Embed Size (px)
Citation preview
Conformer-Specific Ionization Spectroscopy of Bromocyclohexane: Equatorial versus AxialConformers
Songhee Han, Hyun Sik Yoo, and Sang Kyu Kim*Department of Chemistry and KI for Nanocentury, KAIST, Daejeon 305-701, Korea
ReceiVed: June 16, 2010; ReVised Manuscript ReceiVed: August 5, 2010
Ionization of equatorial and axial conformational isomers of the chair-bromocyclohexane is investigated withuse of the vacuum ultraviolet mass-analyzed threshold ionization (MATI) spectroscopic technique. Two distinctionization energies of 9.8308 ( 0.0025 and 9.8409 ( 0.0025 eV are determined for equatorial or axialconformers, respectively. From the conformer-selective vibrational analysis, it is found that the equatorialconformer undergoes a drastic structural change upon ionization especially along the C-Br distortion mode,whereas the axial conformer shows the modest change along the ring-puckering mode with ionization,corresponding to the reaction coordinate for the conformational interconversion. Density functional theory(DFT) calculations with and without considering the spin-orbit coupling provide the appropriate modeassignments for the vibrational bands active in the ionization spectra. Natural bond orbital (NBO) analysis iscarried out to give insights into the contribution of the anomeric effect to the structure-energy relationshipin each conformational isomer.
1. Introduction
Chemical reactivity is strongly dependent on the molecularstructure. The reaction outcomes can thus be manipulated if onecan select the specific molecular structure of the reactant byany means. This structure-selective reaction control has becomereality as some successful experimental studies have beenreported recently. These include the conformer-specific photo-dissociation dynamics of 1-iodopropane ion,1 propanal ion,2 andthiophenols.3 Dissociative ionization of some aliphatic aminoacids has also been found to be conformer-specific.4 In ambientconditions, the molecular structure is often the mixture of manypossible structural isomers especially when the barrier for theinterconversion of different isomers is low. Conformationalisomers belong to this category since the conformational changeis induced by the internal rotation about the single bond.Understanding the conformer-specific chemical reactivity is oneof the most important keys for unraveling the complex biologicalproblems such as enzyme reactions or protein folding. In thesupersonically cooled condition, conformers exist according totheir statistical thermodynamic properties, even though it seemsto be in controversy whether or not the relative population ofthe conformers at room temperature remains constant in thesupersonic cooling process.5 In principle, different conforma-tional isomers are separable according to their distinct physicalproperties, and thereafter, one can further study the chemicalreactivity associated with the specific conformational structure.
Cyclohexane and monosubstituted cyclohexanes have longbeen spotlighted as their equatorial or axial conformationalstructures provide the prototypical model system for theinvestigation of the conformer-specific reactivity.6-16 Bromocy-clohexane is a particularly interesting system, as the energydifference between equatorial and axial conformers is smallenough for both isomers to coexist in significant amounts atthe ambient condition.17-28 In spite of many studies on theconformational isomerism of bromocyclohexane, its conformer-
specific ionization spectroscopy has been little investigated.Recent applications of the vacuum ultraviolet mass-analyzedthreshold ionization (VUV-MATI) spectroscopic technique toa number of systems have been very successful in identifyingand separating conformational isomers, and these include variousconformers of 1-iodopropane, propanal,29 and alkanethiols.30
Here, equatorial and axial conformational isomers of the chair-bromocyclohexane have been investigated with VUV-MATIspectroscopy to give the accurate ionization energy of each
* To whom correspondence should be addressed. E-mail: [email protected]. Fax: (+) 82-42-350-2810. Phone: (+)82-42-350-2843.
