Embed Size (px)
Citation preview

TECHNICAL PAPER
ISSN 1047-3289 J. Air & Waste Manage. Assoc. 44: 48-52
Method Development for VOST FractionatorMargaret E. Wickham St. Germain, Stephen B. Cummins and Gil Radolovich
Midwest Research InstituteKansas City, Missouri
A VOST Fractionator was designed and tested to fractionate an original VOST sample into two samples: one large and one small sample.The device allows for quantitation of high levels of compounds in the small fraction and trace levels in the large fraction. Several preliminaryvalidation samples were prepared, split, and analyzed to test the feasibility of the VOST Fractionator. These validation samples contained40,000 ng of three terpene compounds and 100 ng of 42 other volatile target analytes.
Analyte recoveries ranged from 70 to 130 percent, except for five water-soluble compounds. Recovery for the terpene compoundswas 110 to 118 percent. Precision for triplicate spiked samples was less than 30 percent relative standard deviation (%RSD) for mostcompounds. Results indicate that the VOST Fractionator accurately splits the sample and allows quantitation of extremely high levels ofcompounds without sacrificing sensitivity for trace compounds.
In a conventional volatile organic sampling train (VOST) analy-sis, samples are thermally desorbed, and the analytes are concen-trated on a cooled, analytical sorbent trap, such as Teriax. Subsequentanalysis uses a purge gas and thermal desorption, with the targetanalytes released from the sorbent and transferred directly to ananalytical instrument for quantitation.
When gas streams (i.e., incinerator stack gases) are sampled, theamounts of target analytes and other interfering compounds presentare not predictable. Gas samples collected with the VOST are sorbedin a solid sorbent cartridge and are subsequently desorbed foranalysis; thus, only one analysis is possible on each sample. As aresult, quantitation of compounds becomes a tradeoff between quan-titating high-level analytes without detecting trace-level analytesversus quantitating low-level analytes without accurate quantitationof high-level analytes. Both situations are limited by the linear rangeof the analytical instrumentation.
Alternate methods of splitting VOST samples were reviewed andwere determined to be inadequate options.1 To achieve quantitationfor analytes over a wide concentration range in a single sample, aVOST Fractionator was designed by Midwest Research Institute(MRI) based on two technologies: thermal desorption of compoundsfrom VOST sorbents and differential resistance of gas flows throughdifferent sized tubing.
In MRI's VOST Fractionator, a nitrogen purge gas containing thevolatile organic compounds (VOCs) desorbed from the originalVOST trap is split into two streams (see Figure 1). The resultant flowsof the two streams are controlled by varying the internal diameter of
accurate measurements10 ng upto 1 mg.
the split tubing and hence the flow resistance. The split effluent fromthis primary desorption is collected on two secondary "daughter"traps.
The VOST Fractionator allows for the quantitation of VOCs fromVOST tubes, which are used to sample a gas stream containinganalyte concentrations that range from 10 ng/20 L for some com-pounds to greater than 20,000 ng/20 L for other compounds. Sincethe VOST Fractionator uses the same sorbent media for samplerecollection as the original VOST sample, the original sample maybe spiked with a split surrogate prior to fractionation and analyzed byknown methods.
Experimental Methods and Procedures
New ApparatusNitrogen purge gas fed to a sample trap for desorption is regulated
with an automatic flow controller at 150 mL/min (see Figure 1). TheVOST sample is thermally desorbed via standard methodologyoutlined in EPA Method 5041.2.3
The effluent from this thermal desorption (containing the VOCs)is then split into two branches that are constructed of tubing havingdifferent internal diameters and lengths. The difference in the innerdiameter of the tubings, coupled with the difference in lengths,provides the necessary restriction in flow through one tube versus theother tube to maintain a constant "split ratio." The split ratio chosenfor this study was approximately 120:1, resulting in a flow rate of145 to 150 mL/min to one branch and a flow rate of 1.2 to 1.5 mL/min to the other branch.
The flows through the two branches are recollected onto two, cool(5° to 10°C), clean VOST traps using heat exchangers to maintain aconstant temperature and flow meters to monitor the flows. Therecollected samples can then be analyzed, or re-split again, if neces-sary, using additional stages of thermal desorption.
