célula menor, com núcleo esférico
cobrindo 90% da célula. Tem variação de três tipos de
linfócitos: linfócito T, B e Nk. Os linfócitos NK ou
Natural Killer liberam grânulos citotóxicos que destróem
células tumorais e células virais. Os linfócitos B são
responsáveis por reconhecer o antígeno e liberar
imunoglobulinas ou anticorpos que defenderão o organismo de
infecções no m omento do contato com o antígeno e
reservarão esses anticorpos para toda a vida no caso de
novo contato com o mesmo antígeno. Por fim, os linfócitos T
destróem o antígeno através da apoptose, essas células
ajudam a evitar as doenças auto imune. São encontrados no
leucograma em um valor aproximado de 24 a 32%. (LOURENZI,
1999).
Os Basófilo são encontrados em menor número do
leucograma representando de 0 a 1%, possuem o núcleo com
forma de S com grânulos no citoplasma, produzem histamina e
heparina, podem agir em processos alérgicos, sinusite,
nefrose, anemia, entre outros. E os Neutrófilos tem duas
variações entre eles, os segmentados e os bastonetes. São
células que apresentam o citoplasma rosa claro e estão em
maior quantidade no sangue por volta de 65% considerando os
segmentados, pois os bastonetes se apresentam de 3 a 6%.
(JUNQUEIRA; CARNEIRO, 2004).

8
Os bastonetes se apresentam com um núcleo em forma de
U e o segmentado tem a cromatina do núcleo com invaginações
com três a quatro segmentos. São células especializadas em
fagocitose principalmente de microorganismos. Eosinófilo
possuem citoplasma abundante com grânulos alaranjados e
possuem núcleo bilobulado. Podem aparecer em processos
alérgicos ou em casos de parasitoses. Encontram se em um
leucograma de 2 a 4%., todos podem ser observados na
Fig.01. (FAILACE, 1992).
O leucograma é a parte do hemograma que avalia os
leucócitos. Estes são também conhecidos como série branca
ou glóbulos brancos. São as células de defesa responsáveis
por combater agentes invasores. Os leucócitos são, na
verdade, um grupo de diferentes células, com diferentes
funções no sistema imune. Alguns leucócitos atacam
diretamente o invasor, outros produzem anticorpos, outros
apenas fazem a identificação e assim por diante. A contagem
de leucócitos indica a quantidade de leucócitos em um
microlitro (milímetro cúbico) de sangue total. (FAILACE;
FERNANDES, 2009).
As contagens de leucócitos podem variar até em 2.000,
em qualquer dia em particular, em função de exercício
desgastante, tensão ou digestão. A contagem de leucócitos
pode aumentar ou diminuir significativamente em
determinadas doenças, porém é diagnosticamente útil somente
quando o diferencial de glóbulos brancos e o estado clínico
do paciente são levados em consideração. (VERRASTRO, 2005).

9
O diferencial de leucócitos é usado para avaliar a
distribuição e morfologia dos glóbulos brancos, fornecendo
informação mais específica sobre o sistema imune do
paciente do que a contagem de leucócitos isoladamente. Os
glóbulos brancos são classificados de acordo com os cinco
tipos principais – neutrófilos, eosinófilos, basófilos,
linfócitos e monócitos – sendo determinada a porcentagem de
cada tipo. A contagem diferencial é o valor percentual de
cada tipo de glóbulo branco no sangue. O número absoluto de
cada tipo de glóbulo branco é obtido por meio da
multiplicação do valor percentual de cada tipo pela
contagem total de glóbulos brancos. Os altos níveis desses
glóbulos brancos estão associados com diversas respostas
imunes e anormalidades. Algumas vezes é solicitada uma
contagem de eosinófilos como um teste de acompanhamento,
quando é relatado um nível elevado ou deprimido de
eosinófilos. (VALLADA, 1999).
FIGURA 1: Tipos de leucócitos da espécie humana.
Fonte: iceb.ufop.br.

10
3. Doenças Hematológicas
3.1 Anemias
A anemia é definida pela Organização Mundial da Saúde (OMS)
como a redução da taxa de hemoglobina sanguinea abaixo de
13 g/dL para homens adultos, de 12 g/dL para mulheres
adultas, São vários os tipos de anemia, as quais podem ser
dever à destruição excessiva ou prematura de hemácias, ou
mesmo, em caso de pseudo-anemia, pelo simples aumento do
volume plasmático. Existem outros fatores que podem afetar
a normalidade da proporção dos glóbulos vermelhos e de seus
conteúdos hemoglobínico, como deficiência de nutrientes,
fatores hereditários, doenças crônicas e perda de sangue –
fator muito importante para redução no estoque de ferro do
organismo, visto que não existe um mecanismo de excreção de
ferro que não este. (HOFFBRAND; PETTIT; MOSS,2008).
As anemia de doença crônica representa uma defesa do
organismo contra a proliferação de microrganismos e de
células neoplásicas e pode estar envolvida, juntamente com
a febre, como estratégia complementar que o organismo
emprega para se proteger da doença. O baixo nível de ferro
plasmático inibe o crescimento bacteriano. Desta forma, as
anormalidades no metabolismo de ferro podem representar um
mecanismo evoluído de defesa do hospedeiro contra a invasão
bacteriana, tal fenômeno é denominado imunidade
nutricional. (CANÇADO & CHIATTONE, 2002).

