Embed Size (px)
Citation preview

ON THE SURFACE CHEMISTRY OF SOME RHOMBOHEDRAL CARBONATE MINERALS
IN AQUEOUS SOLUTIONS
Adrián Villegas-Jiménez
Department of Earth & Planetary Sciences
McGill University, Montréal
October 2009
A thesis submitted to McGill University in partial fulfillment
of the requirements of the degree of Doctor of Philosophy
Adrián Villegas-Jiménez, 2009

ii

iii
ABSTRACT
Fundamental aspects of the surface chemistry of calcite, dolomite, magnesite, and
gaspeite in aqueous solutions were examined using different lines of investigation
including experimental, theoretical, and/or computer-assisted modeling approaches (i.e.,
ab initio molecular and surface complexation modeling).
A Genetic Algorithm (GA) was implemented and tested for the calibration of
surface complexation models (SCMs). The GA can successfully optimize numerous
adjustable SCM parameters without incurring convergence problems while minimizing
numerical instability problems, a notable advantage over conventional deterministic, root-
finding, and optimization techniques implemented in codes such as FITEQL. It was
routinely used throughout this thesis for the simultaneous calibration of surface
complexation parameters (e.g., intrinsic constants, capacitances) at carbonate surfaces.
The definition of reactive surface sites at hydrated rhombohedral carbonate
mineral surfaces was critically revisited. Using calcite as the model mineral, a single
generic charge-neutral surface site scheme was proposed for the formulation of surface
equilibria. The resulting molecular representation of surface equilibria is consistent with
experimental and theoretical findings and is compatible with assumptions implicit in
SCMs. Based upon the one-site scheme, new and simplified SCMs for magnesite and
dolomite were formulated. These successfully reproduced published surface charge and
electrokinetic data while yielding surface speciation predictions consistent with available
spectroscopic data.
The acid-base behavior of the gaspeite (NiCO3(s)) surface in NaCl solutions was
investigated for the first time by means of conventional titration techniques and micro-

iv
electrophoresis. Surface protonation and the electrophoretic mobility of gaspeite are
strongly affected by the background electrolyte. Acid-base surface complexation
reactions, formulated according to the one-site scheme, closely reproduced proton
adsorption data and reasonably simulated the electrokinetic behavior of gaspeite
suspensions at I ≤ 0.01 M.
The ground-state structural, energetic properties, and bonding relationships of the
hydrated (10.4) calcite surface were investigated using Roothaan-Hartree-Fock molecular
orbital methods and slab cluster models. A detailed 3D description of the hydrated calcite
surface, including the 1st and 2
nd hydration layers, was derived for the first time at the ab
initio level. Most noteworthy is the distortion of the Ca-O octahedra via the relaxation
and possible rupture of some Ca-O bonds upon hydration, leading to the weakening of the
outermost atomic calcite layer.
Finally, the quantitative characterization of the proton sorptive properties of
calcite in aqueous solutions by a novel surface titration protocol provides evidence for the
following ion-exchange equilibrium between the solution and labile exchangeable cation
sites (“exc”):
(CaCO3)2(exc) + 2 H+
Ca(HCO3)2(exc) + Ca2+
This proposed ion-exchange mechanism has far reaching implications as it directly
impacts the aqueous speciation of closed and partially open (poor CO2 ventilation)
carbonate-rock systems via the buffering of pH and calcite dissolution and CO2(g)
sequestration upon calcite precipitation.

v
RÉSUMÉ
Des aspects fondamentaux sur la chimie surfacique des minéraux carbonatés dans
des solutions aqueuses ont été examinés par des approches expérimentales et théoriques
ainsi que par des méthodes d‟optimisation numérique et de modélisation moléculaire.
Un algorithme génétique (GA, selon son sigle anglais) a été implémenté et testé
pour la calibration de modèles de complexation à la surface (SCMs, selon son sigle
anglais). Le GA peut optimiser de façon stochastique et simultanée des nombreux
paramètres tout en minimisant des problèmes de convergence ou de stabilité numérique.
Cet algorithme est très avantageux par rapport aux techniques déterministiques
conventionnelles adoptées par des codes d‟optimisation de constantes d‟équilibre tel que
FITEQL. Le GA a donc été utilisé de façon routinière dans cette étude, pour estimer les
constantes de formation des espèces chimiques se formant à la surface des minéraux
carbonatés.
En utilisant la calcite comme modèle, nous avons réévalué de façon critique la
définition de sites réactifs à la surface hydratée des minéraux carbonatés rhomboédriques.
Ceci nous a permis de définir un site d‟adsorption générique neutre pour ce type de
minéraux, qui est compatible avec les résultats d‟études théoriques et expérimentales ainsi
qu‟avec des hypothèses associées à la formulation de SCMs. Des nouvelles réactions,
basées sur un seul site générique, ont été formulées pour la magnésite et dolomite et
calibrées en utilisant des données publiées de charge surfacique et, par la suite, testées
avec des données électrocinétiques et spectroscopiques disponibles dans la littérature.
Le comportement acide-base à la surface de la gaspéite (NiCO3(s)), dans des
solutions de NaCl, à été examiné par des techniques conventionnelles de titrage

vi
surfacique et par la micro-électrophorèse. Nous avons trouvé que l‟électrolyte de support
influence, de façon substantielle, la protonation surfacique ainsi que la mobilité
électrocinétique de la gaspéite. Des réactions acide-base ont été formulées en fonction du
site d‟adsorption générique postulé dans cette étude. Celles-ci reproduisent bien les
données d‟adsorption de protons et simulent raisonnablement le comportement
électrocinétique aux forces ioniques ≤ 0.01 M.
Nous avons étudié les propriétés structurales et énergétiques à l‟état fondamental
de la surface (10.4) hydratée de la calcite ainsi que les types de liaisons établies entre les
molécules d‟eau et les atomes à la surface du minéral. À cette fin, nous avons appliqué
des méthodes basées sur la théorie quantique de l‟orbital moléculaire (Roothaan-Hartree-
Fock) en combinaison avec des modèles structuraux tridimensionnels (finis) de la calcite.
Nous proposons, par la première fois au niveau ab initio, un modèle structural détaillé de
la surface (10.4) hydratée de la calcite comprenant la première et la deuxième couche
d‟hydratation. Particulièrement remarquable est la distorsion significative des octaèdres
surfaciques de Ca-O suite à la relaxation (et possiblement rupture) de quelques liaisons
Ca-O. Ceci amène à l„affaiblissement de la couche atomique surfacique de la calcite.
Finalement, nous avons caractérisé de façon quantitative, les propriétés
d‟adsorption de protons par la calcite dans des solutions aqueuses en utilisant une
nouvelle technique de titrage surfacique développée dans la présente étude. Nous
proposons une réaction d‟échange d‟ions entre la solution et des sites cationiques
réactifs de caractère échangeable (“exc”):
(CaCO3)2(exc) + 2 H+
Ca(HCO3)2(exc) + Ca2+

vii
Ce mécanisme a des nombreuses répercussions significatives car il affecte la
spéciation en phase aqueuse des systèmes carbonatés qui sont isolés ou partiellement
isolés (faible ventilation de CO2(g)) de l‟atmosphère, via le tamponnage du pH et de la
dissolution de la calcite et par la séquestration du CO2(g) induite par la précipitation de la
calcite.

viii

ix
TABLE OF CONTENTS
Abstract iii
Résumé v
Acknowledgements xvii
Contribution of Authors xxii
Chapter 1: Introduction 1
REFERENCES 10
Preface to Chapter 2 14
Chapter 2: Estimating Intrinsic Formation Constants of Mineral Surface Species using a Genetic Algorithm 15
ABSTRACT 16
1. INTRODUCTION 18 2. IMPLEMENTATION OF THE GENETIC ALGORITHM 21
3. APPLICATION OF THE GA TO THE FORWARD PROBLEM 25
4. APPLICATION OF THE GA TO THE INVERSE PROBLEM 27
4.1 Estimation of Intrinsic Ionization Constants:
Constant Capacitance Model 27
4.2 Simultaneous Estimation of Intrinsic Ionization Constants
and Adsorption Constants: Constant Capacitance Model 33
4.3 Simultaneous Estimation of Intrinsic Ionization Constants and Adsorption Constants: Triple Layer Model 38
5. CONCLUSIONS 43

x
6. ACKNOWLEDGMENTS 44
7. REFERENCES 45
8. TABLES 51
Table 1 51
Table 2 52
9. FIGURES 53
Figure 1 53
Figure 2 54
Figure 3 55
Figure 4 56
Figure 5 57
Figure 6 58
Figure 7 59
Preface to Chapter 3 60
Chapter 3: Defining Reactive Sites on Hydrated Mineral Surfaces: Rhombohedral Carbonate Minerals 61
ABSTRACT 62
1. INTRODUCTION 64
2. DEFINITION OF PRIMARY SURFACE SITES 67
2.1 Charge Assignment 67
2.2 Elemental Stoichiometry 71
3. RHOMBOHEDRAL CARBONATE MINERALS 72
3.1 Case of the (10.4) Calcite Surface 72
3.1.1 Evidence from Spectroscopic and Molecular Modeling Studies 72
3.1.2 Single Generic Primary Surface Site 75

xi
3.2 SCM Reactions: One-Site vs Two-Site Scheme 77
3.3 Mixed-Metal Carbonate Minerals 79
4. EVALUATION OF THE ONE-SITE SCHEME 81
4.1 Re-calibration of Surface Reactions for Magnesite and Dolomite 81
4.2 Intrinsic Formation Constants and Surface Speciation 91
4.3 Comparison against Spectroscopic Information 93
5. CONCLUSIONS 96 6. ACKNOWLEDGMENTS 98
7. REFERENCES 99
8. TABLES 112
Table 1 112
Table 2 113
Table 3 114
Table 4 116
9. FIGURES 118
Figure 1 118
Figure 2 119
Figure 3 120
Figure 4 121
Figure 5 122
Figure 6 123
Preface to Chapter 4 124
Chapter 4: Acid-Base Behavior of the Gaspeite (NiCO3(s)) Surface in NaCl Solutions 125
ABSTRACT 126
1. INTRODUCTION 127
2. MATERIALS AND METHODS 130

xii
2.1 Preparation and Standardization of Reagents 130
2.2 Chemical Analysis 130
2.3 Gaspeite Synthesis 131
2.4 Surface Titrations 132
2.4.1. pH Electrode Calibration 132
2.4.2. Conditions of Surface Titrations 134
2.5 Computation of Proton Adsorption 136
2.6 Coagulation Experiments 138
2.7 Electrokinetic measurements 139
3. RESULTS AND DISCUSSION 141
3.1 Proton Adsorption on the Gaspeite Surface 141
3.1.1 Acidimetric Titrations 141
3.1.2 Verification of Potential Artifacts 145
3.1.3 Surface Complexation Modeling of Acidimetric Data: One-Site CCM approach 147 3.1.4 Surface Complexation Modeling of Acidimetric Data: One-Site, Multi-Site, BSM, and TLM approaches 153
3.1.5 Alkalimetric Titrations 154
3.2 Electrokinetics 157
4. CONCLUSIONS 160 5. ACKNOWLEDGMENTS 161
6. REFERENCES 162
7. TABLES 166
Table 1 166
Table 2 167
8. FIGURES 168
Figure 1 168
Figure 2 169
Figure 3 171
Figure 4 172

xiii
Figure 5 174
Figure 6 175
Figure 7 176
Figure 8 177
Figure 9 178
Preface to Chapter 5 180
Chapter 5: Theoretical Insights into the Hydrated (10.4) Calcite Surface: Structure, Energetics and Bonding Relationships 181
ABSTRACT 182
1. INTRODUCTION 184 2 METHODS 187
2.1 Computational Methods and Cluster Models 187 3 RESULTS 191
3.1 Structural Details of the Hydrated Clusters 191
3.2 Energies of Adsorption 196
3.3 H2O Interlayer Penetration 198
4 DISCUSSION 200
4.1 Reliability of RHF/6-31G(d,p) Results 200
4.2 Three-D Structural Registry 201
4.3 Bonding Relationships: Geometric and Energetic Criteria 206
5 CONCLUSIONS 214 6 ACKNOWLEDGMENTS 216 7 REFERENCES 217 8 TABLES 227
Table 1 227
Table 2 228

xiv
Table 3 229
9. FIGURES 230
Figure 1 230
Figure 2 231
Figure 3 232
Figure 4 233
Figure 5 234
Figure 6 236
Figure 7 237
Preface to Chapter 6 238
Chapter 6: Proton/Calcium Ion Exchange Behavior of Calcite 239
ABSTRACT 240
1. INTRODUCTION 242
2. MATERIALS AND METHODS 246
2.1 Principle of Calcite Titrations 246
2.2 Description of Reaction Vessel 247
2.3 Surface Titration Conditions 248
2.4 Computation of Sorption Data 250
3. RESULTS AND DISCUSSION 253
3.1 Qualitative Interpretation of Data 253
3.2 Possible Mechanisms of “Proton Uptake/Calcium Release” and “Apparent” Incongruent Calcite Dissolution 260
3.3 Sorption Modeling 264
3.4 Ion-Exchange vs Surface Equilibria 273
3.5 Implications of Proton/Calcium Ion Exchange 275
4. CONCLUSIONS 279

xv
5. ACKNOWLEDGMENTS 281
6. REFERENCES 282
7. TABLES 291
Table 1 291
Table 2 292
Table 3 293
8. FIGURES 294
Figure 1 294
Figure 2 295
Figure 3 296
Figure 4 297
Figure 5 299
Figure 6 300
Figure 7 301
Figure 8 303
Chapter 7: General Conclusions 305
CONTRIBUTIONS TO KNOWLEDGE 305
RECOMMENDATIONS FOR FUTURE RESEARCH 310
REFERENCES 314
Appendices: 340
I. Chapter 1: Gedanken Experiment Data 341
II. Chapter 4: Gaspeite: Acidimetric Titration Data 342
III. Chapter 4: Gaspeite: Alkalimetric Data 345
IV. Chapter 4: Gaspeite: Electrokinetic Data 346
V. Chapter 5: Optimized Small Calcite Cluster 348
VI. Chapter 5: Optimized Large Calcite Cluster 350
VII. Chapter 5: Geometrically-Optimized (CaCO3)9/4H2O cluster 354
VIII. Chapter 6: CaCO3(s) Solubility Product Data 355
IX. Chapter 6: CaCO3(s) Acidimetric Titration Data 356

xvi
X. Chapter 6: CaCO3(s) Calcium Titration Data 358
XI. Chapter 6: Methods and Calculations 359
XII. Chapter 6: Referencing of data to the ZNSRC 365
XIII. Chapter 6: Equilibrium Speciation Calculations involving Ion Exchange 367
XIV. Chapter 6: Tableau-based Formulation: CaCO3(s)-KCl-H2O System 369
XV. Chapters 2, 3, 4, and 6: Matlab© Subroutines 370

xvii
ACKNOWLEDGMENTS
I would like to express my gratitude to my Ph.D. thesis supervisor, Professor Alfonso
Mucci, for giving me the freedom to thoroughly propose, design, and conduct my
doctoral research while according me full intellectual independence to elaborate and test
my scientific hypotheses and formulate my own conclusions. His comments and
constructive criticisms as well as his guidance in anglicizing my prose have undoubtedly
added substantial value to my thesis and are sincerely acknowledged. The participation of
Professor Jeanne Paquette in commenting on crystallographic and editorial aspects of my
thesis and her guidance during my early training on carbonate crystallography is greatly
appreciated.
I acknowledge the hospitality of Dr Oleg S. Pokrovsky and Dr Jacques Schott
during my 6-month visit to their lab (LMTG-CNRS) in Toulouse, France in 2003. Their
scientific contributions to my doctoral work were critical and their financial contribution
during my stay in France is appreciated. I also recognize the invaluable technical
assistance offered by the extremely competent laboratory staff at LMTG and most
particularly by Madame Carole Causserand.
I sincerely thank Emeritus Professor Michael A. Whitehead who granted me
access to his computer facilities and provided guidance over the course of the molecular
modeling work I conducted in his laboratory. I also appreciate the two-month stipend he
provided me with in 2006.
Special recognition goes to Professor Theo van de Ven, Professor David Burns,
and Dr Luuk Koopal for critical and inspiring discussions at early stages of my Ph.D.
residency, which led to significant improvements of my research work. I would also like

xviii
to express my sincere appreciation to Dr Johannes Lützenkirchen for critically reviewing
my thesis and making important remarks on my work.
Thanks also to Dr Nora de Leeuw, Dr Paul Fenter, Dr Kate Wright, Dr Brian L.
Phillips, and Dr Michel J. Rossi who kindly provided additional information on their
published work.
Many thanks to all the staff in the Department of Earth and Planetary Sciences
who kindly offered assistance of various kinds at different stages of my research work. I
am particularly grateful to Brigitte Dionne for her guidance in computer-related issues as
well as to Glenna Keating, Sandra Lalli, and Constance Guignard for technical assistance
in laboratory analyses as well as to Carol Matthews, Kristy Thornton, and Anne
Kosowski for advice and help regarding administrative and academic issues. Critical
advice on laboratory analyses provided by Professor Tariq Ahmedali is truly appreciated.
I am sincerely grateful to Professor Hojatollah Vali for allocating me suitable
office space for nearly two years following the temporary closure of our office/laboratory
facilities in 2005. Special thanks go to Professor Theo van de Ven from the Chemistry
Department who temporarily guaranteed my supply of Milli-Q® water when it was
compromised.
I am also indebted to numerous scientists and professors who, from my early B.Sc.
years in Ensenada (Mexico) to present, have provided me with guidance, encouragement,
or simply with pure scientific inspiration. It is impossible to do justice and accord proper
recognition to all those responsible for triggering my scientific motivations and/or
participating in my training as a science professional. Particularly, wise supervision and
solid guidance from Professor André Tessier and Dr José Vinicio Macías-Zamora during
my M.Sc. and B.Sc. studies, respectively, paved a smooth way towards my Ph.D. studies.

xix
My Ph.D. research was supported financially by a Graduate Student Research
Grant to A. Villegas-Jiménez from the Geological Society of America (GSA), by Natural
Sciences and Engineering Research Council of Canada (NSERC) Discovery grants to A.
Mucci, J. Paquette, and M. A. Whitehead, and by grants from the Centre National de la
Recherche Scientifique (CNRS) to J. Schott. In addition, the following institutions are
deeply acknowledged for kindly awarding me financial support either through fellowships
or by offering “on-campus” work during my Ph.D. residency at McGill:
Consejo Nacional de Ciencia y Tecnología of Mexico (CONACyT):
Excellence doctoral scholarships awarded from 2001 to 2004 (inclusive).
The GEOTOP-UQAM-McGill Research Center: Summer doctoral bursary
(2002).
McGill University: Overseas Alma Mater Student Travel Grants to attend
“The Goldschmidt Conference” held in Davos (2002) and Copenhagen (2004).
The Organizing Committees of the 2004 and 2008 “Goldschmidt Conference”
for providing partial financial support to attend their meetings held in
Copenhagen and Vancouver, respectively.
The National Science Foundation (NSF) for fully supporting my attendance to
the Water-Rock Interactions Symposium held in Saratoga in 2004.
The Department of Earth and Planetary Sciences at McGill University for
providing me with Teaching Assistantships over several years (2001-2005).
The Faculties of Science and Engineering at McGill University for offering me
invigilation work during several examination periods (2002, 2005, and 2006).
In addition, the summer research assistantships (2001-2004) and partial financial
support (2005-2006) offered by my thesis supervisor are sincerely appreciated.
The financial support I received from my parents and my two brothers, Armando
and Omar, from 2006 to 2008 was critical to bring this thesis to a successful end.

xx
This thesis is dedicated to my family, but most particularly to my dearest parents,
Rosa Elvia Jiménez-Rodríguez and Armando Villegas-Bobadilla to whom I am deeply
grateful for their unconditional support, rock-solid encouragement, and sincere
understanding during this rather challenging time of my life. Little doubt remains… they
are the best.
A special dedication goes also to all those fine scientists who, through sound
intuition, solid evidence, vigorous thinking, and pragmatic interpretations, attach
authority to scientific knowledge and do justice to what is known as: “La Force de la
Science”.
Adriano

xxi
Impose ta chance, serre ton bonheur,
et va vers ton risque. À te regarder, ils s’habitueront
René Char (1950)

xxii
CONTRIBUTION OF AUTHORS
This thesis is the outgrowth of the author‟s Ph.D. research work in the Department of
Earth and Planetary Sciences at McGill University under the supervision of Professor
Alfonso Mucci and co-supervision of Professor Jeanne Paquette. The thesis consists of
seven chapters, five of which are scientific research manuscripts whereas the remaining
two are the general introduction and conclusions. Chapter 2 was accepted for publication
by the scientific journal Mathematical Geology and currently awaits publication, Chapter
3 was published in the scientific journal Geochimica et Cosmochimica Acta, Chapter 4
will be submitted to the scientific journal Langmuir, Chapter 5 was published in the
scientific journal Langmuir. Finally, Chapter 6 was published in the scientific journal
Physical Chemistry Chemical Physics. In conformity with the format of the published
articles, all relevant supplementary material associated with each Chapter (e.g., raw data,
computer subroutines, detailed explanations, etc.) can be found in the appendices to this
thesis. With exception of Chapter 4, specifically the gaspeite titration experiment, which
was originally proposed to the author by Dr Oleg S. Pokrovsky and Dr Jacques Schott,
researchers of LMTG, UMR 5563, Université Paul-Sabatier - CNRS in Toulouse, France;
the research presented in this thesis was fully proposed by the author and initiated after
discussions with the author‟s thesis supervisor, Professor Alfonso Mucci and co-
supervisor, Professor Jeanne Paquette.
Theoretical, experimental, analytical, ab initio molecular modeling, Matlab©
computer coding, computer-assisted numerical optimization work, data acquisition as
well as interpretation, speciation calculations, and experimental protocols were entirely
designed and/or carried out by the author. Consequently, the author is responsible for the
content of the thesis and is the lead author of the five associated manuscripts. Professor

xxiii
Alfonso Mucci commented on data evaluation and interpretation, and critically reviewed
the scientific contents and style of all the material presented in this thesis, and therefore,
he co-authors the five associated manuscripts. Professor Jeanne Paquette is the fifth co-
author of Chapter 4 and third of Chapter 6. She commented on the scientific contents and
style of these manuscripts and provided references that helped improve their quality.
Dr Oleg S. Pokrovsky is the third co-author of Chapters 3 and 4. His contributions
to this work include guidance during my laboratory studies conducted at LMTG, UMR
5563, Université Paul-Sabatier - CNRS in France. He also provided constructive
comments, criticisms, and suggestions on the interpretation of experimental data and
modeling results as well as critically reviewed the scientific content and style of the two
associated manuscripts. In addition, he provided novel (unpublished) electrokinetic data
for NiCO3(s) (electrophoretic measurements, series-II) used in Chapter 4 to further
validate the Surface Complexation Model postulated for this mineral. Dr Jacques Schott
is the fourth co-author of Chapters 3 and 4. He provided constructive comments,
criticisms, and suggestions on the interpretation of experimental data and modeling
results as well as critically reviewed the scientific content and style of these two
manuscripts.
Finally, Emeritus Professor Michael Anthony Whitehead is the third co-author of
Chapter 5. He provided guidance on the molecular modeling work I conducted in his
laboratory in the Department of Chemistry at McGill University. He also provided
constructive comments and suggestions on the interpretation of the molecular modeling
results and critically reviewed the scientific content and style of Chapter 5.


1
CHAPTER 1
INTRODUCTION
Under Earth surface conditions, carbonate minerals are among the most chemically
reactive and ubiquitous minerals in the environment. They are found as suspended
particles in aquatic systems (Morse and Mackenzie, 1990) and the atmosphere (Usher et
al., 2003) and as part of the sediment and rock record (Morse et al., 2007). Calcite
(CaCO3(s)) and dolomite (CaMg(CO3)2(s)) are by far the most abundant carbonate
minerals, comprising nearly 20% by volume of Phanerozoic sedimentary rocks. In
modern sediments, aragonite and high-magnesian calcites dominate in shallow water
environments whereas low magnesium calcite (> 99% CaCO3(s)) composes almost all
deep sea carbonate-rich sediments (Morse et al., 2007). These minerals largely impact the
chemistry of aquatic systems by regulating pH and alkalinity through
dissolution/precipitation equilibria, govern the mobility and cycling of hazardous metal
contaminants and radionuclides via ion exchange, adsorption, and co-precipitation
reactions, as well as participate in the long-term biogeochemical cycling of major
elements (Van Cappellen et al., 1993). For instance, CaCO3(s) minerals represent an
important component of the inorganic carbon budget in the ocean where the balance
between continental weathering and biogenic precipitation of calcium carbonates
influence the global carbon cycle (Sarmiento and Sundquist, 1992). CaCO3(s) polymorphs
are also the building blocks of shells and skeletons of various marine invertebrates
(Morse et al., 2007) whereas, in the Earth‟s atmosphere, they constitute a reactive
component of mineral aerosols that regulate the CO2 exchange and influence the

2
chemistry of volatile inorganic and organic acids (Usher et al., 2003; Al-Hosney and
Grassian, 2005). CaCO3(s) polymorphs also have numerous industrial applications that
range from fillers for paints, plastics, rubbers, pharmaceuticals, cosmetics, optical
devices, and paper to raw material in the construction industry, agriculture, as well as in
the production of biomedical scaffolds (e.g., Vanerek et al., 2000 and Tas, 2007).
Given their environmental significance and broad industrial applications,
carbonate minerals have been the subject of extensive research in numerous experimental
and theoretical investigations. It is now well recognized that fundamental reactions at the
carbonate/water interface such as hydration, ion sorption, and development of surface
charge, play a critical role on macroscopic processes such as carbonate mineral
dissolution and growth kinetics, crystal morphology, pathways of carbonate diagenesis,
and particle coagulation (Brady et al., 1996). This realization has stimulated interest about
the surface reactivity of carbonate minerals in aqueous solutions. Accordingly, over the
last few decades, considerable efforts have focused on the experimental characterization
of the ion sorptive properties of carbonate minerals and the derivation of empirical and
semi-empirical relationships to quantitatively interpret ion partitioning between the
aqueous phase and the surface of calcite, aragonite, Mg-bearing carbonates and, to a
lesser extent, other divalent carbonate minerals (Morse and Mackenzie, 1990).
The greater reactivities (i.e., faster reaction rates and larger solubilities) of
carbonates relative to other minerals such as metal oxides, silicates, and clays and the
occurrence of stepwise and/or parallel reactions (e.g., adsorption, surface precipitation,
co-precipitation, dissolution) have made it difficult to experimentally resolve adsorption
processes (Morse, 1986). Furthermore, the interpretation of adsorption data is often
problematic as they may reflect the product of several overlapping reactions. In fact, these

3
data have most commonly been interpreted as a fast initial adsorption and subsequent
slow lattice incorporation (precipitation) of the adsorbate (e.g., Franklin and Morse, 1983;
Davis et al, 1987; Pingitore et al, 1988; Zachara et al, 1991; Tesoriero and Pankow,
1996). These two steps were further decomposed into: 1) diffusion into a hydrated
surface layer (Davis et al., 1987); 2) dehydration and formation of MeCO3 bonds on the
surface (Franklin and Morse, 1983); 3) nucleation (McBride, 1979), and the ultimate
precipitation of a solid solution layer (Lorens, 1981; Davis et al, 1987) or of a pure phase
(McBride, 1979). In addition, it has been suggested that solid-state ion diffusion may
affect the rate and extent of trace metal sorption by calcite (Stipp et al., 1992).
These findings reflect the complexity of ion sorption processes on carbonate
mineral surfaces and explains why carbonate experimentalists must conduct their
adsorption studies within relatively narrow ranges of chemical conditions (e.g., pH,
sorbate/adsorbant ratio) or employ surface-sensitive techniques (e.g., X-ray, electron
diffraction, spectroscopy, chromatography, thermogravimetry, atomic force microscopy)
to characterize the surface structure and obtain quantitative insights on the reactivity of
carbonate mineral surfaces. Nevertheless, despite these efforts, critical aspects on the
surface reactivity of carbonate minerals in aqueous solutions are still not fully understood.
For instance, the nature of the surface reactions that control the (ad)sorption behavior of
potential-determining ions such as H+, OH
-, Ca
2+, CO3
2-, and/or HCO3
- remain
controversial and subject of scientific debate. Consequently, factors that determine the pH
of isoelectric point (pHIEP) of calcite remain ambiguous (e.g., Prédali and Cases, 1973;
Foxall et al., 1979; Cicerone et al., 1992; Moulin and Roques, 2003).
It follows that the design and implementation of experimental approaches for the
rigorous evaluation of adsorption equilibria over expanded ranges of chemical conditions

4
is required. Conventional titration techniques, used in the characterization of the surface
properties of less reactive minerals such as metal oxides or clays (Huang, 1981), are not
suitable for the characterization of highly reactive carbonate minerals that rapidly
respond, via dissolution/precipitation reactions, to minute variations in the solution
chemistry. These considerations drove earlier workers to develop a novel experimental
protocol, based on the use of a fast flow-through reactor, to minimize the contribution of
dissolution and precipitation during acid-base titrations performed on two sparingly
soluble carbonates: siderite and rhodochrosite (Charlet et al., 1990). This protocol was
later used by several researchers to obtain surface charge data for siderite, rhodochrosite
(Van Cappellen et al., 1993), magnesite (Pokrovsky et al., 1999a), and dolomite
(Pokrovsky et al., 1999b; Brady et al., 1999) from which they formulated surface
complexation models (SCMs) for these minerals. Unfortunately, the application of this
approach to highly reactive carbonate minerals such as calcite or aragonite is not feasible
because their fast dissolution kinetics interferes significantly with the computation of
surface charge. Consequently, available SCMs for calcite (Van Cappellen et al., 1993)
were calibrated either to the “generally accepted” (yet ambiguous, given the strong
solution composition-dependency of this parameter) pH of isoelectric point of calcite
recorded under specific solution conditions (pHIEP = 8.2, Mishra, 1978) or against
selected electrokinetic data available in the literature (Wolthers et al., 2008).
Nevertheless, the latter authors concluded that a straightforward validation of the
postulated SCMs was not possible because of the uncertainties associated with the nature
and magnitude of potential artifacts inherent in the electrokinetic data obtained in calcite
suspensions.

5
Despite the success of these SCMs in reproducing the surface charge of FeCO3(s),
MnCO3(s), MgCO3(s), and CaMg(CO3)2(s) (Van Cappellen et al., 1993; Pokrovsky et al.,
1999a,b) and reasonably predicting the electrokinetic behavior of MgCO3(s) and
CaMg(CO3)2(s) (Pokrovsky et al., 1999a,b) in aqueous solutions, the postulated models are
not robust and represent first-order descriptions of the surface chemistry of carbonate
minerals that are amenable to refinement from a theoretical and experimental standpoint.
For instance, in all these studies, the formation constants of surface species were adjusted
simultaneously on a trial and error basis (by arbitrarily varying the values of the
formation constants) until the predicted surface speciation closely reproduced surface
charge data. Hence, the contribution of individual surface reactions (i.e., acid-base and
lattice-derived, constituent, ion adsorption) could not be resolved nor could the formation
constants of surface species be estimated accurately.
Another critical issue is the definition of the reactive sites whereupon surface
reactions are formalized. Based upon spectroscopic evidence (Stipp and Hochella, 1991;
Pokrovsky et al., 1999a; 1999b), two types of vicinal surface hydration sites were
hypothesized to form at the (10.4) surface of rhombohedral carbonate minerals (MeOH0
and CO3H0) and these were assumed to display a distinct reactivity that remained
unaffected by the presence of reacted neighbouring surface species. This scheme yields
complex SCMs defined by at least six (for single-metal carbonate minerals) or twelve (for
mixed-metal carbonate minerals) surface reactions that spawn questionable predictions of
surface speciation which, in turn, may not reflect realistic processes at the
carbonate/water interface. Clearly, to improve our understanding of carbonate surface
reactivity in aqueous solutions we need to: (i) generate representative experimental

6
adsorption data covering wide compositional ranges, (ii) revisit and refine our
quantitative interpretations of old and new data using chemically-sound and
mathematically-tractable ion partitioning models, (iii) test the validity of these models
against additional experimental data acquired by alternate investigative approaches and/or
under conditions beyond the calibration range, (iv) critically evaluate the adequacy of
available experimental and theoretical information to elucidate surface processes at the
carbonate/water interface and, (v) select suitable ion partitioning models for this type of
minerals that reflect an acceptable compromise between the quality of the experimental
data available for model calibration, the compatibility of such model with
physical/chemical constraints, the accuracy of the model predictions, and their
applicability to real-world systems. Some of these issues are addressed in this thesis.
The objectives of this thesis are: (i) to derive a realistic description of the
ionization and lattice ion surface species at the carbonate-water interface by critically
revisiting the definition of primary surface sites (“adsorption centres”) whereupon
mass-action expressions describing adsorption equilibria at hydrated (10.4)
rhombohedral carbonate mineral surfaces are formalized; (ii) to use this description
for the reformulation and calibration of SCMs for magnesite and dolomite, evaluate
their predictive power against published electrokinetic and spectroscopic data for
these two minerals, and compare their results against those of previous SCMs; (iii) to
use gaspeite (NiCO3(s)) as a surrogate carbonate mineral to investigate the acid-base
behavior of rhombohedral carbonate minerals by application of conventional titration
and electrokinetic techniques and interpret proton adsorption data within the
framework of surface complexation theory; (iv) to investigate the structure and
energetics of the 1st and 2
nd hydration layers at the cleavage (10.4) calcite surface

7
using ab initio Roothan-Hartree-Fock molecular orbital techniques and analyze the
bonding relationships between adsorbing water molecules and surface atoms; and (v)
to examine the proton sorptive properties of calcite over a relatively wide range of
chemical conditions using a novel titration protocol.
The contributions of this dissertation include, but are not limited to: (1) the
implementation of a Genetic Algorithm that allows the simultaneous optimization of
numerous adjustable parameters (i.e., intrinsic formation constants of surface species,
capacitances, site densities) for the successful calibration of SCMs; (2) a refined and
simplified formulation of surface equilibria at rhombohedral (10.4) surfaces based upon a
generic single reactive hydration site which reconciles available experimental and
theoretical information and allows reasonable surface speciation predictions; (3) the
quantitative characterization of the acid-base surface properties of gaspeite at different
ionic strengths, the discovery of the important role exerted by the background electrolyte
on the protonation and charge acquisition of the gaspeite surface, and the derivation of
reasonable SCM predictors for the simulation of surface protonation, surface charge, and
electrokinetic behavior of gaspeite at ionic strength ≤ 0.01 M; (4) an improvement of our
theoretical understanding of the structure, energetic, and bonding relationships of H2O
molecules with the (10.4) surface; most noteworthy is the fact that we obtained evidence
of the significant weakening of the outermost calcite layer upon hydration-induced
relaxation, and possible rupture, of surface Ca-O bonds (a process never postulated
before); and (5) a rigorous quantitative characterization of the proton sorptive properties
of calcite in aqueous solutions that strongly suggests the existence of a previously
unreported proton/calcium ion exchange mechanism which, in turn, may have far-
reaching implications on the control of aqueous speciation of carbonate-rock aquatic

8
environments with null (closed system) or restricted (pseudo-closed system) CO2(g)
ventilation.
Chapter 2 investigates the application of a powerful evolutionary optimization
technique, the Genetic Algorithm (GA), to estimate the intrinsic formation constants of
mineral surface species under various scenarios and SCMs. Given the power of the GA
for the simultaneous optimization of numerous adjustable parameters, it was routinely
used throughout this thesis for the calibration of surface complexation reactions at
carbonate surfaces. It was particularly useful for the calibration of multiple surface
complexation reactions for magnesite and dolomite presented in Chapter 3 where we
critically revisit the definition of reactive surface sites at hydrated rhombohedral
carbonate mineral surfaces. In Chapter 3, the formulation of surface reactions for
rhombohedral carbonate minerals based upon a single charge-neutral generic surface site,
(MeCO3)·H2O0, is derived (i.e., one-site binding scheme). Accordingly, new and
simplified SCMs for magnesite and dolomite were formulated, calibrated using published
surface charge data, and qualitatively tested against earlier electrokinetic data acquired
over a wide range of chemical conditions. Available spectroscopic evidence served to
further confirm the viability of the SCMs postulated for these minerals.
Chapter 4 examines the acid-base surface properties of the least reactive of known
naturally-occurring rhombohedral carbonate minerals, gaspeite (NiCO3(s)), in NaCl
solutions by means of conventional titration techniques and micro-electrophoresis. The
acquired proton adsorption data at I ≤ 0.01 M are suitable for the calibration of surface
complexation reactions formulated within the one-site binding scheme (described in
Chapter 3) and can reasonably simulate the electrophoretic mobility of gaspeite

9
suspensions under conditions similar to those from which the data, used for calibrating
the SCM, were acquired.
Chapters 5 and 6 focus on the surface chemistry of calcite, a very reactive
naturally-occurring rhombohedral carbonate mineral, in aqueous solution. Chapter 5
investigates the ground-state structural and energetic properties, and bonding
relationships of the hydrated (10.4) calcite surface using ab initio molecular orbital
techniques. Results of this study are compatible with the generalized one-site scheme
formulated in Chapter 3 for rhombohedral carbonate mineral surfaces. They also reveal
the weakening of the outermost atomic calcite layer following the substantial relaxation
and possible rupture of some Ca-O bonds upon hydration. Chapter 6 evaluates the proton
sorptive properties of calcite in aqueous solutions using a novel surface titration technique
and provides reliable sorption data that substantiate the following ion-exchange
equilibrium:
(CaCO3(exc))2 + 2 H+
Ca(HCO3)2(exc) + Ca2+
(1)
According to our data interpretation, the postulated mechanism possibly masks
other proton and/or calcium ion sorption reactions at the calcite surface and, under certain
chemical scenarios, may lead to a net sequestration of CO2(aq) upon enhanced calcite
precipitation. Finally, a brief discussion on the role exerted by proton/calcium ion
exchange in determining the aqueous speciation of aquatic environments exhibiting poor
CO(2)(g) ventilation concludes Chapter 6.

10
REFERENCES
Al-Hosney H.A. and Grassian V.H. (2005). Water, sulfur, dioxide and nitric acid
adsorption on calcium carbonate: A transmission and ATR-FTIR study. Phys.
Chem. Chem. Phys. 7, 1266-1276.
Brady P.V., Krumhans J.L. and Papenguth, H.W. (1996) Surface complexation clues to
dolomite growth. Geochim. Cosmochim. Acta 60(4), 727-731.
Brady P.V., Papenguth H.W., Kelly J.W. (1999). Metal sorption to dolomite surfaces.
Applied Geochem. 14, 569-579.
Charlet L., Wersin P. and Stumm W. (1990) Surface charge of MnCO3 and FeCO3.
Geochim Cosmochim. Acta. 54, 2329-2336.
Cicerone D.S. Regazzoni A.E. and Blesa M.A. (1992) Electrokinetic properties of the
calcite/water interface in the presence of magnesium and organic matter. J.
Colloid Interface Sci. 154, 423-433.
Davis J.A. Fuller C.C. and Cook A.D. (1987) A model for trace metal sorption processes
at the calcite surface: Adsorption of Cd2+
and subsequent solid solution formation.
Geochim. Cosmochim. Acta 51(6), 1477-1490.
Foxall T. Peterson G.C., Rendall H.M. and Smith A.L. (1979) Charge determination at
calcium salt/aqueous solution interface. J. Chem. Soc. Farad. Trans. 175, 1034-
1039.
Franklin M.L. and Morse J.W. (1983) The interaction of manganese (II) with the surface
of calcite in dilute solutions and seawater. Mar. Chem., 12(4), 241-254

11
Huang C.P. (1981). The surface acidity of hydrous solids in: Adsorption of Inorganics at
Solid-Liquid Interfaces. M.A. Anderson and A.J. Rubin (eds.) Ann Arbor Science,
Ann Arbor, Mich., pp. 183-217.
Lorens R.B. (1981) Sr, Cd, Mn and Co distribution coefficients in calcite as a function of
calcite precipitation rate. Geochim. Cosmochim. Acta 45, 553–561.
McBride M.B. (1979) Chemisorption and precipitation of Mn2+
at CaCO3 surfaces. Soil
Sci. Soc. Am. J. 43, 693–698.
Mishra S.K. (1978) The electrokinetics of apatite and calcite in inorganic electrolyte
environment. Int. J. Miner. Process. 5, 69-83.
Morse J.W. (1986) The surface chemistry of calcium carbonate minerals in natural
waters: An overview. Mar. Chem. 20, 91-112.
Morse J.W. and Mackenzie F.T. (1990) Geochemistry of Sedimentary Carbonates;
Develop. Sedimentol., 48. Elsevier: Amsterdam, 724 p.
Morse J.W., Arvidson R.S. and Lüttge A. (2007) Calcium carbonate formation and
dissolution. Chem. Rev. 2007, 107, 342-381.
Moulin P. and Roques H. (2003) Zeta potential measurement of calcium carbonate. J.
Colloid. Inter. Sci. 261, 115-126.
Pingitore N.E. Jr., Eastman M.P., Sandidge M., Oden K. and Freiha B. (1988) The
coprecipitation of manganese (II) with calcite: an experimental study. Mar. Chem.
25(2), 107-120.

12
Pokrovsky O.S., Schott J. and Thomas F. (1999a) Processes at the magnesium-bearing
carbonates/solution interface. I. A surface speciation model for magnesite.
Geochim. Cosmochim. Acta 63(6), 863-880.
Pokrovsky O.S., Schott J. and Thomas F. (1999b) Dolomite surface speciation and
reactivity in aquatic systems. Geochim. Cosmochim. Acta. 63(19/20), 3133-3143.
Prédali J.-J. and Cases J.-M.J. (1973) Zeta potential of magnesian carbonates in inorganic
electrolytes. J. Colloid Interface Sci. 45(3), 449-458.
Sarmiento J.L. and Sundquist E.T. (1992) Revised budget for the oceanic uptake for
anthropogenic carbon dioxide. Nature 356, 589-593.
Stipp S.L. and Hochella M.F. Jr. (1991) Structure and bonding environments at the calcite
surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy
electron diffraction (LEED). Geochim. Cosmochim. Acta 55, 1723-1736.
Stipp S.L.S., Hochella, F., Parks, G.A. and Leckie J.O. (1992) Cd2+
uptake by calcite,
solid-state diffusion, and the formation of solid-solution: Interface processes
observed with near-surface sensitive techniques (XPS, LEED, and AES).
Geochim. Cosmochim. Acta 56, 1941-1954.
Tas A.C., (2007) Porous, biphasic CaCO3-calcium phosphate biomedical cement
scaffolds from calcite (CaCO3) powder. Int. J. Appl. Ceram. Technol. 4(2), 152-
163.
Tesoriero A. and Pankow J. (1996) Solid solution partitioning of Sr2+
, Ba2+
, and Cd2+
to
calcite. Geochim. Cosmochim. Acta. 60(6), 1053-1063.
Usher C.R., Michel A.E. and Grassian V.H. (2003) Reactions on mineral dust. Chem.
Rev. 103, 4883-4939.