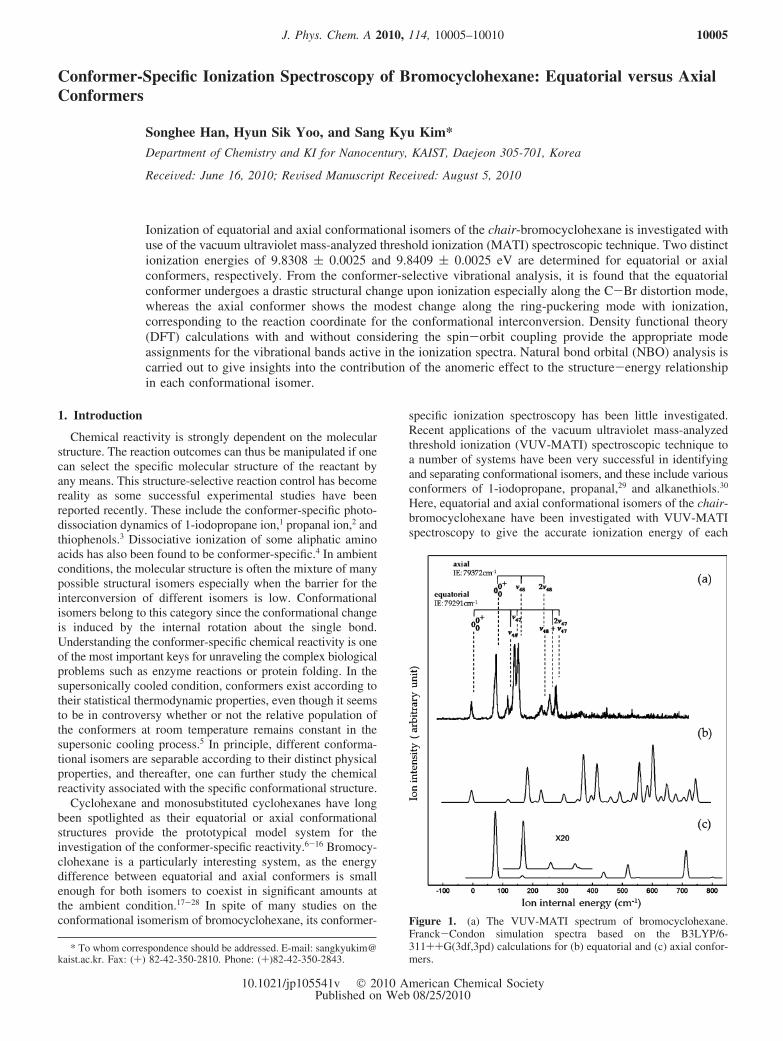
Figure 1. (a) The VUV-MATI spectrum of bromocyclohexane.Franck-Condon simulation spectra based on the B3LYP/6-311++G(3df,3pd) calculations for (b) equatorial and (c) axial confor-mers.
J. Phys. Chem. A 2010, 114, 10005–10010 10005
10.1021/jp105541v 2010 American Chemical SocietyPublished on Web 08/25/2010
conformer. The experimental result has been analyzed with theaid of theoretical calculations to investigate the origin of theconformational preference, which results from the interplaybetween anomeric and steric effects.31
2. Experimental Section
Bromocyclohexane (Aldrich, 98%) was used without furtherpurification, seeded in the Ar carrier gas, and expanded intothe vacuum chamber through a nozzle orifice (General valve,0.5 mm diameter) with a backing pressure of 1 atm. Theresulting supersonic jet was collimated through a 1 mm diameterskimmer (Precision) in a differentially pumped vacuum chamberprior to being overlapped with the counter-propagating VUVlaser pulse, which was generated via the four-wave mixing ina Kr gas cell by combining the UV laser pulse fixed at 212.556nm for the Kr 5p[1/2]0-4p6 transition and the tunable visible(VIS) laser pulse. The dye laser output (Lambda phisik,Scanmate 2) pumped by the third harmonic of a Nd:YAG laser(Continuum, Precision 2) was frequency doubled through aKD*P crystal, rotated by 90° in the linear polarization axisthrough a λ/2 waveplate, and frequency-summed with the dye
laser fundamental on a BBO crystal to generate the UV laserpulse at 212.556 nm. The VIS laser pulse in the 670-710 nmrange for the VUV wavelength tuning was generated by anotherdye laser (Lumonics, HD-500) pumped by the same Nd:YAGlaser. The VUV pulse was spatially separated from UV and VISfundamentals using the edge of a calcium fluoride (CaF2)collimating lens placed at the exit of the Kr cell in which thepressure was maintained at ∼1 Torr. High-n,l Rydberg statesof molecules generated by the VUV laser pulse were pulsed-field ionized (∼10 V/cm) after the delay time of ∼20 µs, whichis long enough for the complete separation of the MATI ionsfrom the prompt ions along the time-of-flight axis. Therefore,no spoil field was used in this scheme. Generated ions wererepelled, accelerated, drifted along the time-of-flight axis, anddetected by MCP. Ion signals were digitized by an oscilloscope(LeCroy, LT584M) and stored in a personal computer that wasalso used for the remote control of all step motors in theexperimental setup.