11
Baixos VCM e CHCM indicam anemias microcíticas
hipocrômicas causadas por anemia por deficiência de ferro,
anemia sideroblástica ou talassemia. Um VCM alto sugere
anemias macrocíticas causadas por anemias megaloblásticas,
devido à deficiência de ácido fólico ou vitamina B12,
desordens congênitas de DNA ou reticulocitose. Em razão de
o VCM refletir o volume médio de muitas células, um valor
dentro da faixa normal pode ocorrer em pacientes cujo
tamanho de glóbulos vermelhos varia, e inclui células
microcíticas e macrocíticas. (FAILACE; FERNANDES, 2009).
FIGURA 2 : Fluxograma de classificação das anemias
Fonte: Revista Adolescência e Saúde, Vol. 6 nº 3 - Jul/Set – 2009.
3.2 Carências Hipoproliferativas

12
A anemia ferropriva é o estágio final de um longo
período de balanço negativo do ferro. Com o início da queda
do nível total de ferro corpóreo, uma sequência
característica de eventos aparece: primeiro, os estoques de
ferro dos hepatócitos e macrófagos do fígado, baço e medula
óssea são detectados; esta fase é denominada de deficiência
de ferro pré-latente ou depleção de ferro. A anemia tem se
destacado mundialmente como uma das carências nutricionais
mais prevalentes, constituindo assim um grave problema de
saúde pública. A Organização Mundial da Saúde (OMS), o
Fundo das Nações Unidas para a Infância (UNICEF) e a
Organização das Nações Unidas (ONU) publicaram dados que
mostram prevalência de 20,1% de anemia em crianças menores
de quatro anos em países industrializados, e de 39,0% em
países em desenvolvimento. (FAILACE; FERNANDES, 2009).
O diagnóstico é geralmente fácil, realizado por meio
de um hemograma que caracteriza a presença de glóbulos
vermelhos com tamanho menor que o normal, em sinal de
microcitose por faltar-lhes conteúdo hemoglobinico no caso
das anemias por deficiência torna se necessário a
realização paralela de exames específicos como no caso de
anemia por deficiência de ferro, deve-se avaliar o perfil
do ferro,com dosagens plasmáticas da ferritina, por
deficiência de Vitamina B12, deve-se avaliar os níveis de
vitamina B12 sérica e acido fólico sérico, o teste de
Schilling para avaliar a suspeita de anemia perniciosa,
conforme observamos na Fig.2 podemos classificar a anemia
com um auxilio de Fluxograma. (ADAMSON; et al, 2006).

13
Habitualmente as anemias hipoproliferativas
apresentam-se como normocíticas e normocrômicas, mas podem
ser observadas células microcíticas hipocrômicas com
anemias por deficiência leve de ferro e inflamação de longa
duração. Ambas apresentam-se com ferro sérico baixo, mas
com diferentes padrões para o restante da cinética do
ferro, Apesar de pouco utilizada na prática clínica com
esse intuito, a coloração para ferro na MO pode elucidar o
diagnóstico. (VENTURELLA; PASQUOLOTO, 2002).
A análise do sangue periférico nos fornece dados
quanto à presença de RDW (variações no tamanho),
pecilocitose (variações na forma) e policromasia, como já
citado. Valiosas informações podem ser obtidas no achado de
células afoiçadas e em alvo, fragmentos eritrocitários,
corpúsculos de Howell-Jolly, alterações em outras séries,
como blastos, e outras alterações que nos sugiram
específicas patologias. (CANÇADO; JESUS, 2007).
Pode-se avaliar a cinética do ferro por meio
da sua dosagem sérica, do valor da capacidade ferropéxica
(TIBC), da porcentagem da saturação da transferrina e da
dosagem de ferritina, lembrando que essa, além de estimar
as reservas de ferro, é também um reagente de fase aguda,
podendo aumentar várias vezes na presença de inflamação
aguda ou crônica. (ADAMSON; et al, 2006).
Outros exames podem ser solicitados conforme as
hipóteses diagnósticas, como dosagem de desidrogenase

14
lática (LDH) e bilirrubinas indiretas (que costumam estar
elevadas em episódios hemolíticos), teste de falcização,
eletroforese de hemoglobina, dosagem de G6PD (lembrando que
logo após o evento hemolítico os valores podem estar
normais, por ter havido destruição da coorte de hemácias
com deficiência enzimática), teste de Coombs direto (para
anemia hemolítica autoimune), dosagens séricas de ácido
fólico e vitamina B12, aspirado e biópsia de MO, entre
outros. (MOURA; WADA; PURCHIO; et al, 2006).
A prevenção deve ser planejada priorizando-se a
educação nutricional e condições ambientais satisfatórias e
envolvendo-se: o incentivo ao aleitamento materno exclusivo
até o sexto mês; a não utilização do leite de vaca no
primeiro ano de vida; a suplementação medicamentosa
profilática; a fortificação de alimentos de consumo
massivo; o controle de infecções; acesso a água e esgoto
adequados; e o estímulo ao consumo de alimentos que
contenham ferro de alta biodisponibilidade na fase de
introdução da alimentação complementar e em fases de maior
vulnerabilidade a essa deficiência, como a adolescência.
(ADAMSON; et al, 2006).
5.3 Anemias Hemolíticas e Hemorrágicas
A anemia falciforme é uma das doenças hereditárias
mais comuns no Brasil e apresenta, já nos primeiros anos de
vida, manifestações clínicas importantes, o que representa