13
Van Cappellen P., Charlet L., Stumm W. and Wersin P. (1993) A surface complexation
model of the carbonate mineral-aqueous solution interface. Geochim. Cosmochim.
Acta 57, 3505-3518.
Vanerek A., Alince B. and van de Ven T.G.M. (2000) Interaction of calcium carbonate
fillers with pulp fibres: Effect of surface charge and cationic polyelectrolytes. J.
Pulp Paper Sci. 26(9), 317-322.
Wolthers M., Charlet L., and Van Cappellen P. (2008) The surface chemistry of divalent
metal carbonate minerals; a critical assessment of surface charge and potential
data using the charge distribution multi-site ion complexation model. Am. J. Sci.,
308, 905-941.
Zachara J.M., Cowan C.E. and Resch C.T. (1991) Sorption of divalent metals on calcite.
Geochim. Cosmochim. Acta 55, 1549-1562.

14
PREFACE TO CHAPTER 2
Derivative-based and simple hill-climbing root-finding numerical techniques such as the
Newton-Raphson approach, frequently implemented in forward and inverse modeling
chemical equilibrium codes such as MINEQL+, HYDRAQL, PHREEQC, MINTEQA2,
and FITEQL, are local in scope and are sometimes plagued by numerical convergence
problems that, in the best case scenario, require the implementation of back-substitution
algorithms for the adequate initialization of the iterative process. For example, FITEQL, a
derivative-based non-linear least squares optimization routine, may face convergence
problems when numerous parameters are adjusted or when extensive data sets are not
available. It follows that an alternative tool that can circumvent these limitations and
allow the simultaneous optimization of the numerous adjustable parameters implicit to
some Surface Complexation Models (SCMs) would be desirable.
This issue is addressed in the following chapter, “Estimating intrinsic formation
constants of mineral surface species using a genetic algorithm”, where we introduce and
evaluate the applicability of a powerful evolutionary programming technique, the Genetic
Algorithm (GA), for the determination of intrinsic equilibrium constants of geologically-
relevant reactions at mineral surfaces under scenarios of varying complexity. This
includes cases where FITEQL fails to converge or yields poor data fits upon convergence.
As shown in Chapter 3, the implementation of the GA approach served to
calibrate, via numerical optimization, the SCMs for magnesite and dolomite, a task that
had not been carried out before due to the lack of a suitable optimization tool for this type
of data. In addition, the GA approach is used in Chapter 4 to calibrate surface
complexation reactions describing the acid-base behavior of the gaspeite surface.

15
CHAPTER 2
ESTIMATING INTRINSIC FORMATION CONSTANTS OF MINERAL SURFACE SPECIES USING A GENETIC ALGORITHM
Adrián Villegas-Jiménez*1 and Alfonso Mucci
1
1 Earth and Planetary Sciences, McGill University, 3450 University Street
Montréal, Qc H3A 2A7, Canada.
*Corresponding Author:
E-mail: [email protected]
Accepted for publication by Mathematical Geosciences

16
ABSTRACT
The application of a powerful evolutionary optimization technique for the estimation of
intrinsic formation constants describing geologically-relevant adsorption reactions at
mineral surfaces is introduced.
We illustrate the optimization power of a simple Genetic Algorithm (GA) for
forward (aqueous chemical speciation calculations) and inverse (calibration of Surface
Complexation Models, SCMs) geochemical modeling problems of varying degrees of
complexity, including problems where conventional deterministic derivative-based root-
finding techniques such as Newton-Raphson, implemented in popular programs such as
FITEQL, fail to converge or incur notable numerical instability problems.
Subject to sound a priori physical-chemical constraints, adequate solution
encoding schemes, and simple GA operators, the GA conducts an exhaustive probabilistic
search in a broad solution space and finds a suitable solution regardless of the input
values and without requiring sophisticated GA implementations (e.g., advanced GA
operators, parallel genetic programming). The drawback of the GA approach is the large
number of iterations that must be performed to obtain a satisfactory solution.
Nevertheless, for computationally-demanding problems, the efficiency of the
optimization can be greatly improved by combining heuristic GA optimization with the
Newton-Raphson approach to exploit the power of deterministic techniques after the
evolutionary-driven set of potential solutions has reached a suitable level of numerical
viability.
Despite the computational requirements of the GA, its robustness, flexibility, and
simplicity make it a very powerful, alternative tool for the calibration of SCMs, a critical
step in the generation of a reliable thermodynamic database describing adsorption

17
reactions. This aspect is key in the forward modeling of the adsorption behavior of
minerals and geologically-based adsorbents in hydro-geological settings (e.g., aquifers,
pore waters, water basins) and/or in engineered reactors (e.g., mining, hazardous waste
disposal industries).
Keywords: Evolutionary programming, heuristic optimization, surface complexation
modeling and calibration, inverse modeling.

18
1. INTRODUCTION
The quantitative characterization of the sorptive properties of minerals and geologically-
based adsorbents is key to the understanding of natural geochemical processes (e.g.,
solute mobility/sequestration) in hydro-geological settings (e.g., aquifers, pore waters,
water basins) and to the optimization of multiple engineering processes (e.g., mining,
hazardous waste disposal) and wastewater treatment technologies. Among the approaches
devised for the quantitative description of adsorption equilibria (e.g., isotherm equations,
partition coefficients), Surface Complexation Models (SCMs) represent, at present, the
most geochemically-sound and powerful theoretical framework for the prediction of
adsorption equilibria. Detailed descriptions of SCMs can be found in most modern
aquatic chemistry/geochemistry textbooks (e.g., Morel and Hering, 1993; Stumm and
Morgan, 1996; Langmuir, 1997).
In the last few decades, a number of computer programs have been developed and
successfully validated to perform SCM-based routine calculations of adsorption equilibria
in heterogeneous systems involving aqueous and adsorbent phases (forward modeling):
MICROQL II (Westall, 1979), WATEQ (Ball et al., 1981), HYDRAQL (Papelis et al.,
1988), SOILCHEM (Sposito and Coves, 1988), MINTEQA2/PRODEFA2 (Allison et al.,
1991), MINEQL+ (Schecher and McAvoy, 1992), EQ3NR (Wolery, 1992), WHAM
(Tipping, 1994), PHREEQC (Parkhurst, 1995), GEOSURF (Sahai and Sverjensky, 1998),
CHESS (van der Lee and de Windt, 1999), and ECOSAT (Keizer and van Riemsdijk,
1999).
In general, the solution to adsorption equilibrium problems can be achieved by
two equivalent approaches: i) the Gibbs Free Energy Minimization of the system (GEM)
and ii) the application of Laws of Mass Action (LMA) and mass balance constraints

19
where chemical species concentrations are relaxed until solution of the derived set of
nonlinear equations (see Zeleznik and Gordon, 1968 for a review of both methods). This
latter approach was exploited by Morel and Morgan (1972) nearly four decades ago to
develop a derivative-based iterative numerical procedure for the solution of aqueous
chemical speciation in homogeneous and heterogeneous systems which was later
extended to the computation of adsorption equilibra (Westall, 1979). This method
typically computes a correct and unique solution, provided the geochemical equilibrium
problem is mathematically defined in terms of adequate chemical components (see for
instance the Tableau approach in Morel and Hering, 1993) and the set of values
initializing the iterative procedure is wisely chosen to prevent convergence problems
(Westall, 1979). Most forward adsorption modeling computer codes are based upon the
LMA-Tableau approach.
Similarly, computer codes may be adapted for the calibration of Surface
Complexation Model (SCM) parameters such as intrinsic formation constant(s),
capacitance(s), and/or site densities from experimental sorption (i.e.,
adsorption/desorption) data (inverse modeling). For example, FITEQL (Herbelin and
Westall, 1996) is a derivative-based non-linear least squares optimization program (also
based upon the LMA-Tableau approach) that is commonly used to obtain best estimates
of intrinsic formation constants of mineral surface species using data from batch or
titration adsorption experiments. As most geochemical equilibrium programs, FITEQL
shows few convergence problems, provided the number of adjustable parameters is not
particularly large (especially when an extensive data set is not available) and the
adjustable SCM parameters are not strongly correlated (Herbelin and Westall, 1996).
Other inverse modeling codes with similar applications either make use of deterministic

20
root-finding approaches (ECOSAT-FIT, Kinniburgh, 1999), heuristic direct search
minimization techniques (Protofit, Turner and Fein, 2006), or hybrid optimization
schemes that combine heuristic Particle Swarm Optimization with deterministic
Levenberg-Marquardt non-linear regression (ISOFIT, Matott and Rabideau, 2008). The
latter two were devised for specific inverse modeling applications: Protofit estimates
proton adsorption/desorption intrinsic (surface complexation) constants using a proton
buffering function whereas ISOFIT fits conditional constants to adsorption isotherms, in
large contrast with FITEQL and ECOSAT-FIT that can extract intrinsic constants
involving any type of sorbate(s) (in addition to protons) within the framework of surface
complexation theory.
Despite the usefulness of these computer codes, it is well known that derivative-
based and simple hill-climbing numerical techniques are local in scope and are plagued
by numerical instability and convergence problems particularly for non-differentiable,
discontinuous, and under-determined (i.e., more unknowns than data points) functions.
Furthermore, non-linear regression techniques are susceptible to excessive parameter
correlation (Essaid et al., 2003). Consequently, these techniques may provide solutions
close to the initial “guess” values, possibly a local well from which the solver may not be
able to emerge, rather than the best solution, or they may not converge at all.
Accordingly, an alternative tool that can circumvent or minimize these limitations and
provide a higher flexibility in the optimization of multiple SCM parameters (including
multi-sorbate adsorption) would be desirable.
Genetic Algorithms (GAs) are efficient and robust heuristic, evolutionary,
exhaustive sampling techniques that have been successfully used in a wide range of
applications (e.g., Holland, 1975; Goldberg, 1989; Mestres and Scuseira, 1995;

21
Michalewicz, 1996; Gen and Cheng, 1997; Sait and Youssef, 1999; Gen and Cheng,
2000). Nowadays, GAs and Simulated Annealing are the preferred stochastic
optimization algorithms (Mosegaard and Sambridge, 2002) and are particularly reliable
for small inverse problems (Mosegaard, 1998). GA optimizations are performed using
probabilistic rather than deterministic rules and, thus, are especially well-suited for ill-
conditioned, non-smooth, discontinuous problems (Fernández Alvarez et al., 2008) and
perform well irrespectively of the number of data points or the error associated with the
data. This contrasts with other conventional root-finding methods such as Newton-
Raphson and Quasi-Newton that require: i) calculation of the local gradient, ii) a
reasonably well-behaved (smooth) objective function with reasonably separated roots,
and iii) extensive data sets (Gans, 1976; Epperson, 2002).
In this paper, we examine the application of a simple GA to the solution of
adsorption equilibrium inverse problems of varying degrees of complexity. We first
verify the ability of GAs to solve several forward aqueous speciation problems subjected
to identical thermodynamic, mass and charge balance constraints to those of the inverse
problems. We then test the performance of the GA on several inverse problems requiring
the optimization of multiple SCM parameters and compare the results against those
returned by FITEQL, the most frequently-used inverse modeling speciation code for SCM
calibration.
2. IMPLEMENTATION OF THE GENETIC ALGORITHM
Matlab©
software (MathWorks, Inc.) was used to write the subroutines in which we
incorporated a modified version of the GA originally written by Ron Shaffer from the
Chemometrics Research Group of the Naval Research Laboratory (USA). Six subroutines

22
are required: (i) EQUIL reads the input file containing all the information relevant to the
definition of the sorption equilibrium problem, (ii) FITGEN defines the GA parameters,
performs the binary-string encoding (see below) and initializes iterations, (iii) FITLOG
decodes the potential set of solutions, performs all calculations defining the objective
function, and computes the weighted squared residuals corresponding to the mass and/or
charge balance equations (see later sections); finally, three additional subroutines: (iv)
EVAL_GA, (v) MUT_GA, and (vi) XOVER_GA, contain the genetic operators:
selection, mutation, and crossover, required by the evolutionary process.
The unknowns for the forward problem are the concentrations of chemical
species. They are treated as optimizing quantities whereas, for the inverse problem, the
fitting parameters are: the intrinsic formation constant(s), the capacitance(s), and the site
densities invoked by the SCM of interest. Each unknown was encoded as a binary string
within a section of the solution chromosome. The length of each section (number of bits,
nj) is proportional to the search domain and constrained to reasonable boundary values
(i.e., maximum and minimum expected values for each parameter). The length of each
section is given by:
nj = log2 · (Vj) (1)
where Vj stands for the boundary value that requires the maximum number of bits for its
encoding. For instance, in the case of the forward problem, the maximum value assigned
to the free chemical component concentrations would correspond to the total analytical
molar concentration (i.e. free plus complexed species) whereas the minimum is assigned
an arbitrary value of 10-50
M, grossly overestimating the degree of interaction with other

23
chemical components in the system. Accordingly, the search domain would be defined by
log2 (10-50
) which, in binary representation, corresponds to 166 bits. To shorten the string
length and, thus, save computational time while maintaining an acceptable numerical
precision of the adjustable parameters, Vj values were expressed as 103 times their
logarithmic values. Accordingly, the modified string length (nj-ext):
nj-ext = log2 ( log10 (Vj )· 103) ) (2)
corresponds to a chromosome section of 16 bits for each chemical component. This
operation requires that the decoded values (extended logarithmic units) be divided by a
factor of 103 at each generation (iteration) to be consistent with units of the objective
function described below. This simple encoding scheme substantially reduces round-off
and truncation error during GA optimization while keeping the chromosome size practical
for GA optimization (see below). In addition, as recognized earlier (Fernández-Alvarez et
al., 2008), logarithmic parameterization linearizes the correlation structure among model
parameters, improving the sampling efficiency by reducing the number of rejected moves
in the algorithm. All fitting parameters involved in the solution of forward and inverse
problems were encoded according to this scheme.
Simple stochastic genetic operators (Gen and Cheng, 2000) were used in all
optimization problems presented in this study. To carry out chromosome selection, the
binary tournament operator was used (Goldberg et al., 1989). Only the fittest
chromosome from each generation was preserved to exploit its entire numerical genotype
(encoded solution) in the next generation. This operation is called elitism and was used in

24
all the equilibrium problems described in this paper. All other selected chromosomes
participate in the crossover and mutation operations to generate a transient set of solutions
to the problem. In this study, we tested four crossover operators: one-cut-point, two-cut-
point, uniform (Gen and Cheng, 2000), and the randomized and/or crossover (RAOC,
Keller and Lutz, 1997). This operator is of special relevance because the robustness of
GA comes from its ability to transmit information (through crossover) and create, after a
number of generations, better fitted individuals. Hence, the search for the best individual
is not blind, as in random walk procedures, but guided (Mestres and Scuseira, 1995).
Similarly, different types of mutation techniques can be carried out (e.g., Sait and
Youssef, 1999) but only the simplest type, the so-called “uniformly distributed random
mutation”, was applied in this study. Low mutation probabilities, equal to the reciprocal
of the length of the chromosomes (Keller and Lutz, 1997), were chosen to avoid pushing
the population towards unfavorable areas of the solution space. Nevertheless, higher
probabilities (0.05, 0.1 and 0.15) were also tested but produced statistically identical
results. In all optimizations performed in this study, all other GA parameters (population
size, number of generations, and type and probability of crossover) were arbitrarily
chosen for each run and were empirically optimized for each type of problem.
In the following sections, we illustrate the application of a simple GA to a number
of forward and inverse problems defined within the LMA-Tableau approach. The fitting
strategy shown in Figure 1 applies in all cases presented here but some adaptations were
made to meet problem-specific requirements and are specified below.

25
3. APPLICATION OF THE GA TO THE FORWARD PROBLEM
In this section, we verified the performance of the GA in the solution of aqueous chemical
speciation (forward problem) which requires the optimization of adjustable parameters
(molar concentrations) varying over several orders of magnitude. We solved a number of
speciation problems and compared the GA results to those returned by commercially-
available programs (MINEQL+, HYDRAQL, WHAM). The forward problem consists of
optimizing the concentration of the chemical components which are constrained by
equilibrium constants, mass and charge balance equations. Hence, the GA searches the
solution that best minimizes the total sum of residuals (Y) between the total experimental
concentrations (free plus complexed) and those estimated from the mass balance
equations of all chemical components in the system as defined by (Herbelin and Westall,
1996):
m
j
n
i
ji Tc)j,i(vY
1
2
1
(3)
where the first term inside the brackets represents the calculated molar concentration of
the jth chemical component, n is the number of species derived from the jth chemical
component, v is the stoichiometric coefficient for the jth chemical component describing
the formation of the ith aqueous species, ci is the molar concentration of the ith species
produced by chemical component jth, and m is the number of chemical components. Tj is
the total experimental molar concentration specified by the modeler (Morel and Morgan,
1972; Westall, 1979).

26
Large differences in the experimental concentrations of the chemical components
may bias the optimization because of the weight carried by the individual residuals (Rj,
term in brackets in Equation 3). Consequently, these residuals were normalized (Rj‟) as
follows:
)jlog10(R)jexp(R
1j'R (4)
In general, for problems with 10 chemical components or less, the GA returned a
suitable solution after 100 generations using a population size of 100 and either the one-
point (Gen and Cheng, 2000) or the RAOC crossover strategy (Keller and Lutz, 1997) at
a 10% of crossover probability. For this type of applications, both crossover operators
appeared to outperform the two-cut-point and uniform operators both in terms of speed
and ability to locate the best solution in the search space. The GA-predicted
concentrations of chemical components for three equilibrium problems (involving 4, 6,
and 10 chemical components and 7, 32, and 39 chemical species, respectively) were
nearly identical (RSD 0.03%) to those obtained using HYDRAQL, WHAM, and
MINEQL+ for ionic strengths 0.01 M. This exercise confirmed the efficiency of the GA
in dealing with optimization problems with numerous adjustable parameters varying over
several orders of magnitude and subjected to similar constraints to those of the inverse
adsorption problems presented below.

27
4. APPLICATION OF THE GA TO THE INVERSE PROBLEM
4.1 Estimation of Intrinsic Ionization Constants: Constant Capacitance Model
Our main objective is to implement a reliable and flexible approach to address specific
inverse problems that cannot be easily handled by conventional, deterministic, derivative-
based, root-finding techniques implemented in popular SCM calibration programs such as
FITEQL.
Because reasonable a priori knowledge is available (physical-chemical
constraints, geochemistry of the adsorbent phase, etc.), the viability of conceptual
adsorption reactions can be evaluated intuitively against specified criteria prior to
optimization, greatly reducing the number of alternative SCMs that deserve systematic
evaluation via inverse modeling. Furthermore, emphasis on conceptual and mathematical
simplicity in the formulation of SCMs is paramount and must be consistent with the
quantity and quality of available data. As emphasized in earlier studies (e.g., Herbelin and
Westall, 1996), when several SCMs fit the data equally, the most parsimonious one must
be preferred unless there is compelling evidence in support of another. Models with many
degrees of freedom incur serious risks among which: (i) fitting of inconsistent or
irrelevant „„noise‟‟ in the data records; (ii) severely diminished predictive power; (iii) the
generation of ill-defined, near-redundant parameter combinations; and (iv) masking of
geochemically-significant behavior derived from data over-fitting (Jakeman et al, 2006).
Irrespectively, the inverse problem should be well determined and, hence, contain more
data points than adjustable parameters. Finally, a posteriori physical-chemical evaluation
of the fitted SCM parameters is key to ascertain their thermodynamic relevance within the
SCM. It follows that within this scheme, the calibration of SCMs, by deterministic or

28
heuristic approaches, is properly constrained and goes well beyond a mere data fitting
exercise.
We illustrate the above approach by testing the GA on various inverse problems.
The first and simplest one calls for the estimation of surface ionization (i.e., proton
adsorption/desorption) constants from proton adsorption data at a mineral surface. For a
generic hydrated reactive surface site or “adsorption center” (e.g., S·H2O) these
reactions can be generalized as follows:
S·H2O S·OH- + H
+ (deprotonation, 5a)
S·H2O + H+ S·H3O
+ (protonation, 5b)
The first step is to define the generalized objective function that applies to all
adsorption studies. This implies the computation of the residuals between the theoretical
and experimental adsorption values, Yk, which, for any adsorbate, k, is defined as:
n
i
kik TM)k,i(vY
1
(6)
where Mi represents the molar concentration of the ith adsorbate-bearing surface species,
Tk is the experimental adsorbed molar concentration of the adsorbate k. For each
chromosome, the GA optimization is subjected to:
2
1 1
p s
)k(
)k(
S
YW SSE (7)

29
where WSSE is the weighted sum of squared errors of an s number of adsorbate
components computed at all titration points, p, and S(k) is the error calculated for Y(k) from
the experimental errors associated with the quantitative determination of the kth
adsorbate. Equation 7 is the generalized objective function used in the optimization of
intrinsic constants (ionization and adsorption) when suitable adsorption data for all
adsorbates under consideration are available.
To extract intrinsic equilibrium constants, reactions must be referenced to a zero
electrical potential surface by taking into account, at each stage of the titration, the
electrostatic work required to transport ions through the interfacial electrical potential
gradient (Dzombak and Morel, 1990) according to:
e
x
xintapp ψexpKK
1RT
ZF- (8)
where Kapp
is the apparent formation constant, Kint
stands for the intrinsic constant, Z is
the net charge transfer of the reaction, F is the Faraday constant, R is the gas constant, T
is the absolute temperature, e is the number of electrostatic planes where explicit
adsorption is assumed to take place, and x is the electrical potential at the adsorbing
plane(s) “x” (e.g., 0-plane, Stern-plane; Davis and Kent, 1990). For brevity, the latter term
will, hereafter, be referred to as potential (ascribed to a specific electrostatic plane). It is
an adjustable parameter that, upon minimization of Equation 7, must satisfy the following
constraint for each adsorbing plane (the surface plane in the case of the CCM):

30
electx
q
i
ii CzAS
F
1
][ (9)
where S is the specific surface area of the mineral (m2 g
-1), A is the mass:volume ratio of
the experimental suspension (g L-1
), zi and [Ci] are, respectively, the charge and molar
concentration of species i adsorbed at plane x, and q is the number of species contributing
to the charge at plane x. The left-hand term gives the net charge density at plane “x” (C
m-2
) from the surface species concentrations computed at each generation, whereas the
right-hand term, xelect
, represents the charge density at plane x (C m-2
) derived from a
theoretical electrostatic model describing the relationship between the surface charge and
surface potential (see Davis and Kent, 1990). The electrostatic correction is specified in
the mathematical definition of the equilibrium problem, according to the method
presented by Westall and Hohl (1980). A d number of “dummy” chemical component(s)
is added to the model, corresponding to the number of adsorbing planes as defined by the
selected electrostatic model.
The GA was initially tested with data taken from Gao and Mucci (2001) who used
FITEQL v. 2.0 to optimize the intrinsic ionization constants of the goethite surface in a
0.7 M NaCl solution considering the following set of surface reactions:
FeOH + H+ FeOH2
+ Ka1 (10)
FeOH FeO- + H
+ Ka2 (11)
To describe the electrostatics at the interface, these authors applied the Constant
Capacitance Model (CCM, Schindler and Kamber, 1968; Hohl and Stumm, 1976) and,

31
thus, this model was implemented in the Matlab©
script to compute the value of x at
each generation according to the following expression (Stumm and Morgan, 1996):
C0
0σ
ψ (12)
where C is the specific capacitance and 0 and 0 are the charge and potential at the
surface (i.e., plane “0”). Given that only ionization reactions were considered to take
place at the surface, experimental surface charge data are available (net proton adsorption
densities are identical to surface charge densities) and, therefore, for a given capacitance
value, these data can be used to compute the surface potential at each titration point and
perform the electrostatic correction using Equations 8 and 12, respectively.
Aqueous equilibrium was solved first using either the GA or the Newton-Raphson
approach (implemented in an additional Matlab©
subroutine) to compute the
concentration of the free chemical components in solution before the intrinsic ionization
constants were optimized with the GA. In contrast to the strategy typically applied by
FITEQL users, whereby the specific capacitance is varied manually in each optimization
and the goodness of fit evaluated on the basis of the WSOS/DOF (“weighted sum of
squares divided by the degrees of freedom”) parameter (Dzombak and Morel, 1990), the
specific capacitance and the intrinsic ionization constants were optimized simultaneously
by the GA. Using the encoding rules described earlier, the capacitance can be
successfully treated as a fitting parameter and added to the chromosome encoding the
solution to the intrinsic ionization constants.

32
Like the chemical component concentrations (see preceding section), the intrinsic
constants were formulated in extended logarithmic units. Conventionally, chromosome
binary-strings represent solutions from -25000 to 25000 (i.e., 10-25
to 1025
M or 50 orders
of magnitude) that approximately cover the range of formation constants typically
reported in thermodynamic compilations (e.g., NIST, 1998). Nevertheless, for specific
adsorption reactions, such a wide range may not necessarily represent
thermodynamically-meaningful quantities and, hence, based upon physical-chemical
concepts, the modeler may select a reasonable solution space for each adjustable
parameter (as explained above), constraining the range of potential solutions and its
compatibility with the physical reality. The specific capacitance was constrained between
0.2 and 2 F/m2, covering the range of values previously reported for oxide mineral
surfaces (e.g., Hiemstra et al., 1999).
SCM parameters optimized with the GA and input values of S and A are presented
in Table 1 and compared to those reported by Gao and Mucci (2001). Because of the
probabilistic nature of the GA optimization, several GA runs were performed using
different GA parameters (e.g., population size, maximum number of generations,
mutation rates) and the associated error was calculated for each adjustable quantity. It
should be noted that this error is associated with the GA-optimization rather than to the
experiment. True uncertainties of the optimized SCM parameters must be determined
from multiple GA runs using independent replicate data sets ( 3). As shown in Table 1,
the optimized values are very reproducible, even when the population size or number of
generations varied significantly, and the mean values are close to those obtained by Gao
and Mucci (2001). Nevertheless, the ionization constants and capacitance values that best

33
describe the experimental data are slightly different than those obtained when fixed
capacitance values are used in the optimization, as in the case of FITEQL. It is
noteworthy that the GA facilitates the search of suitable intrinsic constants and
capacitance values in a single optimization run, in contrast to FITEQL and other available
inverse modeling programs (e.g., ECOSAT-FIT, Protofit). Although the difference is
small in this particular example, in other applications, relaxation of the capacitance(s) and
minimization of round-off and truncation errors via GA optimization may impact
significantly on the optimized SCM parameters. The experimental proton adsorption data
and the GA-predicted proton adsorption densities are shown in Figure 2.
4.2 Simultaneous Estimation of Intrinsic Ionization and Adsorption Constants:
Constant Capacitance Model
Typically, direct, experimental surface charge data are unavailable when adsorption of
adsorbates other than protons occurs. Consequently, surface charge must be computed
from the speciation predicted at each generation to obtain adequate estimates of the
surface potential and apply the appropriate electrostatic corrections (Eqs. 8 and 12).
Within the CCM scheme, this problem can be easily circumvented by initializing the GA
iterative process using a set of “best guess” surface charge values. This serves to pre-
adjust the constants to reasonable estimates of surface potential (related to surface charge
via an electrostatic model, Westall and Hohl, 1980) and leads the chromosome population
towards favorable regions within the solution space. After a number of generations
(iterth), the GA will calculate surface potentials from Equation 12 using the surface charge
computed from the predicted surface speciation (left-hand term in Equation 9) and will
estimate the electrostatic correction in subsequent generations. This procedure guarantees

34
fulfillment of Equation 9 from generation iterth while pushing evolution towards
successful minimization of Equation 7.
Using published phosphate adsorption data on the goethite surface in 0.7 NaCl
solutions (Gao and Mucci, 2001), we re-optimized the phosphate adsorption constants
with the GA using an identical conceptual SCM defined by the following set of surface
reactions:
FeOH + H2PO4- FePO4
2- + H
+ + H2O
(13)
FeOH + H2PO4- FePO4H
- + H2O
(14)
FeOH + H2PO4- + H
+ FePO4H2
+ H2O
(15)
For this application, assigning initial zero potentials (i.e., null electrostatic
corrections) to the surface allowed for a good performance of the GA in combination with
a rather large maximum number of iterations (maxiter 300). Several GA runs indicated
that after 50% of the pre-fixed maxiter (150), the numerical viability of the evolved
chromosome population was suitable for the computation of surface potentials from the
surface speciation and Equation 12 as well as for successful fitting of the data (Figure 3).
The success of the optimization confirms the ability of the GA to extract values of the
intrinsic adsorption constants within the CCM formalism, in the absence of surface
charge data, without explicit fitting of the surface potential.
The optimized values of the intrinsic adsorption constants are reported in Table 2
and compared to those obtained by Gao and Mucci (2001). To see the impact of the GA
optimization on the estimation of phosphate adsorption constants independently of other

35
SCM parameters, the Ka1, Ka2 and capacitance values originally proposed by Gao and
Mucci (2001) were used in this GA optimization exercise. Note that, whereas the intrinsic
constants of reactions 14 and 15 optimized by the GA are, within error, very similar to
those estimated by FITEQL, the GA-optimized intrinsic constant of reaction 13 is nearly
one order of magnitude lower. This is a clear distinction between the output returned by
FITEQL and the GA and is attributable to the better numerical stability displayed by the
latter.
To evaluate the performance of the GA with more complex optimization problems
within the CCM scheme, we optimized ionization and adsorption intrinsic constants using
synthetic data of a gedanken experiment, an acidimetric titration of a reactive mineral
(MeX(s)) displaying moderate pH-promoted dissolution. In this experiment, we considered
that dissolved, lattice-constituent divalent metal ions (Me2+
) reabsorb on the mineral
surface, competing with protons for reactive surface sites and altering surface protonation
and surface charge. The ionization and metal adsorption surface reactions, formulated in
terms of the single-site 2-pK ionization model (Lützenkirchen, 2003), are:
MeOH + H+ MeOH2
+ (16)
MeOH MeO- + H
+ (17)
MeOH + Me2+
MeOMe+
+ H+
(18)
pH, total dissolved Me2+
concentration, total adsorbed proton ([H+]Ads, data set 1), and
total adsorbed metal concentrations ([Me2+
]Ads, data set 2) compose the available data at
each titration point and were used in the minimization of Equation 7 for the optimization

36
of the intrinsic constants describing reactions 16-18. It is assumed that Me2+
does not
form ion pairs or complexes in aqueous solution (i.e., total Me2+
concentrations are equal
to free Me2+
concentrations). The chemical scenario of this gedanken experiment is
similar to classical, competitive ion adsorption studies where the pH and the aqueous
adsorbate(s) concentration are simultaneously monitored at each experimental point.
Using a population of 100 chromosomes and after 500 generations, the GA
successfully reproduces all the experimental data, as shown in Figure 4. In contrast, the
simultaneous optimization of two or three of the constants (reaction 16, 17 or 18) and the
number of available reactive surface sites with a specified capacitance (GA-optimized
value) with FITEQL v. 3.2 resulted in convergence problems and no output. When only
the constant describing reaction 16 was optimized, while the constants for reactions 17
and 18 were fixed at the GA-optimized values, FITEQL converged but gave very poor
fits to the [Me2+
]Ads, and [H+]Ads data (Figure 4), with a WSOS/DOF value of 19.5. This
reveals that, in some cases, successful convergence of FITEQL does not necessarily
return good fits to all the experimental data. In turn, this raises questions about the
validity of the recommended range of WSOS/DOF values (0.1 to 20) for the evaluation of
the goodness of fit (Westall, 1982). It appears that FITEQL converged towards a local
minimum rather than a global one and the relative contribution of reactions 16 and 17
(ionization reactions) versus 18 (divalent metal adsorption reaction) to the proton
adsorption behavior could not be resolved accurately upon fitting of data sets 1 and 2.
Conversely, the GA optimization gradually minimizes the difference between the
experimental and simulated data and, thus, the evolved generation retains critical
information for searching the global minimum. Given an adequate conceptual SCM and
sufficient high quality data, the GA will find the combination of model parameters that

37
best minimizes Equation 7 even in cases that are difficult to tackle by deterministic
approaches.
It has been stressed that an a posteriori analysis of non-uniqueness and uncertainty
of solutions to inverse problems (resolution analysis) is necessary to assess scientific
conclusions based on inverse modeling (Moosegard, 1998). Whereas this is an important
step for inverse problems involving a large number of ill-defined, poorly constrained
parameters, this is not crucial for the problems presented in our study since the selected
SCM parameters are physically-meaningful and well constrained. Hence, they do not
generally yield numerous solutions to a given problem (or selected SCM), unless the
available data are insufficient or unsuitable to quantitatively resolve the contribution of
each adsorption reaction. Nevertheless, a sensitivity analysis should be conducted to
select the best solution among the possible options (several SCMs fitting the data
reasonably well) and prevent over-interpretation of the data. This is typically done by
gradually incorporating adsorption reactions to the simplest physically-meaningful SCM
and by testing its ability to reproduce the experimental data. We performed this analysis
in a recent study (Villegas-Jiménez et al., 2009) where, based upon fundamental
chemistry concepts, several SCMs were formulated and alternatively calibrated via GA
optimization using relatively large experimental data sets for two rhombohedral carbonate
minerals: magnesite and dolomite (Pokrovsky et al., 1999a,b). Whereas more than one
SCM fitted the data within the same tolerance (as shown by ANOVA tests), only one
reasonably complied with all other a posteriori criteria (theoretical constraints and
alternate experimental data) imposed for model validation. Again, emphasis must be
placed on model parsimony.

38
4.3 Simultaneous Estimation of Intrinsic Ionization and Adsorption Constants:
Triple Layer Model
It is often necessary to adopt an electrostatic model other than the CCM to describe the
spatial distribution of charged surface species at the mineral-water interface
(Lützenkirchen, 2003). For multi-layer electrostatic models, such as the Triple Layer
Model (TLM), a specific potential must be defined at each interfacial plane (surface and
Stern planes and diffuse layer). In this case, the GA must find a suitable set of intrinsic
constants, capacitance values, and site densities (intensive variables) that, in combination
with appropriate potentials (extensive variables), minimize Equation 7 while satisfying
Equation 9. Since the surface charge displayed by the mineral varies according to the
chemical conditions of the system (i.e., pH, chemical speciation), individual potentials
must be adjusted at each titration or experimental data point, and for each electrostatic
plane. Given the large number of adjustable extensive variables, some modifications
were made to the optimization approach described earlier.
Sets of potential solutions for these extensive variables are encoded in a 3D
binary-string matrix (Matrix-A) whose dimensions are determined by: (i) the selected
population size, (ii) the number of available data points, and (iii) the number of interfacial
electrostatic planes (“dummy” components), excluding the diffuse region, required by the
selected multi-layer electrostatic model (Westall and Hohl, 1980). To define the size of
the chromosomes in Matrix-A and the solution space, realistic boundary values of surface
potential must be chosen for each electrostatic plane. For this purpose, zeta-potentials
obtained from electrokinetic measurements are useful for the selection of these
constraints (Dzombak and Morel, 1990). The structure of a generalized 3D matrix,
including the “dummy” components, is illustrated in Figure 5.

39
Another matrix (Matrix-B) encodes the solutions for the intensive variables. Its
dimensions are dictated by the selected population size and the number of adjustable
intensive variables. To improve the effectiveness of the evolutionary process, Matrix-A
and Matrix-B were dimensioned and manipulated separately throughout the GA
optimization. In other words, genetic operations are carried out independently for each
matrix, maintaining a good numerical diversity in both matrices and facilitating the search
for the best solution to the optimization problem. Whereas the evaluation of the
chromosomes in Matrix-B is subject to Equation 7, the evaluation of chromosomes in
Matrix-A is based on fulfillment of Equation 9 (for each electrostatic plane where
adsorption occurs) by minimization of:
p eElec
xCalc
xY
1 1
2 (19)
where the total sum of residuals (Y) corresponds to the difference between the charge
calculated from the speciation predicted at plane “x” (xCalc
, in C m-2
) and the charge
computed from electrostatics for plane “x” using appropriate surface-charge-potential
relationships (xElec
, in C m-2
). For the TLM, these are (Davis and Kent, 1990):
)(00
ψψElec
1C (20)
0
)(
d
Elec
ψψ2C (21)
)2/sinh(1174.0 RTZFψI d
Elec
d (22)

40
where, as before, and represent, respectively, charge and potential at each specific
interfacial plane: i) the surface (plane-0), ii) the plane where outer-sphere complexes are
located (plane-), and iii) the diffuse layer (plane-d). C1 and C2 are the integral
capacitance values of the interfacial layers, and F/RT represents the reciprocal of the
Boltzmann factor for charged molecules and ions (0.02569 V-1
). Because the TLM makes
no explicit provision of ion adsorption at the diffuse layer, the calculation of the diffuse
layer charge, d, is obtained from the charge neutrality equation (Davis and Kent, 1990):
d = - 0 - (23)
and hence, the d can be obtained directly from equation 22. In other words, only 0 and
are subjected to optimization via Equation 19, and thus, only two “dummy”
components are required in Matrix A. Additional “dummy” components will be required
by other sophisticated multi-layer electrostatic models such as the Four Layer Model
(Charmas, 1999).
One advantage of this treatment is that the “best” potential computed at each
iteration, the one that best minimizes Equation 19, can be tested against inequality
constraints defined by physically realistic boundary values for each electrostatic plane
(Dzombak and Morel, 1990; Davis and Kent, 1990). This operation serves to determine
whether the calculated potentials are realistic and, thus, can be used in the next iteration,
otherwise the old value is retained for evaluation in the next generation. The performance
of the GA is improved by interrupting the optimization of the extensive variables once the
above-mentioned constraints are achieved. In other words, hereafter, only Matrix-B will

41
be subject to evolution, via Equation 7 using the most realistic potential values (that best
minimize equation 19) to continue the iterative procedure.The strategy employed for each
matrix is schematically represented in Figure 6.
To further improve the performance (i.e., speed and computational requirements)
of the GA optimization in this type of application, an additional implementation must be
made. Given a chromosome population 100 and after a sufficient number of generations
(i.e., n > 100), the GA should have reached a suitable level of numerical viability and can
be interrupted even if the data fit is inadequate. The optimized, intensive variables are
best values and can be used to quickly verify whether they can successfully simulate the
data upon solution of the adsorption equilibrium forward problem (adjustment of
extensive variables) by a derivative-based numerical technique. The implementation of
the Newton-Raphson (NR) technique for these types of problems is straightforward and
was explained in detail elsewhere (e.g., Papelis et al., 1988). In other words, evolutionary
optimization can be switched to a subroutine that calculates the Jacobians corresponding
to all chemical component(s) and electrostatic term(s). Once the Jacobians are obtained,
the matrix is solved by the Gauss-elimination procedure (Nicholson, 1995) and the
iterative procedure carries on until the residual meets a pre-fixed tolerance level (Sahai
and Sverjensky, 1998). This strategy exploits the advantages of the GA to optimize the
intensive variables (with pre-adjusted extensive variables) which, in combination with
deterministically-derived potentials can simulate titration data. The Newton-Raphson
technique is used to fine-tune the estimation of the potentials, ensure minimization of
Equation 19 at the end of the iterative process, and reduce the computational time. If

42
required, additional GA-NR micro-iterations can be implemented to improve the quality
of the fit and further refine the intensive variable estimates.
This “hybrid” GA-NR optimization technique was successfully tested against
published surface titration data for goethite in 0.01 M NaCl solutions (Villalobos and
Leckie, 2001). The authors originally modeled their data using the TLM (Figure 7). In
this formalism, seven intensive variables must be adjusted: two ionization constants
(reactions 10 and 11), two background electrolyte adsorption constants:
FeOH + Na+ FeO
-Na
+ + H
+ (24)
FeOH + Cl- + H
+ FeOH2
+Cl
- (25)
two capacitance values (C1 and C2), and the total number of reactive surface sites.
Because FITEQL convergence is not assured when several parameters are
adjusted, particularly when interdependent reactions (reactions 10, 11, 24, and 25 are pH-
dependent) are fitted simultaneously (Hayes et al., 1991), some criteria are commonly
applied to pre-select either the ionization or electrolyte adsorption constants before
proceeding with the optimization of all other fitting parameters. Accordingly, Villalobos
and Leckie (2001) selected fixed values of the ionization constants (reactions 10 and 11)
and optimized those corresponding to reactions 24 and 25 to fit their data. In contrast, the
hybrid GA-NR technique allows for the simultaneous optimization of all fitting
parameters required by the TLM and for the successful simulation of titration data, as
shown in Figure 7.