Computational Details. All calculations were carried outwith the Gaussian 03W program package.32 The minimumenergy structures and normal modes for ground neutral and
TABLE 1: Some of Vibrational Frequencies (cm-1) of Equatorial and Axial Conformers of the chair-Bromocyclohexane in theNeutral and Cationic Ground States Calculated at the B3LYP/6-311++G(3df,3pd) Level. The Vibrational Frequencies Obtainedby the B3PW91 and PBEPBE Methods with 6-311++G(3df, 3pd) Basis Set are Given in Parentheses (Left: B3PW91, Right:PBEPBE)
chair-equatorial chair-axial
S0 D0 S0 D0
calcd calcd expt description calcd calcd expt descriptiona
ν47 212 187 (179, 149) 145 C-Br distortion 175 163 (162, 154) C(3,5)H2 rockν48 126 122 (123, 117) 122 ring puckering 114 90 (86, 87) 75 ring puckering
a Refer to Table 3 for the atomic labeling.
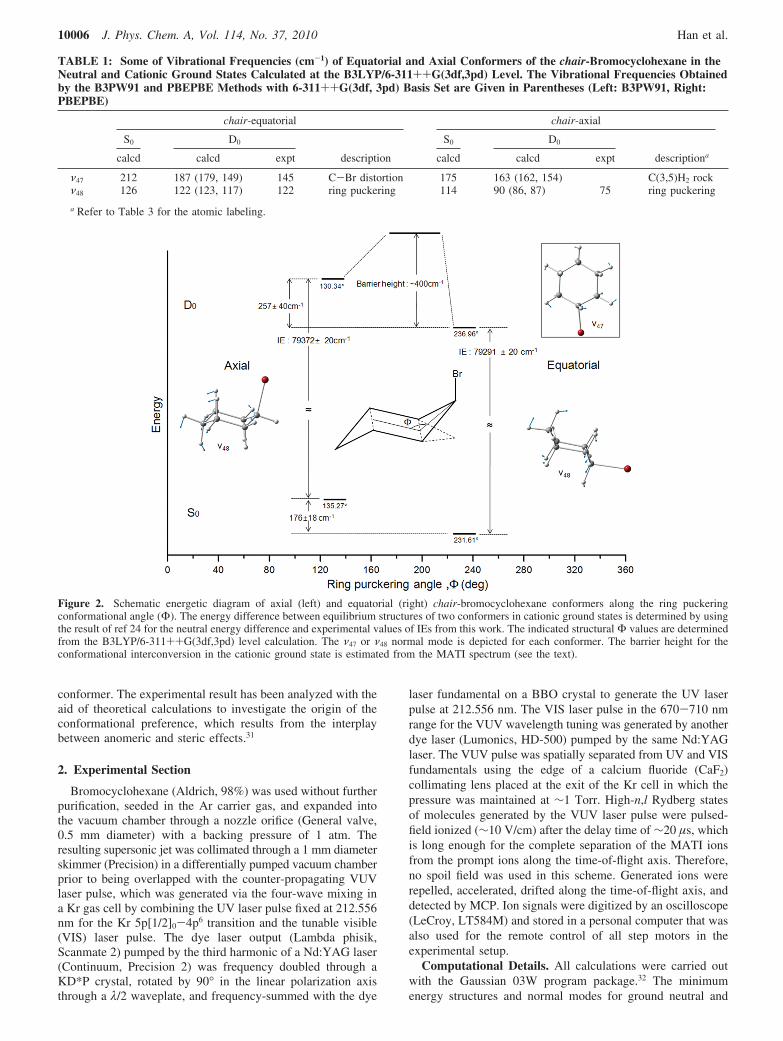
Figure 2. Schematic energetic diagram of axial (left) and equatorial (right) chair-bromocyclohexane conformers along the ring puckeringconformational angle (Φ). The energy difference between equilibrium structures of two conformers in cationic ground states is determined by usingthe result of ref 24 for the neutral energy difference and experimental values of IEs from this work. The indicated structural Φ values are determinedfrom the B3LYP/6-311++G(3df,3pd) level calculation. The ν47 or ν48 normal mode is depicted for each conformer. The barrier height for theconformational interconversion in the cationic ground state is estimated from the MATI spectrum (see the text).