15
um sério problema de saúde pública no país. A doença
falciforme é resultante de alteração genética caracterizada
pela presença de um tipo anormal de hemoglobina denominada
Hemoglobina S (HbS). Ela faz com que as hemácias adquiram a
forma de foice (daí o nome falciforme), em ambiente de
baixa oxigenação, dificultando sua circulação e provocando
obstrução vascular. (CANÇADO; JESUS, 2007).
As hemácias têm a função de carregar oxigênio para os
tecidos, principal combustível para os órgãos. No caso da
doença falciforme, pelo fato de as hemácias apresentarem a
forma de foice são destruídas precocemente, além de se
agregarem e diminuir a viscosidade do sangue nos pequenos
vasos do corpo. Com isso, ocorre lesão nos órgãos
atingidos, causando dor, destruição dos glóbulos vermelhos,
icterícia e anemia. (MOURA; WADA; PURCHIO; et al, 2006).
A forma mais frequente da doença, e também a mais
grave, é a homozigótica, que é denominada Anemia Falciforme
ou Drepanocitose (Hb SS) e ocorre quando a criança herda de
ambos os pais o gene S. Quando a criança herda o gene S de
um dos pais e, do outro, o gene para a Hemoglobina A
normal, ela será apenas portadora do Traço Falciforme
(HbAS). Nesse caso, não apresentará a doença, podendo, no
entanto, transmiti-la aos filhos. (VALLADA, 1999).
A anemia falciforme é caracterizada por uma anemia
hemolítica por toda a vida, ocorrência de exacerbações
agudas ou repentinas chamadas de crises, e diversas
complicações resultantes de uma propensão aumentada a
infecções e dos efeitos deletério do repetido bloqueio

16
vascular. Os indivíduos com o caráter falcêmico exibem
maior resistência à malária do que os sem este caráter, e
esta característica dá conta da prevalência continuada da
anemia falciforme na população. (CANÇADO; JESUS, 2007).
A púrpura trombocitopênica idiopática (PTI), também
conhecida como púrpura trombocitopênica imunonológica,
autoimune ou isoimune, é uma doença adquirida e geralmente
benigna, de causa desconhecida, que se caracteriza por
trombocitopenia (baixas contagens de plaquetas), e o
resultado é um aumento da tendência para as hemorragias.
(FAILACE; FERNANDES, 2009).
Apesar da etiologia desconhecida, reconhecem-se
autoanticorpos, geralmente da classe IgG, direcionados a
antígenos da membrana plaquetária. Uma vez que a plaqueta
apresenta um anticorpo aderido à sua membrana, é
reconhecida por macrófagos localizados no baço e em outras
áreas de tecido reticulo endotelial, onde são destruídas,
levando a um menor tempo de vida médio plaquetário e,
consequentemente, a menores contagens de plaquetas
circulantes, o diagnostico diferenciado da anemia
hemolítica são através dos exames: eletroforese proteica,
LDH sérico, teste de Coombs, entre outros. (VERRASTRO,
2005).
Pode ser classificada, de acordo com a faixa etária
acometida, como infantil ou adulta e, quanto ao tempo de
evolução, como aguda ou crônica. A púrpura trombocitopênica
aguda é mais comum em crianças com uma incidência anual em
torno de 3-8 casos por 100.000, com maior número de casos

17
entre os 2-5 anos de idade e com leve predomínio no sexo
masculino. Em cerca de 75% dos pacientes, o episódio segue
vacinação ou infecção, como varicela ou mononucleose
infecciosa. A maioria dos casos deve-se a ligação de
imunocomplexos inespecíficos. As remissões espontâneas são
usuais, mas em cerca de 5 a 10% dos casos a doença fica
crônica (duração superior a 6 meses). Felizmente a
morbidade e a mortalidade na PTI aguda são muito baixas.
(CANÇADO; JESUS, 2007).
Já na PTI crônica, existem incidências mais altas em
mulheres com idade entre 15 e 50 anos, mas alguns relatos
recentes sugerem haver aumento da incidência com o passar
dos anos. É a causa mais comum de trombocitopenia sem
anemia ou neutropenia. Geralmente é idiopática (por isso a
sigla PTI, também serviria para púrpura trombocitopênica
imunológica), mas pode ser vista em associação com outras
doenças como lúpus eritematoso sistêmico, infecção por
vírus (HIV, HCV), leucemia linfocítica crônica, doença de
Hodgkin e anemia hemolítica auto-imune. O início é quase
sempre incidioso, com petéquias, equimoses fáceis e, em
mulheres, menorragia. Sangramento das mucosas, como
epistaxe ou sangramento gengival, ocorre em casos graves,
mas, felizmente, a hemorragia intracraniana é rara. (MOURA;
WADA; PURCHIO; et al, 2006).
A Hemofilia é um distúrbio de coagulação sanguínea
geneticamente determinado quase que exclusivamente aos
homens (doença recessiva ligada ao cromossomo X). Em 85%
dos casos é causada por deficiência do fator VIII, sendo

18
denominada hemofilia tipo A ou hemofilia clássica. Em
aproximadamente 15% dos casos, ocorre deficiência do fator
IX (hemofilia tipo B). A redução dos níveis funcionais dos
FVIII e FIX resulta em prolongamento do tempo de
sangramento. (VALLADA, 1999).
As hemofilias têm a mesma apresentação clínica, sendo
a dosagem específica dos fatores a única forma de distinção
entre as mesmas. A diferenciação é importante, pois o
tratamento é específico com relação ao tipo do fator a ser
reposto. Historicamente, a mortalidade dos portadores de
hemofilia e outros distúrbios congênitos de coagulação era
devida à hemorragia incontrolada associada à severa
deficiência de fatores de coagulação VII e IX. (VERRASTRO,
2005).
Dependendo dos níveis de fatores mensurados no
plasma, a hemofilia pode ser classificada em leve (6 – 25%
de atividade de fator), moderada (1 – 5% de atividade) e
grave (< 1%). Todos os membros com hemofilia de uma mesma
família têm o mesmo grau de deficiência de fator. Os
pacientes com hemofilia podem apresentar sangramentos
espontâneos ou relacionados a traumas. O sangramento
espontâneo é mais comum na hemofilia grave. As articulações
são geralmente as mais afetadas, mas qualquer outra parte
do corpo, inclusive o sistema nervoso central (SNC), pode
estar sujeito à hemorragia espontânea. (FAILACE; FERNANDES,
2009).
Na infância, um sangramento pode se manifestar como
mancha ou hematoma, mas à medida que a criança vai ficando