43
5. CONCLUSIONS
A powerful evolutionary optimization technique, the genetic algorithm, was applied to the
calibration of SCMs for mineral surfaces. This technique was successfully tested for the
inverse modeling of adsorption equilibria under several scenarios of varying degrees of
complexity using published and synthetic data.
The GA performs an exhaustive probabilistic search in a broad solution space that
is constrained by physically realistic values selected by the modeler. Given suitable
combinations of geochemical (set of adsorption reactions) and electrostatic (charge
distribution at the adsorbent-water interface) models and sufficient quality experimental
data, the GA can successfully optimize numerous parameters without incurring
convergence problems while achieving good numerical stability, a notable advantage over
conventional deterministic, root-finding, and optimization techniques implemented in
popular inverse modeling codes such as FITEQL. At the modeler‟s discretion, multiple
intrinsic ionization and adsorption constants, capacitance(s), and/or reactive surface site
densities can be simultaneously fitted by the GA. Nevertheless, an a posteriori theoretical
evaluation of the fitted SCM parameters must be made to ascertain their thermodynamic
meaningfulness and confirm their relevance within the SCM. This aspect is key for the
construct of sound, yet parsimonious, SCMs.
The drawback of the GA approach is the large number of iterations that must be
performed to obtain a satisfactory solution, particularly for cases involving numerous
fitting parameters. Alternatively, the power of the GA can be more efficiently exploited
when a deterministic technique such as Newton-Raphson is incorporated once the
chromosome population has reached a suitable level of numerical viability. Consequently,

44
we propose the use of a hybrid GA-NR approach for the efficient optimization of intrinsic
constants in complex problems such as those involving the TLM.
We believe that the computational requirements of the GA to the calibration of
SCMs are greatly outweighed by its robustness, simplicity, and potential to generate a
reliable thermodynamic database describing adsorption reactions. This is a critical step to
making reliable predictions of the adsorption behavior of minerals and geologically-based
adsorbents in hydro-geological settings (e.g., aquifers, pore waters, water basins) and/or
in engineered reactors (e.g., mining and wastewater treatment industries). Future work to
test and develop more sophisticated adaptive strategies and hybrid heuristic-deterministic
optimization approaches may improve the GA performance in this type of applications.
6. ACKNOWLEDGMENTS
A.V.-J. thanks Dr. David Burns for critical discussions that inspired this investigation and
EMEA S.C. Environmental Consulting Firm for providing suitable facilities to complete
this work. This research was supported by a graduate student grant to A.V.-J. from the
Geological Society of America (GSA) and Natural Sciences and Engineering Research
Council of Canada (NSERC) Discovery grants to A.M. A.V.-J. benefited from post-
graduate scholarships from the Consejo Nacional de Ciencia y Tecnología (CONACyT)
of Mexico and additional financial support from the Department of Earth and Planetary
Sciences, McGill University and Consorcio Mexicano Flotus-Nanuk.

45
7. REFERENCES
Allison J.D., Brown D.S. and Novo-Gradac K.J. (1991) MINTEQA2/PRODEFA2, a
geochemical assessment model for environmental systems: version 3.0. User‟s
manual, Environmental Research Laboratory, Office of Research and
Development, U.S. Environmental Protection Agency, Athens, GA, 106.
Ball J.W., Jenne E.A. and Norstrom D.K. (1981) WATEQ2- A computerized chemical
model for trace and major element speciation and mineral equilibrium of natural
waters. In: E.A. Jenne (Ed.) Chemical Modeling in Aqueous Systems, Symposium
series 93, American Chemical Society, Washington, D.C. pp 815-836.
Charmas R. (1999) Four-layer complexation model for ion adsorption at energetically
heterogeneous metal oxide/electrolyte interfaces. Langmuir 15(17), 5635-5648.
Davis J.A. and Kent D.B. (1990) Surface complexation modeling in aqueous
geochemistry. In: Mineral-Water Interface Geochemistry. (ed. M.F. Hochella and
A.F. White). Rev. Mineral. 23. Mineral. Soc. Washington, DC. pp 177-260.
Dzombak D.A. and Morel F.M.M. (1990) Surface Complexation Modeling. John Wiley
and Sons, New York, NY, 393.
Epperson J.F. (2002) An Introduction to Numerical Methods and Analysis. John Wiley
and Sons, New York, NY, 556.
Essaid H.I., Cozzarelli I.M., Eganhouse R.P., Herkelrath W.N., Bekins B.A. and Delin
G.N. (2003) Inverse modeling of BTEX dissolution and biodegradation at the
Bemidji, MN crude-oil spill site. J. Cont. Hydr. 67(1), 269-299.
Fernández Alvarez J.P., Fernández Martínez J.L. and Menéndez Pérez C.O. (2008)
Feasibility analysis of the use of binary genetic algorithms as importance samplers
Application to 1-D DC resistivity inverse problem. Math. Geosci. 40, 375-408.

46
Gans P. (1976) Numerical methods for data-fitting problems. Coord. Chem. Rev. 19, 99-
124.
Gao Y. and Mucci A. (2001) Acid base reactions, phosphate and arsenate complexation,
and their competitive adsorption at the surface of goethite in 0.7 M NaCl solution.
Geochim. Cosmochim. Acta 65, 2361-2378.
Gen M. and Cheng R. (1997) Genetic Algorithms and Engineering Design. John Wiley
and Sons, New York, NY, 411.
Gen M. and Cheng R. (2000) Genetic Algorithms and Engineering Optimization. John
Wiley and Sons, New York, NY, 495 pp.
Goldberg D. (1989) Genetic Algorithms in Search, Optimization and Machine Learning.
Addison-Wesley, Reading, MA, 412.
Golberg D., Korb K. and Deb K. (1989) Messy genetic algorithms: motivation, analysis
and first results. Complex Systems 3, 493-530.
Hayes K.F., Redden G., Ela W. and Leckie J.O. (1991). Surface complexation models:
An evaluation of model parameter estimation using FITEQL and oxide mineral
titration. J. Colloid Interface Sci. 142(2), 448-469.
Herbelin A. and Westall J. (1996) FITEQL-. A computer program for determination of
chemical equilibrium constants from experimental data version 3.2: user‟s
manual. Department of Chemistry. Oregon State University, Corvallis, OR,
Report 96-01.
Hiemstra T., Yong H. and van Riemsdijk W.H. (1999) Interfacial charging phenomena of
aluminum (hydr)oxides. Langmuir 15(18), 5942-5955.

47
Hohl H. and Stumm W. (1976) Interaction of Pb2+
with hydrous -Al2O3. J. Colloid
Interface Sci. 55, 281-288.
Holland J. (1975) Adaptation in Natural and Artificial Systems. University of Michigan
Press, Ann Arbor MI, 183.
Jakeman A.J., Letcher R.A. and Norton J.P. (2006) Ten iterative steps in development
and evaluation of environmental models. Environ. Model. Software 21, 602-614.
Keizer M.G. and van Riemsdijk W.H. (1999) ECOSAT, Technical Report of the
departments of Soil Science and Plant Nutrition. Wageningen University,
Wageningen, The Netherlands.
Keller B. and Lutz R. (1997) A new crossover operator for rapid function optimization
using a genetic algorithm. Proceedings of the Eighth Ireland Conference on
Artificial Intelligence (AI-97), Vol. 2, pp. 48-57.
Kinniburgh D.G. (1999) FIT User Guide. British Geological Survey, Keyworth, UK,
WD/93/23.
Langmuir D. (1997) Aqueous Environmental Geochemistry. Prentice Hall, Upper Saddle
River, N.J., 600.
Lützenkirchen J. (2003) Surface complexation models for adsorption: A critical survey in
the context of experimental data. In: Tóth J. (Ed) Adsorption: Theory, Modeling,
and Analysis, Surfactant Science Series 107, New York, NY, Marcel Dekker Inc.,
pp. 631-710.
Matott L.S. and Rabideau A.J. (2008) ISOFIT - A program for fitting sorption isotherms
to experimental data. Environ Model Software 23: 670-676.

48
Mestres J. and Scuseira G.E. (1995) Genetic algorithms, A robust scheme for geometry
optimizations and global minimum structure problems. J. Comput. Chem. 16(6),
729-742.
Michalewicz Z. (1996) Genetic Algorithms + Data Structure = Evolution Programs, 2nd
ed. Springer-Verlag, New York, NY, 387.
Morel F.M.M. and Morgan J. (1972) A numerical method for computing equilibria in
aqueous chemical systems. Environ. Sci. Technol. 6, 58-87.
Morel F.M.M. and Hering J.G. (1993) Principles and Applications of Aquatic Chemistry.
John Wiley and Sons, New York, NY, 588.
Mosegaard K. (1998) Resolution analysis of general inverse problems through inverse
Monte Carlo sampling. Inverse Probl. 14, 405-426.
Mosegaard K. and Sambridge M. (2002) Monte Carlo Analysis of inverse problems.
Inverse Probl. 18, 29-54.
Nicholson K. (1995) Linear Algebra with Applications. PWS Pub. Co., Boston, MD, 540.
NIST (1998) Critically Selected Stability Constants of Metal Complexes, Standard
Reference Database 46, Version 5, National Institute of Standards and
Technology, US Department of Commerce, Gaithersburg, MD, USA.
Papelis C., Hayes K.F. and Leckie J.O. (1988) HYDRAQL: A program for the
computation of chemical equilibrium, composition of aqueous batch systems
including surface-complexation modeling of ion adsorption at the oxide/solution
interface. Report No. 306. Stanford University, Stanford, CA, 130.
Parkhurst D.L. (1995) User‟s guide to PHREEQC. U.S. Geological Survey, Water
Resources Investigations Report 95-4277, 212.

49
Pokrovsky O.S., Schott J. and Thomas F. (1999a) Processes at the magnesium-bearing
carbonates/solution interface. I. A surface speciation model for magnesite.
Geochim. Cosmochim. Acta 63(6), 863-880.
Pokrovsky O.S., Schott J. and Thomas F. (1999b) Dolomite surface speciation and
reactivity in aquatic systems. Geochim. Cosmochim. Acta 63(19/20), 3133-3143.
Sahai N. and Sverjensky D.A. (1998) GEOSURF: A computer program for modeling
adsorption on mineral surfaces from aqueous solution. Comp. Geosci. 24(9), 853-
873.
Sait S.M. and Youssef H. (1999) Iterative Computer algorithms with Applications in
Engineering Solving Combinatorial Optimization Problems. IEEE Computer
Society Los Alamitos, CA, 387.
Schecher W.D. and McAvoy D.C. (1992) MINEQL+: A software environment for
chemical equilibrium modeling. Computers, Environment and Urban Systems 16,
65-76.
Schindler P.W. and Kamber H.R. (1968) Die acidität von silanolgruppen. Helv. Chim.
Acta 51, 1781-1786.
Sposito G. and Coves J. (1988) SOILCHEM: A computer program for the calculation of
chemical speciation in soils. Keamey Found. Soil Sci., Univ. California,
Riverside.
Stumm W. and Morgan J. (1996) Aquatic Chemistry: Chemical Equilibria and Rates in
Natural Waters. John Wiley and Sons Inc., New York, 1022.
Tipping E. (1994) WHAM- A chemical equilibrium model and computer code for water,
sediments and soils incorporating a discrete site/electrostatic model of ion-binding
by humic substances. Comp. Geosci. 20, 973-1023.

50
Turner B.F. and Fein J.B. (2006) Protofit: A program for determining surface protonation
constants from titration data. Comp. Geosci. 32, 1344–1356.
van der Lee J. and de Windt L. (1999) CHESS Tutorial and cookbook. Technical report
No. LHM/RD/99/05. École des Mines de Paris. Fontainebleu, France. 77.
Villalobos M. and Leckie J.O. (2001) Surface complexation modeling and FTIR study of
carbonate adsorption to goethite. Geochim. Cosmochim. Acta 235,15-32.
Villegas-Jiménez A., Mucci A., Pokrovsky O.S. and Schott J. (2009) Defining reactive
sites at hydrated mineral surfaces: rhombohedral carbonate minerals. Geochim.
Cosmochim. Acta 73(15), 4326-4345.
Westall J.C. (1979) MICROQL II: Computation of adsorption equilibria in BASIC.
Technical Report. Swiss Federal Institute of Technology, EAWAG, 8600
Dübendorf, Switzerland.
Westall J.C. (1982) FITEQL: A computer program for determination of chemical
equilibrium constants from experimental data. Version 1.2 Report 82-01,
Department of Chemistry, Oregon State University, Corvallis, OR, USA.
Westall J. and Hohl H. (1980) A comparison of electrostatic models for the
oxide/solution interface. Adv. Colloid Int. Sci. 12, 265-294.
Wolery T.J. (1992) EQ3NR: A computer program for geochemical aqueous speciation-
solubility calculations: theoretical manual, user‟s guide and related documentation
(Version 7.0), Report No. UCRL-MA-110662-PT-III, Lawrence Livermore
National Laboratory, Livermore, CA.
Zeleznik F.J. and Gordon S. (1968) Calculation of complex chemical equilibria. Ind. Eng.
Chem. 60, 27-57.

51
8. TABLES
Table 1. Intrinsic acidity constants of the goethite surface in a 0.7 NaCl solution using the
Constant Capacitance Model. Errors represent confidence intervals at 95%. Results
shown using the GA approach are averaged values obtained from three optimizations
using the following GA parameter sets (population size, number of generations, crossover
rate, mutation rate): [10, 1000, 0.1, 0.1], [50, 500, 0.1, 0.05], and [100, 1000, 0.1, 0.02].
Surface Equilibria
Log10 Kint
Gao and Mucci (2001)
FITEQL 2.0
This study
GA
FeOH + H+ = FeOH2
+ 7.45 7.23 ± (<0.01)
*
FeOH = FeOH2+ H+ -9.60 -9.6 ± 0.04
*
Capacitance (F/m2) 1.86 1.93 ± (<0.01)
*
Experimental conditions: A= 27.7 m2 g
-1, S= 7.93 g L
-1
Site Density: 2.96·10-6
moles m-2
*Reported errors are a measure of the reproducibility of the optimization itself, and thus, do not
provide information on the associated experimental error.
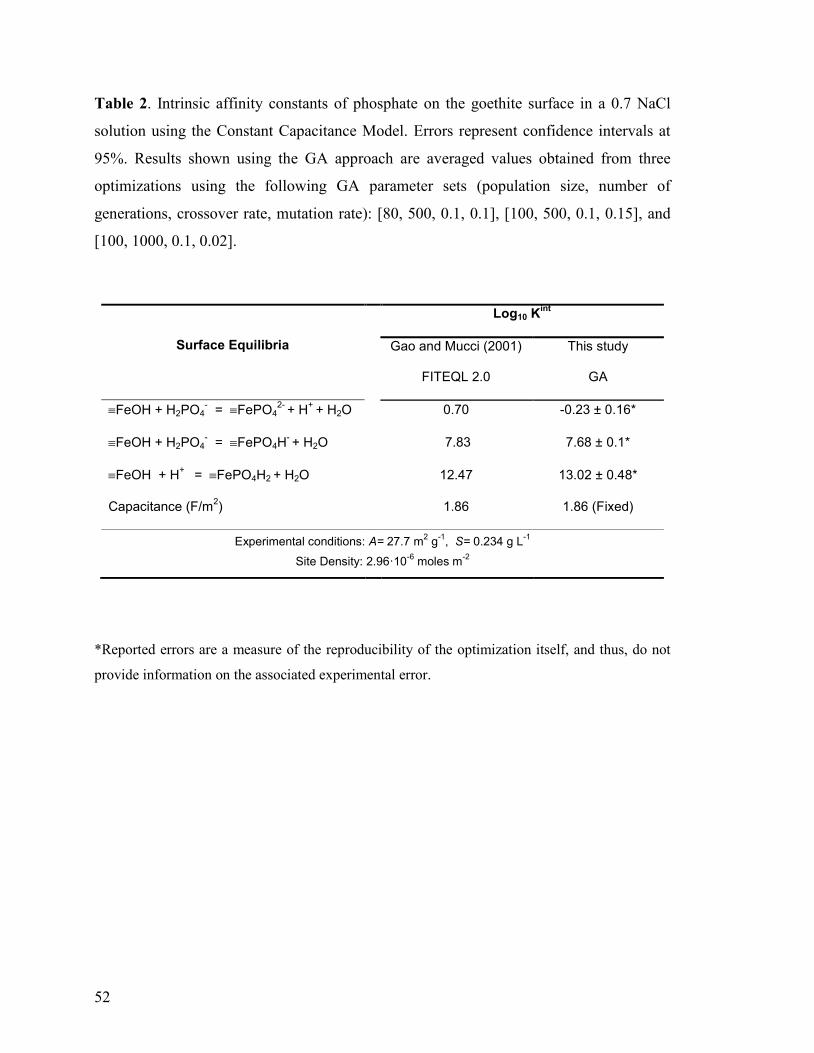
52
Table 2. Intrinsic affinity constants of phosphate on the goethite surface in a 0.7 NaCl
solution using the Constant Capacitance Model. Errors represent confidence intervals at
95%. Results shown using the GA approach are averaged values obtained from three
optimizations using the following GA parameter sets (population size, number of
generations, crossover rate, mutation rate): [80, 500, 0.1, 0.1], [100, 500, 0.1, 0.15], and
[100, 1000, 0.1, 0.02].
Surface Equilibria
Log10 Kint
Gao and Mucci (2001)
FITEQL 2.0
This study
GA
FeOH + H2PO4- = FePO4
2- + H
+ + H2O 0.70 -0.23 ± 0.16*
FeOH + H2PO4- = FePO4H
- + H2O 7.83 7.68 ± 0.1*
FeOH + H+
= FePO4H2 + H2O 12.47 13.02 ± 0.48*
Capacitance (F/m2) 1.86 1.86 (Fixed)
Experimental conditions: A= 27.7 m2 g
-1, S= 0.234 g L
-1
Site Density: 2.96·10-6
moles m-2
*Reported errors are a measure of the reproducibility of the optimization itself, and thus, do not
provide information on the associated experimental error.
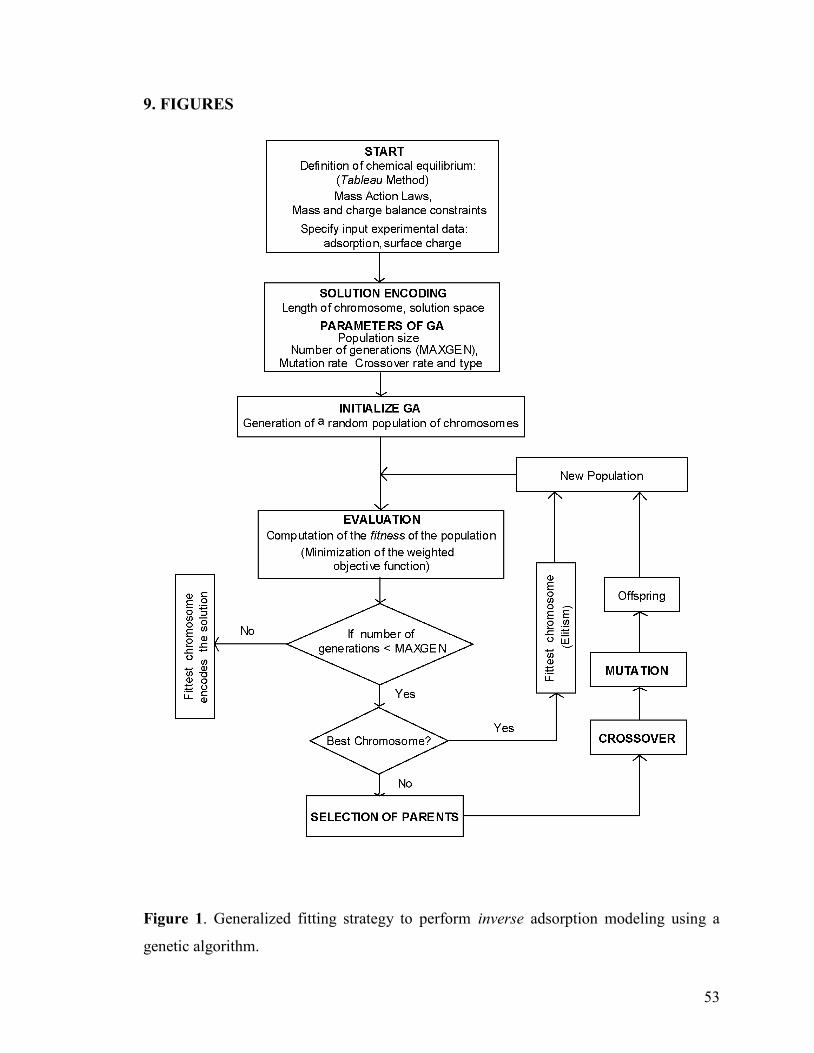
53
9. FIGURES
Figure 1. Generalized fitting strategy to perform inverse adsorption modeling using a
genetic algorithm.
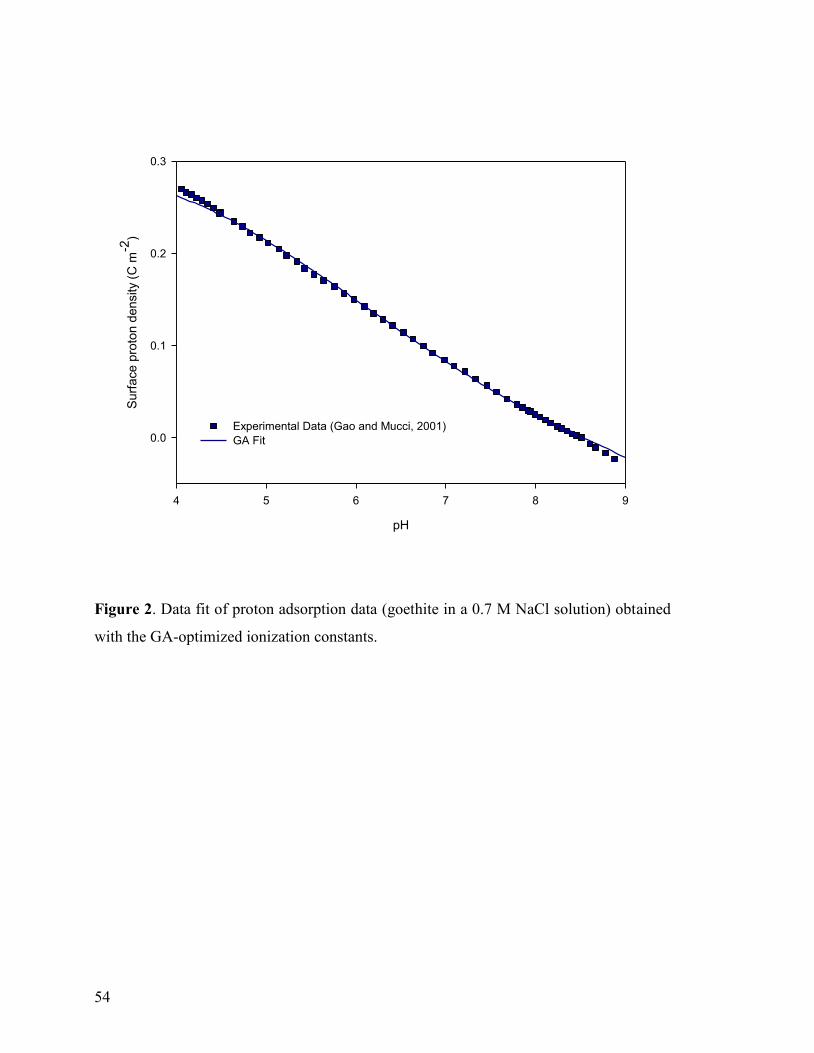
54
pH
4 5 6 7 8 9
Su
rfa
ce
pro
ton d
en
sity
(C m
-2)
0.0
0.1
0.2
0.3
Experimental Data (Gao and Mucci, 2001)
GA Fit
Figure 2. Data fit of proton adsorption data (goethite in a 0.7 M NaCl solution) obtained
with the GA-optimized ionization constants.
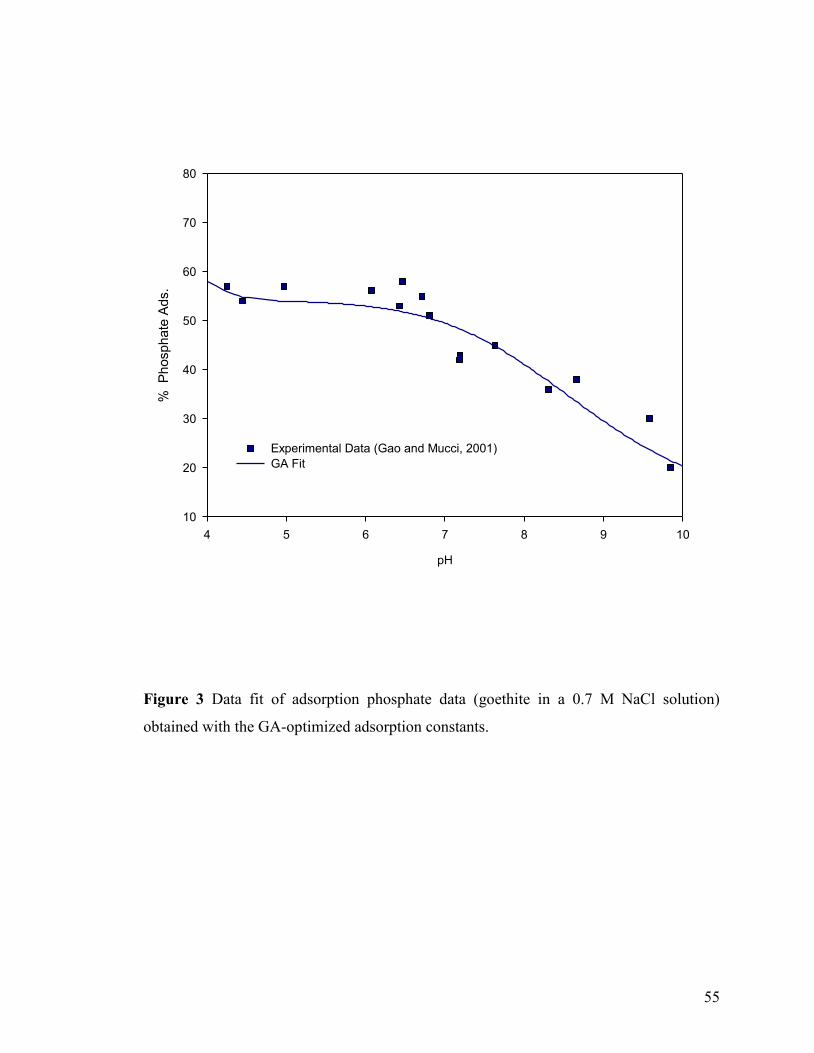
55
pH
4 5 6 7 8 9 10
% P
hosphate
Ads.
10
20
30
40
50
60
70
80
Experimental Data (Gao and Mucci, 2001)
GA Fit
Figure 3 Data fit of adsorption phosphate data (goethite in a 0.7 M NaCl solution)
obtained with the GA-optimized adsorption constants.
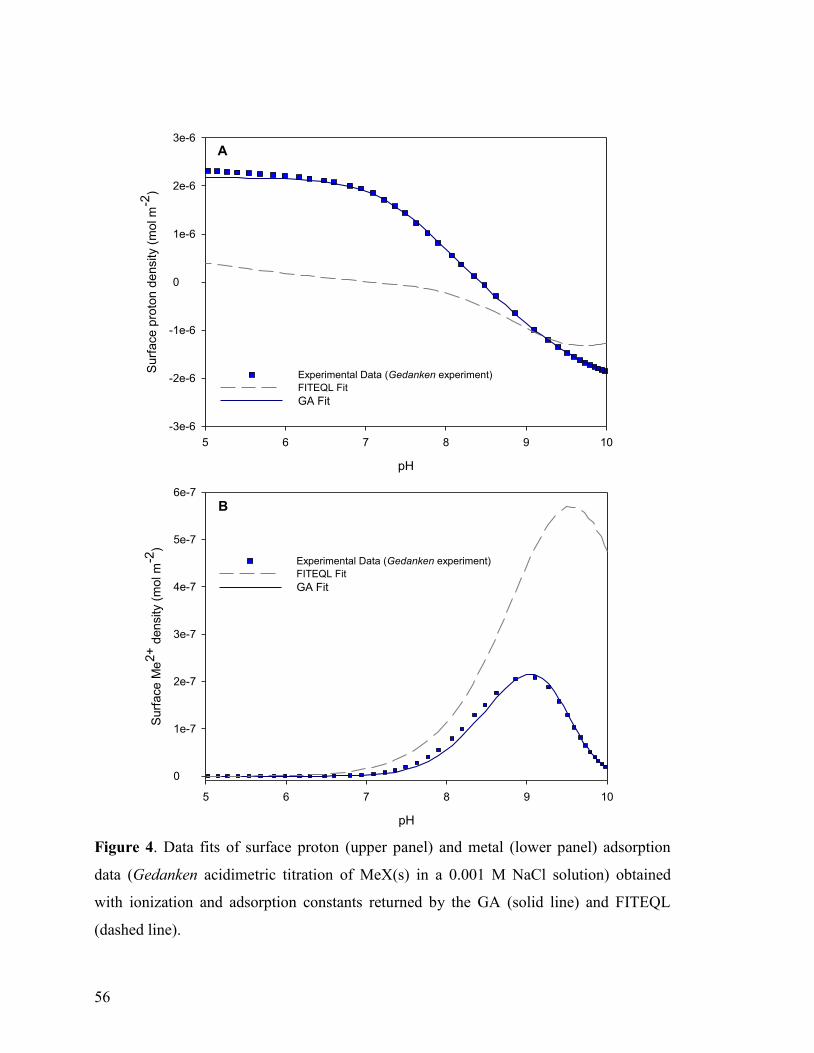
56
pH
5 6 7 8 9 10
Surf
ace M
e2+
density (
mol m
-2)
0
1e-7
2e-7
3e-7
4e-7
5e-7
6e-7
pH
5 6 7 8 9 10
Surf
ace p
roto
n d
ensity (
mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
Experimental Data (Gedanken experiment)
FITEQL Fit
GA Fit
A
B
Experimental Data (Gedanken experiment)
FITEQL Fit
GA Fit
Figure 4. Data fits of surface proton (upper panel) and metal (lower panel) adsorption
data (Gedanken acidimetric titration of MeX(s) in a 0.001 M NaCl solution) obtained
with ionization and adsorption constants returned by the GA (solid line) and FITEQL
(dashed line).
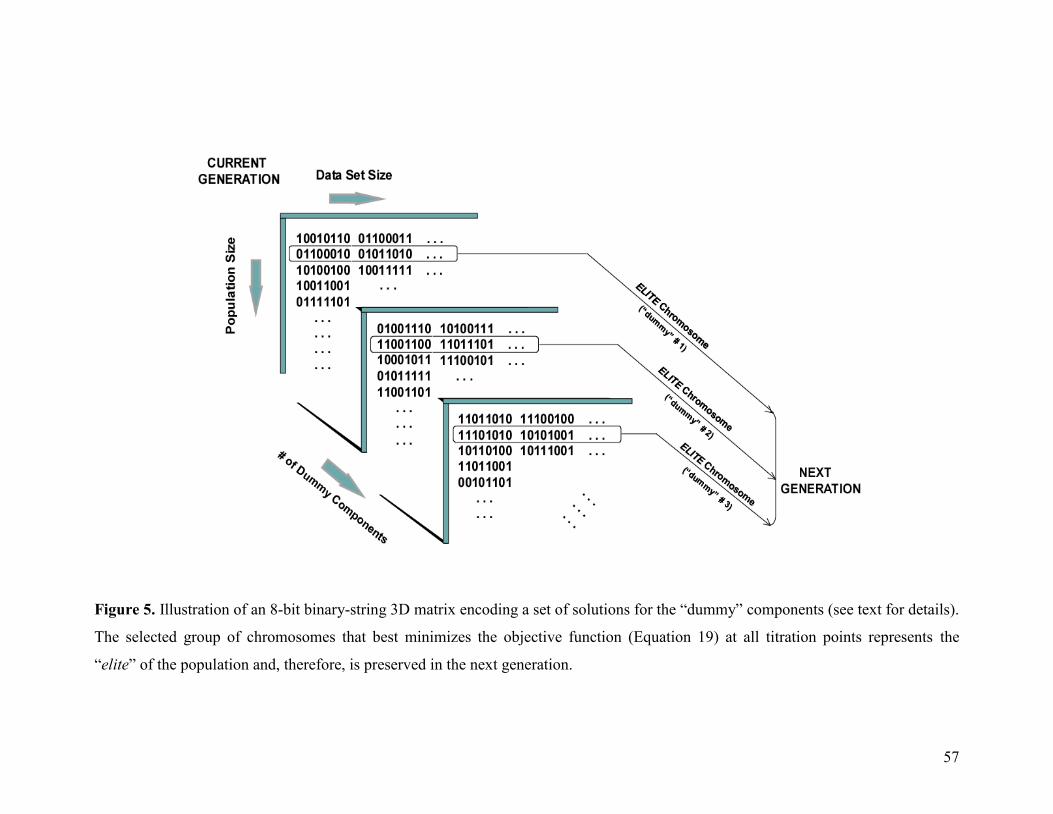
57
Figure 5. Illustration of an 8-bit binary-string 3D matrix encoding a set of solutions for the “dummy” components (see text for details).
The selected group of chromosomes that best minimizes the objective function (Equation 19) at all titration points represents the
“elite” of the population and, therefore, is preserved in the next generation.
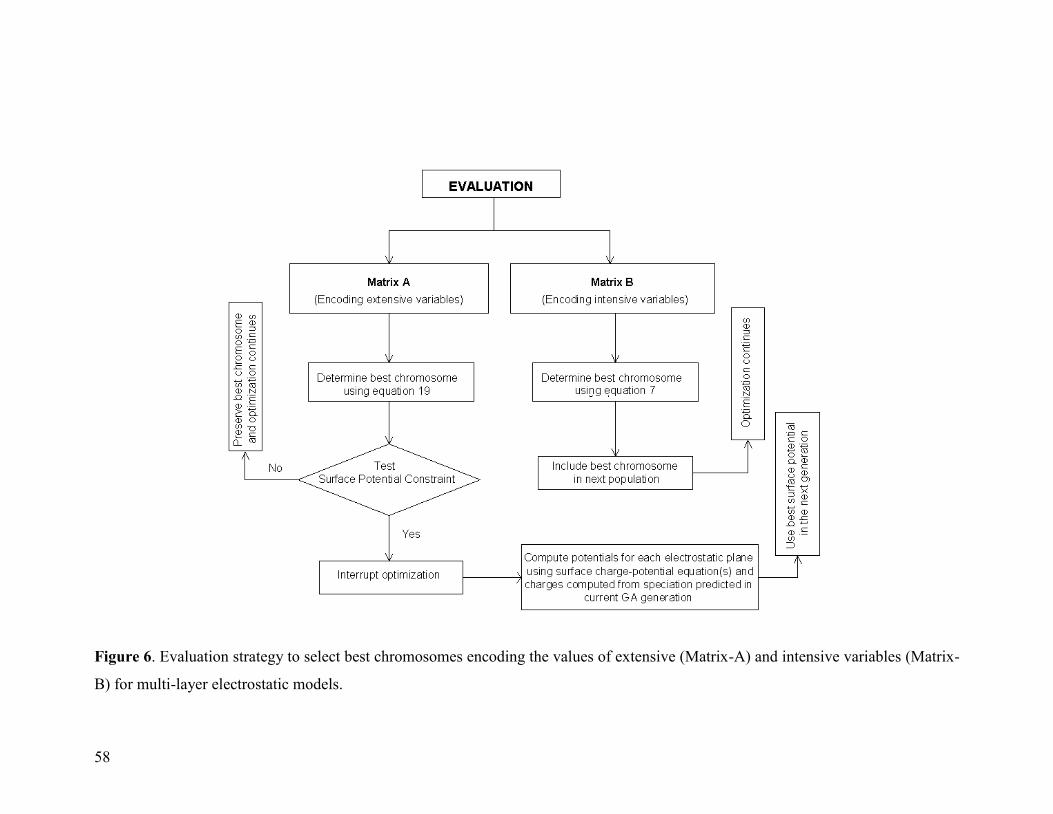
58
Figure 6. Evaluation strategy to select best chromosomes encoding the values of extensive (Matrix-A) and intensive variables (Matrix-
B) for multi-layer electrostatic models.
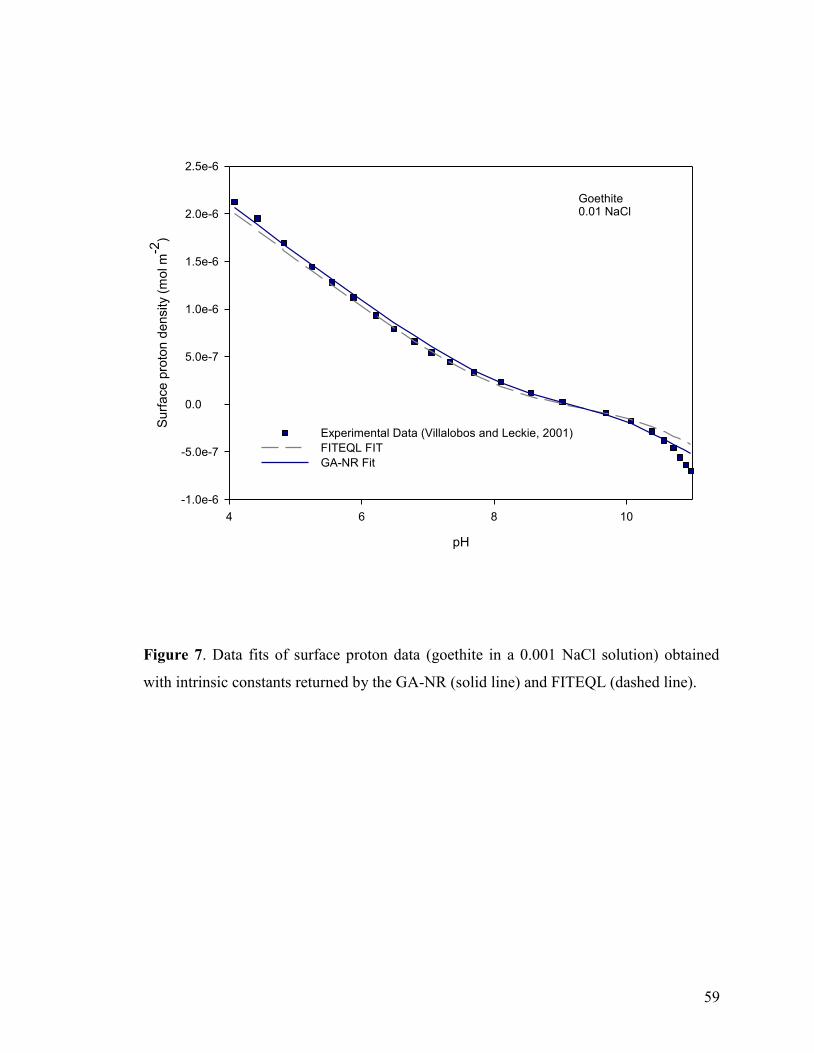
59
pH
4 6 8 10
Su
rfa
ce
pro
ton
de
nsity
(mo
l m
-2)
-1.0e-6
-5.0e-7
0.0
5.0e-7
1.0e-6
1.5e-6
2.0e-6
2.5e-6
Experimental Data (Villalobos and Leckie, 2001)
FITEQL FIT
GA-NR Fit
Goethite0.01 NaCl
Figure 7. Data fits of surface proton data (goethite in a 0.001 NaCl solution) obtained
with intrinsic constants returned by the GA-NR (solid line) and FITEQL (dashed line).

60
PREFACE TO CHAPTER 3
In the early 1990‟s, researchers designed and implemented a novel flow-through titration
reactor to perform acid-base titrations of carbonate mineral surfaces which was used to
generate surface charge data for rhodochrosite, siderite, magnesite, and dolomite. Using
these data, the first SCMs were devised for these minerals in aqueous solutions based
upon a two (siderite, rhodochrosite and magnesite) and four reactive site scheme
(dolomite). To date, these studies provide the only available surface charge data for
carbonate minerals whose quantitative interpretation in terms of surface complexation
reactions represents a first-order description of the chemistry of the carbonate/aqueous
solution interface. No significant refinements or major improvements to these SCMs have
been achieved since, from either an experimental or theoretical standpoint.
In the following chapter: “Defining Reactive Sites on Hydrated Mineral Surfaces:
Rhombohedral Carbonate Minerals”, we critically review the definition of reactive
surface sites whereupon mass-action expressions describing adsorption equilibria at the
hydrated surface of rhombohedral carbonate minerals are formulated. The analysis led to
the derivation of a single generic charge-neutral surface site scheme analogous to the one-
site 2-pKa ionization model employed in the description of the amphoteric behavior of
numerous metal oxide surfaces. This scheme allows for a simplified and generalized
representation of surface equilibria for this type of minerals and is compatible with
experimental and theoretical findings as well as with common assumptions implied by
SCMs. Via the GA approach introduced in the previous chapter, new SCMs were
calibrated, using available data for magnesite and dolomite and evaluated qualitatively
against published electrokinetic for these two minerals.

61
CHAPTER 3
DEFINING REACTIVE SITES ON HYDRATED MINERAL SURFACES: RHOMBOHEDRAL CARBONATE MINERALS
Adrián Villegas-Jiménez*1, Alfonso Mucci
1, Oleg S. Pokrovsky
2 and Jacques Schott
2
1 Earth and Planetary Sciences, McGill University, 3450 University Street
Montréal, Qc H3A 2A7, Canada.
2Géochimie et Biogéochimie Expérimentale,
LMTG, UMR 5563,
Université de Toulouse – CNRS, 14, Avenue Edouard Belin 31400 Toulouse, France
*Corresponding Author
E-mail: [email protected]
Published in Geochimica Cosmochimica Acta
Geochim. Cosmochim. Acta 73(15) 4326-4345

62
ABSTRACT
Despite the success of surface complexation models (SCMs) to interpret the adsorptive
properties of mineral surfaces, their construct is sometimes incompatible with
fundamental chemical and/or physical constraints and, thus, cast doubts on the physical-
chemical significance of the derived model parameters. In this paper, we address the
definition of primary surface sites (i.e., adsorption units) at hydrated carbonate mineral
surfaces and discuss its implications to the formulation and calibration of surface
equilibria for these minerals.
Given the abundance of experimental and theoretical information on the structural
properties of the hydrated (10.4) cleavage calcite surface, this mineral was chosen for a
detailed theoretical analysis of critical issues relevant to the definition of primary surface
sites. Accordingly, a single, generic charge-neutral surface site (CaCO3·H2O0) is defined
for this mineral whereupon mass-action expressions describing adsorption equilibria were
formulated. The one-site scheme, analogous to previously postulated descriptions of
metal oxide surfaces, allows for a simple, yet realistic, molecular representation of
surface reactions and provides a generalized reference state suitable for the calculation of
sorption equilibria for rhombohedral carbonate minerals via Law of Mass Action (LMA)
and Gibbs Energy Minimization (GEM) approaches.
The one-site scheme is extended to other rhombohedral carbonate minerals and
tested against published experimental data for magnesite and dolomite in aqueous
solutions. A simplified SCM based on this scheme can successfully reproduce surface
charge, reasonably simulate the electrokinetic behavior of these minerals, and predict
surface speciation agreeing with available spectroscopic data. According to this model, a

63
truly amphoteric behavior is displayed by these surfaces across the pH scale but at
circum-neutral pH (5.8-8.2) and relatively high CO2 ( 1 mM), proton/bicarbonate co-
adsorption becomes important and leads to the formation of a charge-neutral H2CO3-like
surface species which may largely account for the surface charge-buffering behavior and
the relatively wide range of pH values of isoelectric points (pHiep) reported in the
literature for these minerals.
Keywords: One-site surface complexation, carbonate minerals, primary surface sites,
residual charges, carbonic acid-like surface specie.