Preliminary TestingThree preliminary experimental tests were performed. First, the
lengths of tubing needed were determined; the final selection oftubing was an 8 cm x 0.53 mm i.d. tube on the high-flow side and a
Copyright 1994 - Air & Waste Management Association
48 • January 1994 • Vol. 44 • AIR & WASTE

Divert Valve forFlow Checks
Heated Transfer Line (170°C)
Heated Oven with Capillary Columns (180°C)
Sample Trap at Position B (99% Fraction)
* Megabore Tubing, 0.53 mm ID,8 cm Length
* Capillary Tubing, 0.25 mm ID,24 cm Length
Sample Trap at Position C (1% Fraction)
Water Cooled Trap Holders (5°C)
Figure 1 . VOST Fractionator.
24 cm x 0.25 mm i.d. tube on the low-flow side. This arrangementallowed a 150:1 split on dry VOST traps and approximately a 120:1split on moisture-laden traps. Second, the variability of the split ratiowas established; the split ratio remained constant, while the gas flowrates ranged from 50 to 160mL/min. Third, several traps wereinterchanged; the variability in the traps did not significantly affectthe split ratio onto the two new traps.
VOST Fractionator Split RatioA final test was performed to confirm the split ratio. An original
sample trap was spiked with a surrogate compound prior to insertingit into the VOST Fractionator. A flow rate of 150 mL/min for 5 minwas selected for a total of 750 mL of gas transfer or 600 mL inmoisture-laden traps. By recording the exact flow rate of the nitrogenpurge gas at the exit of each new trap during desorption, the ratio forthe flow split was obtained. The surrogate amount obtained for eachof the daughter VOST traps was in the same ratio as the split flows,which confirmed the split ratio calculated using the flow rates.Table I summarizes triplicate results for split ratios calculated by theflow rates and by the surrogate amounts.
Preparation of Blank Samples and Validation SamplesA blank sample (i.e., a clean VOST tube) was spiked with
2,000 ng of the surrogate, 1,2,3-trifluorobenzene. The sample wasfractionated, and all three traps [the spent blank trap (A), the 99percent fraction (B), and the 1 percent fraction (C)] were spiked withan internal standard and analyzed according to SW-846 Method5041.
Replicate VOST validation samples were prepared by spikingwith 40,000 ng of three terpenes (a-pinene, p-pinene, and cymene).Other target analytes (see Table II) were spiked at 100 ng, and thesurrogate was spiked at 2,000 ng. These three replicate samples werethen split using the VOST Fractionator, which generated nine totalVOST traps to be analyzed. Each VOST trap was spiked with theinternal standard just prior to analysis.
Analytical ProceduresThe samples were analyzed following SW-846 Method 5041.
The target analyte list included three internal standards, five surrogates,up to two split surrogates (1,2,3-trifluorobenzene and 1,3,5-fluorobenzene), and 45 target analytes, including three terpenes. TheGC/MS system was set up according to the method with minormodifications to allow desorption of the terpenes. Specifically, thetransfer line temperatures were increased to 170°C, and the trapdesorption temperature was increased to 220°C. The quality controlprocedures described in the method were followed.
Results
Quality Control Sample ResultsQuality control samples included calibration
curve samples, initial precision and recovery (IPR)samples, continuing calibration standards, systemblank samples, laboratory blank samples, and per-formance audit samples.
Relative response factors for the analytes werecalculated from the five-point calibration curve.System Performance Check Compounds (SPCCs)met the minimum relative response factor (RRF)criterion of 0.300, except for bromoform, whichwas only 0.142. However, these results were con-sistent with the method.
Calibration Check Compounds (CCCs) met thepercent relative standard deviation criteria (%RSD),except for vinyl chloride, which had a 35.6 percent
RSD. Of the remaining compounds, five [acetone, methyl ethylketone (MEK), methyl isobutyl ketone (MIBK), acrylonitrile, andbenzene] had a relative standard deviation (RSD) greater than 30percent which is typical for the method. The benzene backgroundobserved on the traps used for the calibration curve was equivalent to100 ng and was a suspected eluent from degraded Tenax. Thecorrected RRF for benzene (1.402) was consistent with two previouscalibration curves for benzene; it was used for all calculations in thestudy. A single-point calibration for 1,2,3-trifluorobenzene, the splitsurrogate, was performed at the 2,000 ng level, so the amountsidentified for this surrogate are considered to be estimates.
As prescribed in Method 5041, four replicate samples wereanalyzed at the mid-level standard amount of 100 ng on column. Allcompounds, except iodomethane and the water-soluble compounds(acetone, MEK, MIBK, acrolein, and acrylonitrile), had detectedamounts between 69.1 ng and 157 ng with %RSD values less than 21percent. The recoveries for the water-soluble compounds rangedfrom 188 to 403 percent. Iodomethane was recovered at 45.2 percent.
