Embed Size (px)
Citation preview
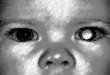
Leukocoria
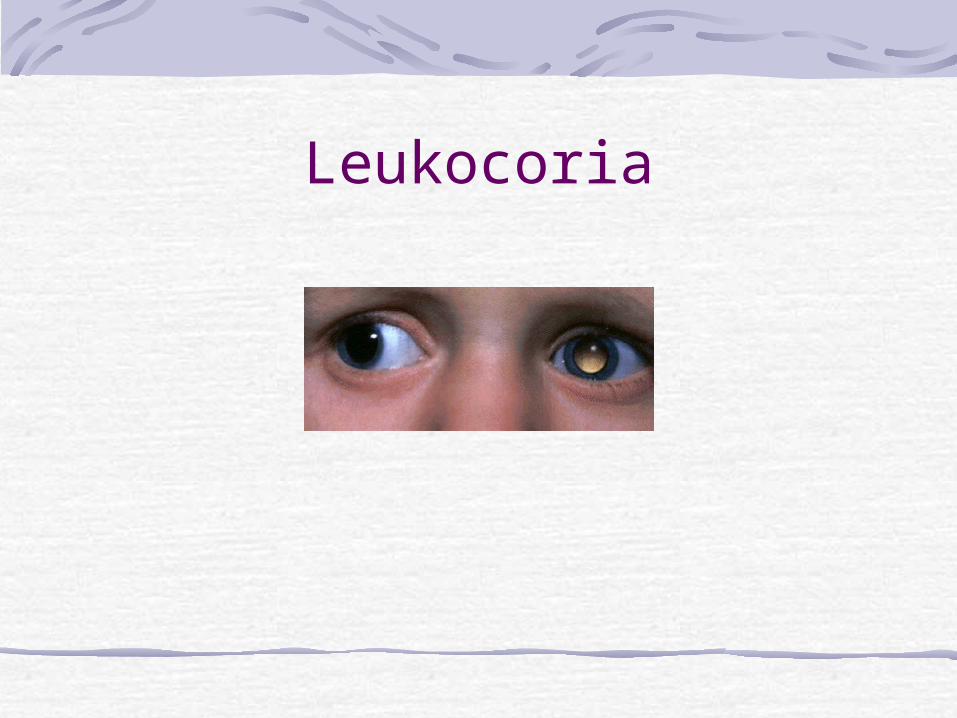
Causes of LeukocoriaDIFFERENTIAL DIAGNOSIS OF LEUKOCORIA
CataractRetinoblastomaToxocariasisCoat´s diseaseROPPHPVRetinal detachmentColobomaRetinal dysplasiaNorrie´s disease

Developmental CataractsNontraumatic unilateral cataracts first detected after 6 months of age also present special concerns.Usually, the precise age of onset is not known. In some cases, particularly those associated with thinning of the posterior lens capsule (posterior lenticonus or lentiglobus), the duration of significant visual deprivation may have been relatively brief. A history of recent-onset strabismus or leukocoria, preservation of good alignment with central steady fixation (even on a light), family photographs documenting symmetrical red fundus reflexes, or pediatrician's records of red reflex observation can help to establish a good visual prognosis.

