Embed Size (px)
Citation preview

www.technology.matthey.comJOHNSON MATTHEY TECHNOLOGY REVIEW
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3), 257–283
257 © 2015 Johnson Matthey
Atomic-Scale Modelling and its Application to Catalytic Materials ScienceDeveloping an interdisciplinary approach to modelling
By Misbah Sarwar, Crispin Cooper and Ludovic BriquetJohnson Matthey Technology Centre, Blounts Court, Sonning Common, Reading, RG4 9NH, UK
Aniekan Ukpong, Christopher Perry and Glenn Jones*Johnson Matthey Research Centre, CSIR, Meiring Naude Road, Brummeria, Pretoria, 0184, South Africa
*Email: [email protected]
Computational methods are a burgeoning science within industry. In particular, recent advances have seen first-principles atomic-scale modelling leave the realm of the academic theory lab and enter mainstream industrial research. Herein we present an overview, focusing on catalytic applications in fuel cells, emission control and process catalysis and looking at some real industrial examples being undertaken within the Johnson Matthey Technology Centre. We proceed to discuss some underpinning research projects and give a perspective on where developments will come in the short to mid-term.
1. Introduction
The use of atomic-scale modelling, whether force-field methods or electronic structure theory calculations (for example density functional theory (DFT) (1, 2)) in chemical and material research within industry is
becoming increasingly commonplace. This is a field that entails linking fundamental chemical properties, for instance electronic and geometric structure, to activity, ultimately providing the basis from which enhanced materials can be predicted (3). A particularly attractive feature of computational approaches is the flexibility and applicability to all divisions of Johnson Matthey’s business. For instance within New Business Development methods to simulate optical properties of materials (for smart glass applications) are being applied; within platinum group metals (pgm) refining we are exploring metal extraction and applying engineering process modelling; and within glass and colour technologies we are using thermodynamic tools to help us understand the formation and properties of glass compositions. Of particular importance to Johnson Matthey is catalyst technology (Chemical Catalysis, Emission Control, Process Technologies and Chiral Catalysis Technologies) where we are working towards a multiscale modelling capability that links the macroscopic engineering world to the atomic-scale chemistry and physical properties of materials.
A key philosophy within the research group and what makes the field so important for industrially relevant applications, is the importance of linking theory with experiment whether through measurement and validation of the computational methods or by actively seeking collaboration with experimentalists who can synthesise, characterise and test interesting materials. This overview article will firstly discuss some of the background to the methods employed, then move on to discuss some recent projects that have been carried
258 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
out with a particular focus on catalytic applications. The aim is to give a feel for the types of application we have been tackling, and the underpinning research we are conducting, before moving on to conclude by highlighting areas where continued research effort will be required in the future.
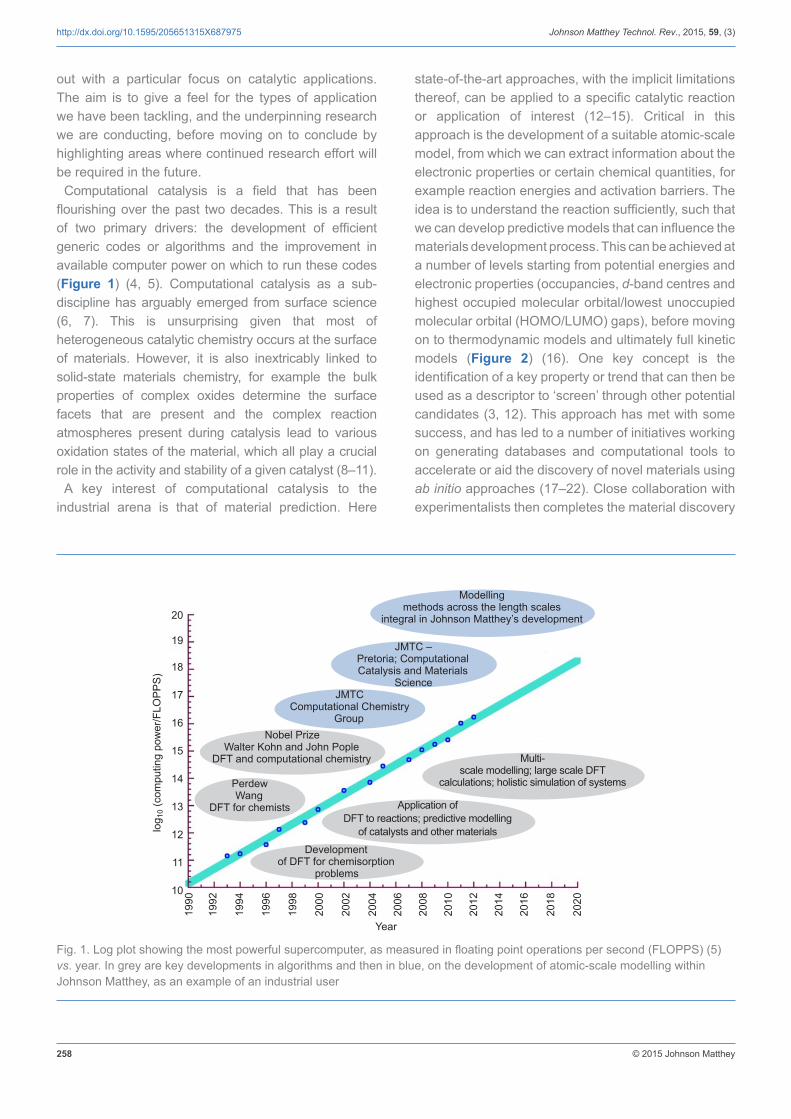
Computational catalysis is a field that has been flourishing over the past two decades. This is a result of two primary drivers: the development of efficient generic codes or algorithms and the improvement in available computer power on which to run these codes (Figure 1) (4, 5). Computational catalysis as a sub-discipline has arguably emerged from surface science (6, 7). This is unsurprising given that most of heterogeneous catalytic chemistry occurs at the surface of materials. However, it is also inextricably linked to solid-state materials chemistry, for example the bulk properties of complex oxides determine the surface facets that are present and the complex reaction atmospheres present during catalysis lead to various oxidation states of the material, which all play a crucial role in the activity and stability of a given catalyst (8–11).
A key interest of computational catalysis to the industrial arena is that of material prediction. Here
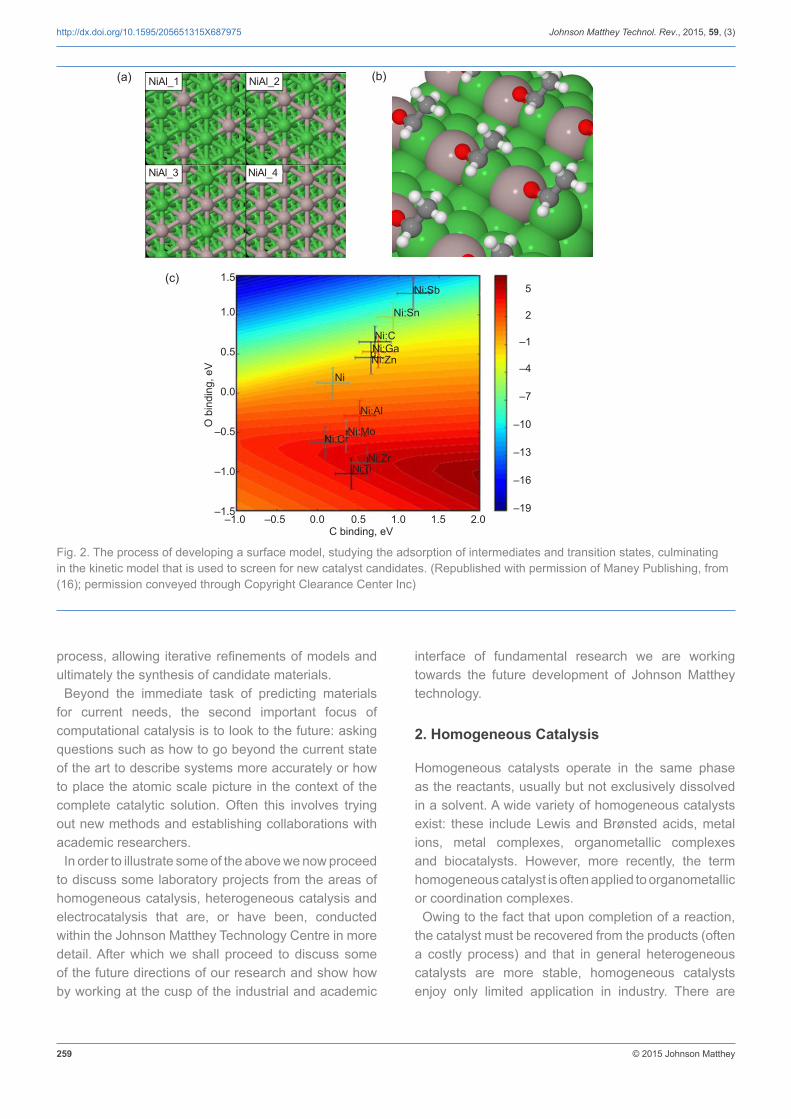
state-of-the-art approaches, with the implicit limitations thereof, can be applied to a specific catalytic reaction or application of interest (12–15). Critical in this approach is the development of a suitable atomic-scale model, from which we can extract information about the electronic properties or certain chemical quantities, for example reaction energies and activation barriers. The idea is to understand the reaction sufficiently, such that we can develop predictive models that can influence the materials development process. This can be achieved at a number of levels starting from potential energies and electronic properties (occupancies, d-band centres and highest occupied molecular orbital/lowest unoccupied molecular orbital (HOMO/LUMO) gaps), before moving on to thermodynamic models and ultimately full kinetic models (Figure 2) (16). One key concept is the identification of a key property or trend that can then be used as a descriptor to ‘screen’ through other potential candidates (3, 12). This approach has met with some success, and has led to a number of initiatives working on generating databases and computational tools to accelerate or aid the discovery of novel materials using ab initio approaches (17–22). Close collaboration with experimentalists then completes the material discovery
Perdew Wang
DFT for chemists
Nobel PrizeWalter Kohn and John Pople
DFT and computational chemistry
JMTC Computational Chemistry
Group
JMTC – Pretoria; Computational Catalysis and Materials
Science
Modelling methods across the length scales
integral in Johnson Matthey’s development
Development of DFT for chemisorption
problems
Application of DFT to reactions; predictive modelling
of catalysts and other materials
Multi-scale modelling; large scale DFT
calculations; holistic simulation of systems
1990
1992
1994
1996
1998
2000
2002
2004
2006
2008
2010
2012
2014
2016
2018
2020
Year
20
19
18
17
16
15
14
13
12
11
10
log 1
0 (c
ompu
ting
pow
er/F
LOP
PS
)
Fig. 1. Log plot showing the most powerful supercomputer, as measured in floating point operations per second (FLOPPS) (5) vs. year. In grey are key developments in algorithms and then in blue, on the development of atomic-scale modelling within Johnson Matthey, as an example of an industrial user
259 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
process, allowing iterative refinements of models and ultimately the synthesis of candidate materials.
Beyond the immediate task of predicting materials for current needs, the second important focus of computational catalysis is to look to the future: asking questions such as how to go beyond the current state of the art to describe systems more accurately or how to place the atomic scale picture in the context of the complete catalytic solution. Often this involves trying out new methods and establishing collaborations with academic researchers.
In order to illustrate some of the above we now proceed to discuss some laboratory projects from the areas of homogeneous catalysis, heterogeneous catalysis and electrocatalysis that are, or have been, conducted within the Johnson Matthey Technology Centre in more detail. After which we shall proceed to discuss some of the future directions of our research and show how by working at the cusp of the industrial and academic
interface of fundamental research we are working towards the future development of Johnson Matthey technology.
2. Homogeneous Catalysis
Homogeneous catalysts operate in the same phase as the reactants, usually but not exclusively dissolved in a solvent. A wide variety of homogeneous catalysts exist: these include Lewis and Brønsted acids, metal ions, metal complexes, organometallic complexes and biocatalysts. However, more recently, the term homogeneous catalyst is often applied to organometallic or coordination complexes.
Owing to the fact that upon completion of a reaction, the catalyst must be recovered from the products (often a costly process) and that in general heterogeneous catalysts are more stable, homogeneous catalysts enjoy only limited application in industry. There are
NiAl_1 NiAl_2
NiAl_3 NiAl_4
Fig. 2. The process of developing a surface model, studying the adsorption of intermediates and transition states, culminating in the kinetic model that is used to screen for new catalyst candidates. (Republished with permission of Maney Publishing, from (16); permission conveyed through Copyright Clearance Center Inc)
(a) (b)
(c)Ni:Sb
Ni:Sn
Ni:CNi:GaNi:Zn
Ni
Ni:Al
Ni:MoNi:Cr
5
2
–1
–4
–7
–10
–13
–16
–19–1.0 –0.5 0.0 0.5 1.0 1.5 2.0
C binding, eV
1.5
1.0
0.5
0.0
–0.5
–1.0
–1.5
O b
indi
ng, e
V
Ni:ZrNi:Ti