A continuing calibration check standard was analyzed at thebeginning of each day to check the performance of the SPCCs, CCCs,and other target analytes. The SPCCs met the minimum RRF, exceptbromoform. The CCCs met the 25 percent difference (D) objectivefor all compounds. Less than 10 compounds did not meet the 30percent D guideline relative to the calibration curve, which is typicalfor VOST-type analyses. The five water-soluble compounds had upto a 90 percent D, while the remaining five compounds had up to a50 percent D.
Two additional standard mixtures were analyzed. The first stan-dard at 100 ng was spiked onto a "parent" trap, split on the VOSTFractionator, and the 99 percent fraction was analyzed. These resultspassed the continuing calibration criteria. The second standard at
Table L Comparison of split ratios, (a)
Ratio for
Replicate 1Replicate 2
Replicate 3
Average
ra/tt\ tinlW ratin - JZ
156:1
149:1
143:1
149:1
te or amount for high
127:1
141:1
142:1
137:1
-level trap
AIR & WASTE • Vol. 44 • January 1994 • 49
TECHNICAL PAPER
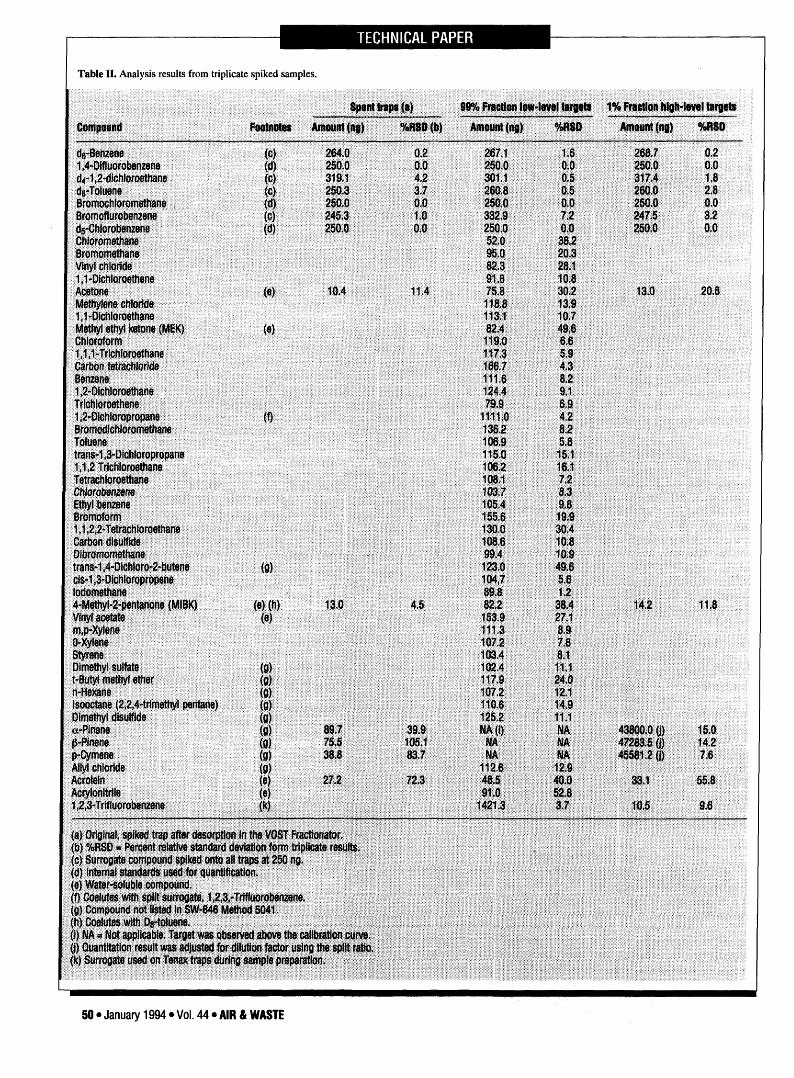
Table I I . Analysis results from triplicate spiked samples.