10006 J. Phys. Chem. A, Vol. 114, No. 37, 2010 Han et al.

cationic states were calculated with the B3LYP,33,34 B3PW91,35
and PBEPBE36 density functional theory (DFT)33,34,36,37 methodswith a basis set of 6-311++G(3df,3pd). The Franck-Condonanalysis was carried out by using the Duschinsky transforma-tion38 with a code developed by Peluso and co-workers.39,40
Natural bonding orbital (NBO) calculations41 were carried outat the DFT and MP242 levels.
3. Results and Discussion
Bromocyclohexane adopts the fully staggered chair form inthe ground electronic state. It is well-known that there existtwo distinct conformational isomers of the chair-bromocyclo-hexane: in the axial conformer, the Br atom is placed at theaxial position whereas it is attached at the equatorial positionin the equatorial conformer. Thermodynamic stabilities of theseconformers have been investigated in many different environ-ments. In the gas-phase electron diffraction study, using therelative abundance of two conformers, it has been found that
the equatorial conformer is more stable than the axial conformerby 176 ( 18 cm-1.24,28 This energy difference is very low, andthus it is nontrivial to separate out the specific conformer inthe ambient condition. In the supersonically cooled condition,however, each conformer is populated with the minimuminternal energy, allowing the clear separation of the conformers.The VUV-MATI spectrum of supersonically cooled bromocy-clohexane in Figure 1(a) represents ionization dynamics of itsequatorial and axial isomers. Apparently, it seems to be notstraightforward to assign the observed bands in the VUV-MATIspectrum to each conformer. The intensity of the origin bandalone cannot be used as a criterion for the judgment of theconformational assignment, since the sum of all band intensitiesbelonging to each conformer should be compared for thedetermination of different conformer populations. We havecarried out the Franck-Condon analysis based on DFT calcula-tions using the B3LYP method with a 6-311++G(3df,3pd) basisset. The geometries and vibrational frequencies in the neutral(S0) and cationic (D0) ground states for each conformer are usedfor the calculation of Franck-Condon factors in the one-photonionization. From theory, it is predicted that the equatorialconformer undergoes the large structural change upon ionization,giving the small intensity for the ionization origin of theequatorial conformer. On the other hand, the origin is expectedto be strongly observed for the axial conformer from theFranck-Condon calculation. These Franck-Condon analysesstrongly indicate that the lowest ionization threshold found atthe VUV photon energy of 79 291 cm-1 in Figure 1(a) shouldbe ascribed to the 0-0+ origin of the equatorial conformer. Theintensity of this origin band is relatively weak, and this isconsistent with the large structural change of the equatorialconformer upon ionization. The second lowest ionizationthreshold observed at 79 372 cm-1 is most likely due to the0-0+ origin of the axial conformer not only because the energydifference of 81 cm-1 is rather low for the vibrational frequencyof the equatorial conformer but also because the spectral bandsat the high energy region cannot be explainable otherwise. Thisassignment for two different conformer origins makes perfectsense in the further assignment of other vibrational modesbelonging to each conformer.