260 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
however, a number of important industrial reactions catalysed by homogeneous catalysts, including hydroformylation, methathesis, carbonylation and polymerisation using Ziegler-Natta catalysts. Perhaps it is in the pharmaceuticals and fine chemicals industries where homogeneous catalysts enjoy their greatest success. Coupling reactions such as the Suzuki-Miyaura reaction, Sonogashira coupling, the Heck reaction, Stille cross-coupling and the Buchwald-Hartwig reaction have long been employed to bring about various organic transformations.
Computational studies with DFT methods on the transmetallation step of the various cross-coupling reactions first appeared just over a decade ago (23–28) and provided useful insight into the energetics and mechanism of the reaction. Within a couple of years, DFT results for the full catalytic cycles were reported (29–34). DFT theoretical studies have also recently been used to investigate cross-coupling reactions involving metals other than palladium; examples include: rhodium-catalysed coupling via C–H bond activation (35, 36), nickel-catalysed cross-coupling reactions (37–42), delineating the mechanism of iron-catalysed cross-coupling reaction (43–47), dialkylzinc-mediated cross-coupling (48), copper-catalysed C–N cyclisation (49) and enantioselective nucleophilic borylation (50).
2.1 Palladium-Catalysed C–H Bond Activation of Heterocycles
Over the past decade, Pd-catalysed direct C–H arylation has emerged as an important alternative to traditional cross-coupling reactions for the synthesis of a wide range of arylated heterocycles (51–56). Compared to traditional Suzuki-type cross-coupling methods, direct arylation has the advantage that it does not require the preparation of stoichiometric quantities of organometallic reagents (typically alkyl or aryl boronic acids), thereby eliminating steps in the synthesis and reducing the amount of toxic metallic waste.
The most widely accepted mechanism for these reactions is the concerted metallation-deprotonation (CMD) pathway, involving simultaneous Pd–C bond formation and aromatic C–H bond cleavage to yield a diaryl Pd(II) species (57–59). A second mechanism, involving a cyclometallated complex, has also recently been proposed to account for the much lower than expected reactivity observed when certain tri-tert-butylphosphine-containing Pd catalysts are reacted with heteroarenes in isolation to the catalytic cycle (60, 61).
In collaboration with experimentalists from Johnson Matthey Catalysis and Chiral Technologies, a modelling study was undertaken to explore some anomalies pertaining to the direct arylation of oxazole and 4-bromotoluene. Oxazole, 1, with three reactive C–H sites, usually displays the reactivity order: C5>C2>C4. However, in certain instances, the reaction at the C5 site appears to be disfavoured, leading to a predominance of C2 product. Table I shows how the product distribution is affected by altering the amount of added phosphine ligand.
Table I Product Distribution for the Reaction of Oxazole with 4-Bromotoluene Using the Pre-Catalyst Palladium Acetate (Pd(OAc)2)
Catalyst Ligand % Mono 5
% Mono 2
% Di 2,5
% Di 4,5
Pd(OAc)2 – 66 0 28 3
Pd(OAc)2 1 × PtBu3 31 4 59 1
Pd(OAc)2 2 × PtBu3 6 35 50 0
In the absence of added phosphine ligand, dimethylacetamide (DMA) solvent is believed to coordinate to Pd. Figure 3 shows the reaction coordinate diagram for the C–H activation step (believed to be rate-limiting) of oxazole at positions C5, C4 and C2, by the well-established CMD mechanism. The results correlate well with the experimental observation that for Pd(OAc)2 in the absence of added phosphine, mono-5- product is formed in preference to both mono-2- and mono-4-substituted product.
Figure 4 shows the reaction coordinate for oxazole arylation, via the CMD mechanism, in the presence of PtBu3 ligand. Formation of the initial intermediate appears to be disfavoured for this mechanism, for both coordination through N3, as well as through the C4=C5 pi bond. The barrier heights for all three intermediates are also fairly large. Our calculations thus suggest that C–H activation at any of the reactive sites via the
Oxazole, 1
O
N
1
234
5

261 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
O H
O PdDMA
NO
Ar
O H
O PdDMA
ON
Ar
O H
O PdDMA
NO
Ar
DMAO Pd Ar
OH ON
ON+
H–OO Pd Ar
DMA
ON+
H–OO Pd Ar
DMA
OO
O
Pd ArDMA
N
OO
O
Pd ArDMA
N
OO
Pd ArDMA
O
N
NO
+
OO Pd
DMAAr
14.0
30.00
25.00
20.00
15.00
10.00
5.00
0.00
–5.00
–10.00
–15.00
ΔG
, Kca
l mol
–1
Fig. 3. Free energy diagram for direct arylation of oxazole with [(DMA)Pd(Ar)(OAc)] via the CMD mechanism
O H
O PdPtBu3
NO
Ar
O H
O PdPtBu3
ON
Ar
PtBu3
O Pd Ar
OH ON
ON+
H–OO Pd Ar
PtBu3
ON+
H–OO Pd Ar
PtBu3
OO
O
Pd ArPtBu3
N
OO
O
Pd ArPtBu3
N
OO
Pd ArPtBu3
O
N
NO
+
OO Pd
PtBu3
Ar
11.0
30.00
25.00
20.00
15.00
10.00
5.00
0.00
–5.00
–10.00
–15.00
ΔG
, Kca
l mol
–1
O H
O PdPtBu3
NO
Ar
Fig. 4. Free energy diagram for direct arylation of oxazole with [(PtBu3)Pd(Ar)(OAc)] via the CMD mechanism

262 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
CMD mechanism in the presence of PtBu3 ligand is unfavourable.
Hartwig et al. (60, 61) have shown that direct arylation of pyridine N-oxide with (PtBu3)Pd(Ar)(OAc) involves cooperative catalysis between two distinct Pd centres. Figure 5 shows the modelling results for oxazole C–H activation via this recently proposed ‘cooperative’ mechanism and is in agreement with our experimental findings. The initial complex that forms has oxazole coordinated to Pd either through N3 or through the C4=C5 pi bond. In this case, however, formation of the latter is unfavourable by around 2 kcal mol–1. C5 activation still has a marginally lower barrier height, but initial intermediate formation is unfavourable. The energy barrier for C2–H activation is still quite large (around 21 kcal mol–1), but not as large as for the DMA ligand. These findings suggest that C5–H activation is not significantly favoured over C2–H activation as is observed in the absence of a phosphine ligand.
This work has shown how modelling can be instrumental in explaining experimental observations.
A better understanding of the factors that control the reactivity and regioselectivity of various heteroaromatic substrates is important in designing reaction conditions that will favour activating a specific C–H bond in a particular substrate. Additionally, it would allow Catalysis and Chiral Technologies to better market their range of Pd-catalysed cross-coupling catalysts towards specific applications. Future work includes investigating the regioselectivities of the di-substituted products, as well as the effect that the amount of PtBu3 has on the product distribution.
3. Heterogeneous Catalysis
Heterogeneous catalytic materials come in several forms. Generically one could say that these are either unsupported (for example zeolites or PdO) or supported (for example nickel/alumina). The majority of our research has focused on the latter type, where either pure, doped or alloy nanoparticles are deposited on a support that may be either inert, acid/base or redox active. Historically, extended infinite surfaces have been used as models corresponding to the limit
ON+
H–OO
tBu
PdPtBu
12.8
30.00
25.00
20.00
15.00
10.00
5.00
0.00
–5.00
–10.00
–15.00
ΔG
, Kca
l mol
–1
tBu
O H
O
ON
PdPtBu
tBu
O
OH ON
PdPtBu
ON+
H–OO
tBu
PdPtBu
O H
O
NO
PdPtBu
tBu
O H
O
NO
PdPtBu
tBu
OO
ON
PdPtBu
tBu
OO
ON
PdPtBu
tBu
OO
tBu
O
N
PdPtBuN
O
+
OO
tBu
PdPtBu
Fig. 5. Free energy diagram for direct arylation of oxazole with [(PtBu3)Pd(Ar)(OAc)] via the cooperative mechanism
263 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
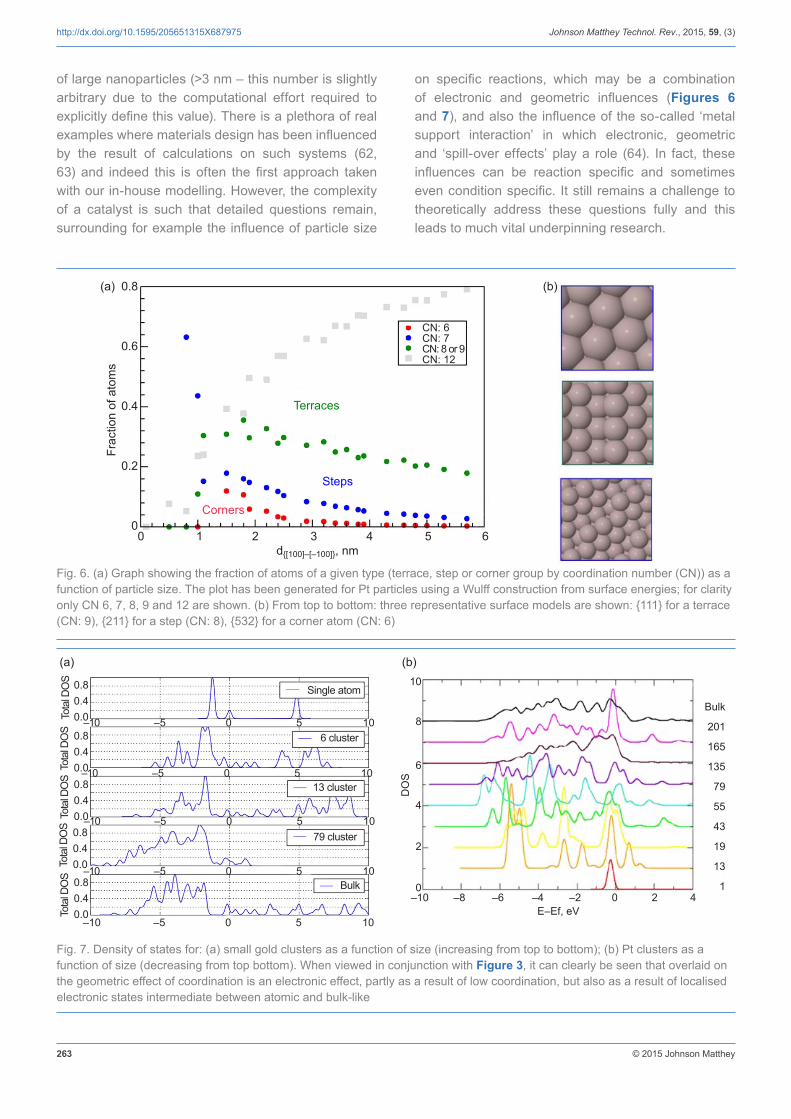
of large nanoparticles (>3 nm – this number is slightly arbitrary due to the computational effort required to explicitly define this value). There is a plethora of real examples where materials design has been influenced by the result of calculations on such systems (62, 63) and indeed this is often the first approach taken with our in-house modelling. However, the complexity of a catalyst is such that detailed questions remain, surrounding for example the influence of particle size
on specific reactions, which may be a combination of electronic and geometric influences (Figures 6 and 7), and also the influence of the so-called ‘metal support interaction’ in which electronic, geometric and ‘spill-over effects’ play a role (64). In fact, these influences can be reaction specific and sometimes even condition specific. It still remains a challenge to theoretically address these questions fully and this leads to much vital underpinning research.
Fig. 6. (a) Graph showing the fraction of atoms of a given type (terrace, step or corner group by coordination number (CN)) as a function of particle size. The plot has been generated for Pt particles using a Wulff construction from surface energies; for clarity only CN 6, 7, 8, 9 and 12 are shown. (b) From top to bottom: three representative surface models are shown: {111} for a terrace (CN: 9), {211} for a step (CN: 8), {532} for a corner atom (CN: 6)
0 1 2 3 4 5 6d{[100]–[–100]}, nm
0.8
0.6
0.4
0.2
0
Frac
tion
of a
tom
s
Terraces
Steps
Corners
CN: 6CN: 7CN: 8 or 9CN: 12
(a) (b)
Fig. 7. Density of states for: (a) small gold clusters as a function of size (increasing from top to bottom); (b) Pt clusters as a function of size (decreasing from top bottom). When viewed in conjunction with Figure 3, it can clearly be seen that overlaid on the geometric effect of coordination is an electronic effect, partly as a result of low coordination, but also as a result of localised electronic states intermediate between atomic and bulk-like
–10 –5 0 5 10
Single atom
6 cluster
13 cluster
79 cluster
Bulk
0.80.40.0To
tal D
OS
Tota
l DO
S 0.80.40.0
Tota
l DO
S 0.80.40.0
Tota
l DO
S 0.80.40.0
Tota
l DO
S 0.80.40.0–10 –5 0 5 10
–10 –5 0 5 10
–10 –5 0 5 10
–10 –5 0 5 10
(a)
–10 –8 –6 –4 –2 0 2 4E–Ef, eV
10
8
6
4
2
0
DO
S
Bulk
201
165
135
79
55
43
19
13
1
(b)