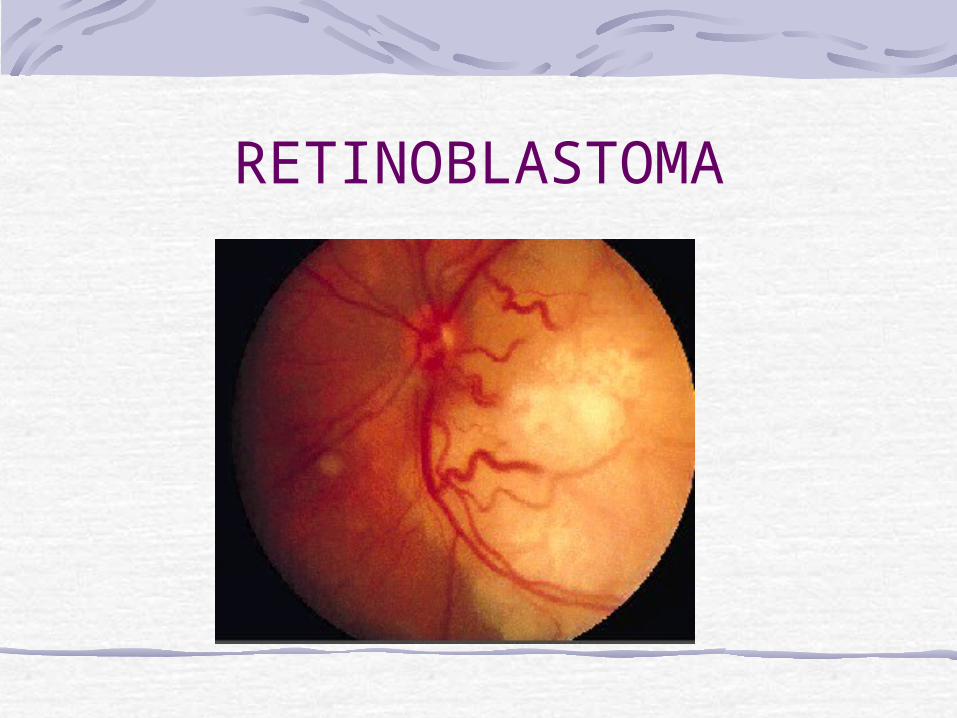
RetinoblastomaRetinoblastoma is the most common intraocular tumor of childhood, accounting for 1% of childhood cancer deaths in the United States and 5% of blindness in children. The incidence is 1 in 15,000 to 1 in 20,000 live births.Overall mortality from retinoblastoma decreased from 95% a century ago. With modern diagnostic