Spent tr
Compound
de-Benzene1,4-Dlffuorobenzene(J4-1,2-dichloroethane
da-TolueneBromochloromethaneBromoflurobenzenedg-ChlorobenzeneCNorometfianeBromomethaneVinyl chloride1,1-DichloroetheneAcetoneMethylene chloride1,1-DicliloroethaneMethyl ethyl ketone (MEK)Chloroform1,1,1-TrichloroethaneCarbon tetrachloride
Benzene1,2-DichloroethaneTrichloroethene1,2-Dichloropropane
BromodlchloromethaneToluenetrans-1,3-Dichloropropane1,1,2 TrichloroethaneTetrachloroethaneChiorobenzeneEthyl benzeneBromoform1,1,2,2-TetrachtoroethaneCarbon disuttideDlbromomethanetrans-1,4-Oichloro*2-buteneCi$-1,3-Dlchlorapropenelodomethane4-Methyl-2-pentanone (MIBKVinyl acetatem.p-XyleneO-XyfeneStyreneDimethyl suifatet-Butyl methyl ether
n-HexaneIsooctane (2,2,4-trimettiyl pe
Dimethyl disulfidea-Pinenep*-Pfoenep-CymeneAltyl chlorideAcroieinAcfyionitrile1,2,3-Trifluorobenzene
(a) Original, spiked trap after 1(b) %RSD » Percent relative $it*\ Citrrrirtato pmnnminrf cnita\V/ OUnUyalB vylII|)UUIIU Splft
(d) Internal standards used tofa\ tA/atatvc/tliihta jvuYtnAimrl(f) Coelutes with split surroga(g) Compound not fisted In $V(h) Coelutes with Dg-toiuene.(i) NA = Not applicable. Targe!(j) Qitantitation result was adji(k) Surrogate used on Tenax t
Footnol
(c)
(c)(c)(rf)(c){&)
(e)
(e)
(t)
(9)
) (e)(n)
(9)(9)(9)
rstane) (Q)
(9)(9)(9)(9)(9)(8)(e)
mjesorption in the VOJtandard deviation for>H ntitn stii trane nt Ofiu uniu «n it ctpii at «*
r quantification.
te( 1,2,3,-TrifluorabeV-846 Method 5041.
was observed abovejsted for dilution factraps during sample p
es Amount (ng)
264.0250.0319.1250.3250.0245.3250.0
10.4
13,0
89.775.538.8
27.2
rr Fracttonator,m triplicate results.ft It/Tu rty.
nene.
the calibration curve,or using the split ratio,reparation.
aps(a)
%RSD(b)
0.20.04.23.70.0100.0
11.4
4.5
39.9105.183.7
72.3
99% Fraction low-le*
Amount (ng)
267.1250.0301.1260.8250.0332.9250.052.095.082.391.876.8118.8113.182.4
119.0117,3106.7111.6124.479.9
1111.0136.2106.9115.0106.2108.1103.7105.4155.6130.0108.699.4123.0104,789.882.2153.9111.3107.2103.4102.4117.9107.2110.6125.2NA(i)NANA
112.648J91.0
1421.3
el targets
%RSD
1.60.00.50.50.07.20.038.220.328.110.830.213.910.749.66,65.94.38.29.16,94.28.25.815.116.17,28.39.819.930.410.810,949,65.61.2
38.427.18.97.88.111,124.012.114.911.1NANANA
12,940.052.837
1 % Fraction hi(
Amount (ng!
288.7250.0317.4260.0250.0247.5250.0
13,0
14.2
43800,0 (j)47283.5(045581.2 (I)
33.1
10.5
Ih-level targets
%RSD
0.20.01.82.80.03.20.0
20.8
11,8
15.014.27.6
55.8
9.6
50 • January 1994 • Vol. 44 • AIR & WASTE

2,000 ng was prepared and analyzed to determine the removalefficiency from the spent trap, to estimate recovery for the high-levelrecovery test, and to check feasibility of the fractionation technique.The water-soluble compounds were the only compounds observedon the spent trap. The low boiling point compounds and water-soluble compounds were the only compounds not meeting a 50 to 150percent recovery. In the daughter samples, all but 14 compounds meta 70 to 130 percent analyte recovery objective, which was compa-rable to the IPR objective. Based on the results, fractionation ofVOST trap samples was deemed feasible using the described appa-ratus and procedure.
Other quality control samples that were analyzed included systemblank samples, unused and new traps, and a performance auditsample. System blank samples were analyzed according to themethod. Two precleaned traps were analyzed before they were usedfor spiking to determine possible contamination in new traps. Theselaboratory-generated, blank samples were found to be free of con-tamination, except for the water-soluble compounds which werequantitated at 8 to 38 ng. The performance audit sample (PAS) wasanalyzed to verify the proper preparation of the standards anddetermine the accuracy of the calibration curve. Only the datareported for bromoform may be questionable because the source ofthe error was not positively identified.