264 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
3.1 Methane Activation
The activation of methane is central to many technologies, from hydrocarbon combustion and methane oxidation in catalytic emission control to the synthesis of hydrogen and syngas as a feedstock for Fischer-Tropsch and ammonia syntheses (65).
In the area of steam reforming catalysis, models have been developed to go beyond understanding the intrinsic activity of steam reforming catalysts and begin to consider the influence of poisons and unwanted side reactions. For instance, building on published models (66), extensions have been made to account for: (a) site blocking and (b) the electronic deactivation caused by the presence of sulfur (67). Using the modified model the influence of sulfur can be explicitly simulated (68), enabling strategies to be developed to mitigate its influence. Figure 8 shows an example of a sulfur overlayer, along with a calculated thermodynamic isostere for sulfur coverage on Ni {111}, as a function of temperature and partial pressure of hydrogen sulfide (H2S). This figure allows direct comparison to published experimental work (69), against which calculated enthalpies (ΔH) and entropies (ΔS) can be validated. This exercise showed excellent agreement (ΔH (800 K): –145 (Exp. –155.2) kJ mol–1, ΔS (800 K) = +38 (Exp. +35.9) J K–1 mol–1), indicating that these more complex reactivity studies result in good trends. Figure 9 (70) illustrates the influence
of sulfur adsorption on the steam reforming reaction over a Ni{211} and the resulting activity trends for a series of alternative alloys.
During the steam reforming reaction the dissociation of methane on a nickel catalyst can lead to the formation of polymeric carbon on the catalyst surface (Figure 10). The carbon filaments so formed are very problematic, ultimately leading to deactivation of the catalyst and shutting down of the reactor. Using DFT modelling, the origin of the deactivation has previously been investigated along with possible mitigation strategies (71–73). Two fundamental approaches have been studied in-house: the first is to examine the role of dopants and alloys on the initial formation and nucleation of the problematic carbon, the second to look at burn-off of any formed carbon using oxygen based species and how this can be promoted.
The work has shown that in general if one inhibits the formation of carbon there is a negative effect on the activity of the catalyst; there is the unfortunate problem that the very surface sites responsible for catalytic activity are also the primary nucleation sites for carbon formation. This leads one to consider the promotion of burn-off as the pragmatic route to deal with it (Figure 10). Through modification of the support or surface composition of the catalyst it is hoped that promotion of this mechanistic route can be achieved and this is the subject of ongoing research.
Ordinarily, the presence of excess carbon is unwanted. However, there are also reactions where the presence
–10
–12
–14
–16
–18
–20
–221.0 1.2 1.4 1.6 1.8 2.0
1000 k/T
(a) (b)
Fig. 8. (a) Optimised structure of 0.25 ml S on a Ni surface; (b) isostere of S calculated from first-principles. Good agreement for saturation coverage (just below 0.25 ml) and entropies is found with the work of McCarty and Wise (69)

265 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
of carbon growth has been found to promote catalytic activity (74). This has inspired a fundamental research project looking at how defective graphene sheets can potentially act as catalysts for converting propane to propene (75). Furthermore, carbon is also used as a support in applications such as fuel cells. These observations highlight that it is not straightforward to classify whether a given element is a poison; as exemplified by the case of carbon in catalysis, it can be both a friend and a foe.
Partial oxidation of methane over yttrium stabilised zirconia (YSZ) has been experimentally shown to be catalytically active for the partial oxidation of methane (76, 77). First-principles electronic structure calculations have been applied to explore the possible mechanism of methane C–H bond activation over the oxygenated YSZ [111] surface (Figure 11) (78). Previous theoretical
studies (79) and experimental evidence (80) suggest that YSZ will favourably adsorb molecular oxygen over intrinsic vacancy sites and partially reduce it creating stabilised superoxide-like species. DFT calculations of molecular oxygen adsorbed over a vacancy site on the YSZ [111] surface showed that the O2 species was partially reduced. Charge distribution analysis showing it to be in an approximately –0.5 oxidation state, with the O–O bond lengthened slightly and large regions of unpaired electron density on each oxygen atom, all demonstrating transfer of electron density from the surface in to the oxygen π* anti-bonding orbital. The adsorption was found to be energetically favourable, the DFT model giving an adsorption energy of –0.47eV.
The oxygenated surface model described above was used to study the methane activation process. The pure zirconia (ZrO2) and unoxygenated YSZ surfaces were
Fig. 10. Illustration of: (a) nucleation of C at step edge (or defect); (b) growth of graphitic-like carbon; (c) reaction of graphitic carbon with surface OH (burn-off mechanism)
(a) (b) (c)
0
–1
–2
–3
–4
–5
log
(θ)
0
–1
–2
–3
–4
–5–9 –8 –7 –6 –5 –4 –3 –2
log (PH2S/Pstan.)
50% drop in activity
TOFθHθCθSθCH2θOθOHθ*
log (TOF/s
–1)
0
–1–9 –8 –7 –6 –5
(a)
1500
1000
500
0
Pric
e, $
km
ol–1
0 0.2 0.4 0.6Activity
Rh
Rh3NiRh3Ru
Ni3Rh
Ni3NiRu3NiNi3Ru
Ru3RhPt3Ru
Pd3RuRu3PdPd3Co
Ru3RuCo3Pd
(b)
Fig. 9. (a) Calculated turn over frequency and coverage of surface species for methane steam reforming in the presence of S over a Ni {211} surface. Kinetic models like this can be used as the basis for theoretical screening studies; (b) Pareto optimal, intrinsic metal cost (2009), vs. activity for methanol steam reforming (MSR) catalyst, oxidised catalysts have been filtered out following the method outlined in (70)

266 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
found to be relatively inert towards methane. However, on the oxygenated surface, methane was found to transfer a hydrogen to a surface lattice oxygen ion in a site neighbouring an adsorbed O2 species to yield a surface OH group and a triangular planar CH3 molecular species in the gas phase. Analysis of the charge density distribution confirms that the CH3 entity is a charge neutral methyl radical and that the H3C–H bond has undergone homolytic cleavage. The overall process is predicted to be energetically favourable (–0.23 eV). Transition state calculations using a constrained algorithm in combination with the nudged elastic band method (81), revealed a relatively low activation barrier of approximately 0.4 eV, suggesting that the reaction should occur readily. Analysis of the changing geometry and electronic structure allowed the prediction of the simplified mechanism given in Figure 11, with the adsorbed oxygen acting as an electron acceptor. It is likely that similar mechanisms may operate over other related metal oxide materials. Further information and analysis of this work is available (78).
4. Electrocatalysis
Proton exchange membrane (PEM) fuel cells offer a promising clean energy source for a range of both stationary and automotive applications. However, there are a number of issues which must be overcome in order for these to be commercialised. These include the high cost of the platinum electrodes, slow kinetics of the oxygen reduction reaction (ORR) and stability of the electrodes under operating conditions (82, 83).
Alloying Pt with another transition metal is one possible way of overcoming some of these issues and this is an area that has been subject to intensive research in the last few years (84–89).
The ORR at the cathode consists of several reaction pathways, the detailed understanding of which is still the subject of ongoing research (90). Generally, the reaction can follow two pathways: the four electron route to water (Equation (i)) and the two electron route to H2O2 (Equation (ii)):
O2 + 4H+ + 4e–à 2H2O (i)
O2 + 2H+ + 2e–à H2O2 (ii)
On Pt and Pt alloy surfaces it is generally agreed that the ORR follows the four electron pathway to water. This reaction can be broken down into the following elementary steps (Equations (iii)–(v)):
½O2à O* (iii)
O* + H+ + e-à OH* (iv)
OH* + H+ + e-àH2O + * (v)
(where * denotes a surface atom or empty surface site).In order for a surface to be an effective catalyst it
should follow Sabatier’s principle (91, 92). This states that a good working catalyst should have the ability to break bonds and generate intermediates. However, it should also have a low enough interaction energy with these intermediates not to stabilise them on the surface, so that they can react further and free up adsorption sites. It has been suggested however that the reason for the slow ORR may be due to the O intermediate binding too strongly to the Pt surface, therefore accumulating on the surface and blocking active sites. The surface d-band centre and oxygen binding energies have been widely used as descriptors for the ORR. Both these show a volcano type relationship with catalytic activity (85).
4.1 Transition Metal Alloy Nanocatalysts
High throughput screening of materials using computational approaches such as DFT has become a powerful and valuable tool in catalyst design. The search for the optimal material, based on predicted activity and stability, among a great number of alloy combinations is both a materials and a combinatorial problem. A high throughput approach was developed and adopted for
Fig. 11. Schematic diagram of the proposed methane activation mechanism over oxygenated YSZ surface. The excess charge balance on the product is delocalised throughout the ZrO2 system (78)
HH
H
O O
HHH
H–O
Zr4+Zr4+O2–
H~0.5–
O O–
DelocalisedYSZ surface YSZ surface