and therapeutic advances, the mortality rate from metastatic or recurrent retinoblastoma has been as low as 5%.
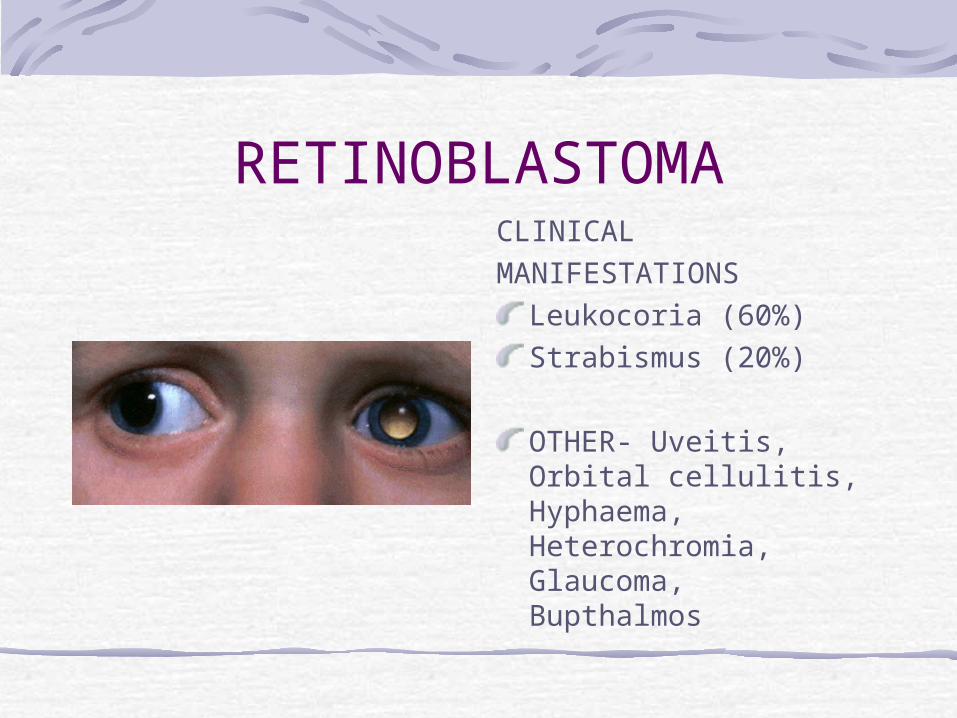
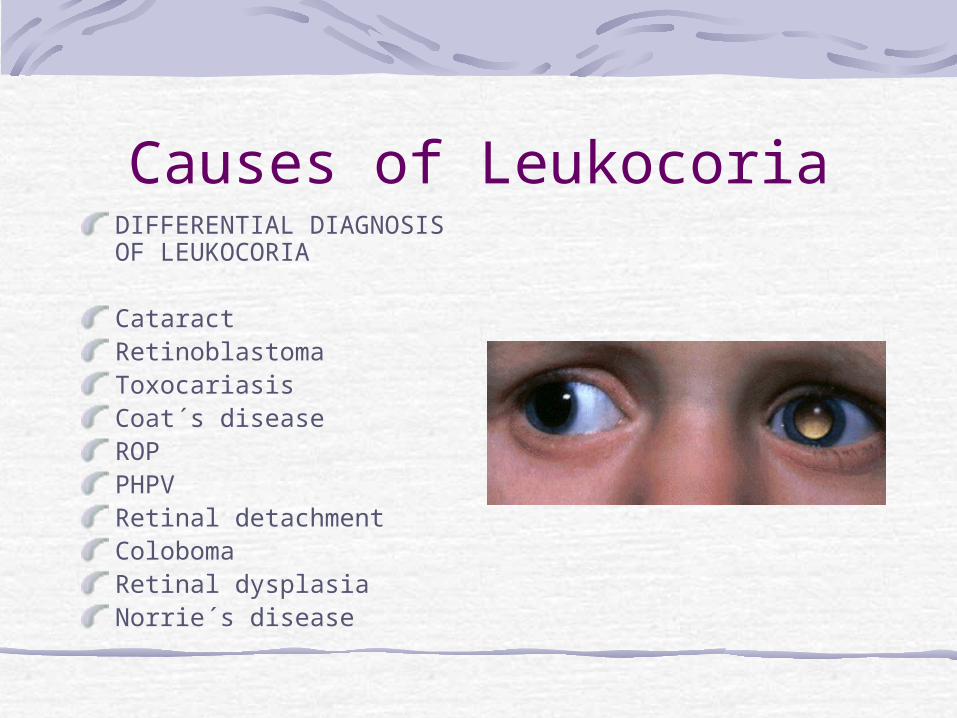
RETINOBLASTOMACLINICALMANIFESTATIONS
Leukocoria (60%)Strabismus (20%)
OTHER- Uveitis, Orbital cellulitis, Hyphaema, Heterochromia, Glaucoma, Bupthalmos
RETINOBLASTOMA