64
1. INTRODUCTION
Surface complexation models (SCMs) have been extensively applied to the interpretation
of adsorption and surface reactivity data on a large number of minerals in aqueous
solutions. Their relative simplicity and capacity to incorporate fundamental concepts of
thermodynamics, crystallography, and inorganic and colloid chemistry make them
suitable tools for the description of the adsorptive properties of minerals under a wide
range of chemical conditions.
Nevertheless, despite the success and refinements achieved by many of these
models, many shortcomings remain to be addressed before their applicability to natural
systems and their validation at the molecular level can be established (e.g., Westall and
Hohl, 1980; Goldberg, 1991; Sahai and Sverjensky, 1997; Kallay and Žalac, 2000; Zuyi
et al., 2000; Lützenkirchen, 2002). Among these, the definition of the surface sites
(hydrated adsorption units) that serve as reference species, hereafter referred to as
“primary surface sites”, for the formulation of surface reactions is at the heart of a
realistic description of adsorption processes (Healy and White, 1978; Pivovarov, 1997;
Kulik et al., 2000; Kulik, 2002). These can be formalized in terms of discrete chemical
units of given chemical composition and charge, in analogy to functional groups of ionic
and molecular aqueous species. However, they differ from their aqueous analogues
because primary surface sites have a fixed density per unit area and cannot be diluted
infinitely on the surface (Kulik, 2002). These properties affect the definition of standard
states for surface species and reflect on the values of the intrinsic formation constants
(Kint
) of surface species (Kulik, 2002; Sverjensky, 2003). Other distinctions include
stereochemical, structural and electrostatic conditions at the mineral/water interface that

65
influence the energetics of the primary surface sites (Sposito, 1989; Zachara and Westall,
1999).
Two major ions binding schemes (or models) have been postulated for the
formulation of SCMs: one-site and multi-site complexation. One-site schemes assign an
average “macroscopic” reactivity to all atoms present at the mineral surface whereas
multi-site schemes formalize the reactivities of individual surface atoms in terms of their
chemical identity, coordination environment, and hydrogen bonding arrangements
(Hiemstra et al., 1989; Barrow et al., 1993; Hiemstra et al., 1996). Despite their generic
nature, one-site schemes are simple, practical, and powerful predictive tools based upon
well established statistical mechanical grounds (Borkovec, 1997) and are, despite their
disregard of the complexities inherent to real-world sorbents (chemical heterogeneity),
the foundation of numerous electrical double-layer models that describe the charge-
potential relationship at the mineral/water interface (Sposito, 1983).
On the other hand, multi-site schemes are more realistic insofar as they reflect,
semi-quantitatively, the compositional “heterogeneity” of the predominant mineral
surfaces (Hiemstra et al., 1989; Hiemstra and van Riemsdijk, 1991; Hiemstra and van
Riemsdijk, 1996; Scheidegger and Sparks, 1996). Nevertheless, the quantitative
characterization of the reactivity of individual primary surface sites is far from trivial
because the proper calibration of these multi-site, multi-reaction models requires: (i)
uniform and/or well-characterized mineral surfaces in terms of chemical composition and
micro-topography (Barrow et al., 1993; Piasecki et al., 2001), (ii) suitable experimental
data arising from various independent sources and carrying sufficient information to
properly resolve the energetic contributions of individual surface sites (Rudziński et al.,
1992, 1998; Piasecki et al., 2001), and (iii) the application of sophisticated mathematical

66
treatments (Chandler, 1987; Jäger, 1991; Borkovec and Koper, 1994). For instance, it is
well known that, because of compensating effects, the composite adsorption (or surface
charge) isotherms obtained from titration experiments that are typically used for the
calibration of adsorption chemical models, are largely insensitive to surface energetic
heterogeneity and, therefore, additional data (e.g., potentiometric, electrokinetic,
radiometric, calorimetric) are required to properly discriminate among potential
heterogeneity models and prevent misleading over-interpretations of available data (van
Riemsdijk et al., 1987; Blesa and Kallay, 1988; Ĉerník et al., 1995; Rudziński et al.,
1992, 1998; Lützenkirchen, 2005). Furthermore, the presence of surface irregularities
(steps, kinks, and dislocations), chemical micro-heterogeneities, and multi-domain
crystal surfaces, difficult to characterize experimentally, add to the complexity of multi-
site models, so that the physical-chemical significance of the derived model parameters
and their application to natural systems is seriously questioned (Lützenkirchen, 1997). It
follows that simpler models are expected to remain prominent in the quantitative
modeling of equilibrium adsorption phenomena (Lützenkirchen, 2002) and kinetic
dissolution processes (Bandstra and Brantley, 2008).
Despite the lack of scientific consensus with regards to the application of one-site
vs multi-site schemes to describe sorption reactions, it is generally agreed that primary
surface sites must contain sufficient information about the sorbent phase for the accurate
description of its surface reactivity while allowing for a simple and realistic
representation of sorption equilibria (Kulik, 2002). Furthermore, because Law of Mass
Action (LMA)-based sorption modeling approaches (Morel and Morgan, 1972; Westall
and Hohl, 1980; Goldberg, 1995), frequently incorporated in widespread computer codes
(e.g., MINEQL, HYDRAQL, PHREEQC, FITEQL etc.), are subjected to charge and

67
mass balance constraints, the definition of primary surface sites in terms of their residual
charges and elemental stoichiometry is critical for the reliable estimation of model
parameters (i.e., intrinsic formation constants of surface species, capacitances, site
densities) and the solution of surface speciation equilibria. This issue is the focus of the
present study within the context of rhombohedral carbonate minerals.
We begin by highlighting critical aspects relevant to the definition of the residual
charges and the elemental stoichiometry of primary surface sites. Later, the discussion
focuses on the (10.4) cleavage calcite surface, as a model for all rhombohedral carbonate
minerals, to rationalize the available theoretical and experimental evidence and formalize
a realistic primary surface site for these surfaces. Accordingly, new surface equilibria are
derived, calibrated and evaluated against published experimental data (i.e., surface
charge, electrokinetic, and spectroscopic) for two common rhombohedral carbonate
minerals: magnesite and dolomite.
2. DEFINITION OF PRIMARY SURFACE SITES
2.1 Charge Assignment
Atomic charges are not physical properties that can be readily defined accurately
(Chandra and Kollman, 1984) and, hence, their assignment to primary surface sites at
mineral surfaces is problematic. Typically, either a “zero residual charge” or a “fractional
residual charge” scheme is assigned to primary surface sites in surface complexation
studies. Whereas the former is based on simple, yet realistic, chemical stability grounds
(charge dissipation upon surface hydration), the latter is based upon Bond Valence
concepts: “Pauling‟s Electrostatic Valence Principle” (Pauling, 1929) and “Bond Valence

68
Theory” (Brown, 1981). These concepts were originally developed and calibrated to bulk
structures and were later applied to idealized, unrelaxed, unreconstructed metal oxide
surfaces (displaying no bond relaxation and/or bond breaking) for the estimation of the
unsatisfied valence of surface atoms which was considered as an approximate measure of
their residual charge (Yoon et al., 1979; Hiemstra et al., 1989; Bleam, 1993; Hiemstra et
al., 1996). It should be noted that Bond Valence Theory, a semi-empirical approach based
upon central atom valences, coordination number and bond lengths, must not be confused
with Valence-Bond Theory that complements Molecular Orbital Theory and involves
fundamental quantum chemistry concepts where bonding is accounted for in terms of
atomic valences and hybridized orbitals (Gallup, 2002).
Depending on the residual charge and relative abundance of the primary surface
sites, the unreacted hydrated mineral surface will carry a neutral, positive, or negative
“reference charge density” (REF) as described by (Hiemstra and van Riemsdijk, 1996):
jjREF NzF (1)
where zj and Nj represent, respectively, the charge and density (in moles m-2
) of the j
primary surface site and F is the Faraday constant. In other words, REF represents the net
charge of the surface when only primary surface sites are present. It is the resultant of
crystal truncation (“dangling bonds”) and mineral hydration which, in turn, may lead to
the re-organization of bonds (e.g., bond relaxation, bond breaking and/or bond making) at
the mineral surface and the establishment of “unknown” residual charges at the primary
surface sites. REF contributes to the net surface charge density (0, C m-2
) as follows:

69
REFPSISH0 (2)
where H is the net proton surface charge density (F(ΓH+ - ΓOH-), ΓH+ and ΓOH- are,
respectively, the net surface H+ and OH
- adsorption densities in mol m
-2), IS is the net
charge density resulting from the total charge of ions (excluding H+ and OH
-) bound by
inner-sphere surface coordination, and PS is the net permanent structural surface charge
arising from isomorphic substitutions exhibited by some minerals (Chorover and Sposito,
1995). For simplicity, we will focus our discussion exclusively on minerals without
permanent structural charge (PS = 0) and only within the context of adsorption at the 0-
plane (surface).
The calibration of intrinsic formation constants (Kint
) by LMA approaches
(Herbelin and Westall, 1996) is constrained by proton and, if applicable, inner-sphere ion
adsorption data (HandIS, respectively) and is subjected to the following charge
equality constraint:
elect0REF0iiz
ΑS
][
F (3)
where A is the specific surface area (m2 g
-1), S is the solid to solution ratio (g L
-1), zi is
the net charge transfer of the reaction producing surface species i, [i0] is the molar
concentration of species i adsorbed at the surface (plane-0), and 0elect
represents the
electrostatically-derived net surface charge density (an a priori unknown) computed from

70
an electrostatic interfacial model (EIM), describing the surface charge-potential
relationship (Westall and Hohl, 1980), and the iteratively-optimized surface potential
(0). The left-hand term in Equation 3 represents the “net surface charge” and requires
definition of REF and computation of the “apparent surface charge” (0app
, first left-hand
term of Eq. 3) from the surface speciation predicted by the iteratively-optimized Kint
values. Because Kint
and 0elect
are interdependent (Eq. 3), their optimization is a function
of two fixed experimentally-accessible quantities (H and IS, although their
measurement may not be trivial for some minerals) and one ill-defined quantity (REF). In
addition, the latter affects the Kint
values via the estimated 0 which depends on the
adjusted 0elect
and the selected EIM. Any modification in the values of 0 is reflected in
the electrostatic correction necessary to reference apparent constants, Kapp
, to a zero
potential standard state for the calibration of Kint
values:
RT
F- 0i ψZexpKK intapp (4)
where R is the gas constant and T is the absolute temperature. The main corollary to this
discussion is that the selected REF may impact the calibration of the intrinsic formation
constants via LMA approaches, and therein lies the importance of assigning appropriate
charges to primary surface sites.

71
2.2 Elemental Stoichiometry
The selection of the elemental stoichiometry of the primary surface sites is also
critical because it influences the molecular representation of surface reactions and
participates in the mass balance constraint imposed by LMA-based approaches (total
crystallographic site density) in the modeling of sorption equilibria (Kulik, 2002).
The simplest scenario is to consider individual surface atoms (hydroxylated or
hydrated) as the primary surface sites. However, electrostatic and steric interactions
between neighboring surface species may arise and affect the energetics of the sorption
processes. For instance, vicinal surface atoms may both interact with the same sorbate
(bidentate adsorption; Ludwig and Schindler, 1995) or, upon reaction with a sorbate,
adjacent surface atoms may be inactivated (Benjamin, 2002). In both cases, two adjacent
surface atoms could be formalized as one surface site.
These premises were championed by Pivovarov (1997) who, based upon
crystallographic considerations, proposed the elemental stoichiometry of two generic
types of charge-neutral surface sites for the hydrated hematite surface, (FeOH)2(OH2)+
and (O3H)2(H2O)-, assuming that H2O molecules physically adsorbed to hydroxylated
vicinal surface metal and oxygen atoms represent a single “adsorption center”
whereupon surface reactions occur. This approach yielded surface OH group densities in
close agreement with experimental values (Morimoto et al., 1969). A similar definition of
the elemental stoichiometry of one generic surface site for all metal oxides (O0.5H) was
postulated by Kulik (2002) under the assumption that primary surface sites at mineral
surfaces comprise H2O molecules from the adsorbed water monolayer. This definition of
surface sites was influenced by results of wet chemical, spectroscopic and molecular

72
modeling studies that confirmed the presence of OH groups at metal oxide surfaces
(Morimoto et al., 1969; Davis and Kent, 1990). The proposed one-site scheme considers
the bidentate coordination of a H2O molecule to one surface metal and one surface
oxygen and yields two vicinal charge-neutral hydroxyl groups (i.e., MeOH and OH) of
which either only one is reactive (Kulik, 2002) or both participate simultaneously in the
adsorption processes and are thus conceptualized as a single site (Pivovarov, 1998). This
scenario allowed the modeling of sorption equilibria by both LMA (Pivovarov, 1998) and
Gibbs Energy Minimization (GEM, Kulik, 2002) approaches and provided a suitable
molecular representation of surface reactions at the metal oxide/H2O interface. It follows
that the application of identical criteria for the definition of elemental stoichiometries for
other mineral surfaces is warranted.
3. RHOMBOHEDRAL CARBONATE MINERALS
3.1 Case of the (10.4) Calcite Surface
3.1.1 Evidence from Spectroscopic and Molecular Modeling Studies
The (10.4) calcite surface has been extensively studied (e.g., Stipp and Hochella, 1991; de
Leeuw and Parker, 1997; Fenter et al., 2000; Wright et al., 2001; Geissbühler et al.,
2004). This surface is of great interest because it represents the most stable and
predominant crystallographic face displayed by this mineral in aqueous solutions and it
serves as a model for other rhombohedral carbonate minerals such as magnesite,
dolomite, siderite, rhodochrosite, and gaspeite. The ideal (10.4) cleavage surface
configuration displays a stoichiometric number of adjacent cations and anions that carry
an equivalent but opposite residual charge per surface unit cell and corresponds to the

73
atomic plane where strictly ionic (metal-oxygen) and no covalent bonds (carbon-oxygen)
are broken. The major stability of this atomic configuration over others was confirmed
using a simple crystal lattice truncation protocol (based upon Bond Valence concepts)
devised to determine the most stable atomic configuration of oxide mineral surfaces
according to charge and bond strength minimization criteria (Koretsky et al., 1998).
Numerous surface-sensitive instrumental techniques have been employed to
characterize the (10.4) calcite surface under wet and/or vacuum conditions such as X-Ray
Photoelectron Spectroscopy (XPS, Stipp and Hochella, 1991; Stipp, 1999), Low Energy
Electron Diffraction (LEED, Stipp and Hochella, 1991; Stipp, 1999), Time-Of-Flight
Secondary Ion Mass Spectroscopy (TOF-SIMS, Stipp, 1999), Infrared Spectroscopy (IR,
Neagle and Rochester, 1990), Fourier Transform Infrared Spectroscopy (FTIR, Kuriyavar
et al., 2000), Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFT,
Pokrovsky et al., 2000), Attenuated Total Reflection-Fourier Transform Infrared
Spectroscopy (ATR-FTIR, Al-Hosney and Grassian, 2005), Atomic Force Microscopy
(AFM, Rachlin et al., 1992; Stipp et al., 1994; Liang et al., 1996; Stipp, 1999), and X-
Ray Reflectivity and Scattering (SXR, Chiarello et al., 1993; Fenter et al., 2000;
Geissbühler et al., 2004). These techniques revealed that the outer-most atomic layer
relaxes and the surface undergoes a certain degree of reconstruction upon hydration. The
presence of OH groups was detected near the cleaved (10.4) calcite surface exposed to
moistened conditions (e.g., Stipp and Hochella, 1991; Fenter et al., 2000) and the
formation of a hydration monolayer was confirmed (e.g., Fenter et al., 2000). These
findings established that water constituents are chemically associated with the surface
allowing for the formation of hydrated surface species. Furthermore, “chemisorption” of
water was also demonstrated by earlier thermogravimetric studies (Morimoto et al., 1980;

74
Ahsan, 1992). Nevertheless, it is not yet possible to ascertain, by any of these analytical
techniques, whether hydration occurs through adsorption of dissociated or undissociated
water molecules because these are unable to detect hydrogen atoms and, thus, hydroxyl
ions cannot be distinguished from adsorbed H2O molecules. In other words, it is not
possible, for instance, to distinguish whether the primary surface site: Ca(H2O) or
CaOH° (or both) form at the calcite surface. Consequently, water dissociation products
cannot be ascribed to specific surface atoms. The only conclusion that can be drawn from
these data is that the internal coordinates (i.e. O-H bond lengths and H-O-H angle) of
undissociated water molecules are possibly modified upon adsorption but it is unknown to
what extent. To ascertain whether H2O dissociates to its hydrolysis products (H+
and OH-)
upon adsorption on the calcite surface, additional information is needed.
Theoretical studies provide information on the structure, energetics, and atomic
bonding relationships at the hydrated mineral surface. Computer-assisted Atomistic
simulations (de Leeuw and Parker, 1997; de Leeuw and Parker, 1998; de Leeuw et al.,
1998; Kuriyavar et al., 2000; Hwang et al., 2001; Wright et al., 2001; Kerisit et al., 2003;
Kerisit and Parker, 2004), Molecular Dynamics (Kerisit et al., 2003; Kerisit and Parker,
2004; Kerisit et al., 2005a; Perry et al., 2007), ab initio Density Functional Theory
(Parker et al., 2003; Kerisit et al., 2003; Archer, 2004; Kerisit et al., 2005b), and Roothan-
Hartree-Fock Molecular Orbital Theory (Villegas-Jiménez et al., 2009a) were used to
investigate the interactions of H2O monomers with the (10.4) calcite surface. All these
studies reveal that the internal coordinates of water monomers remain essentially
unchanged upon adsorption and, that associative adsorption of H2O on the (10.4) calcite
surface is energetically favorable (over dissociative adsorption) where one H2O monomer

75
bonds to one calcium atom and is likely hydrogen-bonded to either one (Archer, 2004;
Villegas-Jiménez et al., 2009a) or two neighboring surface oxygen atoms of carbonate
groups (de Leeuw and Parker, 1997; Wright et al., 2001; Kerisit and Parker, 2004; Perry
et al., 2007). In other words, each adsorbed H2O monomer interacts simultaneously with
one cationic and at least one anionic centre, and hence, charge and mass discretization of
hydrolysis products among individual surface atoms is problematic. Clearly, a suitable
formalism must be adopted for mass and charge assignment of water constituents among
individual surface atoms in the definition of primary surface sites.
Extension of these Atomistic simulations to other hydrated (10.4) carbonate
surfaces, such as magnesite and dolomite, reveals that H2O adsorbs associatively on these
surfaces according to a 1:1 H2O:MeCO3(surface) stoichiometry where each adsorbed H2O
molecule interacts with one metal center (Mg for magnesite and Mg or Ca for dolomite)
and at least one neighboring O surface atom (de Leeuw and Parker, 2001; de Leeuw and
Parker, 2002; Wright et al., 2001; Parker et al., 2003; Kerisit et al., 2005a; Austen et al.,
2005). This information strongly suggests that, regardless of the specific orientation of
adsorbed H2O molecules relative to the mineral surface, all (10.4) single- and mixed-
metal carbonate surfaces are subjected to similar hydration processes where water
remains undissociated upon adsorption.
3.1.2 Single Generic Primary Surface Site
Based upon the results of spectroscopic and molecular modeling studies, a generic
primary surface site for the (10.4) cleavage calcite surface can be defined as:
(CaCO3)·H2O, where the constituents in parentheses represent surface atoms. This

76
reactive surface site reflects the elemental stoichiometry of the (10.4) hydrated surface
unit cell: two neighboring surface atoms, one metal atom and one carbonate group,
interacting with one water (undissociated) molecule (Figure 1a). This scheme is
equivalent to those of Kulik (2002) and Pivovarov (1997) for metal oxides insomuch as
the adsorbed H2O molecules are considered as the reactive elemental units at the surface,
whereupon surface reactions occur.
Because a stoichiometric number of divalent cationic (Ca atoms) and anionic (CO3
groups) are present at the idealized (10.4) cleavage calcite surface (and of all
rhombohedral carbonate minerals to that matter), and regardless of the residual charge
displayed by individual surface atoms (following bond re-organization on hydration),
charge-neutrality should be preserved at the idealized stoichiometric unit, (CaCO3), and
maintained upon adsorption of neutral H2O monomers. Accordingly, a neutral charge can
be assigned to the newly defined primary surface site: (CaCO3)·H2O0.
In contrast to earlier multi-site SCMs that assume the formation of primary surface
sites of type MeOH
and CO3H, (δ = residual charge), the one-site scheme
circumvents the problem of mass and charge discretization allowing for a generic, yet
realistic, mass and charge localization at the primary surface site, rather than at individual
surface atoms. This yields a REF=0, identical to that of earlier multi-site SCMs for single-
and mixed-metal rhombohedral carbonate surfaces (Van Cappellen et al., 1993;
Pokrovsky et al., 1999a,b; Pokrovsky and Schott, 2002; Wolthers et al., 2008) and
consistent with the reference level of zero proton charge typically adopted by 2-pK
models (Sposito, 1998). Detailed structural information of hydrated (relaxed) carbonate
surfaces from independent studies (e.g., molecular modeling, Fitts et al., 2005; Kubicki et

77
al., 2008) is required to determine realistic bond lengths, coordination environments and
hydrogen bonding arrangements of surface atoms as well as derive more accurate δ values
(and REF), as emphasized by earlier workers (Bickmore, 2004; Villegas-Jiménez et al.,
2005; Wolthers et al., 2008).
3.2. SCM Reactions: One-Site vs Two-Site Scheme
The one-site scheme is analogous to the one describing non-overlapping bidentate
adsorption, where pairs of specific neighboring surface sites (rather than pairs of random
neighboring surface sites as for overlapping bidentate adsorption) react with a given
sorbate to produce a bidentate surface complex (Benjamin, 2002), and thus, two
neighboring sites are inactivated for further reaction (see Figure 1b). For carbonate
minerals, polydentate adsorption is well-exemplified by the interaction of aspartate with
calcium ions on the calcite surface (Teng and Dove, 1997), leading to a large perturbation
of the local molecular surface geometry, significant steric effects and the inactivation of
adjacent sites (see also the “Umbrella effect”, Kovačević et al., 1998). This contrasts with
the two-site scheme where, with the exception of synergistic effects related the
development of the macroscopic electrical potential, each site is independent and, thus, it
is assumed that adsorption at one site does not affect the macroscopic reactivity of any of
its neighbors.
Earlier workers described the charging behavior of single-metal carbonate
minerals with six generic reactions (ionization and constituent-ion adsorption reactions)
based upon a two-site (Van Cappellen et al., 1993; Pokrovsky et al., 1999a; Pokrovsky
and Schott, 2002) or a multi-site scheme (Pokrovsky et al., 1999b; Wolthers et al., 2008).
Similarly, analogous reactions can be derived for the one-site scheme (see Table 1).

78
Although reactions are equivalent in terms of charge and mass transfer, the participating
individual species in both schemes carry a distinct stoichiometry and different
“conceptual” mechanisms are implied in each case. Figure 2 illustrates ionization
reactions conceptualized at the molecular-level for the one-site scheme.
The main distinction between the one-site and two-site models is revealed upon
reaction of the primary surface sites. In the case of the protonation reaction for calcite
(reactions 3a and 3b, Table 1), the one-site model conceptually involves the participation
of both CO3H0
and CaOH0 sites to produce the protonated positively-charged specie:
(CaCO3)·H3O+. In contrast, according to the two-site scheme, only one cationic site
reacts to produce the protonated positively-charged specie: CaOH2+ while leaving one
anionic site, CO3H0, available for further reaction. In other words, in both cases, one
positive charge is transferred to the surface per mole of primary surface site reacted but,
whereas only one primary surface site is available for further reaction in the one-site
model, (CaCO3)·H2O0 (on a surface unit cell basis), three sites remain available
according to the two-site model, one CaOH0 and two CO3H
0. Once surface equilibrium
is established, the charge density of the surface unit cell will depend on whether
additional reactions take place at the surface (e.g., constituent ion adsorption). Clearly,
multi-site schemes allow for a multitude of reactions that can lead to surface charge
acquisition and may equally reproduce experimental surface charge data, at the expense
of a larger number of unknown parameters (that must be adjusted or arbitrarily selected,
see below) than for the one-site scheme. This has important consequences in the
calibration of intrinsic formation constants and directly reflects on surface speciation
predictions, as discussed in section 4.2. In Figure 3, the availability of “unreacted”

79
primary surface sites at a generic single-metal (10.4) carbonate surface is illustrated as a
function of the proton occupancy for acid-base reactions formulated for the one-site
(reactions 1a to 3a, Table 1) and two-site (reactions 1b to 3b, Table 1) schemes.
3.3 Mixed-Metal Carbonate Minerals
In mixed-metal rhombohedral carbonate minerals, two different types of constituent
cations (i.e. Me1 and Me2) alternate along the (10.4) surface. Thus, for the one-site
scheme, two types of generic primary surface sites, (Me1CO3)·H2O0 and
(Me2CO3)·H2O0 would be required to formulate equivalent surface reactions to those of
the earlier four-site SCM model for dolomite that requires twelve surface reactions
(Pokrovsky et al., 1999b). The Kint
values describing these reactions, however, would be
difficult to calibrate without a combination of suitable experimental data (e.g., titration,
calorimetric, radiometric, electrokinetic) acquired over a wide range of solution
compositions and use of suitable mathematical strategies that would allow resolution of
the contribution of individual surface reactions (through their intrinsic formation
constants) to the development of surface charge (see Introduction). Unfortunately,
because of their reactivity (fast dissolution/precipitation kinetics, Pokrovsky and Schott,
2002), the experimental characterization of the sorptive properties of carbonate minerals
is typically constrained to a relatively narrow range of solution compositions (pH, Me
and/or CO2, solid:solution ratios) which complicates the quantitative evaluation of these
constants. This type of calibration would be even more problematic within the Charge
Distribution MultiSite Ion Complexation (CD-MUSIC) approach as it requires the fitting
and/or arbitrary selection of a larger number of parameters (Wolthers et al., 2008). Other

80
approaches, such as molecular modeling techniques (Rustad et al., 1996; Wasserman et
al., 1999), may be required to establish critical theoretical constraints for the accurate
evaluation of the individual reactivity of multiple primary surface sites at carbonate
surfaces.
An alternate treatment for mixed carbonate minerals is to formulate surface
reactions in terms of a single charge-neutral surface site that reflects half the
stoichiometry of the hydrated (10.4) surface unit cell: (MeCO3)·H2O0 where MeCO3
represents generically Me1 or Me2 (e.g., Ca or Mg for dolomite). Under this scheme, the
number of adjustable intrinsic formation constants is reduced by at least a factor of two
with respect to previous multi-site models (i.e., dolomite: Pokrovsky et al., 1999b;
Wolthers et al., 2008), which renders the model more mathematically tractable. This
formalism is justified by the inadequacy of the available experimental data for the proper
calibration of multiple surface reactions and largely disregards the attribution of too much
mechanistic meaning (e.g., surface site heterogeneity) to composite charging curves or
adsorption isotherms (see Lützenkirchen, 1997). The newly-formulated surface reactions
can be generalized for single- and mixed-metal carbonate minerals as shown in Table 1,
the only difference being that, for mixed-metal carbonate minerals, one additional cation
adsorption reaction (reaction 4a, Table 1) is required to express the individual affinity of
Me1 and Me2 towards (MeCO3)·H2O0. One corollary to this single charge-neutral
surface site formalism is that an average reactivity is assigned to the generic surface site,
and hence, whether surface reactions formally take place at (Me1CO3)·H2O
or
(Me2CO3)·H2O remains undefined. In other words, the individual reactivities of these
sites are averaged out during model calibration and expressed in terms of the formation

81
constant, a reasonable approximation considering that the individual site reactivities in
mixed-metal carbonate minerals are hard to decouple experimentally. That Ca2+
and Mg2+
ions adsorb in near-stoichiometric ratios on dolomite surfaces over a wide range of pH,
Ca/Mg, ionic strength, and pCO2 (Brätter et al., 1972; Brady et al., 1996) suggests that
both primary surface sites exhibit similar reactivities.
4. EVALUATION OF THE ONE-SITE SCHEME
4.1. Re-Calibration of Surface Reactions for Magnesite and Dolomite
Despite all the arguments provided in favor of the one-site scheme, it is important to test
the relevance of the derived reactions against experimental data. To this end, the
calibration of one-site-based SCMs for magnesite and dolomite was performed using
experimental surface charge data from Pokrovsky et al. (1999a,b). These data were
obtained using a modified limited residence time (LRT) reactor where the pH was varied
by additions of NaOH or HCl. Given the experimental difficulties involved in the
experimental protocol, only a limited number of data points could be obtained for each
acid-base titration ( 13) under a range of chemical conditions selected at the beginning
of each experiment (i.e. magnesite: g = 0.8 to 7 mM and CO2 = 0.9 to 29 mM;
dolomite: g = 0.07 to 3.8 mM, Ca = 0.03 to 5.7 mM; CO2 = 0.6 to 13 mM).
Unfortunately, the one-site scheme cannot be tested for calcite because of the dearth or
lack of reliable data characterizing the proton and constituent ion sorptive properties of
this highly reactive carbonate mineral (see Villegas-Jiménez et al., 2009b).
In earlier multi-site SCMs models for carbonate minerals, (Van Cappellen et al.,
1993; Pokrovsky et al., 1999a,b; Pokrovsky and Schott, 2002) a single set of surface

82
reactions (involving reactions 1b-6b) was used in model calibration. Additional reactions
were considered in a recent SCM to account for the reactivity of primary surface sites at
terraces, corners and edges and consider the formation of a new surface species: ≡CO3H2+
(Wolthers et al., 2008). Given the nature of the available experimental data (composite
surface charge or electrokinetic data rather than adsorption data) and because of the large
number of adjustable parameters (Kint
‟s, capacitances, etc.), numerical optimization using
commercially-available computer codes such as FITEQL (Herbelin and Westall, 1996)
was not attempted by earlier workers (Van Cappellen et al., 1993; Pokrovsky et al.,
1999a,b; Pokrovsky and Schott, 2002; Wolthers et al., 2008). In the present study, we
tested the one-site scheme for both minerals against different sets of reactions (Models)
which were calibrated via stochastic numerical optimization using an in-house Matlab©
subroutine. The latter incorporates a powerful search and optimization stochastic
technique, the genetic algorithm (GA), that can perform the simultaneous optimization of
a large number of parameters within a pre-established solution space and allows tackling
complex optimization problems (Gen and Cheng, 2000). The application of GAs to
estimate intrinsic formation constants of surface species has been described and
successfully tested on a number of cases of varying degrees of complexity where
adsorption data and/or surface charge data are used for calibrating the SCM (Villegas-
Jiménez and Mucci, 2009, see Chapter 2 of this thesis). Because of the stochastic nature
of GA optimizations, the GA parameters (i.e., population size, number of generations,
type and probability of crossover and mutation probability) must be carefully selected and
the optimization repeated to verify the reproducibility of the adjusted quantities. If poor
reproducibility in the optimized values is observed upon multiple optimizations, the
adopted model is incorrect and/or the data are inadequate for model calibration. All GA

83
optimizations described below were run in triplicate with the following GA parameters:
population of 500 chromosomes, 100 generations, a single-point crossover probability of
0.25, and a mutation probability of 0.02. All associated Matlab©
subroutines can be found
in the appendices to this thesis.
The predictive power of each model (selected set of surface reactions) was
evaluated on the basis of three criteria: i) its ability to reproduce surface charge (used to
perform the model calibration), ii) its capacity to simulate, at a semi-quantitative level,
published electrokinetic data acquired over a wide range of solution conditions, and iii)
the compatibility of the predicted surface speciation with available spectroscopic
information. The latter two are a posteriori SCM validation criteria independent of model
calibration, an important step in inverse modeling. Our starting point was to calibrate the
ionization reactions (generic reactions 1a-3a, Table 1) independently (Model I). To this
end and for each mineral, surface charge data obtained from independent titrations at
identical ionic strengths (I = 0.01M for dolomite and I = 0.1 M for magnesite) and
moderatively low CO2 and Me concentrations (magnesite: CO2 < 1.7 mM, Mg < 1
mM; dolomite: CO2 < 1 mM, Ca < 0.5 mM, and Mg < 0.8 mM) were combined into
a single data set for each mineral and used in model calibration. In these data sets, pH was
the master variable controlling the chemical speciation (covering the range: 5 pH 10
for both minerals) while CO2 and Mg were kept at relatively low concentrations
minimizing the influence of constituent ions on surface charge development. This allowed
us to examine the influence of ionization reactions on surface charge development and
obtain initial estimates of their corresponding Kint
values (reactions 1a-3a, Table 1).

84
Following the procedure applied in earlier studies (Van Cappellen et al., 1993;
Pokrovsky et al., 1999a,b; Pokrovsky and Schott, 2002), we used the Constant
Capacitance Model (CCM) to describe the surface charge-potential relationship, where
the surface is assumed to behave as a flat capacitor with the potential varying linearly
away from the surface (Sposito, 1984, Goldberg, 1993):
C0
0σ
ψ (5)
where C is the specific integral capacitance (F m-2
) of the electrified interfacial layer
(EIL). In this model, the capacitance is a function of the ionic strength and was described
earlier by Van Cappellen et al. (1993):
α
I1/2
C (6)
where I is the ionic strength and is an adjustable parameter related to the physical
properties of the EIL that reconciles working units (m2
· mol½
· V · C-1
). In the CCM
formulation, all surface species are assumed to adsorb chemically at the surface plane (0-
plane), allowing for the formation of inner-sphere surface complexes. This is compatible
with the premises implied by the limited residence time (LRT) experimental protocol (or
Flow-through reactor technique originally developed by Charlet et al., 1990) that
allocates ion charge imbalances recorded in solution (excluding the background
electrolyte) at each titration point to the 0-plane (charge imbalance in filtered

85
solution=surface charge). Surface species are treated in mol kg-1
units referenced to the 1
molal standard state whereas aqueous species are given in molar concentrations under the
constant ionic medium convention (Sposito, 1984). Ion pair formation and aqueous
complexation were considered in model calibration and surface complexation using the
formation constants listed in Table 2. Note that because the LRT technique produces
composite adsorption data (it involves protons, hydroxyls and constituents ions), the
computation of net sorption densities upon referencing of apparent sorption densities to
the Point of Zero Net Proton Charge (PZNPC), as recommended by some authors (e.g.,
Chorover and Sposito, 1995; Sposito, 1998), is not applicable to the data used in our
study (Pokrovsky et al., 1999a,b). Unfortunately, to date, no method can unambiguously
characterize the surface charge of a mineral suspension prior to titration (electrokinetic
measurements yield potentials at the shear plane which can only be related to surface
charge by an electrostatic model). Thus, the common assumption is to assign a “zero”
surface charge to the carbonate mineral surface (REF=0, Equation 2) prior to titration,
rendering apparent surface charge densities identical to net surface charge densities (e.g.,
Charlet et al., 1990; Van Cappellen et al., 1993; Pokvrosky et al., 1999a,b). This is based
on the assumption that once a MeCO3 suspension in pure water has reached equilibrium,
the mineral surface must approach the Point of Zero Net Charge, (PZNC, Charlet et al.,
1990).
Intrinsic constants were referenced to a zero potential standard state by performing
the electrostatic correction to the mass law expression as defined by Equation. 4. We
fixed the site densities of both minerals to their respective crystallographic values (9.8·10-
6 for magnesite and 8.9·10
-6 moles m
-2 for dolomite). In all cases, the value of was

86
adjusted simultaneously for capacitance values comprised between 0.1 to 100 F m-2
. In
contrast, a large solution space was chosen for all log10 Kint
values (-25 to 25) to perform
an exhaustive search for the set of Kint
values that best reproduced the experimental data.
All attempts to fit magnesite and dolomite data with the simplest electrostatic model, the
generalized double-layer model (DLM, Davis and Kent, 1990) were unsuccessful. After
the DLM, the CCM is the simplest electrostatic model describing the surface charge-
potential relationship and is, hence, a reasonable framework to rationalize adsorption data
acquired by the LRT experimental protocol. There is little point in testing more
sophisticated electrostatic models (e.g., Basic Stern Model, Triple Layer Model; Davis
and Kent, 1990) and add complexity to our interpretation without having at our disposal
individual, self-consistent sets of proton and constituent ion adsorption data at different
ionic strengths that would serve to better define the affinity and type of interaction (inner-
sphere vs outer-sphere) of constituent and background electrolyte ions with the surface.
Additional surface-sensitive spectroscopic data such as that of Pokrovsky et al. (2000)
will be also key in distinguishing between these types of interaction.
Upon calibration, Model I provided reasonable fits of surface charge data for both
minerals but the optimised Kint
values of magnesite could not simulate the zeta potential
measurements of Pokrovsky et al. (1999a), particularly at high CO2 (> 0.01 M). This
was expected given that Model I does not account for carbonate adsorption. Furthermore,
the pH of isoelectric point (pHiep) of dolomite and magnesite, reported to range from pH 6
to 8.8 and 6.8 to 8.5, respectively (Prédali and Cases, 1973; Pokrovsky et al., 1999a,b;
Chen and Tao, 2004; Gence and Ozbay, 2006), were poorly predicted by the optimized

87
model parameters. These observations led us to perform further calibrations whereupon
additional reactions (i.e., ionization and constituent ion adsorption) were considered.
Given the known dependency of zeta potential values on CO2, and Me
(Pokrovsky et al., 1999a,b), we tested, individually, the influence of constituent ion (Me2+
and CO32-
) adsorption on the development of surface charge. To this end, specific sets of
surface reactions for both minerals and selected data points for each mineral were used in
subsequent optimizations. For the calibration of ionization and constituent anion
adsorption reactions (Model II, reactions 1a-3a, 5a and 6a, Table 1), only data points with
relative low Me concentrations (i.e., Mg < 3 mM for magnesite; Mg and Ca < 1.5
mM for dolomite) were used in the calibration. In contrast, constituent cation adsorption
and ionization reactions (Model III, reactions 1a-4a) were calibrated using data with
moderate to low CO2 concentrations (i.e., < 3.6 mM for magnesite and < 2.7 mM for
dolomite) and the highest Me concentrations available. Although optimized SCM
parameters are different among models (see Tables 3 and 4), all models reasonably
reproduced the titration data of Pokrovsky et al. (1999a,b). One-way Analysis of Variance
(ANOVA) tests (95% confidence) confirmed that all model fits are statistically identical.
A final calibration (using all available surface charge data) including ionization and
constituent ion adsorption reactions (reactions 1a-6a) was performed for comparison
(Model IV).
For both minerals, the estimated Kint
values for the constituent cation adsorption
reactions (Model III and IV) are very small and carry large uncertainties (suggesting that
these are unnecessary to describe the data, see Tables 3 and 4) whereas the uncertainties
of the Kint
values of the ionization reactions are low. In contrast, constituent anion

88
adsorption constants, optimized within Model II, carry relatively low uncertainties and
are therefore believed to be relevant in the description of the data. This suggests that the
available experimental data are adequate to derive reliable Kint
values for ionization and
constituent anion adsorption reactions but may be insufficient to properly calibrate the
Kint
values of constituent cation adsorption reactions and, thus, additional experimental
data (batch adsorption or LRT-based titrations experiments covering higher constituent
cation concentrations) are required to accurately resolve their affinity for these surfaces.
We recognize that at high metal concentrations, adsorption of constituent metals will
affect surface charge development on carbonate minerals but, in the absence of pertinent
data to calibrate this reaction, its inclusion in the model is premature and, in fact,
unnecessary to fit our data.
In all cases and for both minerals, rather high specific capacitances ( 31.6 F m-2
for magnesite and 18.2 to 31.6 F m-2
for dolomite) are needed to reproduce the
experimental surface charge. High capacitance values (30 to 100 F m-2
) were also
required in previous studies to simulate the surface charge and/or the electrokinetic
behavior of these and other divalent carbonate minerals using either monolayer (CCM,
Van Cappellen et al., 1993; Pokrovsky et al., 1999a,b; Pokrovsky and Schott, 2002) or
multi-layer EIMs (Wolthers et al., 2008). All attempts to fit the data with smaller
capacitance values, by further restricting the GA-optimization range of the adjustable
parameter α, significantly decreased the quality of the fit. Although the estimated
capacitance values for both minerals (except from Model IV for dolomite) are lower than
those derived in earlier studies (Pokrovsky et al., 1999a,b; Pokrovsky and Schott, 2002;
Wolthers et al., 2008) and are in better agreement with physical constraints (i.e., thickness

89
of EIL), they lie outside the range typically assigned to metal oxides (0.1 to 2 F · m-2
).
High capacitances at carbonate surfaces were explained by the presence of a thin, highly
structured, non-diffuse EIL by earlier workers (Van Cappellen et al., 1993; Pokrovsky et
al., 1999a,b; Wolthers et al., 2008) but its strict physical interpretation would require a
very high and unrealistic dielectric constant of the interfacial water (Hayes et al., 1991).
Alternatively, they could be interpreted as being related to the large experimental surface
charge densities rather than to the absence/presence of multiple electrostatic layers, and
hence, we prefer to assign a purely operational character to the capacitance. Accordingly,
in conformity with premises of the CCM, all derived model parameters are considered as
reasonable surface speciation predictors (i.e., model fit parameters), applicable only to the
chemical conditions of model calibration (pH, I, etc.).
Surface charge simulations for magnesite, performed at fixed solution conditions
(Mg = 0.002M and CO2 = 0.003 M.) using Models I and II and the two-site SCM of
Pokrovsky and coworkers (1999a), are shown in Figure 4 and compared against
experimental data. It is noteworthy that at pH 8.5, the large range of measured surface
charge cannot be reproduced by any of the models. Under strongly alkaline conditions,
slight differences in CO2 concentrations may induce significant changes in surface
charge via carbonate adsorption which are not properly described at the selected solution
conditions of our simulations. Consideration of the experimental CO2 and Mg
conditions (the latter influencing the aqueous carbonate ion activities upon ion pair
formation) at each titration point is required to improve the agreement between
experimental data displayed in Figure 4 and surface charge predictions of our one-site-
based Model II (or the two-site-based model of Pokrovsky et al., 1999a). In contrast,

90
surface charge predictions of Model I would remain unchanged because no provision for
carbonate and/or metal ion adsorption is made by this Model.
Because the presence of a shear-plane is ill-defined in the CCM, predicted surface
potentials (the potential at the 0-plane) were compared with zeta potentials (-potentials,
measured at the shear-plane), but only at a semi-quantitative level. We found that Model
II is the only one that provides reasonable predictions of surface potential for a range of
chemical conditions (i.e. it follows the trend displayed by zeta potentials) and best
reproduces the pHiep values measured for both minerals (see Figure 5). The predicted
surface potentials are in reasonable agreement with -potentials measured at pHs < 8.5
but, at pH above 9, Model II consistently predicts more negative surface potentials for
both minerals at all CO2 of our SCM simulations (see conditions in Figure 5). This
observation is, nonetheless, compatible with the premise that the absolute potential
measured at the shear-plane must be lower than the surface potential (Davis and Kent,
1990).
Based upon the selected criteria for evaluating the predictive power of our
Models, we believe that the calibrated model parameters for Model II are good predictors
of the surface charge and the electrokinetic behavior and surface speciation (see sections
4.2 and 4.3) of magnesite and dolomite surfaces in chemical systems whose composition
(i.e., pH, ionic strength, Me and CO2) is similar to those under which the experimental
data used for model calibration were acquired. Nevertheless, the optimized parameters
should be used with caution for predictive purposes since adsorption reactions involving
constituent cations may be significant under specific chemical conditions (e.g., high Me)
and may influence the development of charge at the surfaces of some carbonate minerals

91
such as calcite (e.g., Siffert and Fimbel, 1984; Huang et al., 1991; Cicerone et al., 1992).
Further experimental work (e.g., batch constituent ion adsorption experiments) is needed
to verify the self-consistency of these parameters under different chemical conditions and
to carefully evaluate the relevance of other surface reactions (e.g., constituent cation and
background electrolyte adsorption) that may contribute to the development of the surface
charge and the formation of a more sophisticated EIL than envisioned by the CCM.
4.2. Intrinsic Formation Constants and Surface Speciation
Our selected set of log10 Kint
values are significantly different from those derived from
earlier surface complexation models for both minerals (Tables 3 and 4). This divergence
is explained by the application of the one-site scheme in the formulation of surface
equilibra and the different strategies employed in each study to estimate the intrinsic
formation constants. In earlier studies, Kint
values were calibrated manually against
surface charge or electrokinetic data on a trial and error basis using equilibrium constants
of analogous reactions in aqueous solution as their starting point (Van Cappellen et al.,
1993; Pokrovsky et al., 1999a,b; Pokrovsky and Schott, 2002) or by using theoretical
schemes originally developed for metal oxides (Wolthers et al., 2008). It is noteworthy
that the latter authors report Kint
values for a “hybrid” SCM where ionization reactions
were calibrated according to the CD-MUSIC-Triple-Plane approach whereas the Kint
values of constituent ion adsorption reactions were those of earlier CCM-based SCMs
(Van Cappellen et al., 1993; Pokrovsky et al., 1999a,b). Clearly, comparisons between
these Kint
values and those derived in the present study are unwarranted.