267 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
investigating the impact of varying the ratios of metals in the alloy surface layer to determine which compositions are most stable and electrochemically active. In this work, through collaboration with project partners Accelrys and CMR Fuel Cells, a combined theoretical and experimental approach was taken to investigate trends in the stability of Pt-M-Pt and M-Pt-Pt core-shell type catalysts (M = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Au, Ta, Hf, Cr, Nb, V, Y, Sc, Ti and Zr).
The surfaces were modelled using a 2×2 unit cell of the face-centred cubic (fcc) crystal of Pt (lattice constant bulk Pt system: a = 4.010 Å). The Pt-M-Pt configuration was modelled by substitution of the second atomic layer of Pt with M atoms, whereas the M-Pt-Pt configuration consisted of the Pt atoms in the first atomic layer being substituted with M atoms (Figure 12).
The stability of the electrode under fuel cell operating conditions is a significant challenge to understand, as the highly corrosive environment can promote surface segregation of the alloying component, which can then be leached away removing any benefit obtained from alloying (93–95). The adsorption of O and OH on the surfaces of Pt-M-Pt and M-Pt-Pt alloys was investigated, sampling all possible adsorption sites to identify the most stable. The stability of the surface was assessed by calculating the segregation energy, Eseg, firstly in vacuum (Equation (vi)) and then with the O or OH atom adsorbed (Equation (vii)). The segregation energy is defined as the difference in total energy
between the Pt-M-Pt and M-Pt-Pt structures with and without O/OH adsorbed. If the segregation energy is negative the Pt skin structure is favoured, if it is positive a surface of M atoms is favoured (Equation (vi)):
Eseg = E (Pt-M-Pt) – E (M-Pt-Pt) (vi)
and in the presence of adsorbate (O or OH) (Equation (vii)):
Eseg = E (Pt-M-Pt)(O/OH) – E (M-Pt-Pt)(O/OH) (vii)
Figure 13 shows the segregation energies for the structures with and without O and OH adsorbed. The value of the segregation energy indicates how strongly a particular configuration is favoured. A more negative segregation energy indicates that a Pt skin is
Fig. 12. Schematic depicting the under/overlayer structure of core-shell type catalysts: (a) Pt-M-Pt structure; (b) M-Pt-Pt structure
2.00
1.00
0.00
–1.00
–2.00
–3.00
–4.00
–5.00
–6.00
–7.00
PtA
gP
tAu
PtC
oP
tCu
PtF
eP
tIrP
tNi
PtO
sP
tPd
PtR
hP
tRu
PtA
lP
tBe
PtC
rP
tHf
PtM
nP
tNb
PtS
iP
tTa
PtT
iP
tV
Alloy
CleanOads
OHads
Ese
g, e
V
Fig. 13. Calculated segregation energies for clean {111} surfaces of various Pt alloys in vacuum and with ¼ ml of O or OH adsorbed

268 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
strongly favoured. In vacuum it can be seen that the Pt-M-Pt structure is favoured in all cases except Au and Ag (purple bars). However when O is adsorbed the segregation energy is weakened in comparison to the clean surface (blue bars). For the ORR reaction, the materials that retain a Pt skin are most promising. For Pt-Ni-Pt, Pt-Pd-Pt, Pt-Be-Pt and Pt-Y-Pt surface segregation of M is predicted in the presence of adsorbed O. For the remaining alloys a Pt skin is predicted though this is not favoured as strongly as for the clean surface. A reversal in segregation energies is observed for Pt-Ag-Pt and Pt-Au-Pt, which are more stable with a Pt skin in the presence of O.
Adsorption of OH on the surface destabilises the system resulting in a weaker segregation energy compared to the clean surface. This destabilisation is less pronounced than previously seen for O at the surface, with more negative segregation energies obtained for OH. This indicates that the tendency to retain a Pt skin in the presence of OH is stronger than in the presence of O.
The most promising ORR catalysts are ones in which a Pt skin is retained, acting as a protective barrier to prevent the leaching of M from the subsurface. Analysis of the segregation energies in the presence of both O and OH indicate that in terms of stability all compositions except Pt-Be-Pt, Pt-Ag-Pt, Pt-Au-Pt, Pt-Ni-Pt and Pt-Pd-Pt are promising, as a Pt skin is retained in the presence of O and OH when adsorption takes place.
Figure 14 shows the adsorption energy of ¼ ml of oxygen on the surface of each Pt-M-Pt alloy. Alloys to the left of Pt have a lower d-band centre and are
predicted to bind O more weakly than Pt. It is these alloys which bind O slightly more weakly than Pt that are predicted to have enhanced ORR activity. However alloys such as Pt-Ti-Pt and Pt-Al-Pt are predicted to have poor ORR activity as they bind O too weakly.
Using this approach over 2000 alloy combinations were screened and several potential candidates identified to put forward for further experimental testing. This theoretical pre-screening approach allowed the list of combinations for testing to be significantly narrowed down, saving valuable experimental time and resource.
4.2 Carbide Core Nanocatalysts
An alternative strategy to using a transition metal alloy as a core particle is to explore the possibility of using transition metal nanoparticles (96). The use of carbides as non-transition metal cores supporting thin films of Pt has been investigated. Through the study of various carbides and their interaction with Pt overlayers the project has been able to elucidate (a) the geometric and electronic influences on the stability of Pt on these carbides; and (b) how the presence of the different carbide cores influences predicted catalytic activity (Figure 15). Working closely with the synthetic chemists at Johnson Matthey Technology Centre, Sonning Common, the research has created inspiration and directions for research which are being followed in-house.
It is understood that synthesising a material where Pt wets a carbide surface or other non-metal cores may be challenging from a thermodynamic perspective.
–2.9 –2.4 –1.9 –1.4
PtAl PtTiPtSc
PtHf/PtZrPtNb
PtV PtTa
PtCrPtMn
PtCoPtRu
PtOs PtFe
PtIr
PtCu
PtRhPt
PtAg
PtAu
PtPd
Ed–Ef, eV
–2
–2.2
–2.4
–2.6
–2.8
–3
–3.2
–3.4
–3.6
–3.8
–4
E O
ads,
eV
–3.4
Fig. 14. Correlation of d-band centre and O adsorption energy for Pt-M-Pt alloys

269 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
Therefore the work, in its second phase, investigated strategies to promote the stability of Pt, for instance through the use of so-called tie-layers. Figure 15 illustrates the stability of a test set of tie-layer/Pt combinations, showing the propensity for the Pt to remain as a skin or to be sandwiched between the tie-layer and carbide support.
5. Emission Control Catalysis
The use of catalysts in emission control is wide and varied. Along with the supported nanoparticle catalysts discussed to this point, another significant class of catalyst is the zeolite. Zeolites are crystalline, microporous materials in which the atoms are arranged to form a network of molecular sized pores and channels. This unique porous structure combined with their huge internal surface area gives rise to a vast number of applications. By tailoring the pores and channels, molecules can be excluded on the basis of size and shape and catalytic processes can be driven to yield only preferred reaction products. Within Johnson Matthey’s business areas alone, zeolites are key components of selective catalytic reduction (SCR) catalysts, as additives for fluidised catalytic cracking (FCC) catalysts, and as catalysts in the petrochemicals refining process.
Zeolites have been extensively studied over the last decade using a wide range of modelling techniques. The structure of the framework, effects of doping, location of ions and adsorbates within the pores, diffusion of reactants and products within the structure and the reaction pathways leading to the catalytic breakdown of molecules can be readily computed. Atomistic modelling has been used to aid the characterisation of
zeolites used for SCR of nitrogen oxides (NOx) species for emission control – in particular to help elucidate the location of ions within the pores and to understand how these interact with other species located within the voids, such as water, nitrogen oxide (NO) and ammonia (NH3).
Small pore zeolites such as CHA have recently gained a lot of interest due to their very good low-temperature activity and enhanced stability compared to medium and large pore zeolites such as MFI and beta (97–103). An understanding of the nature and location of transition metal ions such as Cu and Fe within the pores and how these interact with key NH3-SCR adsorbates such as NO and H2O is crucial in understanding the mechanism of NOx reduction in these zeolites and this in turn can help in devising strategies to improve the performance of these catalysts. A combined simulation and experimental approach was used to investigate Cu location in CHA using techniques such as energy minimisation based on interatomic potentials and quantum mechanics/molecular mechanics (QM/MM) methods to identify stable cation exchange sites within the pores and study the influence of adsorbate interactions (Figure 16). These results were compared to high resolution X-ray diffraction (HR-XRD) and infrared (IR) measurements of probe molecules to elucidate the ion location and how this is modified by interaction with adsorbates.
Diffusion of reactants and products to and from the active sites within the zeolite pores is a key part of the catalytic cycle and can have a significant impact on a catalyst’s performance. Therefore an understanding of this process is crucial to optimising the use of zeolites as catalysts. An ongoing project is using modelling to understand diffusion of various molecules within these
1.5
0.5
–0.5
–1.5
–2.5
Ir Pd Ga Sn
Ese
g, e
V a
tom
–1
Eseg (100)Eseg (111)Eseg (001)ΔEads (100)ΔEads (111)Eads (001)
(a) (b)
Fig. 15. (a) Stability of Pt forming an outer shell with several tie-layer candidates. Overlaid is the oxygen adsorption energy relative to pure Pt; (b) schematic illustrating the carbide core material with the tie-layer/Pt overlayers

270 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
materials, including NO, NH3, H2O, propane, xylene and CO2. The atoms in a zeolite can be arranged in a variety of ways to form rings, channels and pores with different sizes and dimensionalities, leading to a large number of possible framework structures. Molecular dynamics (MD) simulations are being used to try and understand the influence of ring size, pore volume, dimensionality and chemical composition on diffusion through the framework and to predict which structures and compositions would allow optimal diffusion.
The self-diffusion coefficients calculated using MD simulations can also be measured using experimental techniques such as pulse field gradient-nuclear magnetic resonance (PFG-NMR) and quasi-elastic neutron scattering (QENS) techniques and a project is underway in collaboration with University of Cambridge, UK, and the UK Catalysis Hub at Harwell, UK, to measure diffusion of selected molecules in selected frameworks to compare with MD simulations. Some initial MD simulation results investigating xylene diffusion in USY are presented in Figure 17 for ortho-, meta- and para-xylene. Figure 17 shows diffusion coefficients at temperatures between 300 K and 700 K for each isomer. The diffusion coefficients increase with temperature and indicate that diffusion of the para isomer is faster than meta or ortho isomers. Initial PFG-NMR measurements at University of Cambridge at 318 K gave a diffusion coefficient of 4.6 × 10–10 m2 s–1 which is in good agreement with MD simulated values of 5.92 × 10–10 m2 s–1 at 323 K.
6. Underpinning Research
In addition to the more applied research reported above, a key philosophy is to look at underpinning
research for the methods used to enable continuous improvement in modelling capability. This research is often carried out in collaboration with academic groups. We now proceed to discuss some of the academic collaboration Johnson Matthey has been involved in and how the underpinning knowledge is being used to improve understanding of catalytic materials.
6.1 Redox Active Oxides and Metal Support Interactions
Certain oxides provide a challenge to standard computational methods. Working with academics at Cardiff University, UK, and also via a Royal Society Industrial Fellowship at University College London (UCL), UK, we have been running academic projects looking at pragmatic ways to model ceria and other redox active metal oxides for catalytic activities. A pertinent question in this work was whether trends in metal screening studies need to go beyond standard DFT methods (Figure 18). The inclusion of the so called Hubbard U parameter has been found not only to be vital for obtaining the ‘correct’ electronic structure (105), but also to have a significant influence on obtained formation energies and, importantly for catalysis, shows that reaction energetics can be highly sensitive to the choice of U (Figure 19) (105–107).
Once a reasonable model for the support phase of the catalyst is obtained, the interaction between the nanoparticle and the support must be considered. A crucial question here is: how big are your nanoparticulate catalysts? The answer clearly has a bearing on how to consider modelling the system. For instance Figure 6 shows three separate regimes for catalysts of different sizes: smaller than 2 nm, between 2 nm and 3 nm and beyond 3 nm where the facets of
0 100 200 300 400 500 600 700 800Temperature, K
3.5
3
2.5
2
1.5
1
0.5
0
Ds,
m2 s
–1, ×
10–9
orthometapara
Fig. 17. Diffusion coefficient (Ds) as a function of temperature (T) for ortho, meta and para xylene in zeolite structure USY
Fig. 16. Two low energy sites for Cu+ identified using: (a) interatomic potentials; and (b) QM/MM methods
2.1772.8692.148 2.020
2.562
2.035
2.010
(a) (b)