19
mais velha a hemorragia intra-articular ou a hemartrose
ocorre com maior freqüência. O quadro clínico da hemofilia
mostra que o sucesso do tratamento é a prevenção de
hemorragias associando o tratamento precoce de reposição e
a fisioterapia como parte dos cuidados integrais ao
indivíduo hemofílico. Somente o conjunto desses dois
acompanhamentos pode reduzir a incidência de artropatias
hemofílicas. Esses derrames articulares são sempre
provocados por microtraumas e são mais frequentemente
notados nas grandes articulações, apenas nos casos mais
graves da doença ocorrem hemorragias espontâneas.
(VERRASTRO, 2005)
4.0 Leucemias
A palavra leucemia deriva do grego Leukós (branco) e
haêma (sangue), significando sangue branco. O termo foi
criado em 1847 pelo médico alemão Rudolf Virchow,
considerado hoje como o pai da patologia moderna. É um
termo muito amplo que se refere a um grupo de tumores que
geralmente afetam a medula óssea, o órgão responsável pela
formação das células do sangue. Parecem doenças misteriosas
por não apresentarem uma causa evidente ou comum, pelo
menos na maioria das vezes. Crianças e adultos,
independente do sexo, cor ou grupo social podem ser
afetadas (CAVALCANTI JR; et al, 1997).
As leucemias são neoplasias derivadas de células
hematopoéticas que proliferam a princípio na medula óssea,

20
antes de se disseminarem para o sangue periférico, baço,
linfonodos e outros tecidos.O quadro clínico das leucemias
está relacionado com a queda do número de células
sanguíneas, decorrente da proliferação de células
neoplásicas (blastos) na medula óssea, ou com a
imunossupressão causada pelas drogas citotóxicas no
tratamento (YAMAMOTO, 1997)
As leucemias constituem 30% das doenças hemato-
oncológicas da infância, e na ampla maioria são leucemias
agudas. Resultam da proliferação clonal de precursores
anormais, os linfoblastos, na medula óssea. A
subcategorização das leucemias agudas é necessária para
estabelecer-se uma terapêutica apropriada, fatores
prognósticos e a presença de doença residual mínima
(ANDRIOLO, 2000).
4.1 Tipos de Leucemias
As leucemias agudas são caracterizadas por uma
progressão rápida e afetam a maioria das células imaturas
(ainda não totalmente diferenciadas ou desenvolvidas) da
medula óssea,podendo ser observada na Fig.1, que perde sua
capacidade de produzir células sanguíneas saudáveis. Podem
ocorrer em adultos jovens e crianças e o tratamento deve
ser imediato, pois com o acúmulo, as células leucêmicas
podem se espalhar para outras partes do corpo. Sem
tratamento, a pessoa invariavelmente morre em alguns meses
ou semanas. (ZAGO; FALCÃO; PASQUINI, 2001).

21
A classificação em se tratando de tipos de leucemia é
feita pelo tipo celular especificado (linfóide, mielóide
ou monocítica) e, secundariamente, pelo chamado sub-tipo,
relativo ao quadro clínico de evolução (aguda, crônica ou
subaguda). As leucemias codificadas apenas como aguda,
subaguda ou crônica, sem os tipos celulares explicitados,
são consideradas leucemias de tipo celular não-
especificado. (ZAGO; FALCÃO; PASQUINI, 2001).
Portanto, as leucemias são classificadas em leucemia
linfóide aguda (LLA), afeta células linfoides e agrava-se
rapidamente. É o tipo mais comum em crianças pequenas, mas
também ocorre em adultos. leucemia linfóide crônica (LLC)
afeta células linfoides e se desenvolve vagarosamente. A
maioria das pessoas diagnosticadas com esse tipo da doença
tem mais de 55 anos. Raramente afeta crianças, leucemia
mielóide aguda (LMA), afeta as células mieloides e avança
rapidamente. Ocorre tanto em adultos como em crianças.
leucemia mielóide crônica (LMC) afeta células mieloides e
se desenvolve vagarosamente, a princípio. Acomete
principalmente adultos. (ANDRIOLO, 2000).
Embora a LLA possa ocorrer em qualquer idade, sua
incidência é maior em crianças entre 2 a 5 anos, em uma
porcentagem de cerca de 70%, diminuindo entre adolescentes
e adultos jovens, entre os quais a incidência de leucemias
aguda é de 20%, sendo mais comum entre crianças de cor
branca e do sexo masculino. (LORENZI, 1999).

22
A LMA possui uma célula neoplasia que se espalha
rapidamente no sangue e medula óssea. Devido a origem das
células leucêmicas, a medula óssea produz rapidamente um
grande número de células, que na maioria das vezes não
funcionam corretamente e substituem as células normais.
(ANDRIOLO, 2000).
4.2 Diagnóstico Clínico
Geralmente, o diagnóstico de LMA inicia-se a partir de
uma suspeita clínica e se baseia na avaliação do sangue
periférico e da medula óssea. Embora a morfologia continue
sendo o fundamento para o diagnóstico, técnicas adicionais,
incluindo imunofenotipagem, avaliação citogenética e
estudos de genética molecular, tornaram-se essenciais e, em
alguns casos específicos, são ferramentas complementares
obrigatórias. O uso de procedimentos diagnósticos permite a
identificação do tipo celular envolvido na leucemogênese, o
que é fundamental para orientar a terapêutica e determinar,
até certo ponto, o prognóstico das leucemias. (YAMAMOTO,
1997).
Podem ser detectados outros sintomas como dor nos
ossos ou articulações e febre persistente. Ainda dentro do
quadro de manifestações clínicas das leucemias, pode haver
infltração, por células leucêmicas, de tecidos como
amígdalas, gânglios linfáticos (ínguas), pele, baço, rins e
outros. No entanto, vale lembrar que todos esses
sintomas quase nunca são causados por leucemia já que