RetinoblastomaThe disease is bilateral in approximately 30% of cases. The average age at diagnosis is 18 months and 90% of patients are diagnosed before the age of 3 years. Less than 10% of retinoblastoma suffers have a family history of the disorder, 90% of cases are sporadic. Of the sporadic cases, the responsible mutation is in a germ cell in 25% of cases and in a somatic cell in 75% of cases

GENETICS
Retinoblastoma gene is a recessive oncogene of 180,000 kilobases.Located chromosome- 13q14Knudson two hit hypothesis:-Germinal cells have one defective and one normal RB gene.A somatic mutation results in loss of the normal RB gene and hence retinoblastoma develops (somatic mutations occur frequently enough in the developing retina, therefore lesions usually affect both eyes)
In addition, the first child of a parent who had had a unilateral retinoblastoma has a 4% chance of developing the disease

PATHOLOGY
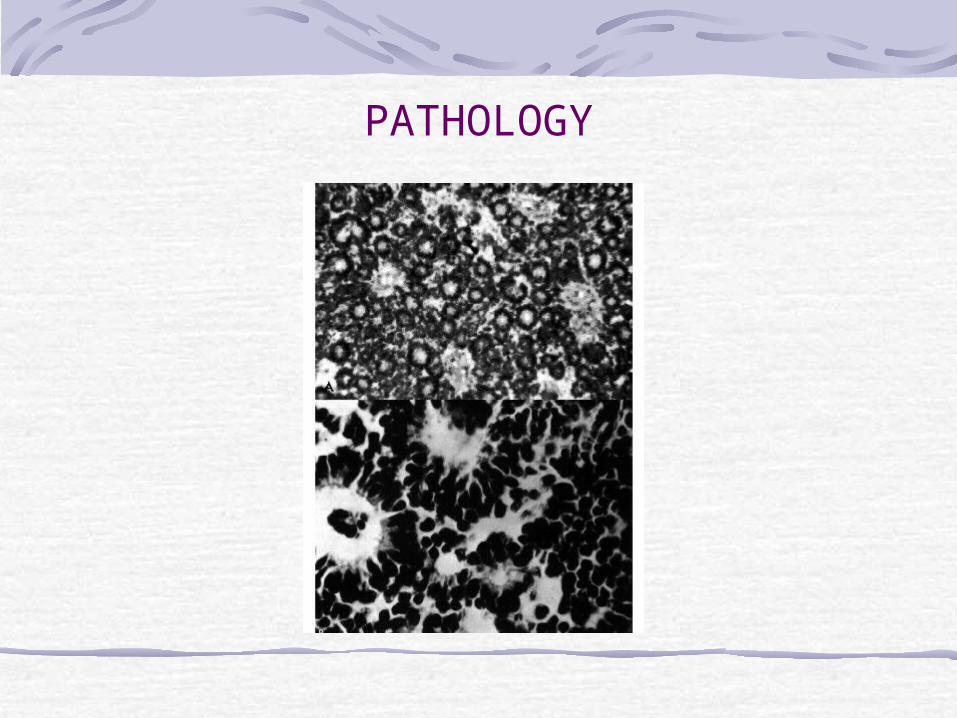
Arise in primitive photoreceptor cells.Characteristic histology:Retinoblastomas are composed of poorly differentiated neuroblastic cells with scanty cytoplasm and prominent basophilic nuclei.The tumour proliferates rapidly, with a tendency to outgrow its blood supply and undergo spontaneous necrosis. Necrotic tumour being eosinophilic stain pink.Characteristic Flexner-Wintersteiner rosettes represent an attempt at retinal differentiation. Histologically, a ring of cuboidal cells is seen surrounding a central lumen. Cuboidal tumour cells with basally oriented nuclei arranged around a central lumen.
Calcification is another feature of retinoblastomas, usually occurring in necrotic areas. Calcium stains with H&E. It is worth identifying calcium in suspect eyes by ultrasound, or CT scan to
differentiate retinoblastomas from other tumours.
PATHOLOGY

Retinoblastoma

MANAGEMENT EMPIRICAL GENETIC COUNSELLING
ENUCLEATIONunilateral, poor visual prognosisPLAQUE4-12mm +/- vitreous seedingEXTERNAL BEAM>12mm, multiple foci, only eyeLASERconsider- indirect, xenon arccryotherapy if <2dd in sizeCHEMOTHERAPY, if intracranial extension

Non-Retinoblastoma Malignancies
Unfortunately, children who have genetic retinoblastoma and survive their primary intraocular cancer have a substantially increased risk of death from one or more nonretinoblastoma malignancies over the course of their lifetimes, up to 35% of children who have had a bliateral retinoblastoma and external beam radiation therapy will develop a second cancer by age 25 years