271 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
the nanoparticle are sufficiently large to begin to think in bulk terms (it should be noted the 3 nm bound is a slightly arbitrary choice of bound for this regime). From Figure 7, bulk-like electronic structure can be observed even before the facets grow significantly large. This has now been confirmed by massively parallel calculations of nanoparticles (108–110). This suggests the following general regimes:
(a) <2 nm: a regime that is modulated by both electronic and geometric effects and significantly different reactivity from the bulk terminated surfaces would be expected
(b) 2–3 nm: a regime where the intrinsic electronic nature of the metal is ‘bulk-like’, but influence of transport or limited numbers of a given facet could be influencing the observed catalysis (for example
Fig. 19. (a) dependence of various properties of PdO as a function of Hubbard U parameter, compared to benchmark results. Vacancy formation energy compared to Heyd-Scuseria-Ernzerhof (HSE) calculations of Delley (106). Band gap compared to (107) and Pd4O4 compared to in-house coupled cluster calculations. The region where the difference in properties and energetics approach zero identifies the required value of U; (b) CeO2 reaction profile adapted from (105), here the acute dependence of reaction profile on the choice of U parameter is illustrated, which necessitates a deeper study of choosing U for catalyst problems (105)
0 1 2 3 4 5 6U, eV
2
1
0
–1
–2
–3
Ene
rgy,
eV
ΔE (Pd4O4)ΔE (band gap)ΔE (vacancy)
(a)
0
–1
–2
–3
–4
Ads
orpt
ion
ener
gy, e
V
CeO2 + CO CO/CeO2 CeO2–d + CO2
0.0 eV2.5 eV4.5 eV5.5 eV6.5 eV7.5 eV8.5 eV
(b)
Fig. 18. Density of states calculated using standard DFT (red) and with the Hubbard U correction (blue): (a) lanthanum oxide (La2O3), wide band gap oxides are reproduced quite well; (b) palladium (II) oxide (PdO), standard DFT predicts PdO to be a metal, the Hubbard U correction is required to obtain the correct band gap (as seen in Figure 13, this also improves the calculated energetics); (c) CeO2, whilst the overall band gap (2p–5d) is reproduced well with standard approaches, the width and location of the Ce-4f is highly sensitive to the choice of Hubbard U parameter, it is this state that is responsible for the facile redox properties of CeO2 (104)
–10 –8 –6 –4 –2 0 2 4Energy, eV
02
5
4
3
2
1
0
Den
sity
of s
tate
s
5
4
3
2
1
0D
ensi
ty o
f sta
tes
–10 –8 –6 –4 –2 0 2 4Energy, eV
–6 –4 –2 0 2 4 6Energy, eV
50
40
30
20
10
0
Den
sity
of s
tate
s
05
05.5
(a) (b) (c)

272 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
the availability of hydrogen in the methanation reaction)
(c) >3 nm: the geometry and electronic influences are sufficiently bulk-like to use extended surfaces as ‘good’ models.
In general the size of a nanoparticle has a profound influence on the observed chemistry and subsequent catalytic behaviour. This has been well documented for Au, however limited attention has been given to other metals (111, 112). In Figure 20, this point is illustrated for the activation of methane over silver. Not only does the small size increase the activity of the particle relative to the extended system, it produces a catalyst that is in a different chemical state, thereby opening up chemical routes that would not otherwise have been present.
For small clusters and nanoparticles, the combination of small size and the presence of surface oxide species opens up even more combinations of chemistry. For the case of a small nanoparticle which can be modelled explicitly, the presence of the support introduces an interfacial region that is critical for the chemistry and for the case of reducible oxides it also changes the availability of oxygen for reaction.
6.2 Metal Support Interactions
The chemical interactions between a metal particle and its metal oxide support are of critical importance
for the chemical industry and have been the focus of numerous computational modelling investigations (113–118). The typical size of these metal particles is of the order of the nanometre. At such scales the interaction of the particle with its chemical surrounding is of great importance as it impacts its stability (with public health and economic impacts) and may impact its catalytic activity as well. As a consequence, a PhD project in collaboration with UCL was initiated with the aim of using DFT computational modelling methods to investigate how transition metals such as Pd, Pt and Ni interact with an α-alumina support.
The initial investigation studied how single metal atoms adsorb on the (0001) and (1102) α-alumina surfaces. On both surfaces, the binding strength follows the trend Pt>Ni>Pd (119). When bonding to the surface, all transition metals promote a charge transfer, moving electron density from a neighbouring surface oxygen to a surface aluminium atom (Figure 21).
In order to take into account alumina’s strong affinity for water, a thermodynamic model of a different surface environment was developed. By including the chemical potential of water in the gas phase it was possible to compute the Gibbs free energy of several water coverages of α-alumina surfaces and to predict the state of the surface at a given pressure and temperature. Figure 22 presents the evolution of the Gibbs free
Reaction coordinate (gas → surface)
3
2.5
2
1.5
1
0.5
0
–0.5
–1
eV
NO*CH3*CH3O*2O*2H*
(a)
Fig. 20. Free energy diagrams for surface species present on: (a) a Ag {111} facet and (b) 13 atom cluster: solid = 423 K, dashed = 623 K, dot-dashed = 823 K. It can be seen for the Ag {111} surface only O is present at low temperature and at higher temperatures the surface is clean. However for the small cluster at 423 K, O, H, NO and CH3O are present, while at 623 K O and NO are present and at 823 K, O is still on the surface. The presence of O has significant influence on the calculated activation barriers. Eact[Ag {211}] = 2.15 eV, Eact[Ag13] = 1.57 eV, Eact[Ag13O6] = 0.74 eV; implying that O covered Ag13 is as active as Ni for the dissociation of methane (8)
Reaction coordinate (gas → surface)
NO*CH3*CH3O*2O*2H*
3
2
1
0
–1
–2
–3
eV
(b)
NO*CH3*CH3O*2O*2H*
Reaction coordinate (gas → surface)

273 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
energy of the clean, low (5 OH nm–2) and high hydrated (10 OH nm–2) states of the (0001) surface. At 3.2 kPa, the (0001) surface gradually changes from a heavy hydration state to a fully clean surface at around 900 K. On the (1102) surface, the transition between the high OH coverage and the clean surface is much more sudden without an intermediate moderate hydration state (120).
Surface hydration has a strong impact on the interaction of the transition metal with its support as the surface aluminium sites are now fully occupied by OH groups and are not available to receive electron density moved from surface oxygen atoms. Instead, upon adsorption, all three investigated transition metal atoms trigger the rupture of a surface OH group, followed by the migration of the hydrogen atom to the metal atom. This mechanism is called ‘spill-over’ and
enables a significant stabilisation of Pt and Ni atoms on the alumina surface (120).
In order to gain some insight into the catalytic activity of supported metal nanoparticles, the interactions of a carbon monoxide probe with a supported Ni6
nanocluster has been studied. CO was chosen for its relative simplicity and its wide use in catalytic processes such as steam reforming, where supported Ni catalysts are also of relevance. The (0001) α-alumina surface has a strong influence on the CO interaction with the Ni6 cluster. The favoured adsorption site on the supported Ni6 cluster is on a hollow site on the side of the particle. On that site, CO is also able to interact with a surface aluminium atom (Figure 23) (121). The CO bond is elongated (1.28 Å vs. 1.13 Å in the gas phase and 1.20 Å on a (111) Ni hexagonal close-packed (hcp) site) and its stretching frequency is lowered (1397 cm–1
vs. 1899 cm–1 on a (111) Ni hcp site). This dramatic activation of the CO molecule illustrates how, even though considered as inert, the catalyst support may play an active role in the reaction cycle.
For small nanoparticles, it is important to provide an explicit computational model to describe mechanistic aspects of sub-nanometre particles supported on oxides. Work in collaboration with David Willock, Cardiff University, has been bringing together studies on redox active supports and small metal nanoparticles in a bid to understand the metal support interaction and its influence on reactivity (122). For the probe reaction of CO oxidation, new mechanistic pathways have been opened up due to the metal/oxide interface that radically changes the rate-determining step (Figure 24) (123). The theme of metal support interaction has also seen some experimental contribution and recent work with Tsang’s group in Oxford has been published on the
200 400 600 800 1000 1200 1400 1600Temperature, K
3.0
2.5
2.0
1.5
1.0
0.5
0.0
Sur
face
ene
rgy,
J m
–2
Fig. 22. Gibbs surface free energy at 3.2 kPa water partial pressure for the clean (blue), 5 OH nm–2 (green), and 10 OH nm–2 (red) hydration coverage of (0001)) α-Al2O3 (Adapted with permission from (120). Copyright (2003) American Chemical Society)
Fig. 23. Side view of CO on the Ni6 (0001)) α-Al2O3 system (Adapted with permission from (121). Copyright (2010) American Chemical Society)
Fig. 21. Top view of an adsorbed Pd single atom on (0001) α-Al2O3 Figure adapted from (119)