23
inúmeras outras doenças podem causá-los. Portanto, o médico
deve sempre ser procurado para investigar a origem de
qualquer sintoma novo. (FARIAS; CASTRO, 2004).
Quando o médico suspeita de leucemia o paciente é
encaminhado ao especialista em Hematologia e geralmente é
feito um exame de mielograma. Neste procedimento que deve
ser feito sob sedação, o hematologista insere uma agulha
através do osso (frequentemente a bacia) até a medula óssea
e então aspira uma pequena parte do seu conteúdo para
análise. No mesmo ato pode ser retirado um pequeno
fragmento de osso e medula óssea, denominado de biópsia. As
análises feitas ao microscópio do fragmento e do conteúdo
líquido permitem frmar na maior parte dos casos um
diagnóstico inicial. Análises pormenorizadas são
necessárias para avaliar prognóstico e inclusive para ditar
o tratamento em alguns casos. (VERRASTRO, 1996).
4.3 Diagnóstico Laboratorial
As colorações citoquímicas é usadas no diagnóstico e
na classificação das leucemias podem ser aplicadas tanto à
medula óssea quanto ao sangue periférico, auxiliando na
confirmação da origem mielóide e/ou monocítica. Apesar dos
progressos da imunofenotipagem, as reações citoquímicas
ainda são úteis no diagnóstico das LMA Os linfoblastos T
revelam atividade paranuclear na esterase inespecífica logo
que realizada em pH ácido, tendo uma atividade maior de 75%
na fosfatase ácida. Na periódica ácida de Schiff (PAS), os

24
linfoblastos da LLA freqüentemente demonstram uma evidente
coloração e forma de anéis concêntricos de grânulos
grosseiros ou blocos maciços. Uma reação PAS negativa é
mais freqüente na LLA de linhagem T que na linhagem B.
(MARTINS; FALCÃO, 2000).
As leucemias agudas o acúmulo de células anormais na
medula óssea gera os principais sintomas da leucemia.
Diante da diminuição da produção de células vermelhas
desenvolve-se um quadro anêmico, que pode estar associado à
falta de ar, palpitações ou batedeira e fadiga ou cansaço.
A deficiência de plaquetas favorece o sangramento excessivo
na pele e mucosas (nariz e gengivas) de pessoas com
leucemia. A supressão dos leucócitos, envolvidos no
combate a agentes patogênicos, coloca o paciente
leucêmico sob risco de graves infecções que podem levar à
morte. (RIZZATTI; ZAGO, 2002).
O mielograma é um dos exames para avaliação da medula
óssea. Como a medula óssea está localizada anatomicamente
no interior dos ossos, o mielograma é realizado através de
uma punção óssea, seguida de aspiração, sendo realizada
sob anestesia local (pode-se também
usar sedação e/ou analgesia sistêmica), O diagnóstico da
LLA fundamenta-se na demonstração de mais de 25% de
linfoblastos na medula óssea A medula encontra-se
hipercelular com substituição dos espaços adiposos e
elementos medulares normais por células leucêmicas, com
precursores mielóides e eritróides residuais de aspecto
25
normal e megacariócitos diminuídos ou ausentes. (BOLUFER,
et al, 2002).
FIGURA 3: Coleta de Mielograma
Fonte: Instituto INCA
O hemograma pode revelar anemias normocítica e
normocrômica e trombocitopenia. A contagem de leucócitos
está ocasionalmente muito alta, mas freqüentemente normal
ou diminuída. Os blastos são raros ou ausentes em pacientes
leucopênicos, mas em casos de leucocitose podem ser
numerosos, chegando a constituir maioria. A maioria dos
pacientes apresenta blastos leucêmicos no sangue
periférico. Podem ser mieloblasto, monoblasto,
megacarioblasto ou uma população mista. (YAMAMOTO, 1997)
5.0 Lupus Eritematoso Sistêmico

26
O lúpus eritematoso sistêmico (LES) é uma doença
inflamatória crônica, multissistêmica, de causa
desconhecida e de natureza autoimune, caracterizada pela
presença de diversos autoanticorpos. Evolui com
manifestações clínicas polimórficas, com períodos de
exacerbações e remissões. De etiologia não esclarecida, o
desenvolvimento da doença está ligado à predisposição
genética e aos fatores ambientais, como luz ultravioleta e
alguns medicamentos.(TEIXEIRA JUNIOR, SILVA, MAGALHAES,
2011).
O lúpus eritematoso sistêmico (LES) é uma doença auto-
imune de etiologia desconhecida, na qual os auto-anticorpos
encontram-se universalmente presentes. Os mecanismos
responsáveis pela produção e pela perpetuação desta
resposta imune aberrante permanecem pouco esclarecidos1. A
doença apresenta distribuição universal, acomete todas as
classes sociais e sua incidência varia entre 2 e 8 por
100.000 habitantes para os Estados Unidos e entre 20 e 60
casos por 100.000 habitantes na Europa, não diferindo entre
a população urbana e rural.(JACOBSON, GANGE, ROSE, et al,
1997).
O LES possui como características epidemiológicas a
predominância de acometimento de indivíduos do sexo
feminino (cerca de 90% dos casos), maior incidência entre
15 e 45 anos de idade, comprome-timento mais grave da raça
negra. Sua etiologia é variada, estando incluídas
alterações imunológicas acarretando a produção de auto-
anticorpos, ação de fatores genéticos, fatores ambientais