92
Interestingly, the one-site-based log10 Kint
values obtained for the one-step
protolysis reaction for both minerals (reaction 1a, Table 1) are in reasonable agreement
with those of analogous reactions in aqueous solutions (NIST, 1998):
CaHCO3 CaCO3 + H+
log10 K = -8.40 (7)
MgHCO3 MgCO3 + H+
log10 K = -8.42 (8)
In Figure 6 we present the surface speciation predicted by Model II for magnesite
and dolomite for the following chemical conditions: ΣCO2 = ΣMe =1 mM, which largely
contrasts with that predicted by multi-site-based SCMs for these minerals (Pokrovsky et
al, 1999a,b; Wolthers et al., 2008). For instance, whereas the one-site scheme predicts the
predominance of protonated and deprotonated species under very different pH regimes
(acid and alkaline, respectively) as it would be expected for a truly amphoteric surface
(analogous to a polyprotic acid in solution), multi-site SCMs predict the simultaneous
predominance of a double-protonated (MeOH2+ for the CCM or MeOH2
+1/3 for terraces
for the CD-MUSIC) and a deprotonated species (CO3- for the CCM and CO3
-1/3 for
terraces for the CD-MUSIC) over a wide pH range (~ 5 to 9). In other words, according to
multi-site SCMs, protonation and deprotonation reactions (1b and 3b, see Table 1)
simultaneously occur over a wide pH range, suggesting that a large number (n) of these
double-protonated species must be neighboring an approximately equal number (m) of
deprotonated species. This is intuitively unrealistic because such a molecular scenario
(i.e., n MeOH2+ m CO3
-) would most likely result in the re-establishment of the
global stoichiometry and charge of primary surface sites (MeOH2+ + CO3
- =

93
MeCO3·H2O0), implying that a negligible net protonation or deprotonation (hence, a
negligible net charge transfer) occurs at the mineral surface under these chemical
conditions. Furthermore, it would be difficult to explain why the anionic primary surface
site, CO3H0 strongly deprotonates at pH ~ 5 whereas the cationic primary surface site,
MeOH0, readily undergoes protonation under identical pH conditions. This is a direct
consequence of assigning individual reactivities (e.g., acidities) to neighboring cationic
and anionic surface sites and performing a simultaneous (unconstrained) adjustment of
their corresponding intrinsic ionization constants. This contrasts with the one-site scheme
that, by assigning an average reactivity to the cationic-anionic primary surface site, allows
for a realistic description of the amphoteric behavior of the carbonate surface, and hence,
yields intuitively reasonable predictions of surface speciation.
According to Model II, the predominance of the charge-neutral H2CO3-bearing
surface species, ≡(MeCO3)·H2CO30, and, to a lesser extent, of the “unreacted” primary
surface site, ≡(MeCO3)·H2O0, at conditions similar to those under which carbonate
mineral studies are typically conducted: ΣCO2 ΣMe 1 mM; pH 5.5-8.5, accounts for
the charge-buffering behavior displayed by magnesite and dolomite surfaces and may
explain the relatively wide range of pHiep values reported in the literature for these
minerals (Prédali and Cases, 1973; Pokrovsky et al., 1999a; Chen and Tao, 2004; Gence
and Ozbay, 2006).
4.3. Comparison against Spectroscopic Information
According to the one-site scheme (Model II), surface speciation and charge acquisition is
dominated only by the protonated species at low pH (< 5) whereas, at circum-neutral pH
(5.8-8.2), the charge-neutral H2CO3-bearing surface species is predominant for both

94
minerals. This is in agreement with results of DRIFT spectroscopic studies (Pokrovsky et
al., 2000) and Knudsen flow reactor-based CO2(g) adsorption studies (Santschi and Rossi,
2006) that revealed the presence of carbonate-bearing species at the dolomite and calcite
surface at pH 5 and CO2 10-3
M (Pokrovsky et al., 2000), and at hydrated calcite
surfaces exposed to CO2(g) atmospheres (Santschi and Rossi, 2006). These findings
dismiss the viability of models that make no provision for carbonate ion adsorption
(Models I and III). Similarly, because of the low Kint
values returned from the
optimization of carbonate adsorption (generic reactions 5a and 6a, Table 1) in Model IV,
this model predicts negligible concentrations of carbonate-bearing species at the above
conditions, in conflict with available spectroscopic information.
Using X-ray Reflectivity, Fenter et al. (2000) investigated the surface speciation
of calcite, under three different chemical scenarios (Ca, CO2, I, pH) which, according
to SCM predictions (Van Cappellen et al., 1993), represented either: i) a “calcium-
terminated” surface (Ca 1.4 M, CO2 = 0.34 mM, pH = 6.83), ii) a “water-terminated”
surface (Ca 0.5 mM, CO2 = 1.33 mM, pH = 8.25) or iii) a “carbonate-terminated”
surface (Ca 0.012 mM, CO2 = 2.27 mM, pH = 12.1). Among these, the solution
composition generating the “water-terminated” scenario most closely reflects the
chemical conditions (Ca = CO2 = 10-3
M) under which our speciation predictions
(Figure 6) were conducted and, thus, is best suited for comparisons. According to our
one-site-based SCM calculations (Figure 6), at a pH ≤ 8.2, the surface speciation of
magnesite and dolomite is dominated by H3O+-, H2O-bearing and/or a carbonate-bearing
species. The abundance of the former and the latter species abruptly drops as pH
increases and, thus, the H2O-bearing and/or the OH-bearing species predominate at

95
slightly higher pH. These two species are undistinguishable from each other given that
protons are not detected by X-ray Reflectivity and thus, under these pH conditions, the
surface speciation predicted by Model II is consistent with Fenter and coworker‟s
conclusion that X-ray Reflectivity data and the implied surface speciation at the three
above-mentioned regimes could be explained solely by protonation/deprotonation
reactions (generic reactions 1a-3a and 1b-3b). In other words, at pH > 8.2, the calcite
surface is essentially dominated by hydroxyl-bearing surface species (be it as OH, OH2
and/or OH3), and at very high pH, by deprotonated species, ≡(MeCO3)·O2-
, which, within
the multi-site scheme, can be interpreted as ≡CO3. Note that the latter is not considered
as an adsorbed carbonate-bearing species but rather as a deprotonated primary surface
site.
According to our one-site SCM calculations (Model II), neither the “carbonate-
terminated” nor the “calcium-terminated” scenario examined by Fenter and coworkers
(2000) are adequate to evaluate carbonate adsorption because the very high pH in the
former (beyond the range of carbonate-bearing surface species) and the very high Ca in
the latter (which substantially decreases CO32-
activities in solution upon ion-pair
formation) are unfavorable to the development of carbonate-bearing surface species.
Hence, under both scenarios, surface speciation would be dominated by H3O+-, H2O-,
and/or OH-bearing species, in agreement with Fenter and coworker‟s results. It should be
noted, however, that the above comparisons should be revised when X-ray Reflectivity-
based surface speciation studies are extended to magnesite and dolomite surfaces.

96
5. CONCLUSIONS
The definition of primary surface sites in terms of their elemental stoichiometry and
residual charge plays a critical role on the molecular representation of reactions at mineral
surfaces and the calibration of surface complexation models via LMA approaches.
Given the abundance of experimental and theoretical information for the (10.4)
cleavage calcite surface, this surface was selected as a case study to revisit the definition
of reactive surface sites on divalent rhombohedral carbonate minerals. A single primary
surface site is proposed for calcite which is compatible with available spectroscopic data
and molecular modeling results as well as with assumptions frequently implied in the
construct of SCMs. In addition, it circumvents the problem of charge and mass
discretization associated with earlier multi-site schemes.
The one-site scheme was extended to the surface of magnesite and dolomite and
published surface charge data for both minerals were used in the calibration of the newly
defined surface reactions. Several sets of one-site-based surface reactions, including
ionization and/or constituent ion adsorption reactions can successfully simulate surface
charge but only one can qualitatively simulate the electrokinetic behavior displayed by
both minerals while yielding intuitively reasonable predictions of surface speciation,
agreeing with available spectroscopic data, and reflecting the behavior of a truly
amphoteric surface. The simplified model for both minerals, involving bicarbonate ion
adsorption and proton/bicarbonate ion co-adsorption reactions (in addition to ionization
reactions), accounts for the surface charge-buffering behavior displayed by these minerals
under circum-neutral conditions and offers a possible explanation to the relatively wide
range of pHiep values typically reported in the literature. This is achieved with a reduced
number of parameters (five log10 Kint
values and one capacitance) which contrasts with

97
more sophisticated multi-site schemes (such as the CD-MUSIC model) that require many
more parameters that must be manually adjusted on a trial and error basis (as borrowed
from earlier CCM-based SCMs) and/or arbitrarily selected on the basis of multiple
theoretical assumptions originally derived for metal oxides. Admittedly, as in earlier
SCMs for carbonate minerals calibrated within single (CCM) or multiple (Triple Plane)
electrostatic layer schemes, the physical interpretation of the adjusted capacitances is
problematic, and hence, we prefer to consider all model parameters as reasonable surface
speciation predictors (i.e., model fit parameters), applicable to the chemical conditions of
model calibration (pH, I, etc.).
Given its simplicity and compatibility with available experimental data, we
propose that the one-site scheme is a convenient approach to use in the construct of SCMs
for other rhombohedral carbonate minerals. As more experimental data of different
sources (adsorption isotherms, calorimetric, radiometric, electrokinetic, etc.) become
available, it might be possible to fine-tune these models and reliably incorporate multi-
layer and multi-site adsorption concepts without the necessity to expand upon numerous
assumptions. Additional theoretical constraints obtained from molecular modeling
techniques and fundamental crystal and colloid chemistry will be key for the proper
calibration of such sophisticated models.

98
6. ACKNOWLEDGMENTS
A.V.-J. thanks Dr. Luuk Koopal for critical discussions that inspired this investigation.
We acknowledge the insightful reviews of Dimitri Sverjensky, Phillipe Van Cappellen,
Mariëtte Wolthers, and two anonymous reviewers. This research was supported by a
graduate student grant to A.V.-J. from the Geological Society of America (GSA) and
Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery
grants to A.M. A.V.-J. also benefited from post-graduate scholarships from the Consejo
Nacional de Ciencia y Tecnología (CONACyT) of Mexico and additional financial
support from the Department of Earth and Planetary Sciences, McGill University and
from Consorcio Mexicano Flotus-Nanuk.

99
7. REFERENCES
Ahsan T. (1992) The surface properties of pure and modified precipitated calcium
carbonate by adsorption of nitrogen and water vapour. Colloids Surf. 64, 167-176.
Al-Hosney H.A. and Grassian V.H. (2005) Water, sulfur dioxide and nitric acid
adsorption on calcium carbonate: A transmission and ATR-FTIR study. Phys.
Chem. Chem. Phys. 7, 1266-1276.
Archer T.D. (2004) Computer simulations of calcite. Ph.D. Thesis, University of
Cambridge. UK 162 p.
Austen K.T., Wright K., Slater B. and Gale J.D. (2005) The interaction of dolomite
surfaces with metal impurities: a computer simulation study. Phys. Chem. Chem.
Phys. 7, 4150-4156.
Bandstra J.Z. and Brantley S.L. (2008) Surface evolution of dissolving minerals
investigated with a kinetic ising model. Geochim. Cosmochim. Acta 72:11, 2587-
2600.
Barrow N.J., Brümer G.W. and Strauss R.G. (1993) Effects of surface heterogeneity on
ion adsorption by metal oxides and by soils. Langmuir. 9, 2606-2611.
Benjamin M. (2002) Modeling the mass-action expression for bidentate adsorption.
Environ. Sci. Technol. 36, 307-313.
Bickmore B.R., Tadanier C.J., Rosso K.M., Monn W.D. and Egget D.L. (2004). Bond-
Valence methods for pKa prediction: critical reanalysis and a new approach.
Geochim. Cosmochim. Acta. 68(9), 2025-2042.
Bleam W.F. (1993) On the modeling proton affinity at the oxide/water interface. J.
Colloid Interface. Sci. 159, 312-318.

100
Blesa M.A. and Kallay N. (1988) The metal oxide-electrolyte solution interface revisited.
Adv. Colloid Interface Sci. 28, 111-134.
Borkovec M. (1997) Origin of 1-pK and 2-pK models for ionizable water-solid interfaces.
Langmuir 13, 2608-2613.
Borkovec M. and Koper G.J.M. (1994) Ising models of polyprotic acids and bases. J.
Phys. Chem. 98, 6038-6045.
Brady P.V., Krumhansl J.L. and Papenguth H.W. (1996) Surface complexation clues to
dolomite growth. Geochim. Cosmochim. Acta 60:4, 727-731.
Brätter P, Möller P. and Rösick U. (1972) On the equilibrium of coexisting sedimentary
carbonates. Earth Planet. Sci. Lett. 14, 50-54.
Brown I.D. (1981) The bond-valence method: an empirical approach to chemical
structure and bonding. In: Structure and Bonding in Crystals. (eds. M. O'Keeffe,
and A. Navrotsky). Academic Press, New York, Vol 2, pp. 1-30.
Cerník M., Borkovec M. and Westall J.C. (1995) Regularized least-squares methods for
the calculation of discrete and continuous affinity distributions for heterogeneous
sorbents. Environ. Sci. Technol. 29, 413-425.
Chandler D. (1987) Introduction to Modern Statistical Mechanics. Oxford University
Press, New York, 288 p.
Chandra S. W. and Kollman P. (1984) An approach to computing electrostatic charges for
molecules. J. Comput. Chem. 5(2), 129-145.
Charlet L., Wersin P. and Stumm W. (1990) Surface charge of MnCO3 and FeCO3.
Geochim. Cosmochim. Acta 54, 2329-2336.

101
Chen G. and Tao D. (2004) Effect of solution chemistry on flotability of magnesite and
dolomite. Int. J. Min. Process. 74, 343–357.
Chiarello R.P., Wogelius R.A. and Sturchio N. (1993) In-situ synchrotron X-ray
reflectivity measurements at the calcite-water interface. Geochim. Cosmochim.
Acta 57(16), 4103-4110.
Chorover J. and Sposito G. (1995) Surface charge characteristics of kaolinitic tropical
soils. Geochim. Cosmochim. Acta 59(5), 875-884.
Cicerone D.S., Regazzoni A.E. and Blesa M.A. (1992) Electrokinetic properties of the
calcite/water interface in the presence of magnesium and organic matter. J.
Colloid Interface Sci. 154, 423-433.
Davis J.A. and Kent D.B. (1990) Surface complexation modeling in aqueous
geochemistry. In Mineral-Water Interface Geochemistry. (ed. M.F. Hochella and
A.F. White). Rev. Mineral. 23. Mineral. Soc. Washington, DC. pp 177-260.
de Leeuw N.H. and Parker S.C. (1997) Atomistic simulation of the effect of molecular
adsorption of water on the surface structure and energies of calcite surfaces. J.
Chem. Soc., Faraday Trans. 93(3), 467-475.
de Leeuw N.H. and Parker S.C. (1998) Surface structure and morphology of calcium
carbonate polymorphs calcite, aragonite, and vaterite: an atomistic approach. J.
Phys. Chem. B. 102, 2914-2922.
de Leeuw N.H. and Parker S.C. (2001) Surface-water interactions in the dolomite
problem. Phys. Chem. Chem. Phys. 3, 3217-3221.

102
de Leeuw N.H. and Parker S.C. (2002) Surface structures, stabilities, and growth of
magnesian calcites: a computational investigation from the perspective of
dolomite formation. Am. Mineral., 87, 679-689.
de Leeuw N.H., Parker S.C., and Hanumantha Rao K. (1998) Modeling the competitive
adsorption of water and methanoic acid on calcite and fluorite surfaces. Langmuir
14, 5900-5906.
Fenter P., Geissbühler P., DiMasi E., Srajer G., Sorensen B. and Sturchio N.C. (2000)
Surface speciation of calcite observed in situ by high-resolution X-ray reflectivity.
Geochim. Cosmochim. Acta 64(7), 1221-1228.
Fitts J.P., Machesky M.L., Wesolowski D.J., Shang X., Kubicki J.D., Flynn G.W., Heinz
T.F. and Eisenthal K.B. (2005) Second-harmonic generation and theoretical
studies of protonation at the water/α TiO2 (110) interface. Chem. Phys. Letters
411, 399-403.
Gallup G.A. (2002) Valence bond methods: Theory and applications. Cambridge
University Press, New York, 238 p.
Geissbühler P., Fenter P., DiMasi E., Sorensen L.B. and Sturchio N.C. (2004) Three-
dimensional structure of the calcite–water interface by surface X-ray scattering.
Surf. Sci. 573, 191-203.
Gen M. and Cheng R. (2000) Genetic algorithms and engineering optimization. John
Wiley and Sons, New York, NY, 495 p.
Gence N. and Ozbay N. (2006) pH dependence of electrokinetic behaviour of dolomite
and magnesite in aqueous electrolyte solutions. Appl. Surface Sci. 252, 8057-
8061.

103
Goldberg S. (1991) Sensitivity of surface complexation modeling to the surface site
density parameter. J. Colloid Interface Sci. 145:1, 1-9.
Goldberg S. (1993) Constant capacitance model: Chemical surface complexation model
for describing adsorption of toxic trace elements on soil minerals. Am. Chem. Soc.
Symp. Ser. 518: 278-307.
Golderg S. (1995) Adsorption models incorporated into chemical equilibrium models. In:
Chemical Equilibrium and Reaction Models. (ed. R. Loeppert, A.P. Schwab and
S. Goldberg), Soil. Sci. Soc. Am. Special Publication 42, 75-95.
Hayes K.F., Redden G., Ela W. and Leckie J.O. (1991) Surface complexation models: an
evaluation of model parameter estimation using FITEQL and oxide mineral
titration data. J. Colloid Interface Sci. 142:2, 448-469.
Healy T.W. and White L.R. (1978) Ionizable surface groups models of aqueous
interfaces. Adv. Colloid Interface Sci. 9, 303-345.
Herbelin A. and Westall J. (1996) FITEQL- A computer program for determination of
chemical equilibrium constants from experimental data; version 3.2: user‟s
manual. Department of Chemistry. Oregon State University, Corvallis, OR,
Report 96-01.
Hiemstra T., van Riemsdijk W.H. and Bolt G.H. (1989) Multi-site proton adsorption
modeling at the solid/solution interface of (hydr)oxides: A new approach. J.
Colloid Interface Sci. 133(1), 91-104.
Hiemstra T. and van Riemsdijk W.H. (1991) Physical chemical interpretation of primary
charging behaviour of metal (hydr)oxides. Colloids Surfaces 59, 7-25.

104
Hiemstra T. and van Riemsdijk W.H. (1996) A surface structural approach to ion
adsorption: The charge distribution (CD) model. J. Colloid Interface Sci. 179,
488-508.
Hiemstra T., Venema P. and van Riemsdijk W.H. (1996) Intrinsic proton affinity of
reactive surface groups of metal (hydr)oxides: The bond valence principle. J.
Colloid Interface Sci. 184, 680-692.
Huang Y.C., Fowkes F.M., Lloyd T.B. and Sanders, N.D. (1991) Adsorption of calcium
ions from calcium chloride solutions onto calcium carbonate particles. Langmuir,
7, 1742-1748.
Hwang S., Blanco M. and Goddard W.A. III (2001) Atomistic simulations of corrosion
inhibitors adsorbed on calcite surfaces I. Force field parameters for calcite. J.
Phys. Chem. B. 105, 10746-10752.
Jäger I. (1991) Adsorption of pairwise interacting atoms on the derivation of the
interaction parameters between first and second neighbours from experimental
data. Surf. Sci. 254, 300-308.
Kallay N. and Žalac S. (2000) Charged surfaces and interfacial ions. J. Colloid Interface
Sci. 230(1), 1-11.
Kerisit S. and Parker S.C. (2004) Free energy of adsorption of water and metal ions on
the (10.4) calcite surface. J. Am. Chem. Soc. 126, 10152-10161.
Kerisit S., Parker S.C. and Harding J.H. (2003) Atomistic simulation of the dissociative
adsorption of water on calcite surfaces. J. Phys. Chem. B. 107, 7676-7682.

105
Kerisit S., Cooke D.J., Spagnoli D. and Parker S.C. (2005a) Molecular dynamics
simulations of the interactions between water and inorganic solids. J. Mater.
Chem. 15, 1454-1462.
Kerisit S., Marmier A. and Parker S.C. (2005b) Ab initio surface phase diagram of the
(10.4) calcite surface. J. Phys. Chem. B, 109:39, 18211-18213.
Koretsky C.M., Sverjensky D.A. and Sahai N. (1998) A model of surface sites types on
oxide and silicate minerals based on crystal chemistry: Implications for site types
and densities, multi-site adsorption, surface infrared spectroscopy and dissolution
kinetics. Am. J. Sci. 298, 349-438.
Kovačević D., Kobal I. and Kallay N. (1998) Adsorption of organic acids on metal
oxides. The umbrella effect. Croat. Chim. Acta 71(4), 1139-1153.
Kubicki J.D., Paul K.W. and Sparks D.L. (2008) Periodic density functional theory of
bulk and the (010) surface of goethite. Geochem. Trans. 9:4.
Kulik D. A. (2002) Gibbs energy minimization approach to modeling sorption equilibria
at the mineral-water interface: Thermodynamic relations for multi-site-surface
complexation. Am. J. Sci. 302, 227-279.
Kulik D.A., Aja S.U., Sinitsyn V.A. and Wood S.A. (2000) Acid–base surface chemistry
and sorption of some lanthanides on K1-saturated Marblehead illite: II. A
multisite–surface complexation modeling. Geochim. Cosmochim. Acta 64:2, 195-
213.
Kuriyavar S.I., Vetrivel R., Hegde S.G., Ramaswamy A.V., Chakrabarty D. and
Mahapatra S. (2000) Insights into the formation of hydroxyl ions in calcium
carbonate: temperature dependent FTIR and molecular modeling studies. J. Mater.
Chem. 10, 1835-1840.

106
Liang Y., Lea A.S., Baer D.R. and Engelhard M.H. (1996) Structure of the cleaved
CaCO3 (104) surface in an aqueous environment. Surf. Sci. 351, 172-182.
Ludwig C. and Schindler P.W. (1995) Surface complexation on TiO2. I Adsorption of
H+and Cu
2+ ions onto TiO2 (anatase). J. Interface Colloid Sci. 169, 284-290.
Lützenkirchen J. (1997) Ionic strength effects on cation sorption to oxides: macroscopic
observations and their significance in microscopic interpretation. J. Colloid
Interface Sci. 195, 149-155.
Lützenkirchen J. (2002) Surface complexation models of adsorption: a critical survey in
the context of experimental data In: Adsorption. Theory, Modeling and Analysis
(ed. Tóth J.), Marcel Dekker Inc., New York. pp 631-710.
Lützenkirchen J. (2005) On derivatives of surface charge of carbonate minerals. J.
Colloid Interface Sci. 2(15), 489-497.
Morel F. and Morgan J. (1972) A numerical method for computing equilibria in aqueous
chemical systems. Environ. Sci. Technol. 6, 58-87.
Morimoto T., Nagao M. and Tokuda F. (1969) Relation between the amounts of
chemisorbed and physisorbed water on metal oxides. J. Phys. Chem., 73(1), 243-
248.
Morimoto T., Kishi J., Okada O. and Kadota T. (1980) Interaction of water with the
surface of calcite. Bull. Chem. Soc. Jpn. 53(7), 1918-1921.
Neagle W. and Rochester C.H. (1990) Infrared study of the adsorption of water and
ammonia on calcium carbonate. J. Chem. Soc., Faraday Trans. 86(1), 181-183.

107
NIST (1998) Critically Selected Stability Constants of Metal Complexes, Standard
Reference Database 46, Version 5, National Institute of Standards and
Technology, US Department of Commerce, Gaithersburg, MD, USA.
Parker S.C., Kerisit S., Marmier A., Grigoleit S. and Watson G.W. (2003) Modeling
inorganic solids and their interfaces: A combined approach of atomistic and
electronic structure simulation techniques. Faraday Discuss. 124, 155–170.
Pauling L.J. (1929) The principle determining the structure of complex ionic crystals. Am.
Chem. Soc. 51, 1010-1026.
Perry T.D., Cygan R.T. and Mitchell R. (2007) Molecular models of a hydrated calcite
mineral surface. Geochim. Cosmochim. Acta 71, 5876–5887
Piasecki W, Rudziński W. and Charmas R. (2001) 1-pK and 2-pK protonation models in
the theoretical description of simple ion adsorption at the oxide/electrolyte
interface: a comparative study of the behaviour of the surface charge, the
individual isotherms of ions, and the accompanying electrokinetic effects. J. Phys.
Chem. B 105, 9755-9771.
Pivovarov S. (1997) Surface structure and site density of the oxide-solution interface. J.
Colloid Interface Sci. 196, 321-323.
Pivovarov S. (1998) Acid–base properties and heavy and alkaline earth metal adsorption
on the oxide–solution interface: Non-electrostatic model. J. Colloid Interface Sci.
206, 122-130.
Pokrovsky O.S., Schott J. and Thomas F. (1999a) Processes at the magnesium-bearing
carbonate/solution interface. I. A surface speciation model for magnesite.
Geochim. Cosmochim. Acta 63(6), 863-880.

108
Pokrovsky O.S., Schott J. and Thomas F. (1999b) Dolomite surface speciation and
reactivity in aquatic systems. Geochim. Cosmochim. Acta 63(19/20), 3133-3143.
Pokrovsky O.S., Mielczarski J.A., Barres O. and Schott J. (2000) Surface speciation
models of calcite and dolomite/aqueous solution interfaces and their spectroscopic
investigation. Langmuir 16, 2677-2688.
Pokrovsky O.S. and Schott J. (2002) Surface chemistry and dissolution of divalent metal
carbonates. Environ. Sci. Technol. 36(3), 426-432.
Prédali J.-J. and Cases J.-M.J. (1973) Zeta potential of magnesian carbonates in inorganic
electrolytes. J. Colloid Interface Sci. 45(3), 449-458.
Rachlin A.L., Henderson G.S. and Goh M.C. (1992) An atomic force microscope (AFM)
study of the calcite cleavage plane; image averaging in Fourier space. Am.
Mineral. 77(9/10), 904-910.
Rudziński W., Charmas R., Partyka S., Thomas F. and Bottero J.Y. (1992) On the nature
of the energetic surface heterogeneity in ion adsorption at a water/oxide interface:
the behaviour of potentiometric, electrokinetic, and radiometric data. Langmuir 8,
1154-1164.
Rudziński W., Charmas R, Piasecki W., Thomas F., Villieras F., Prelot B. and Cases J.
M. (1998) Calorimetric effects accompanying ion adsorption at the charged metal
oxide/electrolyte interfaces: effects of oxide surface energetic heterogeneity.
Langmuir 14, 5210-5225.
Rustad J.R., Felmy A R. and Hay B.P. (1996) Molecular statics calculations of proton
binding to goethite surfaces: A new approach to estimation of stability constants
for multisite surface complexation models. Geochem. Cosmochim. Acta 60(9),
1563-1576.

109
Sahai N. and Sverjensky D.A. (1997) Evaluation of internally consistent parameters for
the triple-layer model by the systematic analysis of oxide surface titration data.
Geochim. Cosmochim. Acta 61(14), 2801-2826.
Santschi Ch. and Rossi M.J. (2006) Uptake of CO2, SO2, HNO3 and HCl on calcite
(CaCO3) at 300 K: Mechanism and the role of adsorbed water. J. Phys. Chem. A
110, 6789-6802.
Scheidegger A.M., and Sparks D.L. (1996) A critical assessment of sorption-desorption
mechanisms at the soil mineral/water interface. Soil Sci. 161, 813-831.
Siffert B. and Fimbel, P. (1984) Parameters affecting the sign and the magnitude of the
electrokinetic potential of calcite. Colloids Surf., 1984, 11, 377-389.
Sposito G. (1983) On the surface complexation model of the oxide-aqueous solution
interface. J. Colloid Interface Sci. 91(2), 329-340.
Sposito G. (1984) The Surface Chemistry of Soils. Oxford University Press, New York,
234 p.
Sposito G. (1989) Surface reactions in natural aqueous colloidal systems. Chimia 43, 169-
176.
Sposito G. (1998) On points of zero charge. Environ. Sci. Technol. 32(19), 2815-2819.
Stipp S. L. (1999) Toward a conceptual model the calcite surface: hydration, hydrolysis,
and surface potential. Geochim. Cosmochim. Acta 63(19/20), 3121-3131.
Stipp S. L. and Hochella M.F. Jr. (1991) Structure and bonding environments at the
calcite surface as observed with X-ray photoelectron spectroscopy (XPS) and low
energy electron diffraction (LEED). Geochim. Cosmochim. Acta 55, 1723-1736.

110
Stipp S.L.S., Eggleston C.M. and Nielsen B.S. (1994) Calcite surface structure observed
at microtopographic and molecular scales with atomic force microscopy (AFM).
Geochim. Cosmochim. Acta 58(14), 3023-3033.
Stumm W. and Morgan J. (1996) Aquatic Chemistry: Chemical Equilibria and Rates in
Natural Waters. John Wiley and Sons Inc., 3rd
Edition New York, 1022 p.
Sverjensky D.A. (2003) Standard states for the activities of mineral surface sites and
species. Geochim. Cosmochim. Acta 67, 17–28.
Teng H.H. and Dove P.M. (1997) Surface site-specific interactions of aspartate with
calcite during dissolution: Implications for biomineralization. Am. Mineral. 82,
878-887.
Van Cappellen P., Charlet L., Stumm W. and Wersin P. (1993) A surface complexation
model of the carbonate mineral-aqueous solution interface. Geochim. Cosmochim.
Acta 57, 3505-3518.
van Riemsdijk W.H., De Wit J.C.M., Koopal L.K and Bolt G.H. (1987) Metal ion
adsorption on heterogeneous surfaces: Adsorption models. J. Colloid Interface
Sci. 116(2), 511-522.
Villegas-Jiménez A. and Mucci A. (2009) Estimating intrinsic formation constants of
mineral surface species using a genetic algorithm. Math. Geosci. (accepted).
Villegas-Jiménez A., Mucci A. and Whitehead M.A. (2005) Ab initio molecular orbital
investigation of the chemical interactions of water with the (10.4) calcite surface.
Proceedings of the First Applied Pulp and Paper Molecular Modeling Symposium.
Montréal, Canada, 227-244.

111
Villegas-Jiménez A., Mucci A. and Whitehead M.A. (2009a) Theoretical insights into the
hydrated (10.4) calcite surface: structure, energetics and bonding relationships.
Langmuir 25(12), 6813-6824.
Villegas-Jiménez A., Mucci A., Paquette, J. (2009b) Proton/calcium ion exchange
behavior of calcite. Phys. Chem. Chem. Phys 39(11), 8895-8912.
Wasserman E., Rustad J.R. and Felmy A.R. (1999) Molecular modeling of surface
charging of hematite: I. The calculation of proton affinities and acidities on a
surface. Surf. Sci. 424, 19-27.
Westall J. and Hohl H. (1980) A comparison of electrostatic models for the oxide/solution
interface. Adv. Colloid Int. Sci. 12, 265-294.
Wolthers M., Charlet L., and Van Cappellen P.V. (2008) The surface chemistry of
divalent metal carbonate minerals; A critical assessment of surface charge and
potential data using the charge distribution multi-site ion complexation model.
Am. J. Sci. 308, 905-941.
Wright K., Cygan R. T. and Slater B. (2001) Structure of the (10.4) surfaces of calcite,
dolomite and magnesite under wet and dry conditions. Phys. Chem. Chem. Phys.
3, 839-844.
Yoon R. H., Salman T. and Donnay G. (1979) Predicting points of zero charge of oxides
and hydroxides. J. Colloid Interface Sci. 70(3), 483-493.
Zachara, J. M. and Westall, J.C. (1999) Chemical modeling of ion adsorption in soils, In
Soil Physical Chemistry, 2nd ed. (ed. D. L. Sparks), CRC Press LLC, Boca Raton,
FL, pp 47–95.
Zuyi T., Taiwei C. and Weijuan L. (2000) On the application of surface complexation
models to ionic adsorption. J. Colloid Interface Sci. 232(1), 174-177.
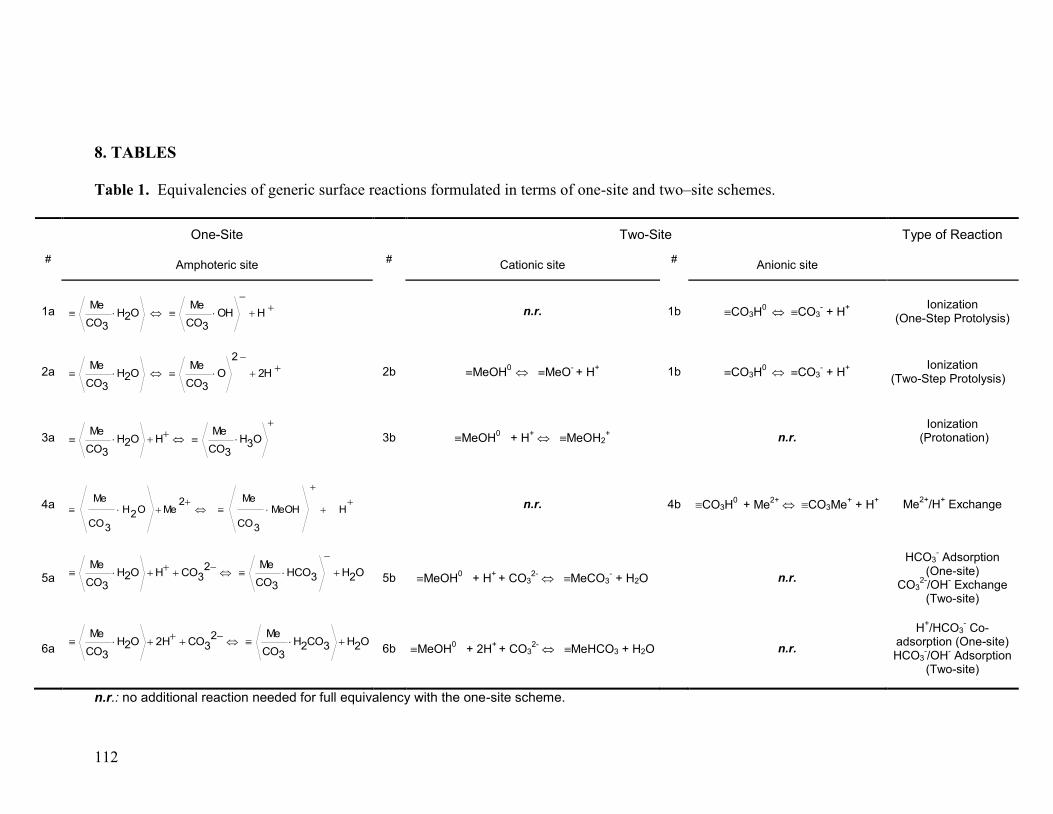
112
8. TABLES
Table 1. Equivalencies of generic surface reactions formulated in terms of one-site and two–site schemes.
One-Site
Two-Site Type of Reaction
# Amphoteric site
# Cationic site
# Anionic site
1a
H OH
3CO
Me O2H
3CO
Me n.r. 1b CO3H
0 CO3
- + H
+
Ionization (One-Step Protolysis)
2a
H2
2
O
3CO
Me O2H
3CO
Me 2b MeOH
0 MeO
- + H
+ 1b CO3H
0 CO3
- + H
+
Ionization (Two-Step Protolysis)
3a O3H
3CO
Me H O2H
3CO
Me
3b MeOH0 + H
+ MeOH2
+
n.r. Ionization
(Protonation)
4a
H MeOH
3CO
Me
2Me O2H
3CO
Me
n.r. 4b CO3H
0 + Me
2+ CO3Me
+ + H
+ Me
2+/H
+ Exchange
5a O2H3HCO
3CO
Me
23COH O2H
3CO
Me
5b MeOH0 + H
+ + CO3
2- MeCO3
- + H2O n.r.
HCO3- Adsorption
(One-site) CO3
2-/OH
- Exchange
(Two-site)
6a O2H3CO2H
3CO
Me
23CO2H O2H
3CO
Me
6b MeOH0 + 2H
+ + CO3
2- MeHCO3 + H2O
n.r.
H+/HCO3
- Co-
adsorption (One-site) HCO3
-/OH
- Adsorption
(Two-site)
n.r.: no additional reaction needed for full equivalency with the one-site scheme.
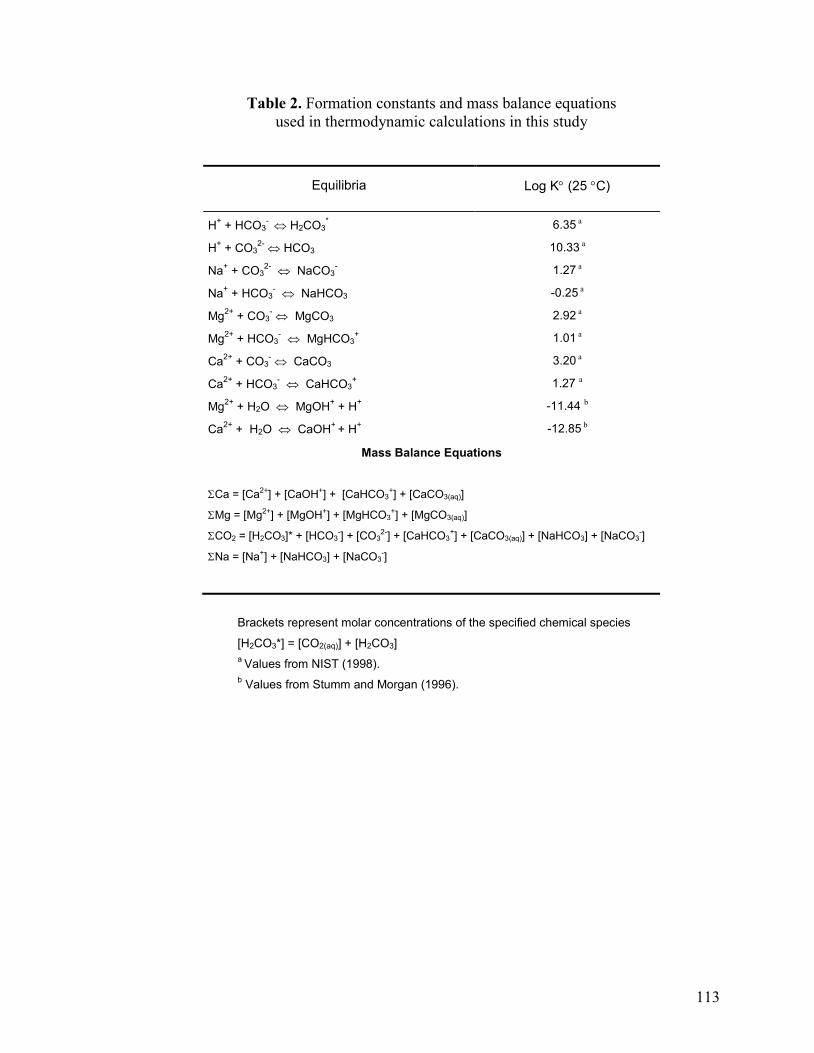
113
Table 2. Formation constants and mass balance equations
used in thermodynamic calculations in this study
Equilibria Log K (25 C)
H
+ + HCO3
- H2CO3
* 6.35 a
H+ + CO3
2- HCO3 10.33 a
Na+ + CO3
2- NaCO3
- 1.27 a
Na+ + HCO3
- NaHCO3 -0.25 a
Mg2+
+ CO3- MgCO3 2.92 a
Mg2+
+ HCO3- MgHCO3
+ 1.01 a
Ca2+
+ CO3- CaCO3 3.20 a
Ca2+
+ HCO3- CaHCO3
+ 1.27 a
Mg2+
+ H2O MgOH+ + H
+ -11.44 b
Ca2+
+ H2O CaOH+
+ H+ -12.85 b
Mass Balance Equations
Ca = [Ca2+] + [CaOH+] + [CaHCO3+] + [CaCO3(aq)]
Mg = [Mg2+] + [MgOH+] + [MgHCO3+] + [MgCO3(aq)]
CO2 = [H2CO3]* + [HCO3-] + [CO3
2-] + [CaHCO3+] + [CaCO3(aq)] + [NaHCO3] + [NaCO3
-]
Na = [Na+] + [NaHCO3] + [NaCO3-]
Brackets represent molar concentrations of the specified chemical species
[H2CO3*] = [CO2(aq)] + [H2CO3]
a Values from NIST (1998).
b Values from Stumm and Morgan (1996).
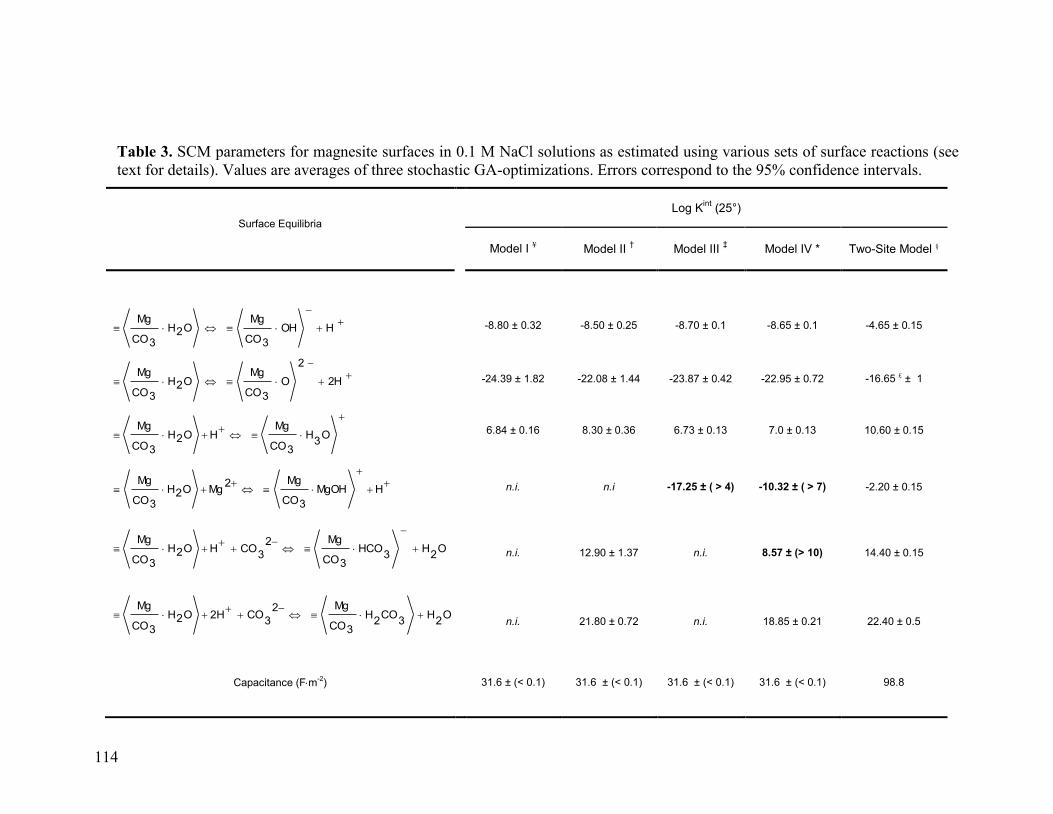
114
Table 3. SCM parameters for magnesite surfaces in 0.1 M NaCl solutions as estimated using various sets of surface reactions (see
text for details). Values are averages of three stochastic GA-optimizations. Errors correspond to the 95% confidence intervals.
Surface Equilibria
Log Kint
(25°)
Model I ¥
Model II
† Model III
‡ Model IV * Two-Site Model §
H OH
3CO
Mg O2H
3CO
Mg
-8.80 ± 0.32 -8.50 ± 0.25 -8.70 ± 0.1 -8.65 ± 0.1 -4.65 ± 0.15
H2
2
O
3CO
Mg O2H
3CO
Mg
-24.39 ± 1.82 -22.08 ± 1.44 -23.87 ± 0.42 -22.95 ± 0.72 -16.65 £ ± 1
O3
H
3CO
Mg H O2H
3CO
Mg
6.84 ± 0.16 8.30 ± 0.36 6.73 ± 0.13 7.0 ± 0.13 10.60 ± 0.15
H MgOH
3CO
Mg
2Mg O2H
3CO
Mg
n.i. n.i -17.25 ± ( > 4) -10.32 ± ( > 7) -2.20 ± 0.15
O2
H3
HCO
3CO
Mg
2
3COH O2H
3CO
Mg
n.i. 12.90 ± 1.37 n.i. 8.57 ± (> 10) 14.40 ± 0.15
O2
H3
CO2
H
3CO
Mg
2
3CO2H O2H
3CO
Mg
n.i. 21.80 ± 0.72 n.i. 18.85 ± 0.21 22.40 ± 0.5
Capacitance (Fm-2)
31.6 ± (< 0.1) 31.6 ± (< 0.1) 31.6 ± (< 0.1) 31.6 ± (< 0.1) 98.8