274 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
role of hydrogen spill-over influence on the reducibility of ceria supported pgm catalysts (64).
Obtaining an explicit description of large metal nanoparticles interacting with an oxide support is still a computational challenge beyond current computing capacity. However, a simple thermodynamic screening model has been developed with the potential to predict the general behaviour of certain transition metal/oxide support combinations, in the limit of large nanoparticles. Taking inspiration from the field of electrochemistry where a chemical reaction is considered in terms of the half-reaction, we can consider the following approximations:(a) metal supported oxides can be split into two
components (metal and oxide)(b) redox catalysts can be considered analogous to
electrochemical half reactions(c) one half reaction occurs on the metal, the other on
the oxide.Taking the example of simultaneous CO oxidation
and NO reduction it can be assumed that NO reduction occurs on the metallic component while CO oxidation occurs on the oxide component. Calculating the free energy profile for a simplified reaction pathway allows firstly existing databases of metal components to be used, secondly new databases to be developed for the oxide component and finally metal/oxide pairings to be identified using calculations that are currently tractable on available resources. Figure 25 illustrates the approach being used in the example of CO and NO. Early results in this area are promising and new
methods are currently being explored to screen for novel catalyst candidates.
Whilst models can be developed either explicitly or indirectly for the metal/support interaction for the two outer-bounds of the nanoparticle regimes under investigation, there is still a significant computational challenge in simulating the intermediate regime. The following section describes work towards developing a capability that will allow the 2–3 nm regime to be tackled explicitly, starting with an electronic structure theory of the metal nanoparticle itself.
6.3 Large-scale Calculations of Metal Nanoparticles
One of the significant barriers to developing complex models of real catalysts is the limitation on system size that can be tackled explicitly. Significant progress can be made with a judicious choice of model and some prior understanding of the system in question. If one is simply interested in geometric structure then arguably, embedded atom or other parameterised potential approaches can be utilised (124, 125). However in catalysis, especially when trying to understand chemical activity, there is often interest in linking the electronic structure to reactivity. This requires methods for conducting large scale electronic structure theory calculations, for instance computer codes with favourable scaling over many computer cores (for example GPAW, Order-N Electronic Total Energy Package (ONETEP) (126, 127)) or semi-empirical approaches (128).
(a)100
500
–50
–100
–150
–200–250–300–350
catalyst + CO (g)
O..CO (ts)
E
C, D
B
A
CO (ads)
CO2 (ads)
Grey: [O] = Au-OBlack: [O] = Fe-O
(b)
Fig. 24. (a) Au_10 nanoparticle adsorbed on iron (III) oxide (Fe2O3) (0001) surface, (blue: Fe, red: surface oxygen, yellow: Au, grey: carbon, green: CO oxygen); (b) reaction profile for the adsorption and reaction of CO with different surface O in the vicinity of the Au cluster. It can be seen there are a several distinct adsorption site Fe2O3, Au and interfacial, that lead to very different adsorption energies, furthermore the different reacting surface oxygens also lead to different activation barriers. (Adapted from Scott Hoh PhD thesis (123))

275 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
One part of this research is looking at exploiting the linear scaling capabilities of ONETEP through collaboration with Chris-Kriton Skylaris, Southampton University, UK. Enhanced functionality of ONETEP has been exploited to help in the simulation of metals (129) and work is now underway benchmarking speed and scaling for the adsorption of molecules such as O2, CO, OH and atomic O. The difference in adsorption energies between constrained but pre-optimised nanoparticles and ligands and between fully geometry-optimised systems of nanoparticles and ligands has been studied by looking at various sizes of cuboctahedral Pt clusters (Pt13, Pt55, Pt147) and comparing to more conventional slab models. The results show significant deviations in binding strength between the fully relaxed and constrained systems.
Another project with the University of Limpopo, South Africa, is developing parameters to describe Pd nanoparticles and their interaction with oxide supports via the semi-empirical, density-functional tight binding (DFTB) approach, which potentially allows thousands of atoms to be simulated using limited computational resources. A model system to understand the usefulness of this approach has been methane oxidation using Pd supported on titania (TiO2). This system has a number of complexities, including the metal/ceramic interface
and the propensity for the oxidation state of the surface to change during reaction. A significant challenge to utilising the DFTB approach is to find truly transferable parameters to describe all of the necessary interaction present in the simulation (130). Figure 26 illustrates geometrical structures obtained from ONETEP, GPAW and DFTB for nanoparticles that are tractable with current computational resources, the future will see this capability extended.
There are several fundamental experimental projects to augment the theoretical work on nanoparticles. The first of these involves the Nellist group from Oxford Materials, UK, who have derived experimental methods using quantitative annular dark-field scanning transmission electron microscopy (ADF-STEM)(131). Pt nanoparticle atomic coordinates have been determined for particles up to Pt943 atoms. The systems were geometry optimised using a damped MD and interatomic force-field approach, before being electronically minimised in ONETEP and studied quantum mechanically. Atomic oxygen adsorption calculations were then performed on the most exposed facet, which exhibits fcc-{111} surface symmetry and the least coordinated sites were found to be the most strongly binding, agreeing with terrace measurements from experimental literature.
4
2
0
–2
–4
–6
–8
ΔG (4
50 K
), eV
Ceria surface
Metal surface
CO (g) +
NO (g)
CO (g) +
NOM *
CO (g) +
NM * +
OM *
CO (g) +
0.5N 2 + O
M *
COOx * +
0.5N 2 + O
M *
CO 2 (g) +
0.5N 2 + O
M *
CO 2 (g) +
0.5N 2
AuAgCuPdPtNiRhCoRuFeReMoW
Fig. 25. Reaction profile for the NO reduction, CO oxidation, occurring on a series of metals left hand side and Ce surface, right hand side. The highlighted dashed line illustrates the interpolation between reactants and products. To a first approximation this interpolation minimises the free energy pathway and shows the region where an optimal catalyst can be found

276 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
The second experimental project is looking at methane oxidation, in order to provide data about nanoparticle surface composition under a reactive gas-phase atmosphere that in turn can be used to validate theoretical predictions. The project is being run with Professor Georg Held, University of Reading, UK, and
methods are being developed to create well defined nanoparticle samples for use in ambient pressure X-ray photoelectron spectroscopy (XPS) synchrotron beamlines (132). The ultimate aim is to bridge the ubiquitous temperature, pressure and materials gaps between surface science and real catalysts. The ambient pressure XPS allows the oxidation of different sized nanoparticles to be observed as a function of reactive atmosphere, which in turn can be correlated back to theoretical predictions. This project is ongoing and in the future will make use of the forthcoming versatile soft X-ray (VERSOX) beamline at Diamond Light Source, Harwell, UK, to develop even further new understanding of catalysis materials.
6.4 Moving Beyond the Atomic Scale
The emerging field of modelling across length scales is ideally suited to modelling catalysis since catalyst solutions rely not only on the intrinsic chemical properties of an active material, but also on the transport properties of the material at different scales. Figure 27 (133) illustrates the case of an automotive monolith, highlighting the levels that need to be modelled to develop a holistic description of the catalytic solution. A complete description of a technical solution includes describing, for example, the atomic or electronic level of matter, the porous structure of the catalyst layer (at two distinct levels: small nanopores and large macropores) and the reactor itself. Furthermore it also
PicosecondFemtosecond
1000
900
800
700
600
500
4000.0 0.5 1.0 1.5
Nanoscale(mesopores)
~100 nm
~10 µm
Inlet gas
macroscale
ck (z), T (z)
Microscale(macropores)
~10 cm
Seconds, minutes
~1 mm
Minutes, hoursDays
0 z l
Fig. 27. Schematic illustrating some of the regimes encountered when modelling a catalyst solution, in this case a monolith reactor for emission control. Starting at the left hand side we have the intrinsic kinetics resulting from the atomic scale interactions, moving across to the meso and macroporous structure where diffusion of the reactant gases is necessary to find the active catalytic sites, finally on the right hand side the macroscale model of the reactor channel in the monolith. (Adapted from (133))
(a) (b)
(c) (d)
Fig. 26. Representations of geometrically relaxed nanoparticles, calculated using the different approaches discussed in the text. (a) Pt309 nanoparticle electrostatic potential and (b) Pt309 electron density, both calculated using ONETEP; (c) Pd321 nanoparticle calculated using the DFTB + approach; (d) 79 atom Pd/Pt core-shell with a monolayer of O adsorbed calculated using GPAW

277 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
requires the study of different time scales ranging from femtoseconds (the timescale on which the chemistry happens) to years (corresponding to the expected lifetime of the catalyst). A combination of physics, chemistry and engineering are required to generate statistical methods and engineering models, which along with chemical insight into mechanisms will provide a more complete picture of the catalytic system.
This project started through a Royal Society Industrial Fellowship held at UCL, Johnson Matthey Technology Centre and the Institute of Chemical Technology, Prague. Its aim is to look at applying the Prague group’s meso-scale methods to simulation of transport and reaction in porous catalysts (133–142). This project has been developing over the last few years and has an experimental component, which is essentially for benchmarking and validating the theoretical methods. Through the careful preparation of active catalyst it has been possible to construct layered catalysts consisting of a uniform active layer overlaid by an inert layer with well-defined morphology (thickness, porosity, particle size) that serves as a diffusion layer (Figure 28) (143, 144).
Through this approach it has been possible, firstly, to validate the computational models by simulating the transport and activity of the precise geometry prepared in the lab; and secondly, to develop a novel approach to determining the diffusivity of gases through porous media. This second aspect is important because it provides insight into the structural properties of the catalyst layer and how they influence its overall performance, as well as providing a simple relationship
for determining diffusivity that can be utilised in simpler models of an entire catalytic reactor (144). With the development of ab initio oxidation models a fully holistic ab initio description of the catalytic system is becoming closer.
7. Research Outlook
This brief review has described some of the projects run within the atomic-scale groups at Johnson Matthey. Looking to the future, the following areas appear as natural progressions. Broadly speaking there are three aspects:
Firstly, theoretical development and understanding. Continued work on model systems aims to enhance the description of nanoparticles and oxides and ultimately bring them together. The description is not just about the structure of the material, it is perhaps more importantly about using thermodynamic models to understand the state of a catalyst under reaction conditions. If one has a good model of the catalyst under reaction conditions, the reaction can be decomposed to its elementary steps and the key bond making and breaking that controls the activity and reactivity of a given material can be understood.
Secondly, further development of the correlation between a theoretical understanding of catalyst structure, activity and stability with experimental characterisation. There has undoubtedly been huge progress in this respect over the last two decades. However, specific challenges remain. For instance the combination of synthesising ideal shapes and
activ
ein
ert
y C
O, %
0.5
0.4
0.3
0.2
0.1
0.0
r CO
, km
ol s
–1
0.08
0.06
0.04
0.02
0.00
20 µm
Fig. 28. (a) Scanning electron micrograph depicting the inert alumina layer, active Pt/alumina layer and substrate material; (b) simulated concentration profile of CO in reconstructed porous media; (c) rate of CO consumption as a function of position in the porous media. Note the activity is purely located in the active region of the catalyst. (Adapted from (144))