27
(vírus, radiação ultravioleta, drogas), fisiológicos
(hormônios) e o stress. (VAUGHAN, 1997)
O LES é uma doença rara, incidindo mais frequentemente
em mulheres jovens, ou seja, na fase reprodutiva, numa
proporção de nove a dez mulheres para um homem, e com
prevalência variando de 14 a 50/100.000 habitantes, em
estudos norte-americanos. A doença pode ocorrer em todas as
raças e em todas as partes do mundo. (FORTE, 2003).
5.1 Diagnóstico
Na prática, costuma-se estabelecer o diagnóstico de
LES utilizando os critérios diagnósticos (também chamados
de critérios de classificação) do Colégio Americano de
Reumatologia (ACR), propostos em 1982 e modificados em
1997, que se baseiam na presença de pelo menos quatro
critérios dentre os onze estabelecidos.(GLADMAN, UROWITZ,
1997).
O LES é uma doença complexa e seu diagnóstico exige
cautela. Deve-se ter em mente que seu diagnóstico é de
exclusão, devendo-se afastar doenças mais prevalentes na
região, como a hanseníase, que é curável, antes de
considerar um indivíduo como lúpico. O LES é uma doença
crônica, sem cura, que apresenta complicações nos mais
variados sistemas, criando diversos temores e expectativas
que acompanharão toda a vida dos que dela são portadores.
(FORTE, 2003).

28
No LES são descritos como sinais e sintomas mais
freqüentes: fadiga, anemia, febre, perda ponderal,
artralgia, artrite, erupção tipo borboleta,
fotossensibilidade, alopécia, linfadenopatia, pleurite,
pericardite, diarréia, anemia, vasculite, síndrome
nefrítica, doenças do sistema nervoso central, distúrbios
da personalidade. São considerados critérios de doença
lúpica: eritema malar, eritema discóide,
fotossensibilidade, úlceras orais, artrites, serosites,
distúrbios renais, neurológicos, hematológicos,
imunológicos e presença de anticorpos antinucleares. No
hemograma de pacientes com doença lúpica podem ser
observados: anemia leve ou moderada, valores de hematócrito
30% abaixo do normal, leucopenia com 4.000 células por
milímetro cúbico, neutropenia, linfopenia e plaquetopenia.
(TAN, COHEN,FRIES, et al, 1982).
Entre os exames encontram-se: presença de
crioglobulina; anticorpos antinucleares em 95% dos casos em
atividade; anticorpos anti-DNA em 50% dos casos em
atividade; anticorpos anti-histona ou anti complexo ácido
desoxiribonucléico-histona em 70% dos pacientes;
complemento sérico total, componentes C2, C3, C4 muitas
vezes diminuídos e exames de atividade inflamatória
alterados nas fases de atividade da doença, sendo
considerados específicos achados de anticorpos anti-DNA de
dupla hélice. (FORTE, 2003).
Durante a evolução do LES, pode ocorrer uma série de
alterações imunológicas que favorecem o agravamento da

29
enfermidade. Como não é possível determinar quando tais
exacerbações ocorrerão, buscou-se relacionar esse evento
com o ciclo viral (latência ou replicação) no interior dos
linfócitos B circulantes de pacientes com LES. Isto foi
feito por meio da análise da avidez dos anticorpos de
classe IgG no soro desses pacientes. (FORTE, ALMEIDA,
1990).
O diagnóstico de Lúpus em atividade é importante, pois
esse diagnóstico é fundamental, uma vez que o prognóstico
depende do tratamento precoce da doença. A falta desse
diagnóstico pode levar a conseqüências graves como
insuficiência renal e até mesmo óbito. Assim, é de
fundamental importância tudo o que possa contribuir para a
detecção de LES em atividade. (FORTE, 2003).
Existem diversas manifestações cutâneas do lupus
eritematoso. Estas podem ser divididas em específicas e
inespecíficas, de acordo com suas características clínicas
e histológicas. As lesões cutâneas específicas do lupus
eritematoso são classificadas como: lesão aguda, subaguda e
crônica. Tal terminologia também reflete a duração e a
gravidade do quadro. Estas lesões ocorrem exclusivamente no
lupus eritematoso, diferentemente das lesões inespecíficas,
que são encontradas também em outras doenças. (TAN; COHEN;
FRIES, et al, 1982).
O diagnóstico de LED pode ser confirmado pela
histologia cutânea, embora esta tenha sido pouco estudada
na infância e adolescência. Os achados histológicos