Congenital retinal telangiectasis (Coats'
disease)Congenital retinal telangiectasis (Coats' disease) is an idiopathic retinal vascular disorder that usually affects young male patients unilaterally in their first or second decade of life. Congenital retinal telangiectasis, however, can affect patients of either gender and become manifest at any age. Up to one third of patients are older than 30 years of age at the time of presentation.There is no defined familial inheritance. Patients may present with decreased vision, as well as strabismus or leukocoria in children. The hallmark feature of congenital retinal telangiectasis is localized fusiform aneurysmal dilations of the retinal vessels reminiscent of tiny light bulbs
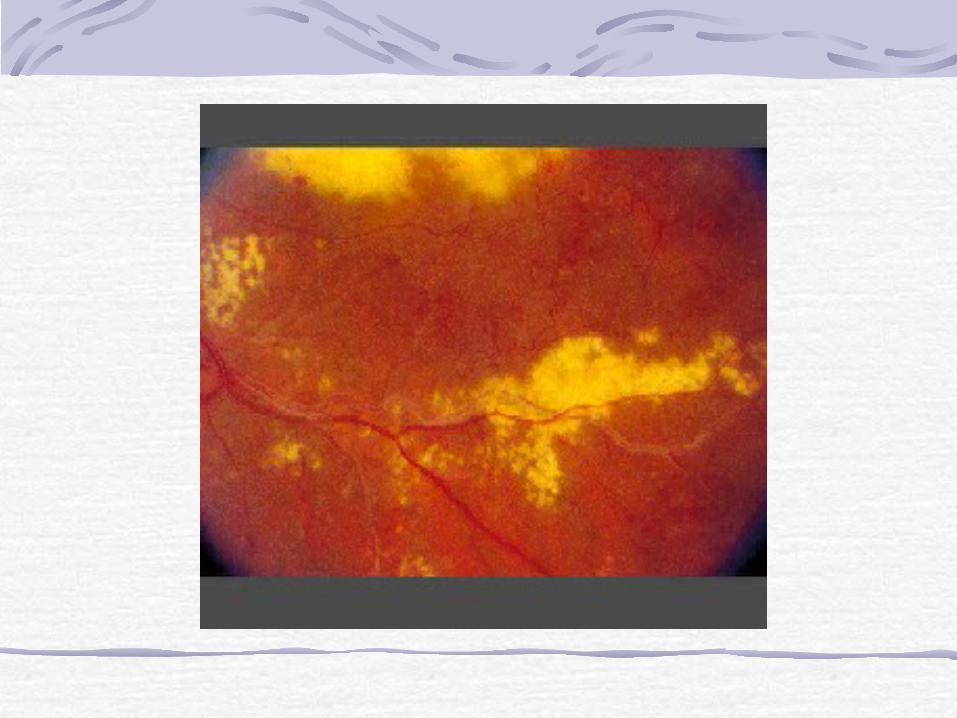

Retinal vascular anomaliesThe vascular anomalies can occur anywhere in the fundus and may involve the capillaries, arteries, and veins. Other findings may include vascular loops and beading, retinal neovascularization, hemorrhagic retinal macrocysts, and segmentally dilated capillaries.Leakage from the incompetent vasculature may lead to retinal edema, lipid deposition, or, in severe cases, an exudative retinal detachment. The extent of retinal involvement is variable. Infants and children often are more severely affected with extensive vascular involvement and massive subretinal lipid exudate.

Persistent hyperplastic primary vitreous (PHPV)
Persistent hyperplastic primary vitreous (PHPV) is a congenital anomaly in which the primary vitreous fails to regress in utero. Highly vascular mesenchymal tissue nurtures the developing lens during intrauterine life. In PHPV, the mesenchymal tissue forms a mass behind the lens. A gray-yellow retrolental membrane may produce leukocoria, with the subsequent suspicion of retinoblastoma. In PHPV, the globe is white and slightly microphthalmic. Patients have no history of prematurity or oxygen administration.

RETINOPATHY OF PREMATURITY (ROP)
Vasoproliferative retinopathy affecting premature infants exposed to high oxygen
INCIDENCEPrematurity (<32/40)Birth weight (30% < 1000gm affected)Oxygen duration90% ROP regresses spontaneously, 5% blindness