278 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
structured nanoparticles for catalyst testing, with advanced characterisation techniques (for example the upcoming VERSOX beamline at Diamond) along with the detailed models from ab initio theory, provides an exciting prospect for developing a deep fundamental understanding of catalyst materials.
Thirdly, the possibility of going beyond catalyst materials problems to include thermodynamic modelling (using the MTData code (145)) and process modelling using tools like gPROMS (146). Furthermore, traditional materials physics problems can also be studied, for instance smart materials where the challenge lies in describing both the thermodynamic and electronic properties adequately to make material predictions. Another burgeoning area is in battery materials technology where simulation and modelling will play a significant role.
8. Conclusions
To conclude, if the last twenty years have seen growth in the capability of computational chemistry and widespread acceptance of the field as a sub-discipline of science, the next two decades hold the prospect of even greater progress. Utilisation of petascale computing, the accessibility of high performance computing and open source code development means that even larger and more complex systems are accessible and can be tackled by more people. Multiscale models that capture not only the fundamental physics and chemistry of the electronic and molecular level process, but also the macroscopic properties of the device in question will also become routine. Automation of simulations coupled with high throughput synthesis will revolutionise the way in which we discover new materials. Atomic-scale modelling and its subsequent expansion to other scales is helping Johnson Matthey be part of this computational revolution that will continue to provide increasing opportunities and will be at the heart of developing catalytic and materials science into the 21st century.
9. Acknowledgements
The work presented herein is a result of a number of internal and external collaborations. It is hard to acknowledge everyone who has been involved in the projects over the last seven years or so, however along with our experimental colleagues within Johnson Matthey, the Engineering and Physical Sciences Research Council (EPSRC) should be acknowledged
for funding of Collaborative Awards in Science and Engineering (CASE) and doctoral training centre students. Misbah Sarwar acknowledges J. L. Gavartin (Accelrys) and the Technology Strategy Board (TSB) for support in the iCatDesign project (DTI Project No: /5/MAT/6/I/H0379C). Glenn Jones acknowledges the EU for supporting the IMPRESS project (Contract NMP3-CT-2004-500635, co-funded by the European Commission in the sixth Framework Programme and European Space Agency) and the Royal Society for supporting his industrial fellowship at UCL (IF090100). Ludovic Briquet acknowledges the EU for supporting his Framework 6 Program Marie-Curie Early Stage Training studentship. Crispin Cooper gratefully acknowledges support from the EPSRC Collaborative Training Account: UCL (GR/T11364/01). Glenn Jones and Crispin Cooper also acknowledge the UK’s super computing facility for time on HECToR, which has now been superseded by ARCHER, through the UK’s HPC Materials Chemistry Consortium (EPRSC: EP/L000202).
References 1 P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136,
(3B), B864
2 W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, (4A), A1133
3 J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Nat. Acad. Sci., 2011, 108, (3), 937
4 F. Göltl and P. Sautet, ‘Density Functional Theory as a Key Approach in Surface Chemistry and Heterogeneous Catalysis’, in “Comprehensive Inorganic Chemistry II: From Elements to Applications”, 2nd Edn., eds. J. Reedijk and K. Poeppelmeier, Volume 7, Chapter 15, Elsevier, The Netherlands, 2013, pp. 405–420
5 ‘Top 500 Supercomputing Sites’, TOP500.org, New Orleans, Louisiana, USA (Accessed on 2nd April 2015)
6 J. L. Whitten and H. Yang, Surf. Sci. Rep., 1996, 24, (3–4), 55
7 B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71
8 C. Chizallet, G. Bonnard, E. Krebs, L. Bisson, C. Thomazeau and P. Raybaud, J. Phys. Chem. C, 2011, 115, (24), 12135
9 J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, (1–2), 83

279 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
10 K. Reuter and M. Scheffler, Phys. Rev. B, 2002, 65, (3), 035406
11 M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Tulhoat, J. Catal., 2002, 211, (1), 1
12 J. K. Nørskov, F. Studt, F. Abild-Pedersen and T. Bligaard, “Fundamental Concepts in Heterogeneous Catalysis”, John Wiley and Sons, Hoboken, New Jersey, USA, 2014
13 A. Walsh, A. A. Sokol and C. R. A. Catlow, “Computational Approaches to Energy Materials”, John Wiley and Sons, Chichester, UK, 2013
14 R. A. van Santen and M. Neurock “Molecular Heterogeneous Catalysis: A Conceptual and Computational Approach”, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany, 2006
15 Chorkendorff and J. W. Niemantsverdriet, “Concepts of Modern Catalysis and Kinetics”, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany, 2003
16 G. Jones, Catal. Struct. React., 2015, 00, 1
17 The White House: About the Materials Genome Initiative: https://www.whitehouse.gov/mgi (Accessed on 20th April 2015)
18 A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Appl. Phys. Lett.: Mater., 2013, 1, (1), 011002
19 J. S. Hummelshøj, F. Abild-Pedersen, F. Studt, T. Bligaard and J. K. Nørskov, Angew. Chem. Int. Ed., 2012, 51, (1), 272
20 Quantum Materials Informatics Project: Publications ht tp: / /www.qmip.org/qmip.org/Welcome.html (Accessed on 20th April 2015)
21 G. Pizzi, A. Cepellotti, R. Sabatini, N. Marzari, B. Kozinsky, ‘AiiDA: Automated Interactive Infrastructure and Database for Computational Science’, arXiv:1504.01163v1 [physics.comp-ph], 5th April, 2015
22 Iowa State University, College of Engineering: Combinatorial Sciences and Materials Informatics Collaboratory (CoSMIC): http://cosmic.mse.iastate.edu/ (Accessed on 20th April 2015)
23 A. A. C. Braga, N. H. Morgon, G. Ujaque and F. Maseras, J. Am. Chem. Soc., 2005, 127, (25), 9298
24 D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, (4), 1066
25 L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, Organometallics, 2006, 25, (1), 54
26 E. Napolitano, V. Farina and M. Persico, Organometallics, 2003, 22, (20), 4030
27 S. Kozuch, C. Amatore, A. Jutand and S. Shaik, Organometallics, 2005, 24, (10), 2319
28 R. Álvarez, O. N. Faza, C. S. López and Á. R. de Lera, Org. Lett., 2006, 8, (1), 35
29 T.-X. Zhang and Z. Li, Comput. Theor. Chem., 2013, 1016, 28
30 A. A. C. Braga, G. Ujaque and F. Maseras, Organometallics, 2006, 25, (15), 3647
31 L. Zhang and D.-C. Fang, J. Org. Chem., 2013, 78, (6), 2405
32 A. Fromm, C. van Wüllen, D. Hackenberger and L. J. Gooßen, J. Am. Chem. Soc., 2014, 136, (28), 10007
33 L. Sikk, J. Tammiku-Taul, P. Burk and A. Kotschy, J. Mol. Model., 2012, 18, (7), 3025
34 M. García-Melchor, M. C. Pacheco, C. Nájera, A. Lledós and G. Ujaque, ACS Catal., 2012, 2, (1), 135
35 D. Zhao, X. Li, K. Han, X. Li and Y. Wang, J. Phys. Chem. A, 2015, 119, (12), 2989
36 S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, (4), 1623
37 Y. Wu, L. Liu, K. Yan, P. Xu, Y. Gao and Y. Zhao, J. Org. Chem., 2014, 79, (17), 8118
38 Q. Ren, F. Jiang and H. Gong, J. Organomet. Chem., 2014, 770, 130
39 L. Liu, Y. Lv, Y. Wu, Gao, X. Zeng, Z. Gao, Y. Tang and G. Zhao, RSC Adv., 2014, 4, (5), 2322
40 D. Liu, Y. Li, X. Qi, C. Liu, Y. Lan and A. Lei, Org. Lett., 2015, 17, (4), 998
41 B. Zheng, F. Tang, J. Luo, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2014, 136, (17), 6499
42 S. K. Sontag, J. A. Bilbrey, N. E. Huddleston, G. R. Sheppard, W. D. Allen and J. Locklin, J. Org. Chem., 2014, 79, (4), 1836
43 A. Hedström, Z. Izakian, I. Vreto, C.-J. Wallentin and P.-O. Norrby, Chem. Eur. J., 2015, 21, (15), 5946
44 A. Bekhradnia and P.-O. Norrby, Dalton Trans., 2015, 44, (9), 3959
45 R. B. Bedford, P. B. Brenner, E. Carter, J. Clifton, P. M. Cogswell, N. J. Gower, M. F. Haddow, J. N. Harvey, J. A. Kehl, D. M. Murphy, E. C. Neeve, M. L. Neidig, J. Nunn, B. E. R. Snyder and J. Taylor, Organometallics, 2014, 33, (20), 5767
46 S. L. Daifuku, M. H. Al-Afyouni, B. E. R. Snyder, J. L. Kneebone and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, (25), 9132
47 Y. Sun, H. Jiang, H. Tang, H. Xu, H. Liu, K. Sun and X. Huang, Mol. Phys., 2014, 112, (16), 2107
48 H. Kato, K. Hirano, D. Kurauchi, N. Toriumi and M. Uchiyama, Chem. Eur. J., 2015, 21, (10), 3895

280 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
49 G.-Y. Ruan, Y. Zhang, Z.-H. Qi, D.-X. Ai, W. Liu and Y. Wang, Comput. Theor. Chem., 2015, 1054, 16
50 K. Kubota, E. Yamamoto and H. Ito, J. Am. Chem. Soc., 2015, 137, (1), 420
51 D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, (1), 174
52 L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem. Int. Ed., 2009, 48, (52), 9792
53 A. Sharma, D. Vacchani and E. Van der Eycken, Chem. Eur. J., 2013, 19, (4), 1158
54 J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, (1), 20
55 N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem. Int. Ed., 2012, 51, (41), 10236
56 R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, (1), 17
57 L. Ackermann, Chem. Rev., 2011, 111, (2), 1315
58 S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, (33), 10848
59 S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77, (1), 658
60 Y. Tan, F. Barrios-Landeros and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, (8), 3683
61 Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, (10), 3308
62 J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nature Chem., 2009, 1, (1), 37
63 F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjaer, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nature Chem., 2014, 6, (4), 320
64 N. Acerbi, S. C. E. Tsang, G. Jones, S. Golunski and P. Collier, Angew. Chem. Int. Ed, 2013, 52, (30), 7737
65 O. R. Inderwildi and S. J. Jenkins, Chem. Soc. Rev., 2008, 37, (10), 2274
66 G. Jones, J. G. Jakobsen, S. S. Shim, J. Kleis, M. P. Andersson, J. Rossmeisl, F. Abild-Pedersen, T. Bligaard, S. Helveg, B. Hinnemann, J. R. Rostrup-Nielsen, I. Chorkendorff, J. Sehested and J. K. Nørskov, J. Catal., 2008, 259, (1), 147
67 F. Abild-Pedersen, O. Lytken, J. Engbæk, G. Nielsen, Ib Chorkendorff and J. K. Nørskov, Surf. Sci., 2005, 590, (2–3), 127
68 G. Jones, ‘Preliminary results for S desorption from Ni{111}’, Internal Report, Johnson Matthey Plc, Sonning Common, UK, 2010
69 G. McCarty and H. Wise, J. Chem. Phys., 1980, 72, (12), 6332
70 T. R. Munter, D. D. Landis, F. Abild-Pedersen, G. Jones, S. Wang and T. Bligaard, Comput. Sci. Disc., 2009, 2, 015006
71 S. Saadi, B. Hinnemann, C. C. Appel, S. Helveg, F Abild-Pedersen and J. K. Nørskov, Surf. Sci., 2011, 605, (5–6), 582
72 S. Saadi, F. Abild-Pedersen, S. Helveg, J. Sehested, B. Hinnemann, C. C. Appel and J. K. Nørskov, J. Phys. Chem. C, 2010, 114, (25), 11221
73 S. Helveg, C. López-Cartes, J. Sehested, P. L. Hansen, B. S. Clausen, J. R. Rostrup-Nielsen, F. Abild-Pedersen and J. K. Nørskov, Nature, 2004, 427, (6973), 426
74 E. H. Stitt, M. J. Watson, L. Gladden, J. McGregor, World Patent 2010/140005
75 A. Ukpong and G. Jones, ‘Ab initio Insight into the Catalytic Dehydrogenation of Propane to Propene over Defective Graphene’, submitted
76 J. Zhu, J. G. van Ommen and L. Lefferts, J. Catal., 2004, 225, (4), 388
77 J. Zhu, J. G. van Ommen, H. J. M. Bouwmeester and L. Lefferts, J. Catal., 2005, 233, (2), 434
78 C. S. Cooper, R. J. Oldman and C. R. A. Catlow, Chem. Comm., 2015, 51, 5856
79 X. Xia, R. J. Oldman and C. R. A. Catlow, J. Mater. Chem., 2012, 22, (17), 8594
80 M. Anpo, M. Che, B. Fubini, E. Garrone, E. Giamello and M. C. Paganini, Top. Catal., 1999, 8, (3–4), 189
81 G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, (2–3), 305
82 ‘The Fuel Cell Today Industry Review 2011’, Johnson Matthey Plc, Royston, UK, 2011
83 ‘Technical Plan — Fuel Cells’, Multi-Year Research, Development and Demonstration Plan, US Government, Washington, DC, USA: http://energy.gov/sites/prod/files/2014/12/f19/fcto_myrdd_fuel_cells.pdf (Accessed on 2nd April 2015)
84 J. Greeley and J. K. Nørskov, J. Phys. Chem. C, 2009, 113, (12), 4932
85 J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, (46), 17886
86 V. Stamenković, T. J. Schmidt, P. N. Ross and N. M. Marković, J. Phys. Chem. B, 2002, 106, (46), 11970
87 I. E. L. Stephens, A. S. Bondarenko, F. J. Perez-Alonso, F. Calle-Vallejo, L. Bech, T. P. Johansson, A. K. Jepsen, R. Frydendal, B. P. Knudsen, J. Rossmeisl, and Ib Chorkendorff, J. Am. Chem. Soc., 2011, 133, (14), 5485
88 V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem. Int. Ed., 2006, 45, (18), 2897
89 Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem.