30
encontrados foram semelhantes aos já descritos no adulto:
graus variados de hiperqueratose, rolha córnea folicular,
atrofia da epiderme, vacuolização das células da camada
basal da epiderme e infiltrado inflamatório de células
mononucleadas ao redor de vasos e anexos na derme, que pode
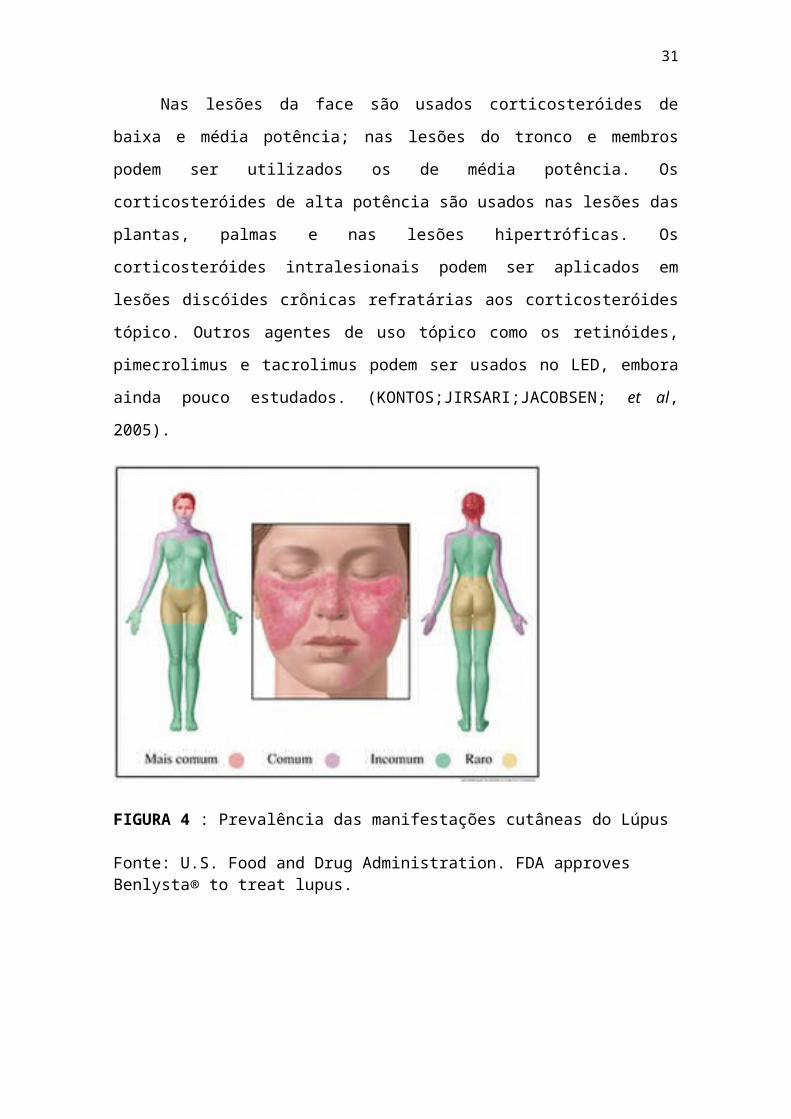
ser observadas na fig.02 em que mostra prevalência das
manifestações cutâneas do Lúpus. (KONTOS; JIRSARI;JACOBSEN;
et al, 2005).
5.2 Tratamento
O tratamento do LED visa melhorar aparência do
paciente e evitar o desenvolvimento de cicatrizes e
discromias. Os fotoprotetores e as medidas de proteção
solar, tais como uso de roupas e chapéus, são essenciais.
Os fotoprotetores precisam ser aplicados diariamente nas
áreas fotoexpostas, devem ser de amplo espectro e com alto
fator de proteção solar, de preferência acima de 15.
Recomenda-se que o paciente os aplique logo pela manhã e os
reaplique a cada duas horas, ou após contato com água ou
sudorese intensa, mesmo em dias nublados. (FREITAS;
PROENÇA; 2003).
Os corticosteróides tópicos são muito utilizados,
sendo sua potência escolhida de acordo com o local e
características da lesão. Eles podem ser aplicados uma a
duas vezes ao dia e, quando necessário, usá-los por tempo
prolongado, devendo-se intercalar intervalos curtos (alguns
dias) sem a medicação para evitar seus efeitos adversos
locais, como a atrofia cutânea. (FORTE, 2003).
31
Nas lesões da face são usados corticosteróides de
baixa e média potência; nas lesões do tronco e membros
podem ser utilizados os de média potência. Os
corticosteróides de alta potência são usados nas lesões das
plantas, palmas e nas lesões hipertróficas. Os
corticosteróides intralesionais podem ser aplicados em
lesões discóides crônicas refratárias aos corticosteróides
tópico. Outros agentes de uso tópico como os retinóides,
pimecrolimus e tacrolimus podem ser usados no LED, embora
ainda pouco estudados. (KONTOS;JIRSARI;JACOBSEN; et al,
2005).
FIGURA 4 : Prevalência das manifestações cutâneas do Lúpus
Fonte: U.S. Food and Drug Administration. FDA approves Benlysta® to treat lupus.

32
CONCLUSÃO
Os procedimentos dos testes devem ser realizados
cuidadosamente, analisando cada passo e observando
possíveis interferentes como sangue hemolisados, lipêmicos,
coagulados, sujidade e calibração dos aparelhos e outros,
os quais podem vir a levar alteração no resultado do exame,
assim faz-se necessário a padronização da execução dos
métodos para um resultado mais seguro e confiável.
O Biomédico deve ter conhecimento dos índices
hematimétricos, Torna-se fundamental o adequado
conhecimento dessa doença, assim como dos diferentes pontos
de corte para o seu diagnóstico, de acordo com o sexo e o
estágio de desenvolvimento Humano. As anemias carenciais

33
são as mais comuns em nosso meio, porém devemos ter em
mente as demais causas de anemia, não se podendo esquecer
de que as anemias muitas vezes representam manifestações de
outras doenças subjacentes presentes nessa população. Cada
tipo de anemia exige um tratamento especial pois depende do
seu tipo, causa e gravidade e isto pode significar mudanças
na dieta, suplementos alimentares, remédios ou
procedimentos médicos.
Os processos anêmicos relacionados à hereditariedade
vêm sendo
esclarecidos por inúmeros estudos, que incluem a base
genética destas
enfermidades, estes processos em geral, apresentam uma
grande dificuldade
quanto ao tratamento. Hoje a terapia com células-tronco
oferece uma perspectiva para melhoria da qualidade de vida
dos portadores de alguns destes distúrbios. Os estudos
relacionados às anemias têm uma importância relevante
para ampliar os conhecimentos sobre os mecanismos
envolvidos no seu desenvolvimento, elucidando os sinais
químicos, a base genética e molecular deste quadro,
proporcionando tratamentos mais adequados para cada tipo
de anemia.
REFERÊNCIAS