115
(Footnote of Table 3)
Intrinsic constants with large uncertainties in bold (see text for details). Optimization performed using data set at following conditions:
(¥)CO2 < 1.7 mM, Mg < 1 mM; (†) 1 < CO2 < 10 mM and 1.2 < Mg < 3 mM (optimization subsequently repeated with full data
set); (‡) 0.9 < CO2 < 3.6 mM and 1 < Mg < 7 mM; (*) Full data set. (§) Log10 Kint values for two-site-based equivalent reactions
taken from Pokrovsky et al., 1999a. (£) Value reflects full surface protolysis (cationic + anionic site). n.i. = reaction not included in the
model. Bold-type identify Log10 Kint with large uncertainties. Note that these values cannot be directly compared with those of
Wolthers et al. (2008) because their Log10 Kint values correspond to a „„hybrid” CCM-CD-MUSIC model. These authors only calibrated
ionization constants (generic reaction 1a–3a, plus a novel reaction involving the doubled-protonated carbonate corner site: >CO3H2+)
according to the CD-MUSIC-Triple-Plane model, whereas all constituent ion adsorption reactions (generic reaction 4a–6a) were taken
from Pokrovsky et al. (1999a), CCM approach) without further adjustment.
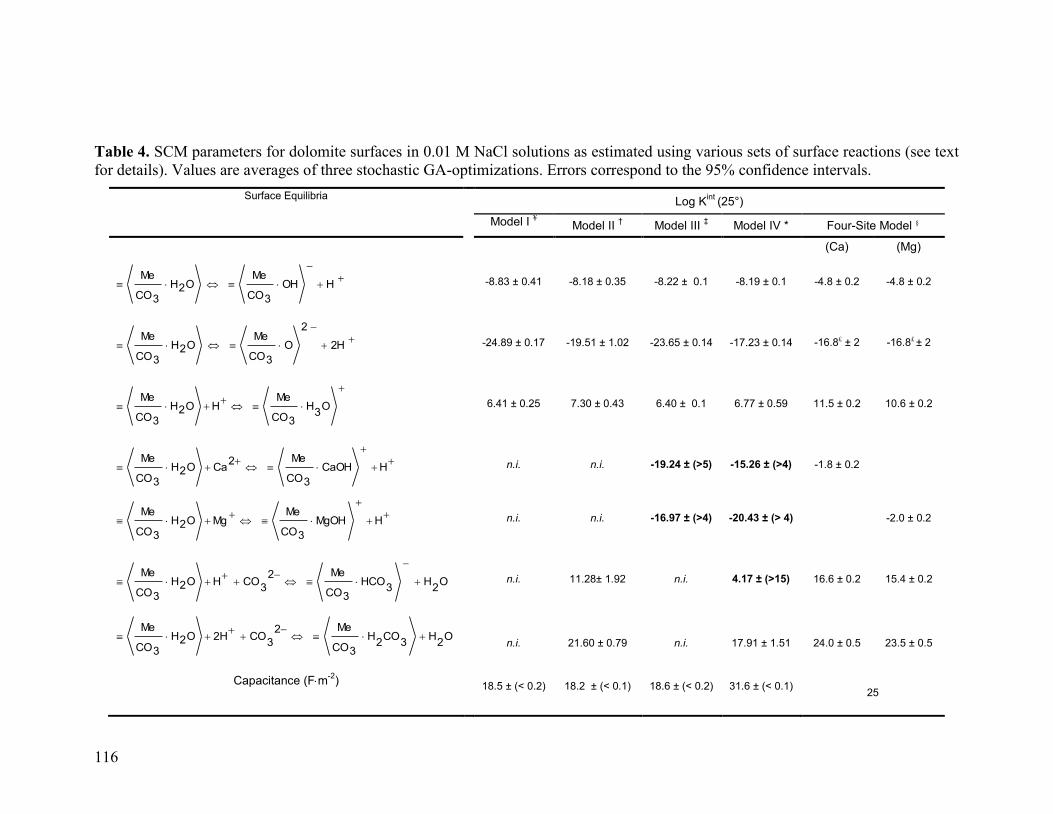
116
Table 4. SCM parameters for dolomite surfaces in 0.01 M NaCl solutions as estimated using various sets of surface reactions (see text
for details). Values are averages of three stochastic GA-optimizations. Errors correspond to the 95% confidence intervals.
Surface Equilibria
Log Kint
(25°)
Model I ¥
Model II
† Model III
‡ Model IV * Four-Site Model §
(Ca) (Mg)
H OH
3CO
Me O2H
3CO
Me
-8.83 ± 0.41 -8.18 ± 0.35 -8.22 ± 0.1 -8.19 ± 0.1 -4.8 ± 0.2 -4.8 ± 0.2
H2
2
O
3CO
Me O2H
3CO
Me
-24.89 ± 0.17 -19.51 ± 1.02 -23.65 ± 0.14 -17.23 ± 0.14 -16.8£ ± 2 -16.8£ ± 2
O3
H
3CO
Me H O2H
3CO
Me
6.41 ± 0.25 7.30 ± 0.43 6.40 ± 0.1 6.77 ± 0.59 11.5 ± 0.2 10.6 ± 0.2
H CaOH
3CO
Me
2Ca O2H
3CO
Me
n.i. n.i. -19.24 ± (>5) -15.26 ± (>4) -1.8 ± 0.2
H MgOH
3CO
Me Mg O2H
3CO
Me
n.i. n.i. -16.97 ± (>4) -20.43 ± (> 4) -2.0 ± 0.2
O2
H3
HCO
3CO
Me
2
3COH O2H
3CO
Me
n.i. 11.28± 1.92 n.i. 4.17 ± (>15) 16.6 ± 0.2 15.4 ± 0.2
O2
H3
CO2
H
3CO
Me
2
3CO2H O2H
3CO
Me
n.i. 21.60 ± 0.79 n.i. 17.91 ± 1.51 24.0 ± 0.5 23.5 ± 0.5
Capacitance (Fm-2
)
18.5 ± (< 0.2) 18.2 ± (< 0.1) 18.6 ± (< 0.2) 31.6 ± (< 0.1)
25

117
(Footnote of Table 4)
Intrinsic constants with large uncertainties in bold (see text for details). “Me” represents generically either Ca2+or Mg2+. Optimization
performed using data set at following conditions: (¥) CO2 < 1 mM, Ca < 0.5 mM, and Mg < 0.8 mM; (†) 0.6 < CO2 < 3 mM, 0.18
< Mg < 1.5 mM, and 0.06 < Ca < 1.5 mM (optimization subsequently repeated with full data set); (‡) 2< CO2 < 2.7 mM, 1.1 < Mg
< 2.7 mM, and 1.1 < Ca < 2.7 mM; (*) Full data set. (§) Log10 Kint values for four-site-based equivalent reactions taken from
Pokrovsky et al., 1999b. (£) Value reflects full surface protolysis (cationic + anionic site). n.i. = reaction not included in the model.
Bold-type identify Log10 Kint with large uncertainties. Note that these values cannot be directly compared with those of Wolthers et al.
(2008) because their Log10 Kint values correspond to a „„hybrid” CCM-CD-MUSIC model. These authors only calibrated ionization
constants (generic reaction 1a–3a, plus a novel reaction involving the doubled-protonated carbonate corner site: >CO3H2+) according
to the CD-MUSIC-Triple-Plane model, whereas all constituent ion adsorption reactions (generic reaction 4a–6a) were taken from
Pokrovsky et al. (1999b), CCM approach) without further adjustment.
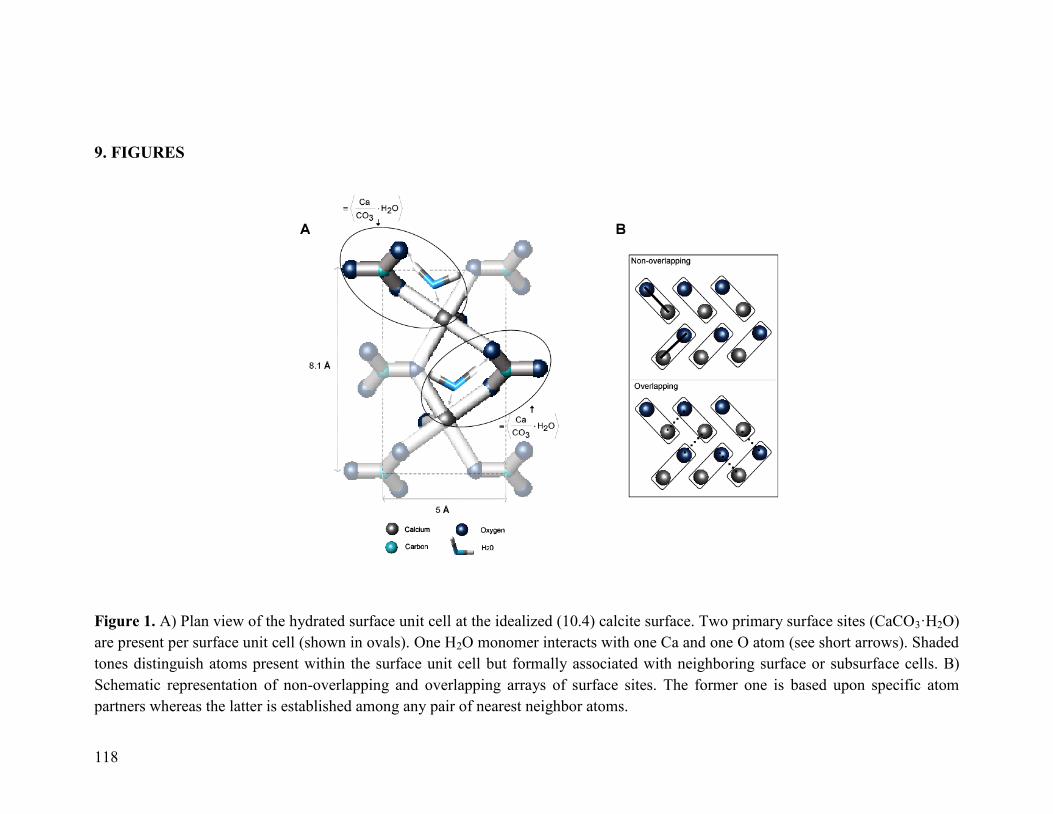
118
9. FIGURES
Figure 1. A) Plan view of the hydrated surface unit cell at the idealized (10.4) calcite surface. Two primary surface sites (CaCO3·H2O)
are present per surface unit cell (shown in ovals). One H2O monomer interacts with one Ca and one O atom (see short arrows). Shaded
tones distinguish atoms present within the surface unit cell but formally associated with neighboring surface or subsurface cells. B)
Schematic representation of non-overlapping and overlapping arrays of surface sites. The former one is based upon specific atom
partners whereas the latter is established among any pair of nearest neighbor atoms.
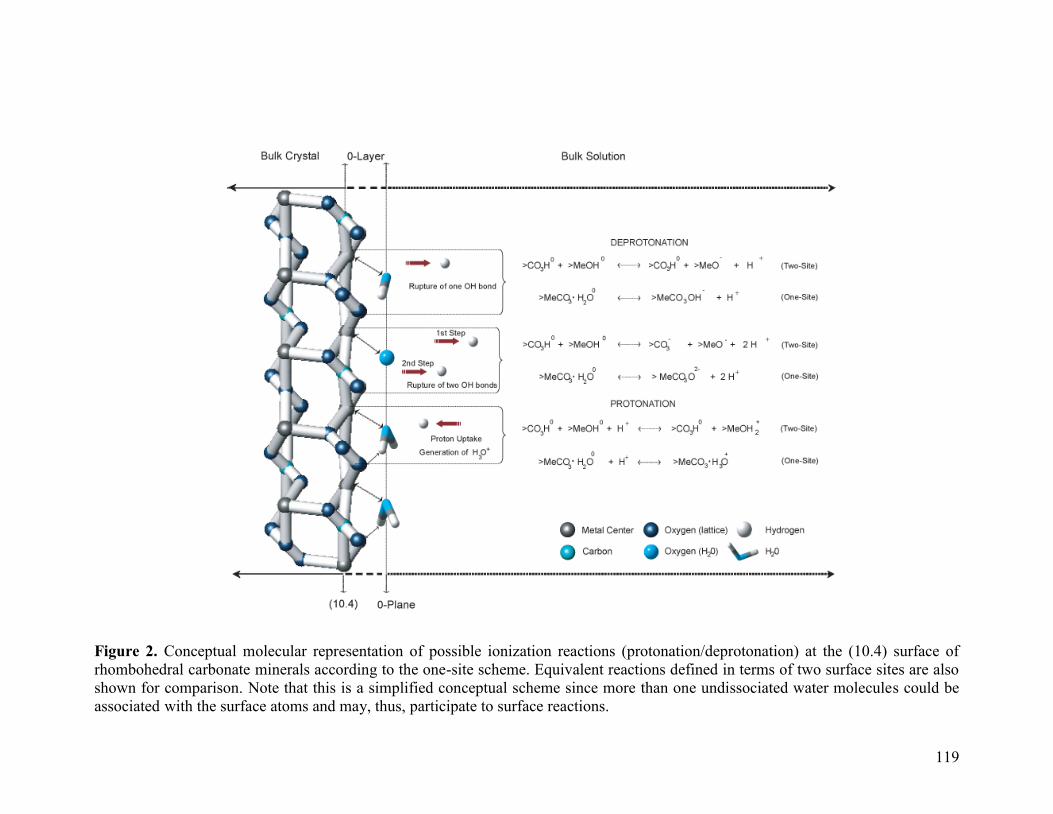
119
Figure 2. Conceptual molecular representation of possible ionization reactions (protonation/deprotonation) at the (10.4) surface of
rhombohedral carbonate minerals according to the one-site scheme. Equivalent reactions defined in terms of two surface sites are also
shown for comparison. Note that this is a simplified conceptual scheme since more than one undissociated water molecules could be
associated with the surface atoms and may, thus, participate to surface reactions.
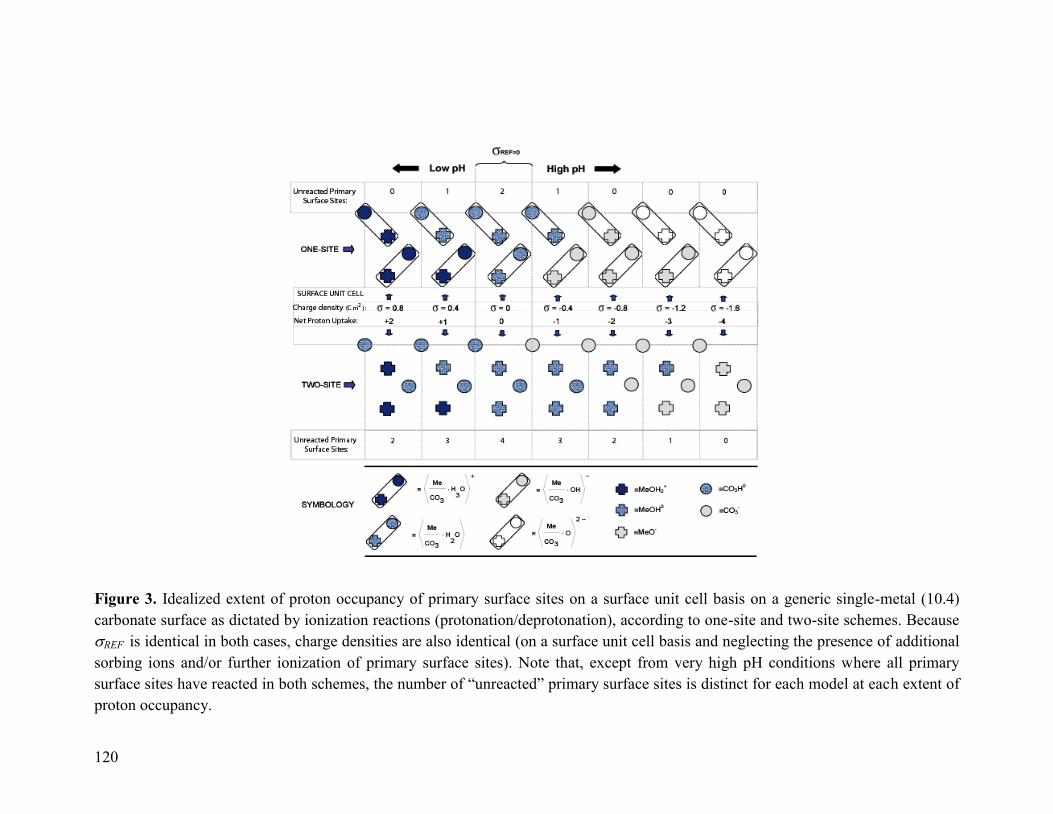
120
Figure 3. Idealized extent of proton occupancy of primary surface sites on a surface unit cell basis on a generic single-metal (10.4)
carbonate surface as dictated by ionization reactions (protonation/deprotonation), according to one-site and two-site schemes. Because
REF is identical in both cases, charge densities are also identical (on a surface unit cell basis and neglecting the presence of additional
sorbing ions and/or further ionization of primary surface sites). Note that, except from very high pH conditions where all primary
surface sites have reacted in both schemes, the number of “unreacted” primary surface sites is distinct for each model at each extent of
proton occupancy.
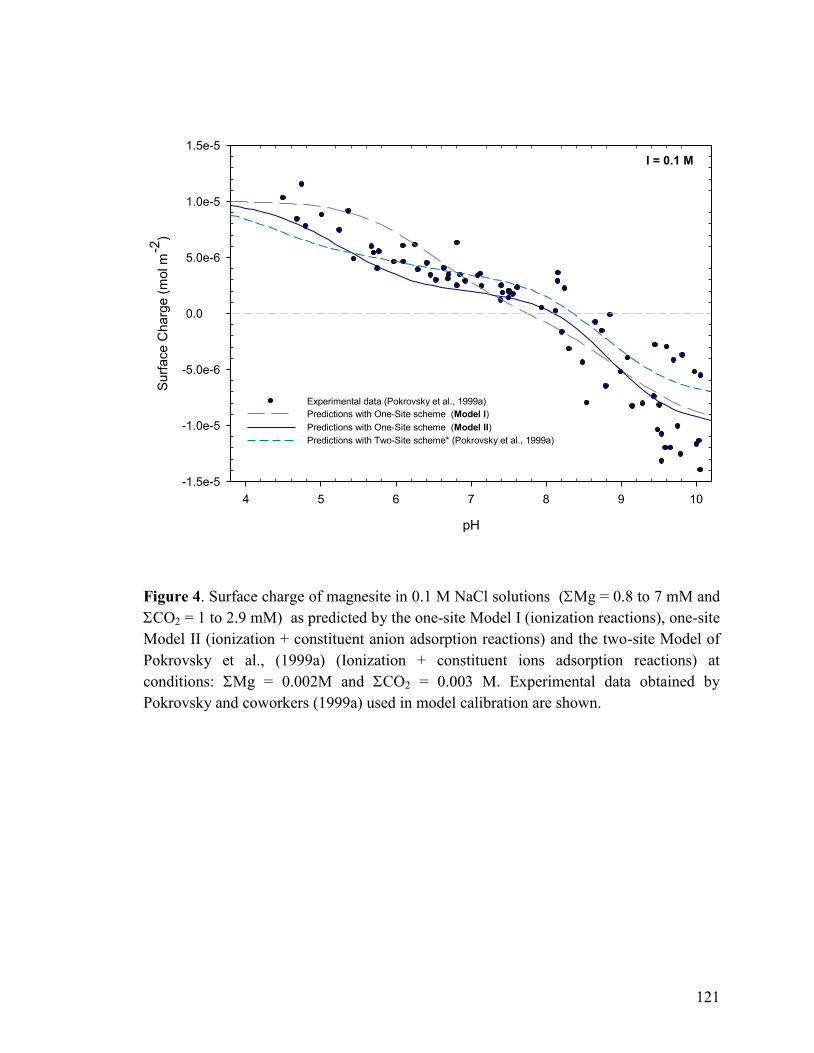
121
pH
4 5 6 7 8 9 10
Surf
ace C
harg
e (
mol m
-2)
-1.5e-5
-1.0e-5
-5.0e-6
0.0
5.0e-6
1.0e-5
1.5e-5
Experimental data (Pokrovsky et al., 1999a)
Predictions with One-Site scheme (Model I)
Predictions with One-Site scheme (Model II)
Predictions with Two-Site scheme* (Pokrovsky et al., 1999a)
I = 0.1 M
Figure 4. Surface charge of magnesite in 0.1 M NaCl solutions (Mg = 0.8 to 7 mM and
CO2 = 1 to 2.9 mM) as predicted by the one-site Model I (ionization reactions), one-site
Model II (ionization + constituent anion adsorption reactions) and the two-site Model of
Pokrovsky et al., (1999a) (Ionization + constituent ions adsorption reactions) at
conditions: Mg = 0.002M and CO2 = 0.003 M. Experimental data obtained by
Pokrovsky and coworkers (1999a) used in model calibration are shown.
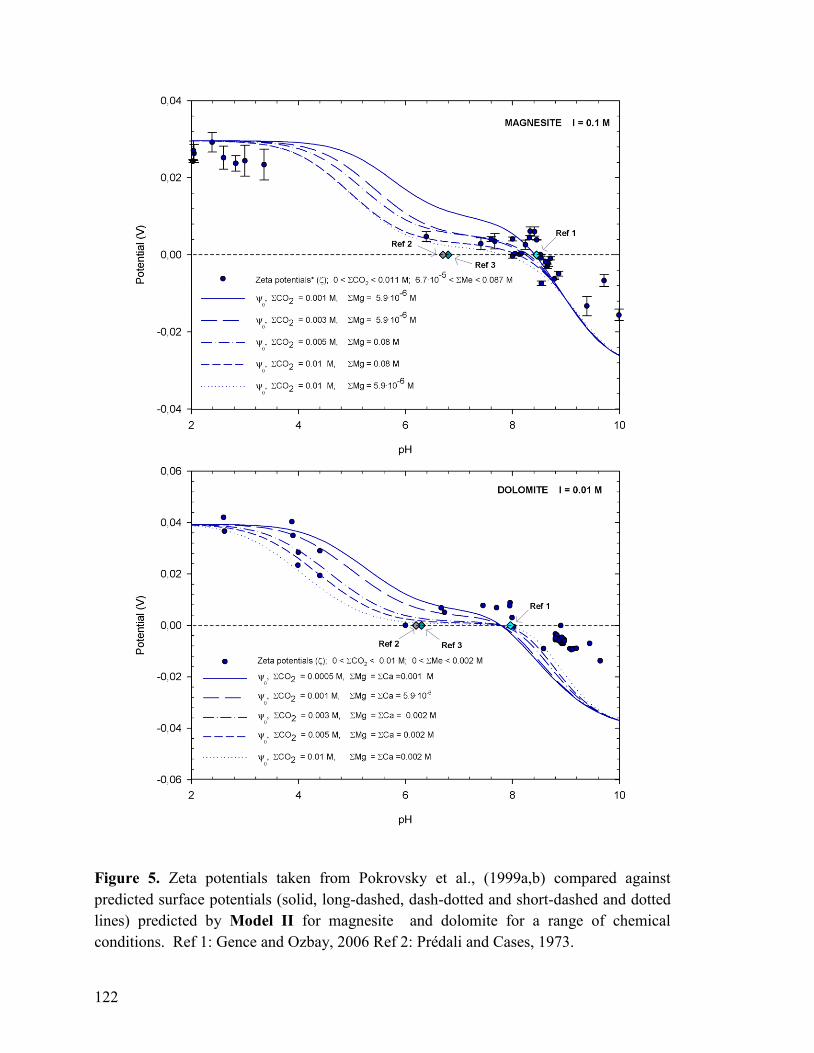
122
Figure 5. Zeta potentials taken from Pokrovsky et al., (1999a,b) compared against
predicted surface potentials (solid, long-dashed, dash-dotted and short-dashed and dotted
lines) predicted by Model II for magnesite and dolomite for a range of chemical
conditions. Ref 1: Gence and Ozbay, 2006 Ref 2: Prédali and Cases, 1973.
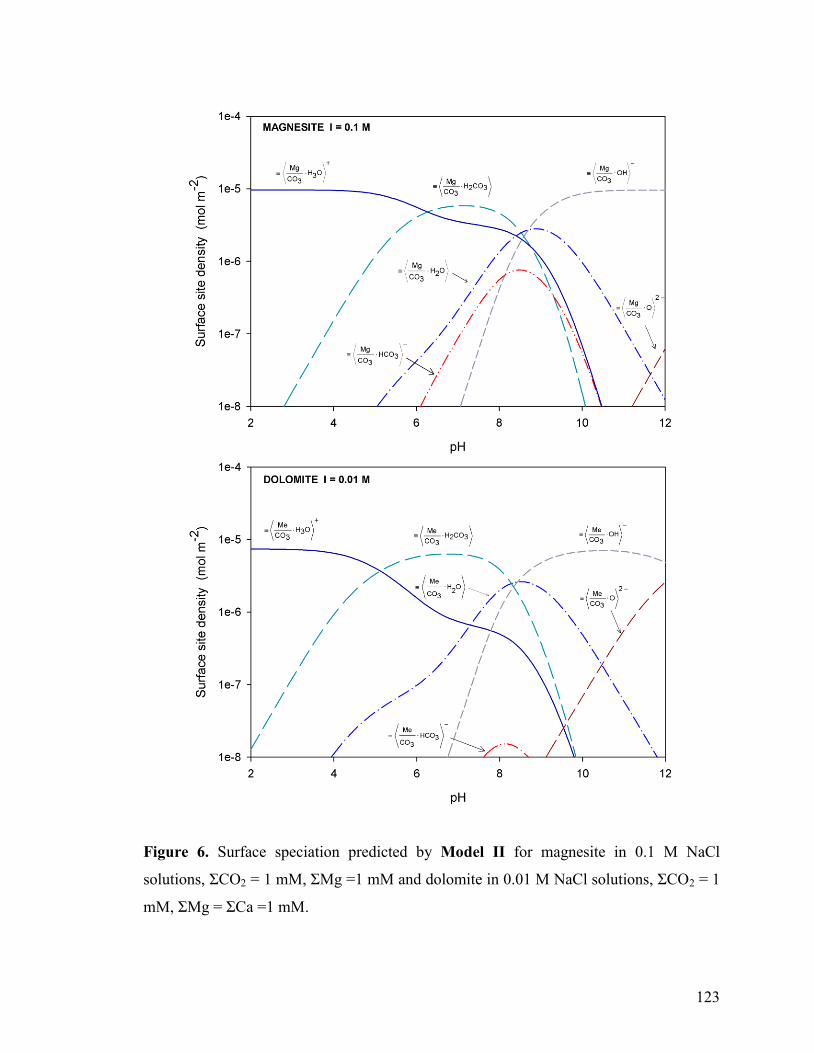
123
Figure 6. Surface speciation predicted by Model II for magnesite in 0.1 M NaCl
solutions, ΣCO2 = 1 mM, ΣMg =1 mM and dolomite in 0.01 M NaCl solutions, ΣCO2 = 1
mM, ΣMg = ΣCa =1 mM.

124
PREFACE TO CHAPTER 4
Having developed and successfully tested the single surface site formalism for magnesite
and dolomite, this scheme was tested on other rhombohedral carbonate minerals to
generalize its application to the construct of SCMs for this type of minerals. This aspect is
addressed in the following chapter, “Acid-Base Behavior of the NiCO3(s) Surface in NaCl
Solutions”.
Because NiCO3(s) (gaspeite) is the least reactive of known naturally-occurring
rhombohedral carbonate minerals in aqueous solutions, it was selected as a surrogate to
obtain additional experimental information on the surface reactivity of calcite-type
minerals in NaCl solutions by means of conventional titration techniques and micro-
electrophoresis. In this study we found that surface protonation of NiCO3(s) is strongly
affected by the background electrolyte beyond what is typically observed in most mineral
surfaces such as metal oxides, silicates and clay surfaces.
A simple one-site-based SCM is postulated that successfully simulates proton
adsorption and electrokinetic data acquired at I ≤ 0.01 M and outperforms the predictive
power of more sophisticated SCMs tested in this study. The most important insights
obtained in this study is that the background electrolyte affects the properties of the
gaspeite surface (surface protonation and the development of surface charge) possibly
through modification of the structure of the electrified interfacial layer, perturbation of
the solvent structure dynamics and the affinity of water molecules and adsorbing ions
towards the mineral surface. These observations challenge earlier conceptions on
carbonate mineral surfaces that traditionally considered surface charge acquisition
processes on these minerals as chemically indifferent to background electrolyte ions.

125
CHAPTER 4
ACID-BASE BEHAVIOR OF THE GASPEITE (NiCO3(S)) SURFACE IN NaCl SOLUTIONS
Adrián Villegas-Jiménez*1, Alfonso Mucci
1, Oleg S. Pokrovsky
2, Jacques Schott
2 and
Jeanne Paquette1
1 Earth and Planetary Sciences, McGill University, 3450 University Street
Montréal, Qc H3A 2A7, Canada.
2Géochimie et Biogéochimie Expérimentale,
LMTG, UMR 5563,
Université de Toulouse – CNRS, 14, Avenue Edouard Belin 31400 Toulouse, France
*Corresponding Author
E-mail: [email protected]
To be submitted to: Langmuir

126
ABSTRACT
The acid-base properties of the gaspeite surface in NaCl solutions were investigated at
nearly-ambient conditions (25 3 C and 1 atm) by means of conventional acidimetric
and alkalimetric titration techniques and microelectrophoresis. Dissolution-corrected
proton adsorption densitites and electrokinetic data were obtained over a pH range of 5 to
10 under CO2-free conditions at three ionic strengths (0.001, 0.01 and 0.1 M). Over the
entire pH range investigated in this study, surface protonation and electrokinetic mobility
are strongly affected by the backgrund electrolyte leading to a significant shift in the pH
of Zero Net Proton Charge (pHznpc) and the pH of isoelectric point (pHiep) towards lower
pH with increasing ionic strength. This is conceptually explained by the role exerted by
the background electrolyte which affects in more than one way the properties of the
gaspeite surface (surface protonation and the development of surface charge) possibly
through modification of the structure of the electrified interfacial layer, perturbation of
the solvent structure dynamics, and the affinity of water molecules and adsorbing ions
towards the mineral surface. These observations challenge earlier conceptions that
traditionally considered surface charge acquisition processes on carbonate minerals as
chemically indifferent to background electrolyte ions. Although no self-consistent
interpretation was found to explain all data, a simple SCM involving ionization reactions
closely simulates proton adsorption data and reasonably predicts the electrokinetic
behavior of gaspeite supensions at low (I=0.001 M) and intermediate (I=0.01 M) ionic
strengths. Nevertheless, the influence of the background electrolyte on the development
of surface charge and surface protonation must be further investigated.
Keywords: Nickel carbonate, gaspeite acid-base behavior, surface ionization, sodium
adsorption

127
1. INTRODUCTION
Carbonate minerals are of considerable environmental significance due to their ubiquity
and high chemical reactivity in natural aquatic systems. In aqueous solutions, their
macroscopic properties are controlled by multiple homogeneous and heterogeneous
equilibria among which, surface reactions (i.e. ionization and adsorption) are recognized
to play a critical role (Van Capellen et al., 1993). This realization has stimulated
numerous scientific studies on the surface reactivity of hydrated carbonate surfaces
(Pokrovsky et al., 1999a,b; Brady et al., 1999; Pokrovsky and Schott, 2002; Jordan et al.,
2001; Duckworth and Martin, 2003; Kendall and Martin, 2005). Particular attention has
been paid to the derivation of empirical and semi-empirical relationships to represent ion
partitioning between the aqueous phase and the surface of calcite, aragonite, magnesium-
bearing carbonates and, to a lesser extent, other divalent carbonate minerals (Morse and
Mackenzie, 1990).
Despite these efforts, the quantitative characterization of most carbonate surfaces
has lagged behind that of other mineral surfaces such as metal oxides and silicates (Davis
and Kent, 1990). This task is a sizable challenge because of the higher reactivities (i.e.,
faster reaction rates and greater solubilities) of carbonates relative to other minerals, and
the occurrence of stepwise and/or parallel reactions (e.g., adsorption, surface
precipitation, co-precipitation, dissolution) that are difficult to resolve experimentally
(Morse, 1986). Dissolution and precipitation reactions, in particular, interfere with the
characterization of surface equilibria. These considerations drove earlier workers to
develop a novel experimental protocol, based upon the use of a fast flow-through reactor,
to minimize the contribution of dissolution and precipitation, during acid-base titrations

128
performed on some sparingly soluble carbonates (Charlet et al., 1990). This protocol was
used by several researchers to obtain surface charge data for siderite, rhodochrosite
(Charlet et al., 1990; Van Capellen et al., 1993), magnesite (Pokrovsky et al., 1999a) and
dolomite (Pokrovsky et al., 1999b; Brady et al., 1999). On the basis of their results, they
proposed surface complexation models (SCMs) for these minerals that include ionization
and lattice constituents adsorption reactions in analogy to acid-base and complexation
equilibria in solution for the CO2(s)-H2O system. Unfortunately, the application of this
approach to highly reactive carbonate minerals such as calcite or aragonite is not feasible
because their fast dissolution kinetics interferes significantly with the computation of
surface charge (Van Cappellen et al., 1993).
Conversely, gaspeite, a nickel-bearing carbonate with a calcite-type structure
displays the slowest dissolution kinetics of all naturally-occurring rhombohedral
carbonate minerals in aqueous solutions (Pokrovsky and Schott, 2002), and thus, it is
amenable to investigations by experimental protocols commonly applied to metals oxides
but unsuitable for other carbonate minerals. Natural gaspeite specimens typically contain
intermediate to high amounts of magnesium which closely reflect the physical properties
of the hypothetical solid solution: Ni0.5Mg0.5(CO3)2 (Kohls and Rodda, 1966). In contrast,
only a small degree of Ca2+
substitution by Ni 2+
ions in the calcite structure has been
experimentally confirmed (Hoffmann and Stipp, 2001), which may explain why a NiCO3-
CaCO3 solid solution series has not be found in nature.
Regardless of their purity, NiCO3(s)-bearing specimens display a rhombohedral
structure predominantly bounded by the (10.4) domain (Bermanec et al., 2000), a
common feature of other carbonate isomorphs such as calcite, magnesite and dolomite
(Reeder, 1990). This characteristic makes nickel carbonate a suitable surrogate to obtain

129
information on the surface reactivity of calcite-type minerals. Although the formation of
hydrated NiCO3(s) phases, NiCO32Ni(HO)2 and NiCO34H2O, is possible at room
temperature (Hoffmann and Stipp, 2001), pure hydrothermally-synthesized gaspeite
rhombohedral crystals are thermodynamically stable at room temperature and, are
therefore, suitable for experimental investigations at ambient conditions.
To our knowledge, a single investigation of the surface reactivity of gaspeite has
been carried out to date (Pokrovsky and Schott, 2002), which included a study of its
electrokinetic behavior in aqueous solutions over a range of pH (6 to 9) and a fairly
constant aqueous composition (CO2 = 510–3
M, 10–6
Ni 210–6
and I = 0.005 M).
No systematic investigation on the acid-base properties of this mineral, however, has been
conducted under a wide range of chemical conditions.
In this study, a series of acidimetric and alkalimetric titrations are performed on
NiCO3(s) suspensions at three ionic strengths within a wide pH range (5 to 10) where
dissolution effects can be quantitatively accounted for in the computation of proton
adsorption. In addition, numerous electrophoretic measurements are performed under a
wide range of solution conditions (pH and ionic strength). These data are used to calibrate
and test surface complexation reactions for gaspeite formulated according to the one-site
scheme previously introduced for the hydrated (10.4) calcite surface (Villegas-Jiménez et
al., 2009a) and successfully extended to other rhombohedral carbonate minerals as
discussed in Chapter 3 of this thesis.