281 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
Soc., 2004, 126, (14), 471790 V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K.
Nørskov, J. Phys. Chem. Lett., 2012, 3, (20), 2948 91 P. Sabatier, Ber. Deut. Chem. Gesell., 1911, 44, (3),
198492 T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C.
H. Christensen and J. Sehested, J. Catal., 2004, 224, (1), 206
93 Y. Ma and P. B. Balbuena, Surf. Sci., 2008, 602, (1), 107
94 G. E. Ramirez-Caballero and P. B. Balbuena, Chem. Phys. Lett., 2008, 456, (1–3), 64
95 J. S. Hummelshøj, F. Abild-Pedersen, F. Studt, T. Bligaard and J. K. Nørskov, Angew. Chem. Int. Ed., 2012, 51, (1), 272
96 J. Yates, G. H. Spikes and G. Jones, Phys. Chem. Chem. Phys., 2015, 17, (6), 4250
97 D. W. Fickel and R. F. Lobo, J. Phys. Chem. C, 2010, 114, (3), 1633
98 A. Godiksen, F. N. Stappen, P. N. R. Vennestrøm, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, J. Phys. Chem. C, 2014, 118, (40), 23126
99 F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2014, 319, 1
100 F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. F. Peden, J. Catal., 2013, 300, 20
101 E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, (1), 548
102 U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, ACS Catal., 2013, 3, (3), 413
103 S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Chem. Commun., 2011, 47, (2), 800
104 Z.-P. Liu, S. J. Jenkins and D. A. King, Phys. Rev. Lett., 2005, 94, (19–20), 196102
105 L. J. Bennett and G. Jones, Phys. Chem. Chem. Phys., 2014, 16, (39), 21032
106 M. K. Bruska, I. Czekaj, B. Delley, J. Mantzaras and A. Wokaun, Phys. Chem. Chem. Phys., 2011, 13, (35), 15947
107 J. Waser, H. A. Levy and S. W. Peterson, Acta Cryst., 1953, 6, (7), 661
108 J. Kleis, J. Greeley, N. A. Romero, V. A. Morozov, H. Falsig, A. H. Larsen, H. Lu, J. J. Mortensen, M. Dułak, K. S. Thygesen, J. K. Nørskov and K. W. Jacobsen, Catal. Lett., 2011, 141, (8), 1067
109 L. Li, A. H. Larsen, A. H. Romero, V. A. Morozov,
C. Glinsvad, F. Abild-Pedersen, J. Greeley, K. W. Jacobsen and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, (1), 222
110 A. H. Larsen, ‘Efficient Electronic Structure Methods Applied to Metal Nanoparticles’, PhD Thesis, Department of Physics, Technical University of Denmark, 2011
111 T. Jiang, D. J. Mowbray, S. Dobrin, H. Falsig, B. Hvolbæk, T. Bligaard, and J. K. Nørskov, J. Phys. Chem. C, 2009, 113, (24), 10548
112 H. Falsig, J. Shen, T. S. Khan, W. Guo, G. Jones, S. Dahl and T. Bligaard, Top. Catal., 2014, 57, (1–4), 80
113 C. Verdozzi, D. R. Jennison, P. A. Schultz and M. P. Sears, Phys. Rev. Lett., 1999, 82, (4), 799
114 Z. Łodziana and J. K. Nørskov, J. Chem. Phys., 2001, 115, (24), 11261
115 J. R. B. Gomes, Z. Lodziana and F. Illas, J. Phys. Chem. B, 2003, 107, (26), 6411
116 M. C. Valero, P. Raybaud and P. Sautet, J. Phys. Chem. B, 2006, 110, (4), 1759
117 R. Grau-Crespo, N. C. Hernández, J. F. Sanz and N. H. de Leeuw, J. Phys. Chem. C, 2007, 111, (28), 10448
118 C. Chizallet and P. Raybaud, Catal. Sci. Technol., 2014, 4, (9), 2797
119 L. G. V. Briquet, C. R. A. Catlow and S. A. French, J. Phys. Chem. C, 2008, 112, (48), 18948
120 L. G. V. Briquet, C. R. A. Catlow and S. A. French, J. Phys. Chem. C, 2009, 113, (38), 16747
121 L. G. V. Briquet, C. R. A. Catlow and S. A. French, J. Phys. Chem. C, 2010, 114, (50), 22155
122 S. W. Hoh, L. Thomas, G. Jones and D. J. Willock, Res. Chem. Intermediat., 2015, doi: 10.1007/s11164-015-1984-7
123 S. W. Hoh, ‘Oxidation Catalysis Using Transition Metals and Rare Earth Oxides’, PhD Thesis, School of Chemistry, Cardiff University, UK, 2014
124 A. P. Sutton and J. Chen, Phil. Mag. Lett., 1990, 61, (3), 139
125 K. W. Jacobsen, J. K. Norskov and M. J. Puska, Phys. Rev. B, 1987, 35, (14), 7423
126 J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B, 2005, 71, (3), 035109
127 C.-K. Skylaris, P. D. Haynes, A. A. Mostofi and M. C. Payne, J. Chem. Phys., 2005, 122, (8), 084119
128 P. Koskinen and V. Mäkinen, Comput. Mater. Sci., 2009, 47, (1), 237
129 Á. Ruiz-Serrano and C.-K. Skylaris, J. Chem. Phys., 2013, 139, (5), 054107

282 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
130 M. H. Chuma, H. R. Chauke, G. Jones and P. E. Ngoepe, ‘Parameterization and Validation of Pd and Pd Nanoclusters Using SCC-DFTB’, in “Proceedings of SAIP2013: the 58th Annual Conference of the South African Institute of Physics”, eds. R. Botha and T. Jili, The 58th Annual Conference of the South African Institute of Physics, University of Zululand, South Africa, 8th–12th July, 2013, pp. 8–12
131 L. Jones, K. E. MacArthur, V. T. Fauske, A. T. J. van Helvoot and P. D. Nellist, Nano Lett., 2014, 14, (11), 6336
132 R. Price, T. Erlap-Erden, E. Crumlin, S. Garcia, S. Rani, R. Smith, G. Jones, L. Deacon, C. Euarusakul and G. Held, ‘The Partial Oxidation of Methane with Catalytic Pd/Al2O3 Nanoparticles, Studied in-situ by Near Ambient-Pressure X-Ray Photoelectron Spectroscopy’, submitted
133 V. Novák, P. Kočí, F. Štěpánek and M. Marek, Ind. Eng. Chem. Res., 2011, 50, (23), 12904
134 V. Novák, P. Kočí, F. Štěpánek, M. Kubíček and M. Marek, Comput. Chem. Eng., 2011, 35, (5), 964
135 J. Kosek, F. Štěpánek and M. Marek, Adv. Chem. Eng., 2005, 30, 137
136 P. Kočí, V. Novák, F. Štěpánek, M. Marek and M. Kubíček, Chem. Eng. Sci., 2010, 65, (1), 412
137 V. Novák, F. Štěpánek, P. Kočí, M. Marek and M. Kubíček, Chem. Eng. Sci., 2010, 65, (7), 2352
138 P. Kočí, F. Štěpánek, M. Kubíček and M. Marek, Chem. Eng. Sci., 2006, 61, (10), 3240
139 M. Marek, Z. Grof, P. Kocí, M. Kohout, J. Kosek and F. Štepánek, J. Chem. Eng. Jpn, 2007, 40, (11), 879
140 F. Štěpánek, M. Marek, J. Hanika and P. M. Adler, Catal. Today, 2001, 66, (2–4), 249
141 P. Kočí, F. Štěpánek, M. Kubíček and M. Marek, Chem. Eng. Sci., 2007, 62, (18–20), 5380
142 P. Kočí, F. Štěpánek, M. Kubíček and M. Marek, Mol. Simul., 2007, 33, (4–5), 369
143 V. Novák, P. Kočí, M. Marek, F. Štěpánek, P. Blanco-García and G. Jones, Catal. Today, 2012, 188, (1), 62
144 M. Dudák, V. Novák, P. Kočí, M. Marek, P. Blanco-García and G. Jones, Appl. Catal. B: Environ., 2014, 150–151, 446
145 R. H. Davies, A. T. Dinsdale, J. A. Gisby, J. A. J. Robinson and S. M. Martin, Calphad, 2002, 26, (2), 229
146 gPROMS, Process Systems Enterprise Ltd, London, UK, 1997–2014: www.psenterprise.com/gproms (Accessed on 2nd April 2015)
The Authors
Misbah Sarwar is currently a Principal Scientist at Johnson Matthey Technology Centre (JMTC), Sonning Common, UK. She has an MSci in Chemistry from University College London (UCL), UK, and a PhD from UCL and the Royal Institution (RI). Misbah joined Johnson Matthey in 2007 to work on the Technology Strategy Board (TSB) funded project iCatDesign. In April 2014 Misbah joined the Emission Control Technology research group at JMTC.
Crispin Cooper is a computational chemist working in the JMTC Emission Control research group. He has a BSc (hons) in Chemistry for Drug Discovery from the University of Bath, UK, and an EngD in Molecular Modelling and Materials Simulation from UCL. He has recently joined Johnson Matthey following an industrial postdoctoral project modelling complex metal oxide catalyst materials.
Ludovic Briquet is a Senior Scientist at JMTC, Sonning Common. He gained his PhD in Chemistry at UCL, UK, in 2010 and specialised in molecular modelling applied to various systems. His main interests however lie in surface science and catalysis.

283 © 2015 Johnson Matthey
http://dx.doi.org/10.1595/205651315X687975 Johnson Matthey Technol. Rev., 2015, 59, (3)
Aniekan Ukpong is a Senior Scientist at JMTC, Pretoria, where he has been working since 2013. He completed his PhD in Physics at the University of Cape Town, South Africa, in 2008. His research focuses on the fundamental study of materials from theory to applications.
Christopher Perry originally trained as a bio-inorganic chemist. He became interested in modelling whilst lecturing at the University of the Witwatersrand, South Africa. Prior to that, he completed two postdoctoral research projects: one in nuclear magnetic resonance (NMR) spectroscopy at Manchester University, UK, and a second in organic synthesis at the University of the Witwatersrand. He has been working at Johnson Matthey since 2013 and is a Senior Scientist at JMTC, Pretoria.
Glenn Jones has extensive experience in the fields of surface science, catalysis and computational materials chemistry. He gained his PhD from the University of Cambridge, UK, before moving to the Technical University of Denmark as a post-doctoral student. He joined JMTC, Sonning Common, UK, in 2008 and was awarded a Royal Society Industrial Fellowship in 2010 which he held jointly between JMTC and UCL. He moved to Pretoria, South Africa, in 2013 to initiate JMTC’s new modelling laboratory in South Africa, where he is currently Research Manager.





