34
ADAMSON JW. Deficiência de ferro e outras anemias
hipoproliferativas. In: Kasper DL, Fauci AS, Longo DL,
Braunwald E, Hauser SL, Jameson JL. Harrison Medicina
Interna. 16. ed. Rio de Janeiro: McGraw-Hill Interamericana
do Brasil Ltda. 2006.p.615-30.
ANDRIOLO, A. Diagnóstico laboratorial em pediatria. São
Paulo: Sarvier, 2000.
CANÇADO, R. D.; JESUS, J. A.A. Revista Brasileira de
Hematologia e Hemoterapia, São José do Rio Preto, v. 29, n.
3, p. 203-206, 2007.
CAVALCANTI Jr., G. B. et al. Importância da aplicação de
anticorpos monoclonais no diagnóstico laboratorial das
leucemias linfóides agudas. Revista Brasileira Analise
Clinica, v. 29, n. 3, p. 159-67, 1997.
BOLUFER, P. et al. Rapid quantitative detection of TEL-AML1
fusion transcrips in pediatric acute lymphoblastic leukemia
by real-time reverse transcription polymerase chain
reaction using fluorescently labeled probes.Haematologica,
v. 87, p. 23-32, 2002.
FAILACE, R.; FERNANDES, F.B. Hemograma: Manual de
Interpretação. Ed.Artmed. Porto Alegre, ed.5, p.424, 2009.
FAILACE; R.: “Hemograma – manual de interpretação”, Segunda
Edição corrigida, Artes Médicas, Porto Alegre, 1992.

35
FARIAS, Mariela Granero and CASTRO, Simone Martins
de.Diagnóstico laboratorial das leucemias linfóides
agudas. Jornal Brasileiro Patologia Medica Lab.[online].
2004, vol.40, n.2, pp. 91-98., Disponível em:
<http://dx.doi.org/10.1590/S1676-24442004000200008>. Acessa
do dia 01 de Outubro de 2014.
Forte WCN, Almeida A, Leão, RC. Resposta fagocitária eatividade quimiotática em crianças eutróficas. Rev HospClin Fac Med Univ São Paulo 1990; vol.45, p.256-259
Freitas THP, Proença NG. Lupus eritematoso cutâneocrônico: estudo de 290 pacientes. An Bras Dermatol 2003;vol.78, p.703-712.
Gladman DD, Urowitz MB. Systemic lupus erythematosus:
clinical and laboratory features. In: Klippel JH, editors.
Primer on the rheumatic diseases. Atlanta: Arthritis
Foundation; 1997. p. 251-257.
HOFFBRAND, A.V.; PETTIT, J.E; MOSS, P.A.H. Fundamentos em
Hematologia. Ed. Artmed. Porto Alegre, ed.5,p.400, 2008.
Jacobson DL, Gange SJ, Rose NR, Graham NMH. Epidemiologyand estimated population burden of selected autoimmunediseases in the United States. Clin Immunol Immunopathol1997; vol. 84, n.2, p.223-243.
Junqueira LC, Carneiro C. Histologia básica. 10 ed. Rio deJaneiro:Guanabara Koogan; 2004. p.223-37.

36
Kontos AP, Jirsari M, Jacobsen G, Fivenson DP.Immunoglobulin M predominance in cutaneous lupuserythematosus. J Cutan Pathol 2005; vol.32, p.352-355.
LIMA, G.A.F.M; GROTTO, H.Z.W. Avaliação das medidas de
Ferro Sérico e capacidade de ligação do Ferro a
Transferrina (TIBC). Revista NewsLab, ed. 65, pag.84-96,
2004.
LORENZI, T. F. Manual de Hematologia – Propedêutica e
Clínica. 2 ed. Rio de Janeiro: Medsi, 1999.
Martins SLR, Falcão RP. A importância da imunofenotipagem
na leucemia mielóide aguda. Revista Associação Médica
Brasileira, n.4, v.1, p.57-62, 2000.
MOURA, Robeto de Almeida. WADA, Carlos S. PURCHIO, Adhemar.
ALMEIDA, Therezinha Verrastro de. Técnicas de Laboratório.
3 ed. São Paulo: Editora Atheneu, 2006.
RIZZATTI, E. G.; ZAGO, M. A. Aplicações da biologia
molecular às leucemias agudas. Ser Monog Esc Brasileira de
Hematatologia, n. 9, p. 1-14, 2002.
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield
NF, et al. The 1982 revised criteria for the classification
of systemic lupus erythematosus. Arthritis Rheum 1982;
vol.25, p.1271-1277.

37
TEIXEIRA JUNIOR, Gilson José Allain; SILVA, Cláudia Elis eFerraz e MAGALHAES, Vera. Aplicação dos critériosdiagnósticos do lúpus eritematoso sistêmico em pacientescom hanseníase multibacilar. Rev. Soc. Bras. Med. Trop. [online].2011, vol.44, n.1, p. 85-90.
TERRA, P. Coagulação – Interpretação Clínica dos Testes
Laboratoriais de Rotina. São Paulo: Atheneu, 2000.
Vaughan JH. The Epstein-Barr virus and systemic lupuserythematosus. J. Clin Invest 1997; vol.100, n.12, p.2939-2940.
VALLADA, E.P. Manual de técnicas hematológicas. São Paulo:
Atheneu, 1999.
Venturella RB, Santos GC, Pasquoloto AC. Contagem de
reticulócitos. In: Soares JLMF, Pasqualotto AC, Rosa DD,
Leite VRS. Métodos diagnósticos: consulta rápida. Porto
Alegre: Artmed Editora. 2002.p.534
VERRASTRO, T. Hematologia e hemoterapia: fundamentos de
morfologia, fisiologia, patologia e clínica. São Paulo:
Atheneu, p.487-507, 1996.
Zago AM, Falcão RP, Pasquini R. Hematologia: fundamentos e
prática. Rio de Janeiro: Atheneu, p.433-447, 487-507,2001.
Yamamoto M. Leucemia linfóide aguda da infância. In: Prado
FC, Ramos JA, Valle JR, organizadores. Ramos OL, Rothschild
HA, editores. Atualização terapêutica.São Paulo: Artes
Médicas; 1997. p. 418-21.





