Accordingly, the ionization energies (IEs) of the equatorialand axial conformers of bromocyclohexane are determined tobe 9.8308 ( 0.0025 and 9.8409 ( 0.0025 eV, respectively.Rather large uncertainties of IE values here are ascribed to thebroad ionization energy window, due to the strong electric fieldof 10 V/cm employed in the pulsed field ionization.43 Thesevalues are higher than previously reported theoretical values of9.75 and 9.78 eV, predicted for respective equatorial and axialconformers.26 However, the higher IE of the axial conformerthan that of the equatorial conformer is consistent with thecurrent experiment. Assignments for vibrationally excited bandsare quite straightforward, Figure 1. The peaks located at 145and 284 cm-1 above the origin of 79 291 cm-1 are assigned tothe fundamental and overtone bands of the C-Br distortionmode (ν47) of the equatorial conformer, respectively. It is quiteinteresting to note that the B3LYP calculation predicts 187 cm-1
for ν47 whereas B3PW91 and PBEPBE methods give rise to179 and 149 cm-1, respectively (Table 1). The PBEPBEcalculation is especially in good agreement with the experiment,and this should be the consequence of the inclusion of theexchange-correlation energy in the corresponding functional.This demonstrates that the spin-orbit coupling should becarefully considered for the appropriate prediction of the vi-brational frequency especially for the mode involving the heavy
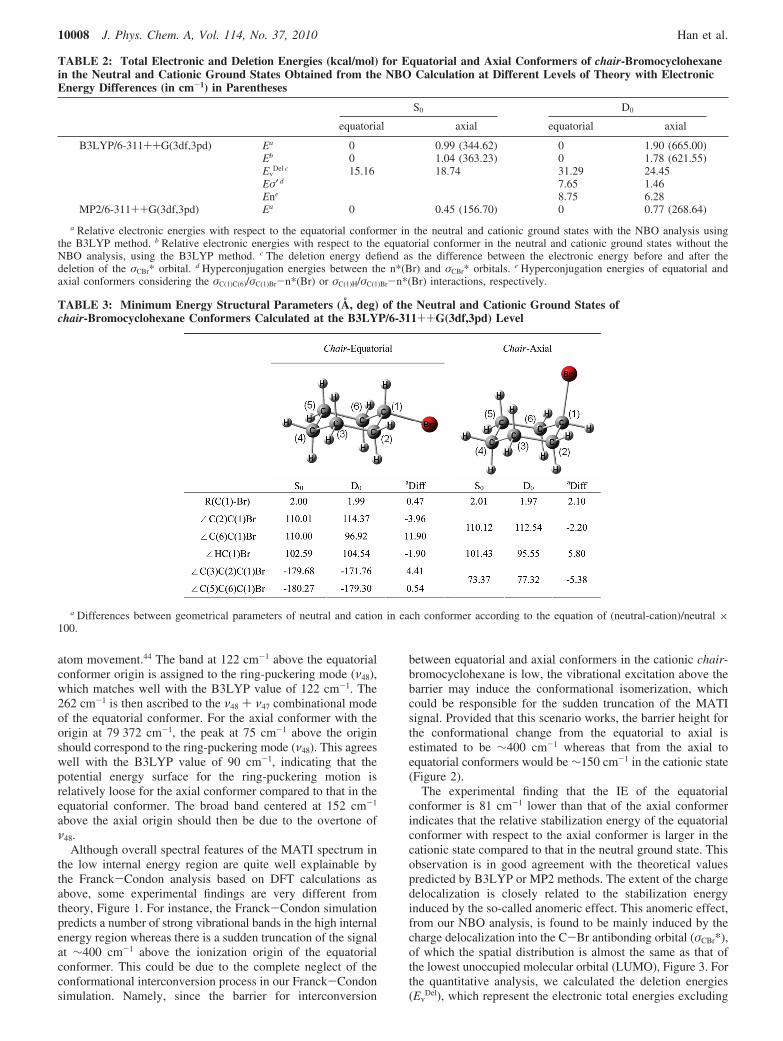
Figure 3. The (a) HOMO, (b) LUMO, and (c) singly occupiedmolecular orbital (SOMO) of the equatorial conformer are shown with(d) other orbitals showing significant hyperconjugation interactions inthe cationic ground state. The (e) HOMO, (f) LUMO, and (g) SOMOof the axial conformer are depicted with (h) hyperconjugating orbitalsin the cationic ground state. All orbitals are obtained with NBOcalculations at the B3LYP level with the basis set of 6-311++G(3df,3pd).The donor-to-acceptor charge delocalization directions are depicted asarrows.
Ionization of Bromocyclohexane J. Phys. Chem. A, Vol. 114, No. 37, 2010 10007
atom movement.44 The band at 122 cm-1 above the equatorialconformer origin is assigned to the ring-puckering mode (ν48),which matches well with the B3LYP value of 122 cm-1. The262 cm-1 is then ascribed to the ν48 + ν47 combinational modeof the equatorial conformer. For the axial conformer with theorigin at 79 372 cm-1, the peak at 75 cm-1 above the originshould correspond to the ring-puckering mode (ν48). This agreeswell with the B3LYP value of 90 cm-1, indicating that thepotential energy surface for the ring-puckering motion isrelatively loose for the axial conformer compared to that in theequatorial conformer. The broad band centered at 152 cm-1
above the axial origin should then be due to the overtone ofν48.
Although overall spectral features of the MATI spectrum inthe low internal energy region are quite well explainable bythe Franck-Condon analysis based on DFT calculations asabove, some experimental findings are very different fromtheory, Figure 1. For instance, the Franck-Condon simulationpredicts a number of strong vibrational bands in the high internalenergy region whereas there is a sudden truncation of the signalat ∼400 cm-1 above the ionization origin of the equatorialconformer. This could be due to the complete neglect of theconformational interconversion process in our Franck-Condonsimulation. Namely, since the barrier for interconversion
between equatorial and axial conformers in the cationic chair-bromocyclohexane is low, the vibrational excitation above thebarrier may induce the conformational isomerization, whichcould be responsible for the sudden truncation of the MATIsignal. Provided that this scenario works, the barrier height forthe conformational change from the equatorial to axial isestimated to be ∼400 cm-1 whereas that from the axial toequatorial conformers would be ∼150 cm-1 in the cationic state(Figure 2).