The surrogate recoveries for all quality control samples rangedfrom 92 to 127 percent and met recovery objectives (50 to 150percent).
Validation Sample ResultsAll surrogate recoveries for the three validation samples met the
50 percent to 150 percent recovery objective. MIBK was observed tocoelute with d8-toluene. Because an attempt to identify uniquemasses for MIBK failed, values for MIBK below 20 ng are consid-ered to be false positives due to the interference of rf8-toluene.
The triplicate split sample results are presented in Table II.
Columns 1 and 2 in Table II summarize the average amounts and
RSD values from the spent traps (i.e., after desorption). Three water-
Table III. Laboratory split blank sample.
Compound
de-Benezene1,4-Difluorobeiizenecl4-1,2 DichloroethanedrTolueneBromochlorometnaneBramofluorobenzeftedg^ChlorobenzeneAcetoneMethyl ethyl ketone(M1,2-DicWoropropane4-Methyf-2-pentanonep-CymeneAHyl chlorideAcrolein1,2,3-Trifluorobenzerie
(a) Originai, spiked traf.(rj) Surrogate compour(c) Internal standard m
(e) Coelutes with «pitt;(f)CoeluteswithdB-toli
Footnotes(b)(e)(b)
(c)
(e)
£K} (d)
(•)(MIBK) (4)0)
(0)(9)
(9)
> atter desorption in the \id spiked onto airdaugh«d for quantification.)oundjurrogate, 1,2,3-trifluoroljene.
1 % Fraction
Z65.0250,0313.5241.8250.0209.8250.Q34.113.9NO
12.40.16.335.1117
fOST Fract ionalW traps at 250 ng,
)en*ene.
Spent trap (1
262.3250,0306.0264.8250.0233.4250.028.8147NO
12.5NOND
33.2NO
1) 99%Fractior
261.4250.0304.2273.3250.0239.6250.0287167
1050713.533.3ND
41.814337
soluble compounds (acetone, MIBK, and acrolein) were observed onthese spent traps. The observed amount of terpenes (a-pinene, |3-pinene, and cymene) remaining on the spent traps was less than 0.2percent of the original amount spiked. No other compounds wereobserved on the spent traps, indicating efficient removal of analytesfrom the original sample trap.
Columns 3 and 4 in Table II summarize the average amounts andRSD values from the 99 percent fraction traps. Most compoundswere recovered efficiently (i.e., 100 ng) even though the terpenesheavily saturated the analytical system. Surrogate recoveries rangedfrom 70 percent to 134 percent. 1,2-Dichloroethane coeluted with1,2,3-trifluorobenzene, which made quantitation difficult. Only sixcompounds were outside of a 70 percent to 130 percent recoveryguideline, and seven different compounds had RSD greater than 30percent.
Columns 5 and 6 in Table II summarize the average amounts andRSD values from the 1 percent fraction traps. The surrogate recov-eries ranged from 70 to 135 percent. The split surrogate, 1,2,3-trifluorobenzene, showed recoveries from 72 to 91 percent. Threewater-soluble compounds (acetone, MIBK, and acrolein) were ob-served at the same level as the laboratory blank samples. The terpeneswere observed at 267 to 353 ng, yielding average recoveries for theterpenes (a-pinene, (3-pinene, and cymene) of 110, 118, and 114percent, respectively.
A blank sample was spiked with the split surrogate (1,2,3-trifluorobenzene) and split on the VOST Fractionator. All three traps(original spent trap and two daughter traps) were analyzed (seeTable III). Only the water-soluble compounds, which were presenton the laboratory blank samples, were also observed on the blank splitfractions. The split surrogate recovery was 92 percent for the 1percent fraction and 72 percent for the 99 percent fraction.
Discussion of ProblemsDuring this brief validation study of the VOST Fractionator, four
issues or problems were identified.First, the VOST Fractionator was used
on several actual field samples, whichwere laden with moisture. This moisture,which was transferred through the VOSTFractionator and caused partial pluggingin the capillary tubing, was the likelycause of the change in split ratio. A 5-minnitrogen purge of the system after eachsplit had been completed appeared to re-move the moisture.