RETINOPATHY OF PREMATURITY (ROP)
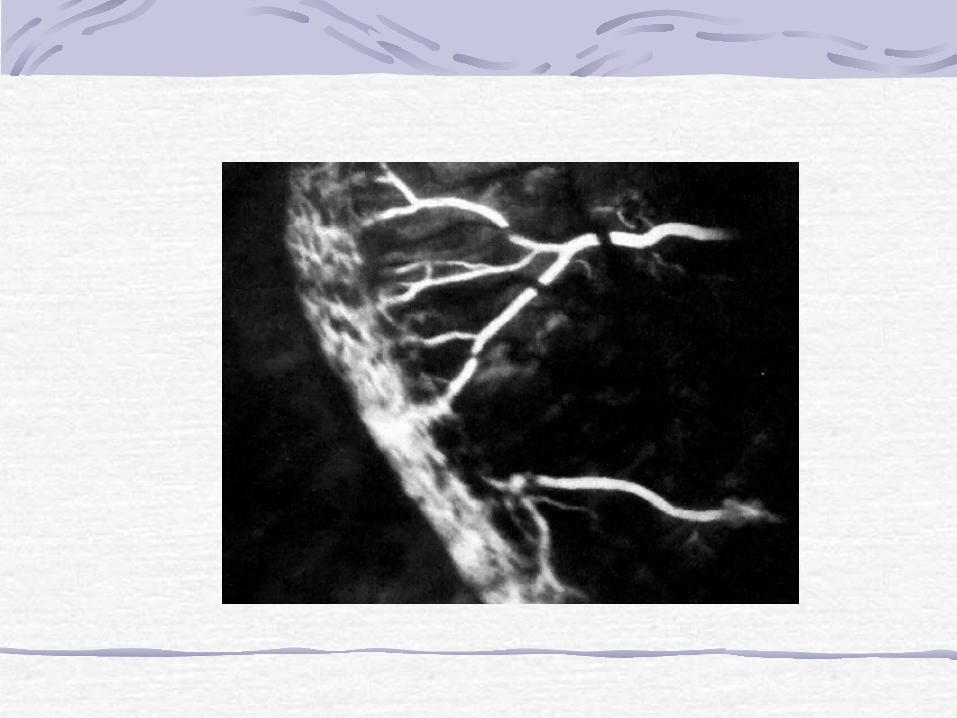
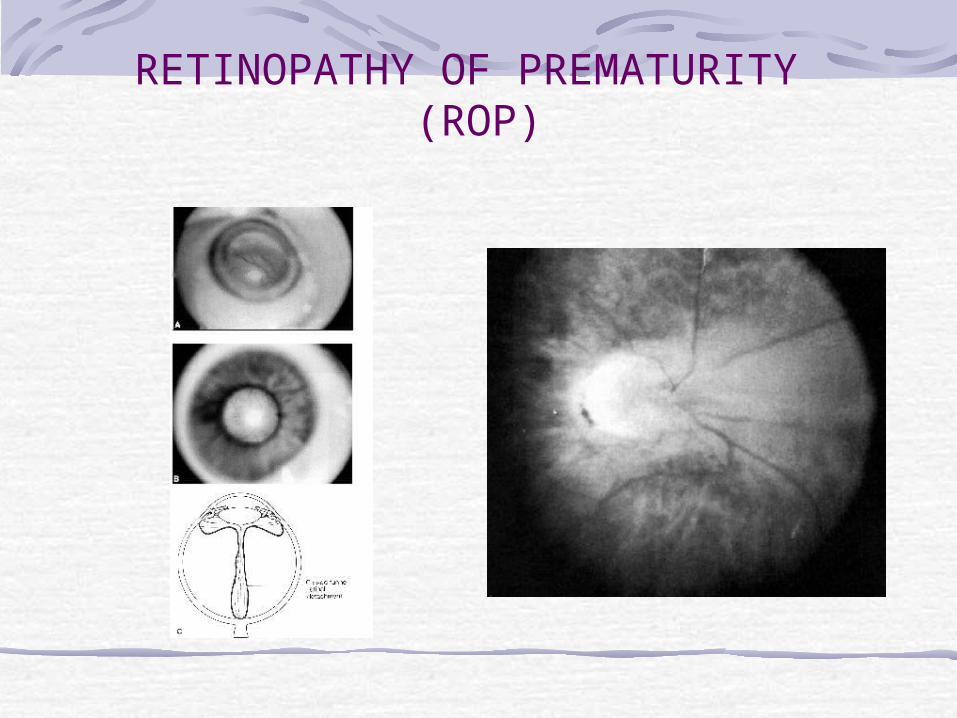
In the early active stages of ROP, a band of glomeruloid capillaries proliferates at the junction between the peripheral nonperfused and the posterior perfused retina. The proliferating vessels break through the internal limiting membrane and invade the vitreous, inciting fibrosis and contraction. In the later cicatricial stages of ROP, the retina is folded on itself by the organized vitreous, forming a fibroneural mass that drags the macula and optic disc temporally. The end stage of the disease is marked by total retinal detachment, leukocoria, blindness, and phthisis bulbi.
RETINOPATHY OF PREMATURITY (ROP)
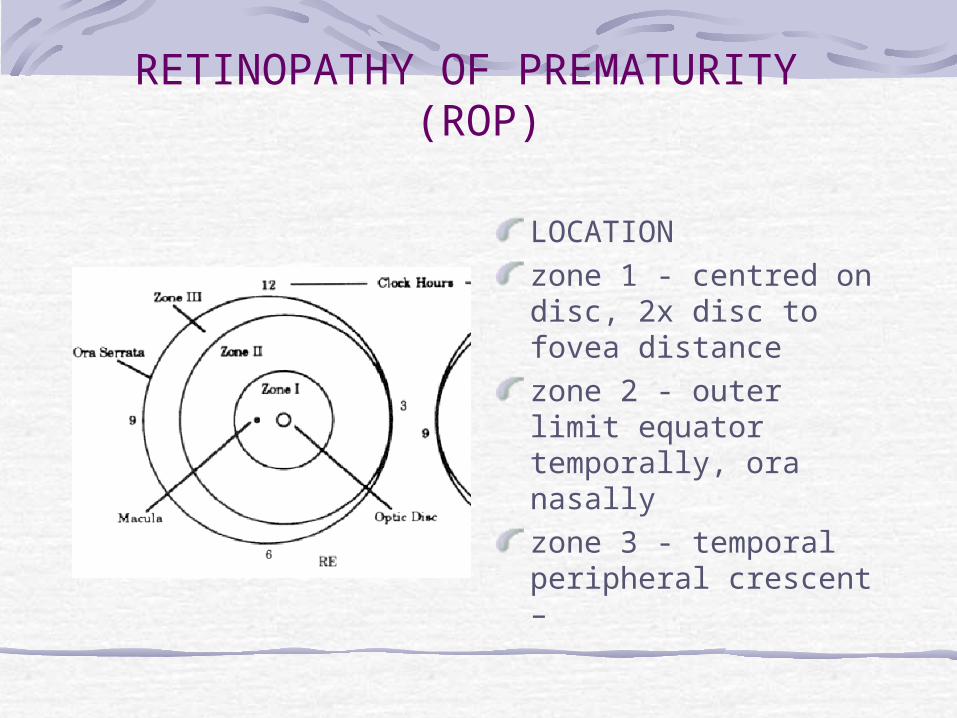
LOCATIONzone 1 - centred on disc, 2x disc to fovea distancezone 2 - outer limit equator temporally, ora nasallyzone 3 - temporal peripheral crescent – in clock hoursrush disease- SI-SV in 2/52
CLASSIFICATION - STAGINGSI- flat demarcation line with branching blood vessels up to lineSII- ridge with volume, blood vessels enter ridgeSIII- ridge + extraretinal fibrovascular proliferationSIV- retinal detachment- a (not involving the fovea), b (involving the fovea)SV- total RD, open or closed funnelplus disease- dilated tortuous vessels in posterior pole, vitreous haze and poor mydriasis
RETINOPATHY OF PREMATURITY (ROP)
LOCATIONzone 1 - centred on disc, 2x disc to fovea distancezone 2 - outer limit equator temporally, ora nasallyzone 3 - temporal peripheral crescent –
RETINOPATHY OF PREMATURITY (ROP)