130
2. MATERIALS AND METHODS
2.1 Preparation and Standardization of Reagents
All solutions were prepared using analytical grade reagents and high purity deionized
(MilliQ, ~ 18 Mohm cm-1
) water. Hydrochloric acid solutions were prepared from 32%
HCl and standardized using gravimetrically prepared tris(hydroxymethyl)methylamine
(TRIS) solutions that were kept refrigerated. NaOH solutions were prepared every two to
three days from NaOH pellets using MilliQ water from which CO2 had been removed by
boiling and bubbling of ultrapure N2 for at least three hours. These solutions were titrated
against standardized HCl solutions before use and were kept under a N2 atmosphere to
minimize CO2 absorption. The ionic strength of the solutions was adjusted (i.e., 0.001,
0.01 or 0.1 M) using a 1 M NaCl solution.
2.2 Chemical Analyses
Alkalinity measurements were carried out using a Radiometer TTT85 titration system
using standardized HCl. The end-point of the titration was identified by the first-
derivative method (APHA-AWWA-WPCF, 1998). The precision of the analysis was ±
0.05 % and the limit of detection was 0.6 mmol kg-1
. Nickel concentrations were
measured by Graphite Furnace Atomic Absorption Spectrophotometry (GFAAS,
AAnalystTM
800 Perkin Elmer) using external standards (i.e., diluted from a 1000 ppm
Certified Standard). The detection limit of this analysis was 0.8 μg L-1
with a
reproducibility of ± 4 %. Free carbonate ion concentrations were determined using a
combination ELIT Ion 8091 ion selective electrode calibrated against NaHCO3 standards
covering a relatively wide range of CO32-
ion concentrations (5 M to 1 mM), as
calculated from thermodynamic equilibrium calculations performed iteratively using the

131
Newton-Raphson method implemented in an in-house computer Matlab©
routine using
alkalinity and pH measurements as input. Equilibrium constants and mass balance
equations used in all the thermodynamic calculations of this study are given in Table 1.
2.3 Gaspeite Synthesis
Gaspeite was hydrothermally synthesized during 2 months at 250 °C in titanium reactors
from analytical reagent grade hydrous nickel carbonate. Synthesis was performed in
solutions of pH (25 °C) ~ 4 and pCO2 of about 40 atm, achieved by addition of ~10 grams
of solid CO2 per 200 mL of distilled water in the reactor before the synthesis.
The precipitated powder was oven-dried at 50°C, dry-sieved, and the 0.1-50 m
size fraction isolated for the surface titrations. Its mineralogy and crystallinity was
confirmed by X-ray diffraction analysis using a G3000 INEL diffractometer. All major
peaks of gaspeite were revealed, and no trace of other phases or impurities were found.
The specific surface area of the < 50 m size fraction, obtained by sieving, was
determined by the multiple-point Ar-BET method (Brunauer et al., 1938) before and after
the titrations to check for variations resulting from the dissolution of the smaller particles.
Note that to minimize the surface irregularities, no grinding of synthetic powder was
performed. The surface area of freshly prepared powder was of 0.38 m2
g-1
; but after the
first titration, it decreased to 0.30 m2 g
-1, probably due to complete dissolution of ultrafine
particles. After repeated titrations (more than two) of the same powder, the specific
surface area remained constant: 0.23 m2 g
-1 within the uncertainty of the measurements (±
0.005 m2 g
-1). For the sake of consistency and considering the limited amount of available
gaspeite powder, only data obtained from surface titrations using recycled powders (n >
2) were used in the computation of proton adsorption. After each titration, the powder

132
was extensively rinsed with Milli-Q® water to remove surface impurities arising from
previous titrations (i.e. adsorbed protons, nickel or background electrolyte ions), filtered,
and oven-dried at 70°C before being re-used. The excellent reproducibility of titration
data acquired at all ionic strengths examined in this study (see below) confirms the
absence of significant amounts of recalcitrant contaminants in the recycled powder (in
contrast to what it is sometimes observed for metal oxide surfaces) and justifies its re-use
in the experiments presented in this study.
2.4 Surface Titrations
2.4.1 pH Electrode Calibration
Surface titrations were conducted with a Radiometer Titralab 865 titrator equipped with a
Schott N6980 pH electrode, suitable for pH measurements in concentrated suspensions,
and a Teflon-coated suspended stir bar to minimize grinding of the powder. The
Nernstian behavior of the electrode was checked before and after each experiment against
four NIST-traceable pH buffers (4, 7, 10 and 11) at 25 0.5°C and it was, in all cases,
very similar ( 0.8 mV) to the theoretical value at 25°C (i.e. 59.2 mV).
The pH electrode was calibrated on the total proton molar concentration scale
according to:
j
0 E EE
][H log
F
RT 2.303 (1)
where E and E° are the observed and the standard potential values for a given ionic
strength, F is the Faraday Constant, R is the universal gas constant, T is the absolute
temperature, [H+] is the total proton molar concentration and Ej is the junction potential.

133
For accurate pH determinations, the effect of the background electrolyte must be
properly accounted for in the calibration of the pH electrode (Wiesner et al., 2006). To
this end, we followed the method described by Pehrsson et al (1976). Briefly, this method
consists of estimating the junction potential which is defined by: Ej = jH [H+] for the
acidic regime (pH < 7) and by: Ej = jOH [OH-] for the alkaline regime (pH > 7). The
coefficients jH and jOH are characteristic of the electrode and the ionic medium. They were
estimated independently by titration of 1 mM HCl solutions with standardized equimolar
NaOH (acidic regime) or of 1 mM NaOH solutions with standardized equimolar HCl
(alkaline regime). In all cases, the ionic strength of the solutions was fixed with NaCl at
the values of interest (i.e. 0.001, 0.01 and 0.1 M). The coefficients jH and jOH were readily
obtained from a linear regression between E – 59.2 log [H+] against [H
+] (or [OH
-] for the
alkaline regime) which yielded jH (or jOH) as the slope and E° as the intercept. Using these
coefficients and Equation 1, a more accurate estimate of E° was computed from blank
titrations of the background electrolyte in the absence of solids conducted at both, the
acid and the alkaline regimes under identical conditions as for the suspension titrations
(i.e. pH range, ionic strength). For each pH regime and ionic strength, the mean of
multiple E° determinations were adopted for pH determination. As the same reaction
vessel was used for both blank and suspension titrations, wall effects (i.e. proton
adsorption) are implicitly considered in the estimation of E0. For a given set of E
0, jH and
jOH values, the proton molar concentration was calculated numerically via Equation 1
using the Newton-Raphson iterative method incorporated in an in-house Matlab©
subroutine (available in the appendices to this thesis). The Davies equation was used to
estimate activity coefficients and compute the pH from the calculated molar
concentrations (Stumm and Morgan, 1996).

134
2.4.2 Conditions of Surface Titrations
Preliminary titrations revealed that highly concentrated gaspeite suspensions are required
to resolve the contribution of adsorption reactions in determining the bulk solution
equilibria. Consequently, all titrations were conducted at a solid/solution ratio of 50 g L-1
.
The stability criterion of the automatic titrator for the pH electrode was set at 0.3 mV min-
1 to minimize the duration of the titration (and minimize dissolution effects and electrode
drift) and to maintain an acceptable accuracy. Two different types of titrations were
conducted at three ionic strengths (0.001, 0.01, 0.10 M NaCl). Type-I experiments, run in
triplicate to verify their reproducibility, were acidimetric titrations performed within the
pH range of 5 to 10. The suspensions were first prepared in CO2-free Milli-Q
water (as
described in section 2.1) and allowed to equilibrate for about 10 minutes before the pH
was adjusted by the addition of a known amount of the standardized NaOH solution and
the titration initiated shortly after. A short pre-equilibration time is critical to allow for the
hydration of the surface while keeping dissolution to a minimum. The duration of these
titrations varied from 8 to 10 hours.
The dissolved nickel concentration was monitored throughout these experiments
in order to determine the consumption of protons due to gaspeite dissolution, as described
by the following reaction:
NiCO3(s) + 2 H+ Ni
2+ + H2O + CO2 (g) (2)
This was achieved by carrying out numerous additional titrations (n > 10), under
the same experimental conditions (i.e., pH range, ionic strength, duration of titration, etc),
that were interrupted at critical pH values along the titration curve. Aliquots of the

135
decanted solution were withdrawn and syringe-filtered through a 0.45 m Milipore HA-
type filter into HDPE bottles. The solutions were acidified with a 1% equivalent volume
of concentrated HCl and stored for later nickel analysis by GFAAS. These data were fit to
logarithmic expressions that served to interpolate nickel concentrations at given pH
values.
Type-II experiments were alkalimetric titrations of gaspeite suspensions prepared
in CO2–free Milli-Q
water with no pre-addition of NaOH and a pre-equilibration time of
less than 10 minutes. Given the enhanced dissolution kinetics of gaspeite at low pH
(Pokrovsky and Schott, 2002), these experiments were initiated at circumneutral pH and
ended at a pH of about 10. The duration of these titrations was between 3 to 4 hours.
To prevent carbonate adsorption reactions from competing with the acid-base
equilibria at reactive adsorption sites, suspensions were maintained CO2-free by bubbling
ultrapure N2 in the suspensions throughout the titrations. In preliminary experiments, the
absence of significant amounts of inorganic carbon in the system was verified by
alkalinity determinations and by direct measurements of the carbonate ion concentration
using the carbonate ion selective electrode at pre-determined points along the titration
curve. Evaporation of the experimental solution was minimized by keeping a positive
pressure of H2O vapor-saturated N2 in the headspace overlying the reaction vessel. In all
cases, the extent of evaporation was determined gravimetrically to be less than 1% over
the course of whole titrations (up to 10 hours).

136
2.5 Computation of Proton Adsorption
In contrast to minerals containing protons and/or hydroxyls groups within their lattices
(e.g., kaolinite, goethite, lepidocrocite, hydroxylapatite), pure divalent metal carbonate
minerals (MeCO3(s)) do not contain protons or hydroxyls groups within their lattices, and
hence, no proton imbalance develops at their dry surfaces. In other words, no proton
excess (or deficit) is introduced into the mineral-H2O system upon MeCO3(s) immersion
in water. Thus, in the case of gaspeite, and provided CO2(g) exchange with the atmosphere
(affecting the proton and carbon balance in solution upon H2CO3(aq) formation) is
prevented or properly accounted for (as in our study, see below), net proton adsorption
densities, HNet
, can be computed by subtracting the experimentally-measured proton
balance in solution from the theoretical proton balance following incremental acid and/or
base additions:
H - OH = (1/AS) · (CA – CB – [H+] + [OH
-] – [H
+]diss) (3)
where H and OH are the calculated adsorption densities of H+ and OH
-
(mol m-2
), CA and CB are the total molar concentrations of the added acid and base, [H+]
and [OH-] are the estimated molar concentrations obtained from pH measurements, A is
the specific surface area (m2 g
-1), S is the solid concentration (g L
-1), and [H
+]diss
represents the net concentration of protons consumed by the dissolution of gaspeite.
Because, for gaspeite, HNet
data are directly obtained with Equation 3, no referencing to
the Point of Zero Net Proton Charge (PZNPC) is required, in contrast to H- and/or OH-
containing minerals that require a pre-determination of the PZNPC by suitable

137
approaches to compute net proton adsorption densities from apparent proton adsorption
data, Happ
(Anderson and Sposito, 1992, Chorover and Sposito, 1995). It follows that the
PZNPC determined across the pH scale for a given ionic strength (i.e., pHpznpc,I) can be
directly obtained from adequate analysis of HNet
data vs pH plots (see below).
According to reaction 2, two protons are needed for the release of one Ni2+
ion
which, in turn, can form a number of hydroxo-complexes depending on the pH of the
solution. Upon the formation of these complexes, protons will be released to the solution,
and thus, will contribute to the computed proton balance. This can be accounted for by
the following expressions:
1
)(
3
321
)(
2
211
3
2
2
22
2
)()()(1
OHNiH
Ni
OHNiH
Ni
NiOHH
Ni
a
KKK
a
KK
a
KNi
(4)
NiOHH
NiNi
a
KNiOH
22 1 (5)
2
2
)(
2
2
)( OHNiH
NiOHNiOH
a
KNiOH
(6)
3
22
3
)(
3
)(3
)(OHNiH
OHNiNiOH
a
KNiOH
(7)
where i and i represent, respectively, the ionization factors and the activity coefficients
of the aqueous species identified by the subscripts, K1-K3 are the thermodynamic stability
constants of the nickel hydroxo-complexes (see Table 1), and aH+ stands for the proton
activity. Thus, the net contribution of dissolution to the observed proton balance can be
computed from:

138
)NiOHNiOHNi
32diss (2 ]2[Ni ][H (8)
where [Ni2+
] is the total nickel molar concentration interpolated from the logarithmic fit
for a given pH and ionic strength (see preceding section).
Consideration of carbonate equilibria is, in principle, not necessary in the
computation of proton adsorption since our experimental set-up should allow for the
removal of most inorganic carbon evolved from gaspeite dissolution while preventing
contamination from atmospheric CO2(g). Nevertheless, the influence of undetected levels
of dissolved carbonate species (below the detection limits of alkalinity and CO32-
ion
selective electrode measurements) on the computation of proton adsorption is discussed
later.
Under the conditions of our titrations, the formation of freshly precipitated
Ni(OH)2(s), potentially affecting the proton and Ni2+
ion concentrations in solution, is not
expected to occur. Thermodynamic calculations using stability constants listed in Table 1
reveal that, in all cases, the experimental systems were kept below saturation with respect
to Ni(OH)2 throughout the titrations (saturation indices < 0.29).
2.6 Coagulation Experiments
To check whether electrolyte-induced coagulation occurred during our titrations and
could affect (upon reduction of the number of available reactive surface sites) the
computation of proton adsorption, we performed a semi-quantitative coagulation test. A
series of polypropylene centrifuge tubes containing aqueous gaspeite suspensions at
solid:solution ratios identical to those of the titrations (i.e. 50 g L-1
) were prepared at

139
varying concentrations of NaCl, which ranged from 100 μM to 1 M, including those
selected in our titrations. This test was run in duplicate. Tubes were shaken vigorously for
15 minutes using a Wrist Action Shaker (Burrell Model 75) followed by a settling period
of 2 minutes, a second stirring of 10 minutes and allowed to stand for 2 hours. A blank
(containing NiCO3(s) but no NaCl) was also tested for comparison (Hunter, 2001). A
slight coagulation effect (<1%) was observed for particles with a radius 0.6 μM
(representing less than 0.5% of the particle population) at ionic strengths equal or higher
than 0.01M, based on the turbidity of the supernatant of these suspensions measured by
UV spectrophotometry (Spectronic 601, Milton Roy Company). The packing volume of
the settled particles at all ionic strengths was nearly identical suggesting that no
significant aggregation of the larger particles took place (loose particles display larger
packing volume (Huang et al., 1991). Consequently, it is considered that the surface area
available for reaction was not significantly reduced by coagulation in any of the systems
investigated in this study.
2.7 Electrokinetic Measurements
The electrophoretic mobility of gaspeite particles in aqueous suspensions was measured
with a micro-electrophoremeter (Zeta-phoremeter IV 4000, CAD Instrumentation) at 25
4 °C under a range of conditions (i.e. pH, CO2, Ni and I). The NiCO3(s) suspensions
were prepared using the 0.5-1 m size fraction of the synthesized powder. According to
Stokes‟ law, this size fraction corresponds to the particle population remaining in
suspension after at least 12 (but less than 48 hours) of decantation in a 1 L Nalgene bottle.
Two series of electrokinetic experiments were carried out: Series-I consisted of individual
-potential measurements acquired in systems of variable composition where the pH was

140
varied with NaOH, CO2 was controlled with additions of NaHCO3 or Na2CO3 solutions
and Ni was varied by additions of supernatant from pre-equilibrated gaspeite
suspensions. In Series-II experiments, -potential measurements were performed in
systems of similar composition as those of the surface titrations, and therefore, only pH
and ionic strength were varied. To this end, several gaspeite suspensions were prepared in
CO2-free solutions at three ionic strengths (0.001, 0.01 and 0.1 M, prepared with NaCl)
and the pH of each suspension was varied by additions of HCl or NaOH. The -potentials
were measured for each selected pH value. Throughout these experiments, N2(g) was
constantly bubbled through the suspensions to prevent contamination from atmospheric
CO2(g).
The measurements were performed in triplicate on new suspensions in a quartz
cell connecting two Pd electrode chambers. The suspended particles were illuminated by
a 2 mW He/Ne laser and an electric field of 80 V (DC) cm-1
was applied. The particle
trajectories were followed by a CCD camera and data transmitted to a computer. The
electrophoretic mobility was derived from a time-lapsed image analysis and the
potentials were estimated using the Smoluchowski equation. This equation is applicable
to the ionic strengths and range of particle size used in this study (Hunter, 2001).
The pH of the suspensions was measured for each replicate using a Schott
Blueline 18 pH combination glass electrode. Aliquots of the supernatant were withdrawn
with a syringe immediately after the electrokinetic measurements, filtered through 0.45
m Millipore HA-type membranes and stored in HDPE bottles for later nickel and
alkalinity analyses (see section 2.2).

141
3. RESULTS AND DISCUSSION
3.1 Proton Adsorption on the Gaspeite Surface
3.1.1 Acidimetric Titrations
To illustrate the influence of NiCO3(s) dissolution on the computation of proton adsorption
for the acidimetric titrations, corrected (see section 2.5) and uncorrected surface proton
density measurements acquired at I = 0.01 M are presented in Figure 1. At low pH (pH <
6.5), dissolution significantly affects the proton balance in solution and the computation
of proton adsorption. Consequently, only corrected data computed from Equation 3 are
presented in the following discussion and were used to construct HNet
data vs pH plots at
each ionic strength, HNet
(pH, I), from which the pHpznpc,I values were obtained by fitting
the complete HNet
(pH, I) dataset to a polynomial function of pH and the resultant
function solved for pH at the condition H=0 (Chorover and Sposito, 1995).
The corrected proton adsorption curves generated at the three ionic strengths
investigated in this study are shown in Figure 2. Results from three titrations conducted at
each ionic strength are shown and highlight the good reproducibility of the
measurements. Inspection of these curves reveals four major common features: i) a clear
asymmetry across the pHznpc,I ii) a distinct “surface-charge buffer” region, within a pH
range of 6.5 to 9, for each ionic strength, iii) a strong dependency on the ionic strength
(i.e., position of the pHznpc,I), and iv) a lower number of maximum protonated and
deprotonated adsorption sites per square meter than the theoretical Ni or CO3 site density
predicted by crystallography (9.99 mol·m-2
). In addition, titration curves at the
intermediate and high ionic strengths (0.01M and 0.1 M) show a much larger maximum

142
number of negatively-charged sites at high pH than the maximum number of positively-
charged sites registered at low pH.
To account for the observed features of the titration curves, and in conformity with
the formalism adopted for the calcite surface (Villegas-Jiménez et al., 2009a), we elected
to explain surface protonation on the basis of surface reactions formulated on the basis of
a single primary surface site, (NiCO3)H2O0. Accordingly, ionization reactions were
postulated as follows:
O)3(H3CO
Ni H O2H
3CO
Ni (9)
H OH3CO
Ni O2H
3CO
Ni (10)
2H
2
O3CO
Ni O2H
3CO
Ni (11)
Reactions 10 and 11 describe, respectively, the step-wise and global deprotonation
reactions of the adsorbed H2O monomer(s). Whereas these reactions may account for the
asymmetry across the pHznpc,I, because two protons may be released at alkaline conditions
and only one proton is adsorbed under acidic conditions, they cannot explain the observed
ionic strength dependency.
Our data show that the background electrolyte has a strong effect on the gaspeite
surface as reflected by the shifting position of the pHznpc,I values and the extent of surface

143
protonation registered at each ionic strength (Figure 2). This effect is more than could be
generated by purely electrostatic interactions between background electrolyte ions and the
charged mineral surface as postulated by the Stern and the Triple-Layer models (Davis
and Kent, 1990). These models postulate that electrolyte ions are adsorbed via
electrostatics at a given distance from the mineral surface (-plane) increasing the extent
of surface protonation or deprotonation (away from the pHznpc) as a result of surface
charge neutralization. In other words, electrolyte adsorption decreases the electrostatic
work required to move protons through the electrified interfacial layer (EIL) affecting the
overall sorption processes:
elecint0 ΔGΔGΔG [12]
where ΔG0 is the Gibbs free energy of adsorption, ΔG
int is the intrinsic free-energy term:
ΔGint
= RT ln Kint
(where Kint
representing the intrinsic formation constant), and ΔGelec
is
the electrostatic work: ΔGelec
=ZFx, a function of the net charge transfer (ΔZ)
associated with the adsorption reaction, the Faraday constant, and the potential recorded
at the adsorbing plane “x” (x).
According to the Stern and the Triple-Layer models, in the absence of the
electrostatic effect (null charge), the electric potential vanishes, and thus, proton titration
curves must intersect at a certain pH commonly referred to as the common point of
intersection (CIP) or the point of zero salt effect (pHpzse). In contrast, our titration curves
do not yield a CIP as they do not intersect at their respective pHznpc,I. To reconcile this
observation, we propose that sodium ions compete with protons for available adsorption

144
sites, possibly adsorbing at the surface plane via chemical interactions and modifying the
extent of protonation of the gaspeite surface.
There is, in fact, some evidence that at high NaCl concentrations, the surface
reactivity of some carbonate minerals is severely affected. For instance, surface charge,
zeta potential, and dissolution kinetics of magnesite are also affected by NaCl (Pokrovsky
et al., 1999a; Gence and Ozbay, 2006) whereas Na+ ions significantly affect the extent of
Ca and Mg ion adsorption on dolomite at high ionic strengths (Brady et al., 1999).
Furthermore, specific adsorption of Na+ was proposed to account for the ionic
strength-dependency of the surface charge on wollastonite (Xie and Walther, 1994)
whereas unequal adsorption affinity of Na+ and Cl
- ions was postulated to explain the
electrokinetic behavior of gibbsite (Rowdlands et al., 1997). In addition, it was recently
shown that NaI background electrolyte concentrations exceeding 0.1 mol L-1
induce a
shift in the pHiep values of hematite and rutile towards higher pH than the “pristine” pHiep
values of these minerals measured at low ionic strengths. Hence, this behavior was
explained in terms of specific adsorption of sodium ions on the mineral surfaces
(Kosmulski et al., 2002). It follows that similar mechanisms may operate at the gaspeite
surface affecting proton adsorption equilibria.
If background electrolyte sodium ions adsorb specifically at the gaspeite surface
(0-plane) according to:
H NaOH3CO
Ni Na O2H
3CO
Ni (13)

145
then surface protolysis would be further promoted at high pH and high ionic strengths,
this may, at least qualitatively, explain why the extent of surface protonation decreases
upon an increase in the ionic strength.
3.1.2 Verification of Potential Artifacts
Before calibration of the model, it is important to evaluate some potential artefacts
associated with our experimental protocols and experimental conditions that may have
affected the computation of proton adsorption data (via Equation 3) and may account, to
some extent, for the atypical acid-base behavior observed at all ionic strengths. For
instance, the presence of undetected amounts of carbonate species (< 5M) could affect
the free proton concentrations and activities in solution upon the formation of protonated
carbonate species (see Table 1). Similarly, the formation of surface carbonate species may
impact the proton concentration in solution as well as the availability of primary surface
sites according to the following equilibria which is analogous to that previously
postulated by earlier workers for the gaspeite surface (Pokrovsky and Schott, 2002):
O2H3HCO
3CO
Ni
23CO H O2H
3CO
3NiCO
(14)
O2H3CO2H
3CO
Ni
23CO 2H O2H
3CO
3NiCO
(15)
To test this, we considered the presence of hypothetical concentrations of total
inorganic carbon (CO2) in solution in proportions relative to the total nickel
concentrations predicted at each titration point (see section 2.4.2). In other words, we

146
assumed that a fraction of inorganic carbon in solution arising from the dissolution of
NiCO3(s) is not completely purged from the system upon bubbling of N2 and remains
undetected upon alkalinity and carbonate ion activity measurements. This may be
particularly true for alkaline conditions where CO2(aq) and H2CO3(aq) are not the
predominant carbonate species, and thus, carbon evacuation from the system via CO2(g)
purging could be difficult (see Equation 3).
To account for the proton consumption/release in solution by carbonate equilibria
in the computation of proton adsorption, the following correction, [H+]CO2, was added to
Equation 3:
[H+]CO2 = f ·[Ni
2+] ·(2 CO3 + HCO3 + NaHCO3 + NiCO3) (16)
where i represent the ionization factors (analogous to Eqs. 4-7) of the carbonate species
identified by the subscripts, brackets represent molar concentrations and f stands for the
hypothetical fraction of CO2 arising from gaspeite dissolution (see reaction 2) that is
assumed to remain in solution despite constant purging with N2(g). All reactions
associated with the dissolution products of gaspeite and their contributions to proton
balance in solution are illustrated in Figure 3.
In Figure 4, we illustrate the effect of aqueous carbonate equilibria on the
computed proton adsorption density for the following values of f : 0.1, 0.25, 0.5, 0.75 and
1. At all ionic strengths, increasingly positive proton adsorption densities are observed in
the pH range between about 6 to 8.5 following a rise in dissolved CO2. This provides an
idea of the potential error carried by the computed surface proton density values used for

147
model calibration (see Figure 2). Clearly, this error is only significant within a relatively
narrow pH range (6.5 to 7.5) and at CO2 / [Ni]T ratios greater than 0.75. The pHznpc.I
values are shifted towards higher pH upon an increase of CO2. This shift is greater at I =
0.1 M because the pHznpc lies within the pH range (6.5 to 7) where the CO2 effect is
greatest.
In addition, using the intrinsic formation constants of the following reactions
(equivalent to reactions 14 and 15) originally proposed by Pokrvosky and Schott (2002):
NiOH + H+ + CO3
2- NiCO3
- + H2O log10 K
int = 14 (17)
NiOH + 2 H+ + CO3
2- NiHCO3 + H2O log10 K
int = 19.5 (18)
we confirmed that carbonate adsorption on gaspeite is negligible at all hypothetical
inorganic carbon concentrations (0.1< f > 1) and experimental conditions of our titrations.
Hence, we can confidently ascertain that whereas residual inorganic carbon in solution
could have slightly affected the computation of proton adsorption via the formation of
aqueous carbonate species, it cannot account for the peculiar surface protonation behavior
of NiCO3(s) observed at different ionic strengths.
3.1.3 Surface Complexation Modeling of Acidimetric Data: One-Site CCM Approach
The optimization of the intrinsic constants was achieved with an in-house Matlab©
subroutine which is provided in the appendices to this thesis. The code uses a powerful
search and optimization stochastic technique, the genetic algorithm (GA), which has
proved efficient in tackling complex optimization problems including a large number of
parameters within a pre-established solution space (Gen and Cheng, 2000) and is

148
described in detail for this type of applications in Chapter 2 of this thesis (see Villegas-
Jiménez and Mucci, 2009). All GA optimizations described below were run in triplicate
using the dissolution-corrected proton adsorption data sets with the following GA
parameters: population of 500 chromosomes, 100 generations, a single-point crossover
probability of 0.25, and a mutation probability of 0.02. The reproducibility of the
optimization is reflected in the error associated with the log Kint
values (see details in Gen
and Cheng, 2000).
Intrinsic constants are referenced to a zero potential standard state by taking into
account the coulombic contribution to the apparent formation constant, Kapp
:
RT
ZF-
exp0ψ
intKappK (19)
where Kapp
is the apparent constant, Kint
stands for the intrinsic constant.
Following the track of earlier workers (Van Cappellen et al., 1993; Pokrovsky et
al., 199a,b; Pokovsky and Schott, 2002) and in consistency with our previous work on
magnesite and dolomite (presented in Chapter 3 of this thesis), our first step was to use
the Constant Capacitance Model (CCM) to describe the surface charge-potential
relationship but other, more sophisticated electrostatic models (i.e., Basic Stern and Triple
Layer, see below) were also tested under the one-site and multi-site scheme scenarios.
In the CCM, the surface is assumed to behave as a flat capacitor with the potential
varying linearly away from the surface (Sposito, 1984):

149
C0
0σ
ψ (20)
where 0 is the experimental surface charge density (C m-2
) and C is the specific integral
capacitance (Farad m-2
) of the EIL. In this model, the capacitance is a function of the
ionic strength as described by:
α
I1/2
C (21)
where I is the ionic strength and is an adjustable parameter related to the physical
properties of the EIL that reconciles working units (m2
· mol½
· V · C-1
). In the CCM
formulation, surface species are treated in mol kg-1
units referenced to the 1 molal
standard state whereas aqueous species are given in molar concentrations under the
constant ionic medium convention (Sposito, 1984). Note that although this definition of
the standard state yields intrinsic constants that depend on the properties of the solid
sorbent such as the site density and surface area, available analytical relationships
between the standard states, on the basis of site occupancy and the usual standard state
definition, allows simple conversion of equilibrium constants from one standard state to
the other (Sverjensky, 2003).
Multiple combinations of reactions 9-11 and 13 (Models) were used in a series of
GA optimizations for the simulation of the titration data at all ionic strengths investigated
in this study. Given that the computed HNet
values reflect a varying number of maximum
charged sites, the total number of adsorption sites was treated as an adjustable parameter.

150
Attempts to fit the data using the theoretical crystallographic number of sites (or
theoretical lattice site density), 9.99 mol m-2
, were unsuccessful for all chemical models
at all ionic strengths investigated in this study. The GA approach allows for all unknowns
quantities (intrinsic constants, capacitance and site densities) to be optimized
simultaneously. Accordingly, the value of was adjusted simultaneously for capacitance
values comprised between 0.1 to 15 F · m2, site densities were adjusted within the range
from 2 to 10 mol m-2
, whereas a large solution space was chosen (-25 to 25) to perform
an exhaustive search for the set of log10 Kint
values that best reproduced the experimental
data.
We found that ionization reactions (9-11) can closely simulate titration data at
ionic strengths of 0.001 and 0.01M (Model I) with capacitances ranging from 10 to 12.5 F
m-2
(Figure 2). Consideration of sodium adsorption (reaction 13), in addition to
ionization reactions (Model II), did not improve the quality of the fits and resulted in
small variations of the estimated log Kint
, capacitance and site density values (see Table
2). Modification of the GA parameters (1000 chromosomes, 200 generations) to extend
the search space of the optimization of this particular data set provided statistically
identical results. In contrast, titration data at high ionic strength (I = 0.1 M) could not be
reproduced with any combination of these reactions. To fit these data, it was necessary to
consider the specific adsorption of nickel ion on the NiCO3(s) surface:
HNiOH3CO
Ni 2Ni O2H
3CO
3NiCO (22)

151
Nickel arising from NiCO3(s) dissolution may re-adsorb on the surface and modify
the extent of surface protonation. According to reaction 22, nickel adsorption promotes
surface protolysis and results in a net increase of the surface charge. Optimization of this
constant was constrained by the predicted activity of Ni2+
and the pH measured at each
titration point (see section 2.4.2). Consideration of this reaction and ionization reactions
(Model III) for the description of data at ionic strengths of 0.001 and 0.01 M did not
improve the quality of the fit, did not significantly affect the values of the ionization
constants and yielded a very low Ni2+
adsorption constant (log10 Kint
= 7) indicating that
reaction 22 is unnecessary to successfully simulate the data at these ionic strengths. In
contrast, a rather high, rather unrealistic, constant for reaction 22 (log10 Kint
= 1.5) was
required, in combination with ionization reactions and a very high capacitance value (
73 F · m-2
), for simulation of data at I = 0.1 M. That data at I 0.1 M did not require
consideration of reaction 22 for the succesful fitting of the data casts serious doubts on
the validity of the estimated log10 Kint
values. Consequently, Model III can be dimissed
and will not be discussed any further.
It is noteworthy that the calibration of this constant is constrained by the pH-
dependency of the reaction (22) and the free nickel ion concentration in solution rather
than by nickel adsorption data. Thus, rigorously speaking, batch nickel adsorption
experiments are needed to properly calibrate this constant. Furthermore, that titration data
at low and intermediate ionic strengths could be fitted using solely the ionization
reactions casts doubt on the reliability of the optimized constant describing Ni2+
adsorption at high ionic strength. Inclusion of this reaction in the modeling of these data
is required because of the additional positive charge that is brought to the surface upon

152
Ni2+
adsorption that influences the electrostatics of the adsorption process. In other words,
the estimated log10 Kint
value for reaction 22, in combination with a high capacitance, are
possibly a mathematical artefact imposed by the GA, via the electrostatic factor (Equation
19), on all mass action laws along the titration curve to modulate surface charge and
successfully fit the data at I = 0.1 M.
Optimized model parameters for Model I and Model II are presented in Table 2.
Although the intrinsic formation constants yielded by Models I and II are very similar,
within their respective uncertainties, they display a clear ionic strength dependency,
consistent with the premises of the CCM: the estimated model parameters are considered
as reasonable surface speciation predictors (i.e., model fit parameters), rather than
thermodynamic quantities, applicable only to the chemical conditions of model
calibration (pH, I, etc.).
The log10 Kint
values for reaction 13 (Model II) are consistently small and, are
thus, clearly not required to successfully fit the data. Furthermore, the optimized intrinsic
constants for this reaction are somewhat questionable because, as in the case of reaction
22, adsorption data are unavailable, and thus, the calibration of this constant is only
constrained indirectly via its pH-dependency. Consequently, the derived constants must
be considered only as first-order estimates of the sodium affinity towards the gaspeite
surface. Additional experimental work such as batch sodium adsorption experiments is
needed to obtain reliable estimates of this constant.
The surface speciation, surface charge and surface protonation as predicted by
Model I and Model II for systems at ionic strengths of 0.001 and 0.01 M are presented in
Figures 5 and 6 respectively. A slight discrepancy of the predicted speciation is observed

153
between models for each ionic strength. It results from different intrinsic constants for
reactions 9 and 10 and capacitance values optimized from each data set.
Model II predicts identical surface charge and surface protonation densities for
systems at 0.001 and 0.01 M from pH=5 until pH values of about 8.2 and 7.5
respectively. Beyond these values, surface charge densities become more positive than
surface protonation densities following a gradual increase of the relative abundance of the
neutral sodium-bearing species over the singly-deprotonated species forming from
reaction 10. Thus, sodium specific adsorption would result in a slight buffering of the
surface charge (more significant at higher ionic strength) and a small shift of the Point of
Zero Net Charge (pHpznc) towards higher pH values. In contrast, Model I, postulates that
pHpznpc is identical to pHpznc for NiCO3(s)-NaCl systems at I 0.01 M.
3.1.4 Surface Complexation Modeling of Acidimetric Data: One-Site, Multi-Site, BSM,
and TLM Approaches
In an attempt to offer a better interpretation to our data, we considered more sophisticated
descriptions of the EIL than envisioned by the CCM. To this end, we applied the Basic
Stern Model (BSM) and the Triple Layer Model (TLM, Davis and Kent, 1990) to the
calibration of multiple sets of reactions (Models or SCMs) which included: i) acid-base
(ionization) reactions (analogous to reactions 9-11), ii) inner-sphere and/or outer-sphere
cation and anion electrolyte binding reactions (e.g., Davis and Kent, 1990, Villalobos and
Leckie, 2001) and, iii) nickel adsorption reactions (analogous to reaction 13). Within the
BSM and/or the TLM, numerous combinations of these reactions (Models), initially
formulated within the one-site scheme, were subjected to numerical optimization using
proton adsorption data generated in our acidimetric titrations. Exhaustive modeling work

154
using these Models, revealed that none of them could yield a self-consistent set of
parameters that could succesfully fit data at all ionic strengths. In addition, fits of data at
I=0.1 M were consistently unsatisfactory. These aspects violate the premises under which
the BSM and TLM are grounded since model parameters must be independent of the
composition of the system, and hence, must remain constant at different ionic strengths.
Reformulation of these reactions within a multi-site scheme (i.e., multiple generic
primary surface sites of type: NiCO3H2O0 but exhibiting distinct reactivities) generated
additional, more complex Models that could not offer a better interpretation to the data
upon calibration.
It follows that one-site-based Models formulated within the CCM described in
section 3.1.3) are the simplest models that can reasonably account for most data without
the necessity to invoke numerous adjustable parameters. With these considerations in
mind, the validity of these models can be better assessed qualitatively against
electrokinetic data as discussed in section 3.2.
3.1.5 Alkalimetric Titrations
Our nickel analyses indicate that a certain amount of nickel is removed from the solution
as the titration proceeds to higher pH (Figure 7) that, according to our calculations, cannot
be attributed to gaspeite precipitation. For this reason, we believe that nickel adsorption
takes place during the titrations (acidimetric and alkalimetric) but is only revealed in the
alkalimetric titrations. This is because proton-promoted dissolution of gaspeite
(Pokrovsky and Schott, 2002), at the beginning of the alkalimetric titrations (around
circumneutral pH), allows for relatively high levels of Ni2+
( 10-6
M) which, in
combination with decreasing proton activities in solution (reaction 22), favor the

155
formation of Ni-bearing surface species possibly revealing the role exerted by the mineral
surface on the Nickel concentrations in solution. In contrast, during acidimetric titrations,
NiCO3(s) dissolution is promoted as the titration proceeds (reaction 2) whereas adsorption
is unfavorable. In other words, in acidimetric titration experiments dissolution controls
the nickel concentrations in solution largely masking the effects of adsorption.
If nickel adsorption is governed by reaction 22, the amount of protons consumed
in this reaction affects the computation of proton adsorption and must be added to
Equation 3 as follows:
H - OH = (1/AS) [CA – CB – [H+] + [OH
-] – [H
+]diss+ [Ni
2+]ads] (23)
where [Ni2+
]ads stands for the amount of nickel removed from the solution and is
equivalent to the amount of protons released upon adsorption. Semi-quantitative estimates
of nickel adsorption were obtained by subtracting the measured nickel concentrations at a
given pH (> 7) from the maximum total nickel concentration recorded at a pH of about
8.5 (slightly higher than the measured Ni at the beginning of the titration because of the
time elapsed) where adsorption is expected to start taking over dissolution (Figure 7).
Log normal fits of these data, also shown in Figure 7, served to predict the amount of
nickel adsorbed at a given pH and revealed that Ni2+
adsorption becomes significant (>
0.5 M) at pH values above 9.
Proton adsorption densities computed for all ionic strengths using Equation 23 are
presented in Figure 8. The adsorption behavior is similar to the one observed from the
acidimetric titrations. Although the alkalimetric titrations cover only the proton-deficient

156
end of the curves, the ionic strength dependency observed in the acidimetric titrations is
reproduced in the alkalimetric plots. There is a small discrepancy between the acidimetric
and alkalimetric titrations with respect to the number of negatively-charged sites recorded
at the alkaline (systems at I = 0.001 M and I = 0.1 M) or acid end (systems at I = 0.01 and
I = 0.1 M) of the titration. This discrepancy is higher for systems at I=0.01 and 0.1 M (~
14 %, at high pH, and ~ 60 % at circumneutral pH) than for systems at I=0.001 M (~ 3 %
at high pH and ~ 30 % at circumneutral pH). The observed discrepancy cannot be easily
explained because of the absence of a clear trend in the data. As stated earlier, Ni2+
adsorption is unaccounted for in the computation of proton adsorption data from
acidimetric titrations, and hence, according to Eq. 23, proton adsorption may be slightly
underestimated data at the alkaline end. Whereas this could explain why higher densities
are computed for alkalimetric data at I=0.1 M in this pH range, it does not explain results
at lower ionic strengths. Reaction kinetics hysteresis, in both directions of the titration, is
possible and could affect the pH measurements acquired under identical instrumental
stability criteria (see above) which may, in turn, partly account for the observed
discrepancies.
Surface complexation modeling of the alkalimetric data using identical sets of
reactions to those used for acidimetric data were tested within the CCM. Intrinsic
constants derived from Model I for reactions 10 and 11 are in good agreement (< 6 %
discrepancy within their respective uncertainties) with those optimized from the
acidimetric data at I=0.001 M and 0.01 M. Whereas good fits to the data acquired at the
low and intermediate ionic strengths were achieved with Model I, no Model tested
allowed a reasonable fit to the high ionic strength data.

157
Alkalimetric data comprise about half of the pH range covered by the acidimetric
titrations, and thus, reflect conditions where surface protolysis dominates. It is unlikely
that these data can fully resolve the contribution of reaction 9 on proton adsorption
densities as in the acidimetric data. To test this, we performed a final optimization with
data at I = 0.001 M, that included reactions 10 and 11 only. As expected, the data could
be reproduced equally well as with Model I, but with small (~ 8 %) discrepancies of the
optimized intrinsic constants, which proves that proton uptake is not predominant within
this pH range. These result suggest that acidimetric data is best suited to calibrate the set
of surface complexation reactions postulated in this study.
3.2 Electrokinetics
Firstly, it is important to point out that none of the Models calibrated within either the
BSM or the TLM that succesfully reproduced proton adsorption data at I 0.1 M, could
also reasonably simulate the electrokinetic behavior of gaspeite suspensions at identical
ionic strengths. In other words, predictions returned by these Models were not consistent
with both types of data. This contrasts with the one-site CCM Models presented in section
3.1.3. that show reasonable consistency with proton adsorption and electrokinetic data for
systems at I 0.1 M.
Unlike sophisticated electrostatic models, the CCM neglects the existence of a
diffuse layer at the EIL (Davis and Kent, 1990). Consequently, no direct relationship can
be established between zeta-potentials and surface potentials. Nevertheless, electrokinetic
data are useful to test, qualitatively, the predictive power of the calibrated SCM. In
Figure 9 we compare the -potential values measured in this study (raw data and solution
conditions for data of Series-I and Series-II are given in the appendices to this thesis) as

158
well as those measured by Pokrovsky and Schott (2002) against the surface potentials
predicted by Model I and Model II (I = 0.001 and I = 0.01 M). For the chemical
conditions selected by the latter authors (pH=6.08-9.26, [Ni2+
]T = 1·10-6
to 2·10-6
,
CO2=5·10-3
M, I=0.005 M), the -potentials follow the trend displayed by surface
potential predictions (I=0.01 M) but the former are consistently more negative than the
surface potentials predicted by both Models. This could be explained by the rather high
CO2 (510-3
M) characterizing this data set which would favor carbonate ion adsoprtion
(reactions 14 and 15) shifting the -potentials towards more negative values.
In contrast, some electrokinetic data for Series-I at pH > 9 (data points 9-14) are
consistently more positive than the predicted surface potentials. Other reactions (e.g.
lattice constituents adsorption), unaccounted for by Model I, may affect the -potential at
the alkaline end. For instance, as noted earlier, nickel adsorption may be important at
these pH values and the additional positive charge brought to the surface by this reaction
(reaction 22) may explain why -potentials shift towards more positive values than
surface potentials. Conversely, the agreement between -potentials and predicted surface
potentials at I=0.01 M significantly improves at pH < 9 (data points 1 to 8) for this
electrokinetic data set.
On the other hand, Series-II experiments confirm the ionic strength-dependency of
surface protonation observed in our titration experiments. Whereas -potentials at I=0.001
and 0.01 M are in very good agreement with the surface potentials predicted by Model I,
data at I=0.1 M could not be simulated becasuse of the lack of a suitable Model
describing surface protonation at this ionic strength. It is noteworthy, however, that -
potentials measured at all ionic strengths (including I=0.1 M) reasonably follow the trend

159
displayed by proton adsorption data at circum-neutral and low pH where surface
protonation and surface potential decrease with increasing ionic strength. In addition, the
pH of isoelectric point (pHiep) shifts towards lower pH as a function of ionic strength
(from 8.9 at I=0.001 to 6.2 at 0.1 M), an identical behavior to that shown by the pHpznpc
(from 8.8 at I=0.001 to 6.6 at 0.1 M).
Admittedly, other mechanisms than those envisioned by Models I and II must be
considered to properly describe the surface protonation and electrokinetic behavior of
NiCO3(s) suspensions at high ionic strengths. For instance, ionic strengthpromoted
dissolution effects, observed for other carbonate minerals (Pokrovsky and Schott, 1999;
Gence and Ozbay, 2006), may explain the decreasing number of charged sites with
increasing ionic strength at low pH whereas, at alkaline conditions, sodium adsorption
may explain the observed differential extent of surface protolysis at different ionic
strengths. However, we think that additional mechanisms, unaccounted for by traditional
SCMs, must also exert a role on the development of surface charge and the electrokinetic
behavior of gaspeite, an effect that is likely accentuated at high ionic strength. Despite
these considerations, the model parameters postulated by Model I at intermediate and low
ionic strengths (I=0.001 and 0.001 M) can be considered, as reasonable predictors of the
surface charge and the electrokinetic behavior of gaspeite for systems at conditions
similar to those of model calibration.