The experimental finding that the IE of the equatorialconformer is 81 cm-1 lower than that of the axial conformerindicates that the relative stabilization energy of the equatorialconformer with respect to the axial conformer is larger in thecationic state compared to that in the neutral ground state. Thisobservation is in good agreement with the theoretical valuespredicted by B3LYP or MP2 methods. The extent of the chargedelocalization is closely related to the stabilization energyinduced by the so-called anomeric effect. This anomeric effect,from our NBO analysis, is found to be mainly induced by thecharge delocalization into the C-Br antibonding orbital (σCBr*),of which the spatial distribution is almost the same as that ofthe lowest unoccupied molecular orbital (LUMO), Figure 3. Forthe quantitative analysis, we calculated the deletion energies(Ev
Del), which represent the electronic total energies excluding
TABLE 2: Total Electronic and Deletion Energies (kcal/mol) for Equatorial and Axial Conformers of chair-Bromocyclohexanein the Neutral and Cationic Ground States Obtained from the NBO Calculation at Different Levels of Theory with ElectronicEnergy Differences (in cm-1) in Parentheses
S0 D0
equatorial axial equatorial axial
B3LYP/6-311++G(3df,3pd) Ea 0 0.99 (344.62) 0 1.90 (665.00)Eb 0 1.04 (363.23) 0 1.78 (621.55)Ev
Del c 15.16 18.74 31.29 24.45Eσ′ d 7.65 1.46Ene 8.75 6.28
MP2/6-311++G(3df,3pd) Ea 0 0.45 (156.70) 0 0.77 (268.64)
a Relative electronic energies with respect to the equatorial conformer in the neutral and cationic ground states with the NBO analysis usingthe B3LYP method. b Relative electronic energies with respect to the equatorial conformer in the neutral and cationic ground states without theNBO analysis, using the B3LYP method. c The deletion energy defiend as the difference between the electronic energy before and after thedeletion of the σCBr* orbital. d Hyperconjugation energies between the n*(Br) and σCBr* orbitals. e Hyperconjugation energies of equatorial andaxial conformers considering the σC(1)C(6)/σC(1)Br-n*(Br) or σC(1)H/σC(1)Br-n*(Br) interactions, respectively.
TABLE 3: Minimum Energy Structural Parameters (Å, deg) of the Neutral and Cationic Ground States ofchair-Bromocyclohexane Conformers Calculated at the B3LYP/6-311++G(3df,3pd) Level
a Differences between geometrical parameters of neutral and cation in each conformer according to the equation of (neutral-cation)/neutral ×100.
10008 J. Phys. Chem. A, Vol. 114, No. 37, 2010 Han et al.

the σCBr* orbital, for equatorial and axial conformers in neutraland cationic ground states. The B3LYP value of Ev
Del with a6-311++G(3df,3pd) basis set is smaller by 3.58 kcal/mol forthe equatorial conformer compared to that of the axial conformerin the neutral ground state, whereas the deletion energy of theequatorial conformer in the cationic ground state is calculatedto be quite larger than that of the axial conformer by 6.84 kcal/mol (Table 2). This big difference of the deletion energy stronglyindicates that the anomeric effect through the charge delocal-ization into the σCBr* orbital is mainly responsible for the largerstabilization energy of the equatorial with respect to the axialconformer in the cationic state, compared to that in the neutralground state, giving the smaller IE for the equatorial conformer.
The ionization-driven structural change is also conformer-specific. As indicated in the MATI spectrum, the C-Brdistortion mode (ν47) is strongly activated for the equatorialconformer whereas the ring-puckering mode (ν48) is enhancedas the axial conformer is ionized. This conformer-specificity isrelevant to the hyperconjugation energy (Eσ′), of which the maincontribution is the anomeric interaction between n*(Br) andσCBr* orbitals, Figure 3. The Eσ′ value for the equatorialconformer in the ground cationic state is calculated to be 7.65kcal/mol, whereas the axial conformer cation is stabilized bythe hyperconjugation energy of 1.46 kcal/mol in terms of theorbital overlap of n*(Br) and σCBr*, Table 2. It is interesting tonote that the n*(Br) orbital of the equatorial conformer isasymmetrically oriented with respect to the C-Br bond axis,and this is probably responsible for the strong activation of theC-Br distortion mode in the corresponding MATI spectrum.