Toxoplasmosis
Toxoplasmosis gondii is an obligate intracellular protozoa causing up to 50% of cases of posterior uveitis.Ocular infection is characterised by focal necrotising retinochoroiditis with vitritis.In congenital infection the eye may also be affected by cataract, microphthalmos, and optic atrophy

Chorioretinitis and congenital toxoplasmosis
The main clinical manifestations of the symptomatic form of toxoplasmosis are microcephaly or hydrocephaly, cerebral palsy, epilepsy, mental retardation, cerebral calcification, and chorioretinitis.The most important signs in the diagnosis of congenital toxoplasmosis are the three Cs: convulsions, calcification (intracranial), and chorioretinitis. Chorioretinitis is present in 80% of children with congenital toxoplasmosis and is most often bilateral; toxoplasmosis is considered one of the most common causes of chorioretinitis.

Congenital Toxoplasmosis
Highest transmission occurs in the IIIrd trimester90% of congenital infections have no clinical signsEarlier infection occurs in pregnancy - worse potential outcomeTriad:- convulsions,cerebral calcificationand chorioretinitisEye - chorioretinitis, cataracts, microphthalmos, panuveitis, optic atrophy

Investigation of Toxoplasmosis
ELISA IgM in neonates, rising IgG in adults (although not that helpful in adults).Fluorescein angiography (hypofluorescence in the early stages and then progressive leakage).Indocyanine angiography - multiple small dark spots may be seen around the visible lesions implying the affected retina is greater than apparent initially. This sign may be useful in assessing the effect of treatment.

Some indications for active treatment of toxoplasmosis
Lesions that involve the macula, papillomacular bundle or optic disc
Large, active lesions should be treated.
Immunocompromised patients should be treated.

Ocular toxocariasisOcular toxocariasis is a unilateral disorder that presents as strabismus, leukocoria or decreased vision. Retinal damage is the result of the host's inflammatory response to the single infection nematode, which must usually be dead before the uveitis can develop. The posterior uveitis may be of severe intensity.

Toxocariasis subretinal granuloma
Ocular toxocariasis may present with decreased vision, strabismus, leukocoria, or uveitis.Most commonly a subretinal granuloma is present in the posterior pole in an otherwise quiet eye. In the early stages, it is elevated above the retina and may resemble a neoplasm.

Retinal detachment in childhood
Retinal detachment in childhood can be confused with retinoblastoma, and vice versa. The possibility of an underlying retinoblastoma should always be considered when a child presents with retinal detachment and vitreous hemorrhage, even when a history of trauma is obtained. Appropriate preoperative studies (ultrasonography or computed tomography) are indicated; if vitrectomy is performed, the specimen should be submitted for cytologic examination.

Retinal detachment in childhood
Retinal detachment in childhood can be confused with retinoblastoma, and vice versa. The possibility of an underlying retinoblastoma should always be considered when a child presents with retinal detachment and vitreous hemorrhage, even when a history of trauma is obtained.

Norrie diseaseNorrie disease, or the progressive oculoacousticocerebral degeneration of Norrie, is a rare, X-linked recessive heritable disorder characterized by bilateral leukocoria caused by retinal detachment. Affected boys classically have a triad of blindness, deafness, and mental retardation. Apparent at birth or in early infancy, the ocular findings usually progress to phthisis bulbi. An identical disorder in a Maltese kindred is called Episkopi blindness.

Retinal dysplasiaRetinal dysplasia and PHPV are characteristic ocular findings in trisomy 13; in fact, trisomy 13 was called retinal dysplasia before the chromosomal defect was identified. The multitude of systemic and ocular findings found in patients with trisomy 13 may include bilateral leukocoria. Rarely, retinal dysplasia occurs unilaterally in the congenitally malformed eyes of otherwise healthy persons.

COLOBOMAOPTIC DISC COLOBOMA
Due to failure of closure of foetal fissure inferiorlyMay be isolated disc or associated chorioretinal colobomaISOLATED DISC COLOBOMARare, Usually sporadic, some ADCan be bilateralVisual acuity varies from normal to NPL.Associated- optic disc pit, hyaloid artery remnant, myopia, posterior lenticonus,transphenoidal encephalocoele, cardiac defects, VII palsyRETINOCHOROIDAL COLOBOMA
ASOCIATIONSColoboma of iris, aniridia, PHPV, microphthalmosAssociated CVS, CNS and ear malformations

CHARGE !CHARGE (For diagnosis at least 4 of the highlighted abnormalities are required).Colobomas,
Heart defects,
Choanal Atresia,
Retarded growth,
Genital abnormalities,
Ear abnormalities
CHARGE is also associated with facial palsy, micrognathia, cleft palate, pharyngeal incompetence, tracheo-oesophageal fistula, renal and cardiac abnormalities.Note many other syndromes have colobomata.