Second, the terpenes (a-pinene, p-pinene, and a-cymene), which have boil-ing points greater than 130°C, requiredspecial handling procedures to reduce sys-tem carryover. By raising transfer linetemperatures to 170°C, the analytical sys-tem carryover was minimized.
Third, the results for the water-solublecompounds (acetone, MEK, MIBK, acro-lein, and acrylonitrile) were not reproduc-ible at low concentrations. These con*-pounds were observed in the blanksamples, and were not as efficiently re-moved from the original sample traps asother compounds. The source of this con-tamination was tentatively identified asbeing the precleaned VOST traps.
Fourth, the use of two selected splitsurrogates allowed verification of flowratios; however, coelution with other tar-get analytes, such as 1,2-dichloropropane,
AIR & WASTE • Vol. 44 • January 1994 • 51

TECHNICAL PAPER
produced masses that interfered with quantitation. Investigation ofadditional surrogates could include other fluorinated congeners ofbenzene.
ConclusionsSeveral conclusions were made after reviewing the results of this
initial VOST Fractionator study: (1) the original sample amounts areefficiently recollected in the high-flow fraction (i.e., 99 percent),while a small amount of sample (i.e., 1 percent) is collected in thelow-flow fraction; (2) the original sample is quantitatively removedfrom the original VOST tube; (3) the split ratios can be monitored foreach new daughter sample, either by monitoring flow rates in eachbranch, or by using a surrogate compound added prior to samplesplitting; (4) the data are reproducible within method criteria; and(5) the original amounts can be saved in the high-flow fraction forrepeat analysis.
The final conclusion from this initial performance evaluation wasthat the VOST Fractionator does successfully split samples, basedprincipally on the fact that all VOCs except for the water solubles metthe SW-846 Method 5041 objectives. Additional validation is re-quired to test its ruggedness and performance.
Once the VOST Fractionator has been fully validated and ap-proved by EPA, the method should provide well-defined and accu-rate results for all target analytes, regardless of the wide range ofanalyte concentrations. The most important benefit would be theelimination of data losses, or resampling costs, due to unexpectedlyhigh levels of some compounds.
AcknowledgmentI would like to acknowledge Dennis Holzschuh and Gary
McAlister, both from the Emissions Monitoring Branch of the U.S.Environmental Protection Agency, for presenting the problem ofdetermining both low- and high-level volatile concentrations fromVOST traps. I would also like to acknowledge the support andassistance of Ms. Barbara Osterman, Ms. Melisande Skillicorn, andMs. April Carender.
References1. J. D. Evans, D. Halsell, and J. Hawkins, "Quality Assurance for
an Alternate Analytical Method for Highly Concentrated VOSTSamples," presented at 1992 U.S. Environmental ProtectionAgency/Air and Waste Management Association Air ToxicsSymposium.
2. U.S. Environmental Protection Agency, SW-846 Method 5041,Revision 0, November 1990.
3. Handbook, Quality Assurance/Quality Control (QA/QC)Procedures for Hazardous Waste Incineration, EPA/625/6-89/023, January 1990, Cincinnati, Ohio.
About the AuthorsM.E. Wickham St. Germain is a senior chemist in the Mass
Spectrometry Section at Midwest Research Institute, 425 VolkerBoulevard, Kansas City, M0 64110. She specializes in trace organicanalysis using GC/MS for environmental samples, QA/QC aspectsof GC/MS analyses, and data interpretation for multimedia samples.She has managed GC/MS work for several projects, includingvolatile organic analysis for incinerator tests and permits. Shereceived her B.S. degree in Chemistry in 1978 from St. MaryCollege, Leavenworth, Kansas. S.B. Cummins is an assistant massspectrometrist at Midwest Research Institute, specializing in vola-tile organic analysis using GC/MS. His activities have includeddesign, preparation, setup, and operation of source testing andambient sampling equipment. He received his B.S. in Agronomy in1980 from the University of Missouri-Columbia. G. Radolovich issection head of the Mass Spectrometry Section at Midwest Re-search Institute. He manages MRI's complex analytical massspectrometry laboratories and the teams of mass spectrometristsconducting numerous ongoing R&D programs. Mr. Radolovichreceived an M.S. in Mathematics (1969) and an M.S. in Physics(1973) from the University of Missouri-Kansas City. This manu-script was submitted for peer review on August 6, 1993. Therevised manuscript was received on November 1,1993.
52 • January 1994 • Vol. 44 • AIR & WASTE





