It is noteworthy that the C(1)-Br bond lengths are slightlydecreased upon ionization for both equatorial and axial con-formers, Table 3. As depicted in Figure 3, the highest occupiedmolecular orbital (HOMO) has a nodal plane along the C(1)-Brbond and the deficiency of the electron in HOMO should resultin the decrease of the antibonding character, thus shorteningthe C(1)-Br bond upon ionization for both conformers. TheHC(1)Br bent angle, ∠HC(1)Br, is increased upon ionizationfor the equatorial conformer whereas it is decreased as the axialconformer is ionized. The hyperconjugation energy between σbonding and n*(Br), En, seems to be most responsible for thisparticular conformer-specific structural change. Namely, ac-cording to our NBO analysis, the hyperconjugation energy dueto the interaction of σC(1)C(6)/σC(1)Br and n*(Br) is calculated tobe 8.75 kcal/mol for the equatorial conformer. On the otherhand, the hyperconjugation energy of the σC(1)H/ σC(1)Br andn*(Br) interaction is calculated to be 6.28 kcal/mol for the axialconformer. This conformer-dependent hyperconjugation energydifference suggests that the charge density localized in theC(1)-Br bond might increase relatively more in the equatorialconformer compared to that in the axial conformer. In thisrespect, the ∠HC(1)Br increases upon ionization for theequatorial conformer due to the increased steric effect, whereasit is the opposite case for the axial conformer ionization. Fromour NBO analysis, it is found that the n*(Br) orbital plays animportant role in the structural change upon ionization with theconformer-specificity in terms of the structure-sensitive anomericinteraction.
4. Summary
In this work, using the VUV-MATI spectroscopic technique,the ionization energies of equatorial and axial conformers ofthe chair-bromocyclohexane are accurately determined to be9.8308 ( 0.0025 and 9.8409 ( 0.0025 eV, respectively. Fromthe vibrationally resolved MATI spectrum in which each band
is classified into a specific conformer, it has been found thatthe equatorial conformer undergoes a drastic structural changeupon ionization whereas only a modest structural distortion isobserved for the axial conformer. The detail NBO analysisprovides the quantitative explanation for the experiment in termsof the anomeric and steric effects. Conformer-specific spectro-scopic and dynamic features provide quite valuable informationfor the understanding of the structure-reactivity relationship,and need to be intensively investigated.
Acknowledgment. This work was supported by the NationalResearch Foundation (2010-0015031, 2010-0000068, 2010-0001635, 2009-0082847).
References and Notes
(1) Park, S. T.; Kim, S. K.; Kim, M. S. Nature 2002, 415, 306.(2) Kim, M. H.; Shen, L.; Tao, H.; Martinez, T. J.; Suits, A. G. Science
2007, 315, 1561.(3) Lim, J. S.; Lee, Y. S.; Kim, S. K. Angew. Chem., Int. Ed. 2008,
47, 1853.(4) Choi, K.-W.; Ahn, D.-S.; Lee, J.-H.; Kim, S. K. Chem. Commun.
2007, 9, 1041.(5) Florio, G. M.; Christie, R. A.; Jordan, K. D.; Zwier, T. S. J. Am.
Chem. Soc. 2002, 124, 10236.(6) Zheng, C.; Subramaniam, S.; Kalasinsky, V. F.; Durig, J. R. J. Mol.
Struct. 2006, 785, 143.(7) Wilson, M. A.; Chandler, D. Chem. Phys. 1990, 149, 11.(8) Strauss, H. L.; Pickett, H. M. J. Am. Chem. Soc. 1970, 92, 7281.(9) Sirjean, B.; Glaude, P. A.; Ruiz-Lopez, M. F.; Fournet, R. J. Phys.
Chem. A 2006, 110, 12693.(10) Pierce, L.; Nelson, R. J. Am. Chem. Soc. 1966, 88, 216.(11) Pierce, L.; Beecher, J. F. J. Am. Chem. Soc. 1966, 88, 5406.(12) Kuharski, R. A.; Chandler, D.; Montgomery, J. A.; Rabii, F.; Singer,
S. J. J. Phys. Chem. 1988, 92, 3261.(13) Kakhiani, K.; Lourderaj, U.; Hu, W. F.; Birney, D.; Hase, W. L. J.
Phys. Chem. A 2009, 113, 4570.(14) Jensen, F. R.; Bushweller, C. H.; Beck, B. H. J. Am. Chem. Soc.
1969, 91, 344.(15) Fernandez-Alonso, M. d. C.; Canada, J.; Jimenez-Barbero, J.;
Cuevas, G. Chem. Phys. Chem. 2005, 6, 671.(16) Damiani, D.; Ferretti, L. Chem. Phys. Lett. 1973, 21, 592.(17) Reeves, L. W.; Stromme, K. O. Can. J. Phys. 1960, 38, 1241.(18) Eliel, E. L.; Martin, R. J. L. J. Am. Chem. Soc. 1968, 90, 682.(19) Reisse, J.; Stien, M. L.; Gilles, J. M.; Oth, J. F. M. Tetrahedron.
Lett. 1969, 1917.(20) Holly, S.; Jalsovszky, G.; Egyed, O. J. Mol. Struct. 1982, 79, 465.(21) Woldbaek, T. Acta. Chem. Scand. A 1982, 36, 641.(22) Damiani, D.; Scappini, F.; Caminati, W.; Corbelli, G. J. Mol.
Spectrosc. 1983, 100, 36.(23) Caminati, W.; Damiani, D.; Scappini, F. J. Mol. Spectrosc. 1984,
104, 183.(24) Shen, Q.; Peloquin, J. M. Acta Chem. Scand., Ser. A 1988, 42,
367.(25) Kolodziejski, M.; Waliszewska, G.; Abramczyk, H. Chem. Phys.
1996, 213, 341.(26) Tian, S. X.; Kishimoto, N.; Ohno, K. J. Electron Spectrosc. Relat.
Phenom. 2002, 125, 205.(27) Carrera, A.; Mobbili, M.; Moriena, G.; Marceca, E. Chem. Phys.
Lett. 2008, 467, 14.(28) Durig, J. R.; El Defrawy, A. M.; Ward, R. M.; Guirgis, G. A.;
Gounev, T. K. J. Mol. Struct. 2009, 918, 26.(29) Choi, S.; Kang, T. Y.; Choi, K.-W.; Han, S.; Ahn, D.-S.; Baek,
S. J.; Kim, S. K. J. Phys. Chem. A 2008, 112, 5060.(30) Choi, S.; Kang, T. Y.; Choi, K.-W.; Han, S.; Ahn, D.-S.; Baek,
S. J.; Kim, S. K. J. Phys. Chem. A 2008, 112, 7191.(31) The Anomeric Effect and Associated Stereoelectronic Effects;
Thatcher, G. R. J., Ed.; American Chemical Society: Washington, DC, 1993.(32) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb,
M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.;Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.;Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.;Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.;Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li,X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.;Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.;Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.;Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich,S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.;Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.;
Ionization of Bromocyclohexane J. Phys. Chem. A, Vol. 114, No. 37, 2010 10009

Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz,P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.;Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson,B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03W;Gaussian, Inc., Wallingford, CT, 2004.
(33) Lee, C.; Yang, W.; Parr, R. G. Phys. ReV. B 1988, 37, 785.(34) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.(35) Perdew, J. P.; Wang, Y. Phys. ReV. B 1992, 45, 13244.(36) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. ReV. Lett. 1996, 77,
3865.(37) Becke, A. D. Phys. ReV. A 1988, 38, 3098.(38) Duschinsky, F. Acta Physicochim. URSS 1937, 7, 551.
(39) Peluso, A.; Santoro, F.; Re, G. D. Int. J. Quantum Chem. 1997,63, 233.
(40) Borrelli, R.; Peluso, A. J. Chem. Phys. 2003, 119, 8437.(41) Glendening, E. D. R., A. E.; Carpenter, J. E.; Weinhold, F. QCPE
Bull. 1990, 10, 58.(42) Moller, C.; Plesset, M. S. Phys. ReV. 1934, 46, 618.(43) Merkt, F. Annu. ReV. Phys. Chem. 1997, 48, 675.(44) Lee, M.; Kim, H.; Lee, Y. S.; Kim, M. S. Angew. Chem., Int. Ed.
2005, 44, 2929.
JP105541V
10010 J. Phys. Chem. A, Vol. 114, No. 37, 2010 Han et al.





