160
4. CONCLUSIONS
The acid-base behavior of gaspeite was examined using titration techniques never applied
before in the study of the surface properties of carbonate minerals. After consideration of
dissolution and potential artefacts, reliable proton adsorption data were obtained within a
pH range of 5 to 10.
Surface protonation is strongly affected by NaCl over the entire pH range
investigated in this study. The background electrolyte plays a critical role in determining
the extent of surface protonation and leads to a shift of the pHpznpc and the pHiep towards
lower pH values with increasing ionic strength. The protonation and electrokinetic
behavior observed at different ionic strength conditions contrasts with what is typically
observed for other mineral-solution interactions. No self-consistent interpretation to this
has been found in terms of background electrolyte binding to the gaspeite surface (inner-
sphere or outer-sphere binding), and thus, we believe that the background electrolyte
affects in more than one way the surface properties of the gaspeite surface (surface
protonation and the development of surface charge) possibly through modification of the
structure of the electrified interfacial layer, perturbation of the solvent structure dynamics
and the affinity of water molecules and adsorbing ions towards the mineral surface. These
observations challenge earlier conceptions on carbonate mineral surfaces that traditionally
considered these minerals as being chemically inert to background electrolyte ions. These
effects should be carefully examined in future studies through alternative experimental
approaches and/or using different background electrolytes.
Ionization reactions formulated in terms of the one-site scheme (Model I) and
calibrated within the Constant Capacitance Model can reproduce titration data at low and
intermediate ionic strengths (0.001 and 0.01 M) but simulation of data at I=0.1 M was

161
unsuccessful probably because of the enhanced ionic strength artifacts that are not fully
accounted for by SCMs. Qualitative agreement with electrokinetic data lends support to
Model I as a useful conditional predictor (i.e., applicable to specific chemical conditions)
of the surface charge of gaspeite for ionic strengths 0.01 M and a pH range from 5 to
10. Model I parameters calibrated at I=0.001 M are likely to best represent the intrinsic
acid-base chemistry of the gaspeite surface because the influence of the electrolyte is low.
Nevertheless, the self-consistency of these values must be verified beyond the calibration
conditions and the effect of the background electrolyte and dissolved lattice ions on the
development of surface charge and surface protonation must be quantified separately.
5. ACKNOWLEDGEMENTS
A.V.-J. thanks the hospitality of Dr Oleg S. Pokrovsky and Dr Jacques Schott during his
visit to LMTG. This research was supported by a student grant to A.V.-J. from the
Geological Society of America (GSA), by Natural Sciences and Engineering Research
Council of Canada (NSERC) Discovery grants to A.M. and by the Centre National de la
Recherche Scientifique (CNRS). A.V.J. acknowledges Consejo Nacional de Ciencia y
Tecnología of Mexico (CONACyT) by the post-graduate scholarships received during his
Ph.D. tenure. A.V.-J. also benefited from additional financial support from the
Department of Earth and Planetary Sciences, McGill University and from Consorcio
Mexicano Flotus-Nanuk.

162
6. REFERENCES
Anderson S.J. and Sposito G. (1992) Proton surface-charge density in soils with structural
and pH-dependent charge. Soil Sci. Soc. Am. J. 56, 1437- 1443.
APHA-AWWA-WPCF (1998). Standard Methods for the Examination of Water and
Wastewater. 20th ed., American Public Health Association, 1015 Fifteenth Street, N.W.,
Washington, D.C., USA, 981 p.
Bermanec V., Sijarić, Kniewald G. and Mandarino J.A. (2000). Gaspeite and associated
Ni-rich minerals from veins in altered ultrabasic rocks from Duboštica, Bosnia
and Herzegovina. Can. Mineral. 38, 1371-1376.
Brady P.V., Papenguth H.W., Kelly J.W. (1999). Metal sorption to dolomite surfaces.
Applied Geochem. 14, 569-579.
Brunauer S., Emmet P.H. and Teller E. (1938) Adsorption of gases in multimolecular
layers. J. Phys. Chem. 60, 309-316.
Charlet L., Wersin P. and Stumm W. (1990). Surface charge of MnCO3 and FeCO3.
Geochim. Cosmochim. Acta. 54, 2329-2336.
Chorover J. and Sposito G. (1995). Surface charge characteristics of kaolinitic tropical
soils. Geochim. Cosmochim. Acta 59(5), 875-884.
Davis J.A. and Kent D.B. (1990). Surface complexation modeling in aqueous
geochemistry. In: Mineral-Water Interface Geochemistry. (ed. M.F. Hochella and
A.F. White). Rev. Mineral. 23. Mineral. Society of America. Washington, DC. pp
177-260.

163
Duckworth O.W. and Martin S.T. (2003). Connections between surface complexation and
geometric models of mineral dissolution investigated for rhodochrosite. Geochim.
Cosmochim. Acta 67, 1787-1801.
Gen M. and Cheng R. (2000) Genetic Algorithms and Engineering Optimization. John
Wiley and Sons, New York, NY, 495 p.
Gence N. and Ozbay N. (2006). pH dependence of electrokinetic behavior of dolomite
and magnesite in aqueous electrolyte solutions. Appl. Surf. Sci. 252, 8057-8061.
Jordan G., Higgins S.R., Eggleston C.M., Knauss K.G. and Schmahl, W.W. (2001).
Dissolution kinetics of magnesite in acidic aqueous solution, a hydrothermal
atomic force microscopy (HAFM) study: Step orientation and kink dynamics.
Geochim. Cosmochim. Acta. 65, 4257-4266
Hoffmann U. and Stipp S.L.S. (2001). The behavior of Ni2+
on calcite surfaces. Geochim.
Cosmochim. Acta 65(22), 4131-4139.
Huang Y.C., Fowkes F.M., Lloyd T.B. and Sanders, N.D. (1991) Adsorption of calcium
ions from calcium chloride solutions onto calcium carbonate particles. Langmuir
7, 1742-1748.
Hunter R.J. (2001). Foundations of Colloid Science. Oxford University, Oxford, 806 p.
Kendall T.A. and Martin S.T. (2005) Mobile ions on carbonate surfaces. Geochim.
Cosmochim. Acta. 69, 3257-3263.
Kohls D.W. and Rodda J.L. (1966). Gaspeite, (Ni, Mg, Fe) (CO3), a new carbonate from
the Gaspé peninsula, Québec. Am. Mineral. 51, 677-684.

164
Kosmulski M., Maczka E. and Rosenholm J.B. (2002). Isoelectric points of metal oxides
at high ionic strengths. J. Phys. Chem. B 106, 2918-2921.
Morse J. W. (1986). The surface chemistry of calcium carbonate minerals in natural
waters: An overview. Mar. Chem. 20, 91-112.
Morse J.W. and Mackenzie F.T. (1990) Geochemistry of Sedimentary Carbonates;
Develop. Sedimentol., 48. Elsevier: Amsterdam, 707 p.
NIST (1998) Critically Selected Stability Constants of Metal Complexes, Standard
Reference Database 46, Version 5, National Institute of Standards and
Technology, US Department of Commerce, Gaithersburg, MD, USA.
Pehrsson L., Ingman F. and Johanssson A. (1976). Acid-Base titrations by stepwise
additions of equal volumes of titrant with special reference to automatic titrations-
I. Theory, discussion of the Gran functions, the Hofstee method and two proposed
methods for calculating equivalence volumes. Talanta. 23, 769-780.
Pokrovsky O.S. and Schott J. (2002). Surface chemistry and dissolution of divalent metal
carbonates. Environ. Sci. Technol. 36(3), 426-432.
Pokrovsky O.S., Schott J. and Thomas F. (1999a). Processes at the magnesium-bearing
carbonates/solution interface. I. A surface speciation model for magnesite.
Geochim. Cosmochim. Acta. 63(6), 863-880.
Pokrovsky O.S., Schott J. and Thomas F. (1999b). Dolomite surface speciation and
reactivity in aquatic systems. Geochim. Cosmochim. Acta. 63(19/20), 3133-3143.
Stumm W. and Morgan J. (1996). Aquatic Chemistry: Chemical Equilibria and Rates in
Natural Waters. John Wiley and Sons Inc., 3rd
Edition New York, 1022 p.
Sverjensky D.A. (2003) Standard states for the activities of mineral surface sites and
species. Geochim. Cosmochim. Acta 67, 17–28.

165
Van Cappellen P., Charlet L., Stumm W. and Wersin P. (1993). A surface complexation
model of the carbonate mineral-aqueous solution interface. Geochim. Cosmochim.
Acta. 57, 3505-3518.
Villalobos M. and Leckie J.O. (2001) Surface complexation modeling and FTIR study of
carbonate adsorption to goethite. Geochim. Cosmochim. Acta 235,15-32.
Villegas-Jiménez A. and Mucci A. (2009) Estimating intrinsic formation constants of
mineral surface species using a genetic algorithm. Math. Geosci. (accepted).
Villegas-Jiménez A., Mucci A., Pokrovsky O.S. and Schott J. (2009a) Defining reactive
sites at hydrated mineral surfaces: rhombohedral carbonate minerals. Geochim.
Cosmochim. Acta 73(15), 4326-4345.
Wiesner A.D, Katz L.E. and Chen C. (2006) The impact of ionic strength and background
electrolyte on pH measurements in metal ion adsorption experiments. J. Colloid
Interface Sci. 301, 329-332.
Xie Z. and Walther J. (1994) Dissolution stoichiometry of alkali and alkaline earth
elements to the acid-reacted wollastonite surface at 25ºC. Geochim. Cosmochim.
Acta. 58, 2587-2598.

166
7. TABLES
Table 1. Formation constants used in thermodynamic calculations
perfomed in this study
Equilibria Log K (25 C)
H
+ + HCO3
- H2CO3 6.35 a
H+ + CO3
2- HCO3 10.33 a
Na+ + CO3
2- NaCO3
- 1.27 a
Na+ + HCO3
- NaHCO3 -0.25 a
Ni2+
+ CO3- NiCO3(aq) 3.57 a
Ni2+
+ HCO3- NiHCO3
+ 1.59 a
Ni(OH)2(s) Ni2+
+ 2 OH- -15.2 a
Ni2+
+ Cl- NiCl
+ 0.6 b
Ni2+
+ H2O NiOH+ + H
+ -9.9 b
Ni2+
+ 2 H2O Ni(OH)2 + 2H+ -19 b
Ni2+
+ 3 H2O Ni(OH)3- + 3H
+ -30 b
Mass Balance Equations
Ni = [Ni2+
] + [NiCO3(aq)]+ [NiHCO3+
)] + [NiOH+] + [Ni(OH)2]+ [Ni(OH)3
-] + [NiCl
+]
CO2 = [H2CO3]* + [HCO3-] + [CO3
2-] + [NaHCO3
+] + [NaHCO3
+] + [NiCO3(aq)]+ [NiHCO3
+)]
Na = [Na+] + [NaHCO3] + [NaCO3
-]
Brackets represent molar concentrations of the specified chemical species
[H2CO3*] = [CO2(aq)] + [H2CO3]
a Values were taken from NIST (1998)
b Values were taken from Stumm and Morgan (1996)
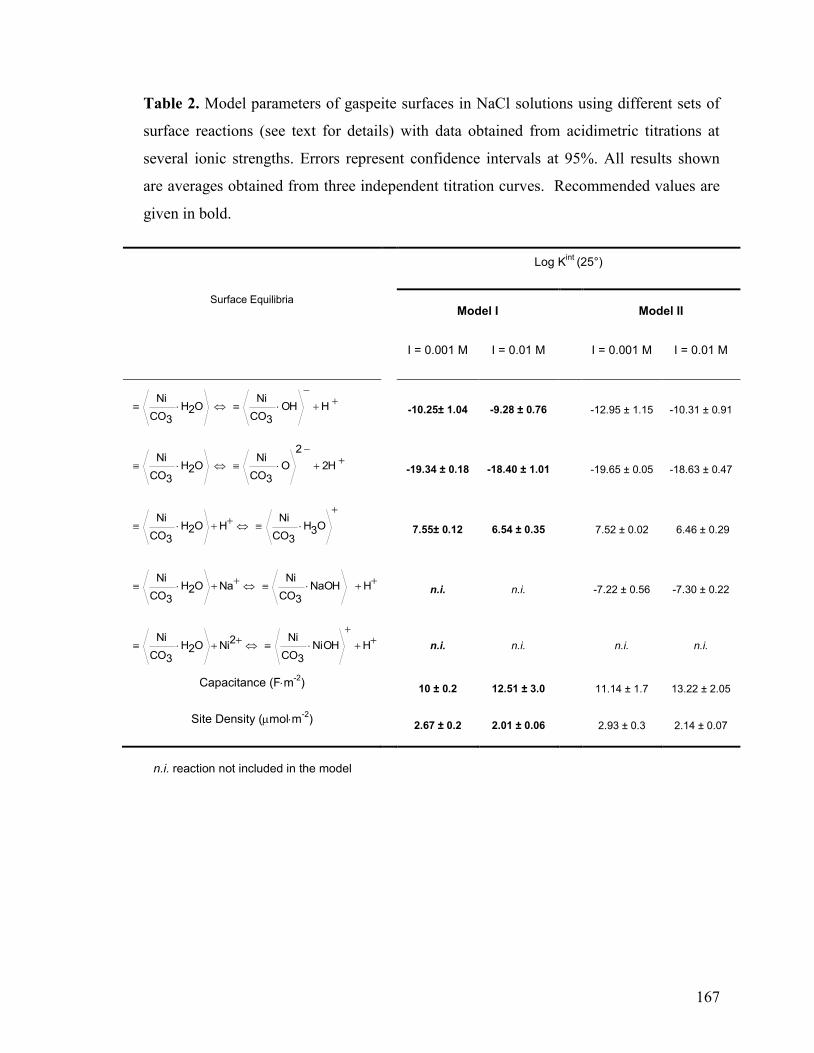
167
Table 2. Model parameters of gaspeite surfaces in NaCl solutions using different sets of
surface reactions (see text for details) with data obtained from acidimetric titrations at
several ionic strengths. Errors represent confidence intervals at 95%. All results shown
are averages obtained from three independent titration curves. Recommended values are
given in bold.
Surface Equilibria
Log K
int (25°)
Model I
Model II
I = 0.001 M I = 0.01 M I = 0.001 M I = 0.01 M
H OH
3CO
Ni O2H
3CO
Ni
-10.25± 1.04 -9.28 ± 0.76 -12.95 ± 1.15 -10.31 ± 0.91
H2
2
O
3CO
Ni O2H
3CO
Ni
-19.34 ± 0.18 -18.40 ± 1.01 -19.65 ± 0.05 -18.63 ± 0.47
O3H
3CO
Ni H O2H
3CO
Ni
7.55± 0.12 6.54 ± 0.35 7.52 ± 0.02 6.46 ± 0.29
H NaOH
3CO
Ni Na O2H
3CO
Ni
n.i. n.i. -7.22 ± 0.56 -7.30 ± 0.22
H NiOH
3CO
Ni
2Ni O2H
3CO
Ni
n.i. n.i. n.i. n.i.
Capacitance (Fm-2
)
10 ± 0.2 12.51 ± 3.0 11.14 ± 1.7 13.22 ± 2.05
Site Density (molm-2
)
2.67 ± 0.2 2.01 ± 0.06 2.93 ± 0.3 2.14 ± 0.07
n.i. reaction not included in the model
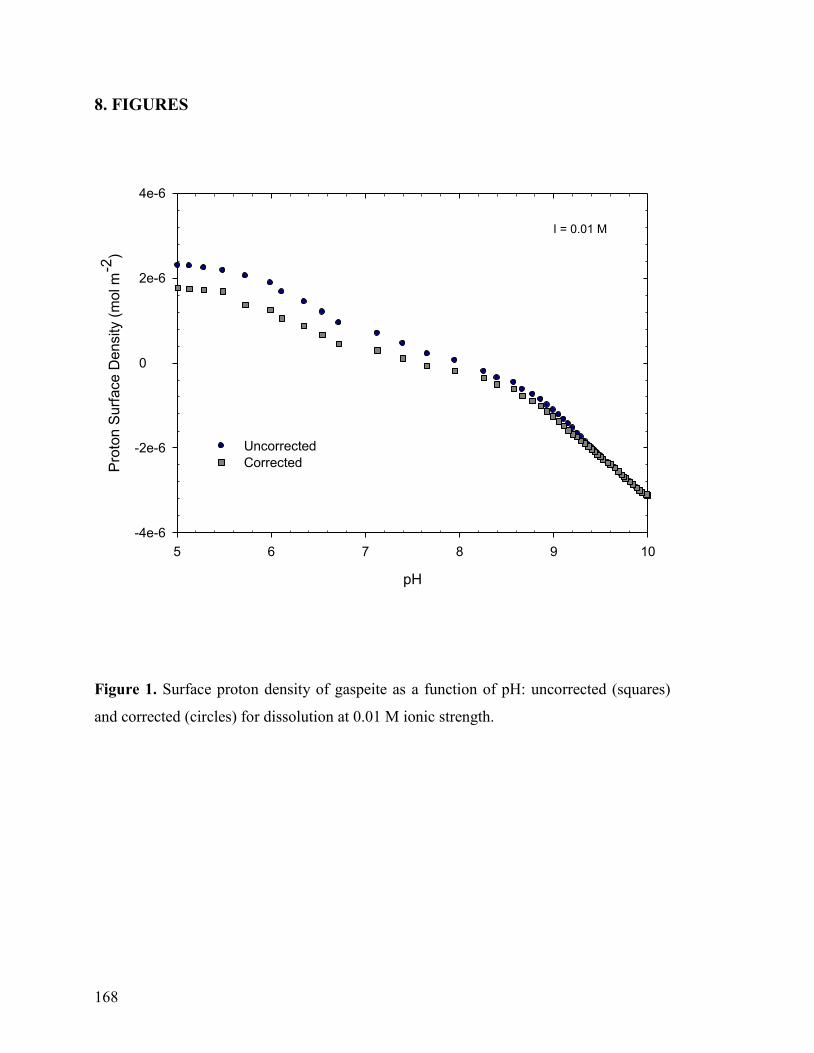
168
8. FIGURES
pH
5 6 7 8 9 10
Pro
ton S
urf
ace D
ensity (
mol m
-2)
-4e-6
-2e-6
0
2e-6
4e-6
Uncorrected
Corrected
I = 0.01 M
Figure 1. Surface proton density of gaspeite as a function of pH: uncorrected (squares)
and corrected (circles) for dissolution at 0.01 M ionic strength.
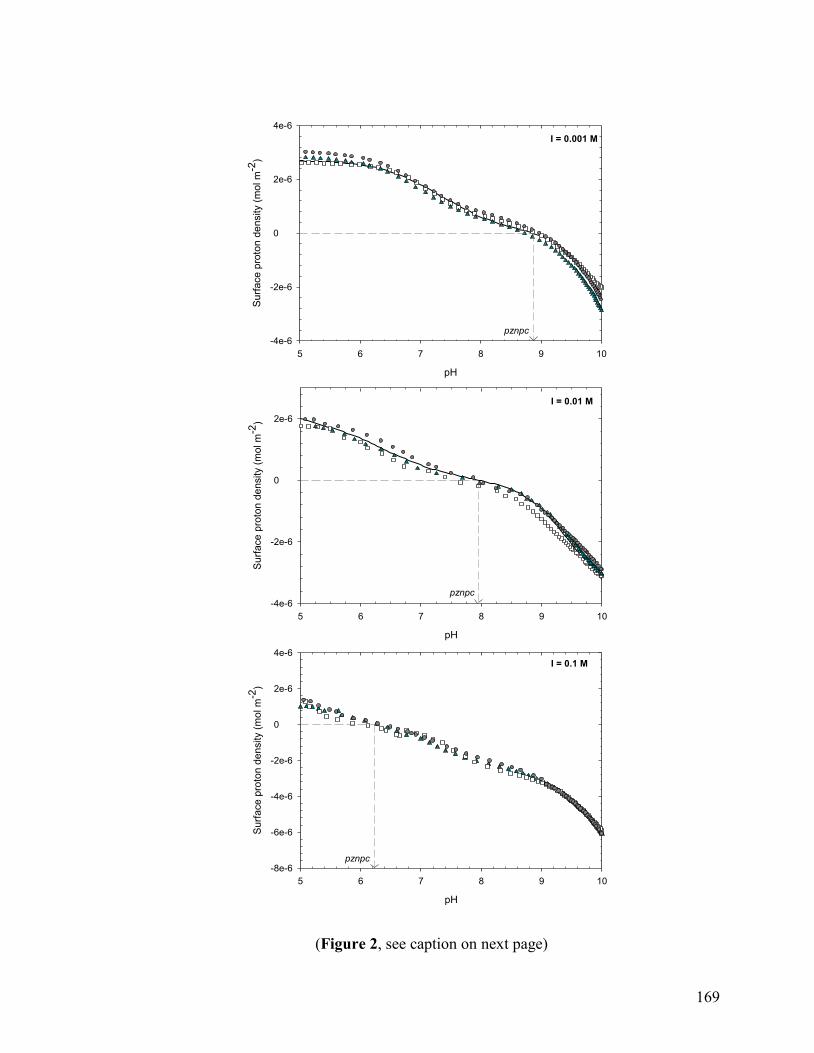
169
I=0.001M
pH
5 6 7 8 9 10
Su
rfa
ce
pro
ton
de
nsity (
mo
l m
-2)
-4e-6
-2e-6
0
2e-6
4e-6
I = 0.001 M
I=0.001M
pH
5 6 7 8 9 10
Su
rfa
ce
pro
ton
de
nsity (
mo
l m
-2)
-4e-6
-2e-6
0
2e-6
I = 0.01 M
Col 1 vs Col 2
Col 1 vs Col 3
Col 1 vs Col 4
Col 1 vs Col 11
I=0.001M
pH
5 6 7 8 9 10
Su
rfa
ce
pro
ton
de
nsity (
mo
l m
-2)
-8e-6
-6e-6
-4e-6
-2e-6
0
2e-6
4e-6I = 0.1 M
pznpc
pznpc
pznpc
(Figure 2, see caption on next page)

170
Figure 2. Corrected surface proton density of gaspeite derived from acidimetric titrations
carried out at three ionic strengths (0.001 M, 0.01 M , 0.1 M). Three independent
titrations (are shown at each ionic strength. Solid lines represent predictions by Model I
for the I = 0.001M and I = 0.01M regimes.
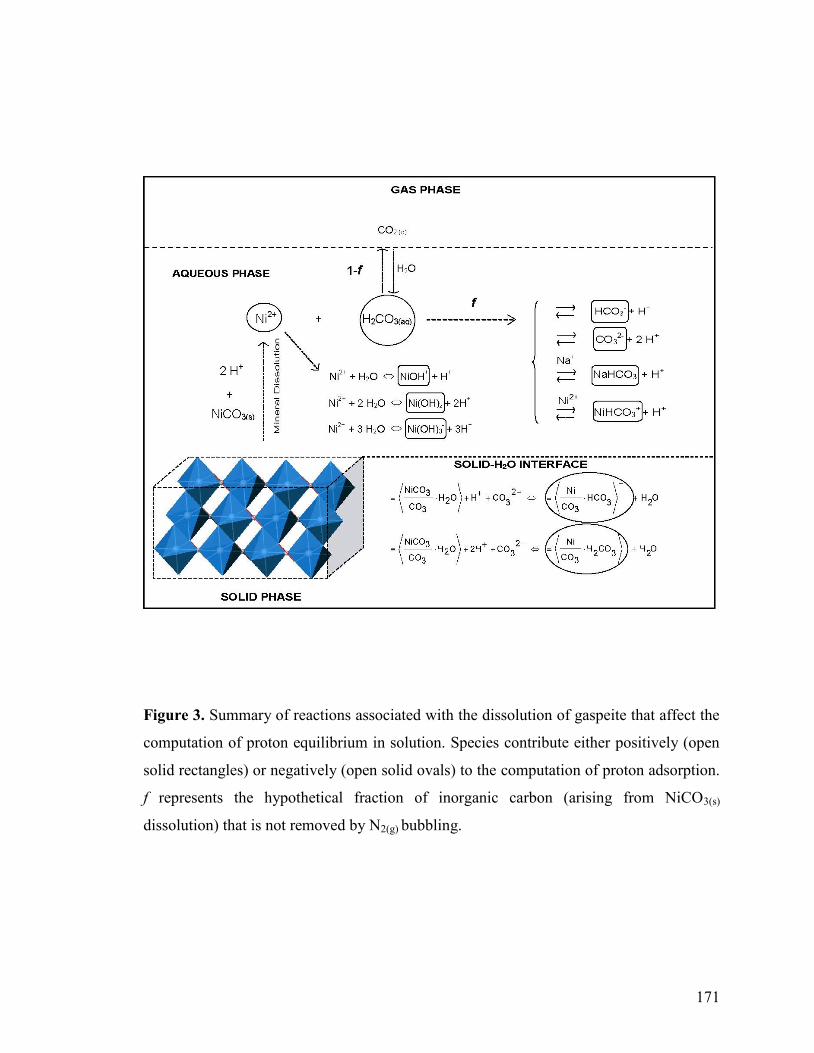
171
Figure 3. Summary of reactions associated with the dissolution of gaspeite that affect the
computation of proton equilibrium in solution. Species contribute either positively (open
solid rectangles) or negatively (open solid ovals) to the computation of proton adsorption.
f represents the hypothetical fraction of inorganic carbon (arising from NiCO3(s)
dissolution) that is not removed by N2(g) bubbling.
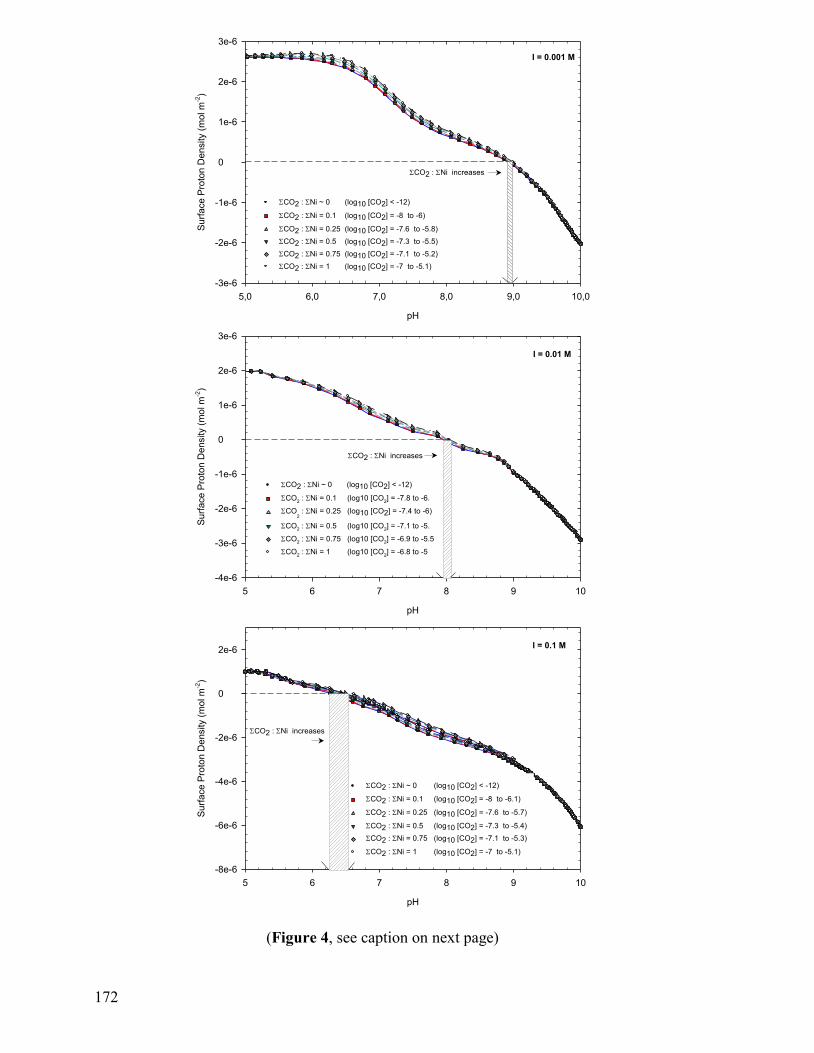
172
(Figure 4, see caption on next page)
I=0.001 M
pH
5 6 7 8 9 10
Su
rfa
ce
Pro
ton D
ensity (
mo
l m
-2)
-8e-6
-6e-6
-4e-6
-2e-6
0
2e-6
CO2 : Ni ~ 0 (log10 [CO2] < -12)
CO2 : Ni = 0.1 (log10 [CO2] = -8 to -6.1)
CO2 : Ni = 0.25 (log10 [CO2] = -7.6 to -5.7)
CO2 : Ni = 0.5 (log10 [CO2] = -7.3 to -5.4)
CO2 : Ni = 0.75 (log10 [CO2] = -7.1 to -5.3)
CO2 : Ni = 1 (log10 [CO2] = -7 to -5.1)
pH
5 6 7 8 9 10
Su
rfa
ce
Pro
ton D
ensity (
mo
l m
-2)
-4e-6
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
CO2 : Ni ~ 0 (log10 [CO2] < -12)
CO2 : Ni = 0.1 (log10 [CO
2] = -7.8 to -6.
CO2
: Ni = 0.25 (log10 [CO2] = -7.4 to -6)
CO2 : Ni = 0.5 (log10 [CO
2] = -7.1 to -5.
CO2 : Ni = 0.75 (log10 [CO
2] = -6.9 to -5.5
CO2 : Ni = 1 (log10 [CO
2] = -6.8 to -5
I = 0.01 M
I = 0.1 M
pH
5,0 6,0 7,0 8,0 9,0 10,0
Su
rfa
ce
Pro
ton D
ensity (
mo
l m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
CO2 : Ni ~ 0 (log10 [CO2] < -12)
CO2 : Ni = 0.1 (log10 [CO2] = -8 to -6)
CO2 : Ni = 0.25 (log10 [CO2] = -7.6 to -5.8)
CO2 : Ni = 0.5 (log10 [CO2] = -7.3 to -5.5)
CO2 : Ni = 0.75 (log10 [CO2] = -7.1 to -5.2)
CO2 : Ni = 1 (log10 [CO2] = -7 to -5.1)
I = 0.001 M
CO2 : Ni increases
CO2 : Ni increases
CO2 : Ni increases

173
Figure 4. Surface proton density plots computed with equation 3 plus additional
corrections to account for the presence of protonated carbonate species in solution
(equation 16) at various concentrations. The shaded area indicate the pH range where the
point of net zero proton charge would appear depending on the concentration of total
inorganic carbon in solution.
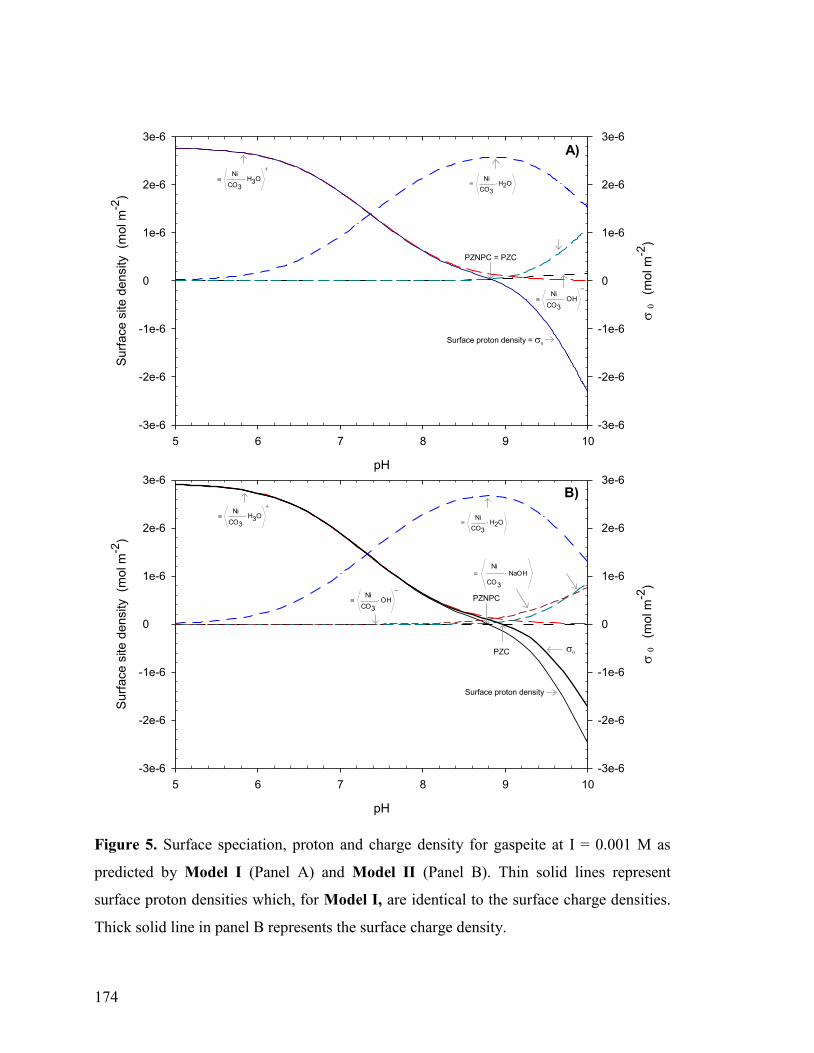
174
pH
5 6 7 8 9 10
Surf
ace s
ite d
ensity
(mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
(
mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
A)
pH
5 6 7 8 9 10
Surf
ace s
ite d
ensity
(mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
(
mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
Surface proton density
Surface proton density =
B)
PZNPC = PZC
PZNPC
PZC
2
O
3CO
Ni
NaOH
3CO
Ni
O2H
3CO
Ni
O3H
3CO
Ni
OH
3CO
Ni
O3H
3CO
Ni
O2H
3CO
Ni
2
O
3CO
Ni
OH
3CO
Ni
Figure 5. Surface speciation, proton and charge density for gaspeite at I = 0.001 M as
predicted by Model I (Panel A) and Model II (Panel B). Thin solid lines represent
surface proton densities which, for Model I, are identical to the surface charge densities.
Thick solid line in panel B represents the surface charge density.
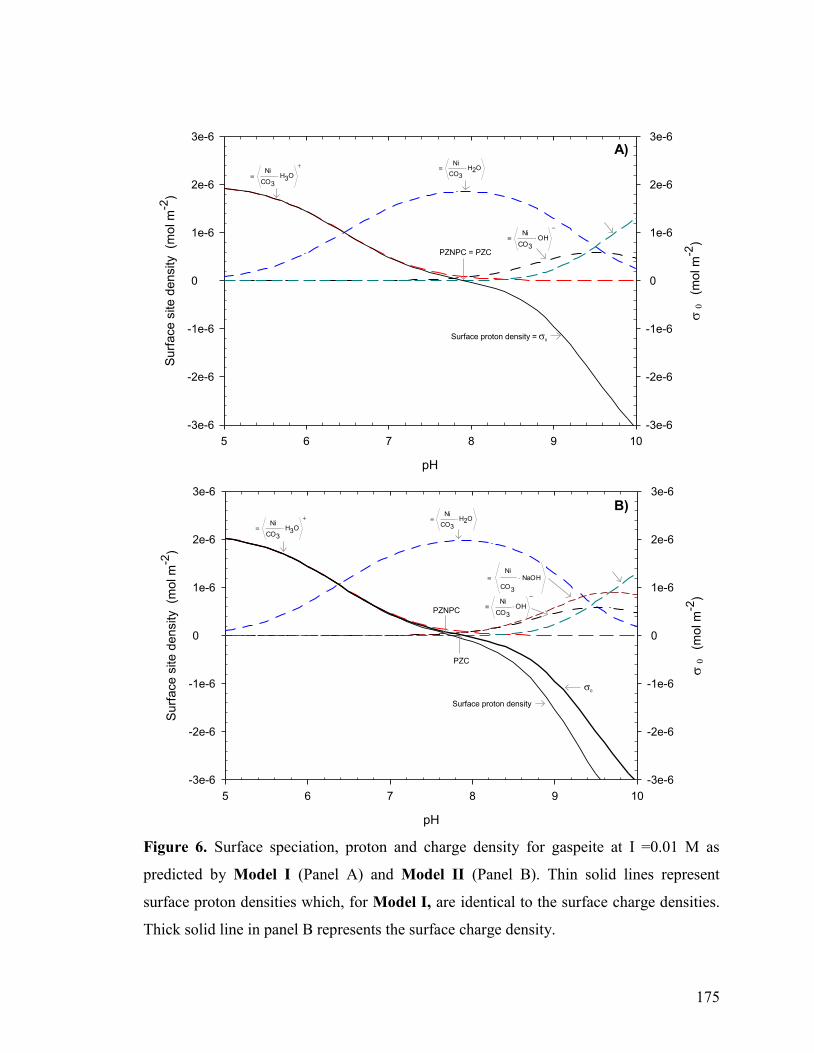
175
pH
5 6 7 8 9 10
Surf
ace s
ite
den
sity
(mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
(
mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
Surface proton density =
A)
O3H
3CO
Ni
O2H
3CO
Ni
OH
3CO
Ni
PZNPC = PZC
pH
5 6 7 8 9 10
Surf
ace s
ite
den
sity
(mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
(
mol m
-2)
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
3e-6
Surface proton density
B)
PZNPC
PZC
2
O
3CO
Ni
NaOH
3CO
Ni
O2H
3CO
Ni
2
O
3CO
Ni
O3H
3CO
Ni
OH
3CO
Ni
Figure 6. Surface speciation, proton and charge density for gaspeite at I =0.01 M as
predicted by Model I (Panel A) and Model II (Panel B). Thin solid lines represent
surface proton densities which, for Model I, are identical to the surface charge densities.
Thick solid line in panel B represents the surface charge density.
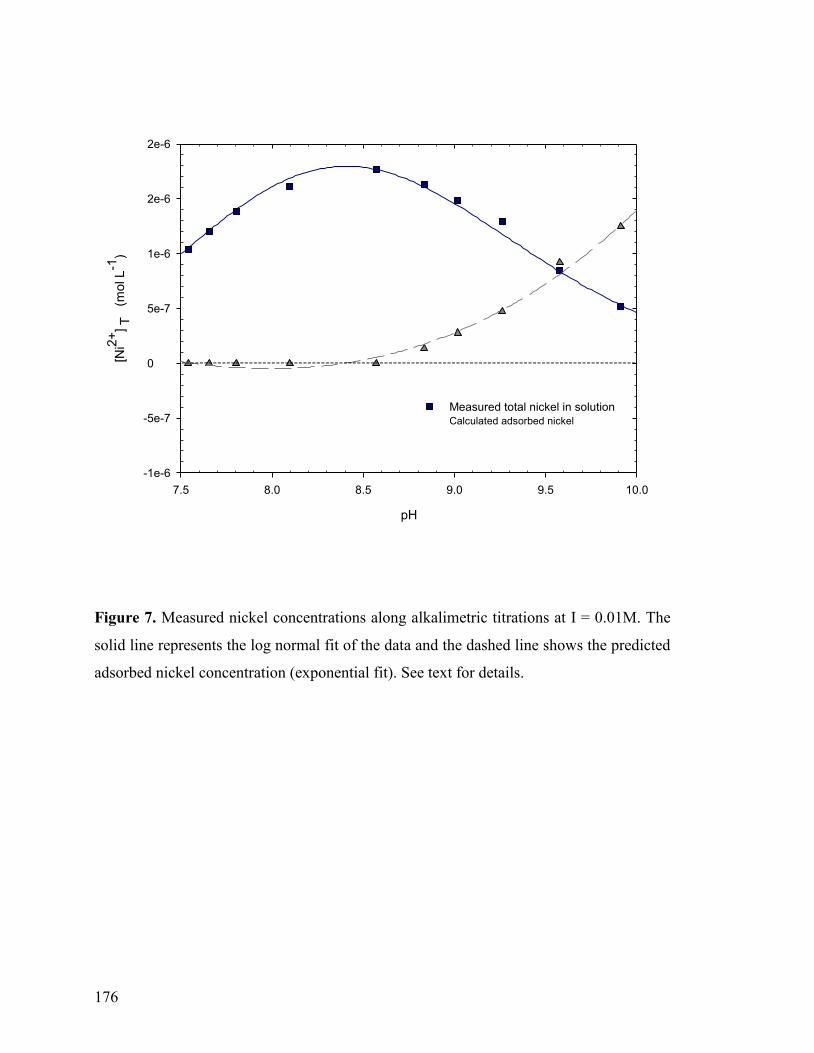
176
pH
7.5 8.0 8.5 9.0 9.5 10.0
[Ni2
+] T
(
mol L
-1)
-1e-6
-5e-7
0
5e-7
1e-6
2e-6
2e-6
Measured total nickel in solutionCalculated adsorbed nickel
Figure 7. Measured nickel concentrations along alkalimetric titrations at I = 0.01M. The
solid line represents the log normal fit of the data and the dashed line shows the predicted
adsorbed nickel concentration (exponential fit). See text for details.
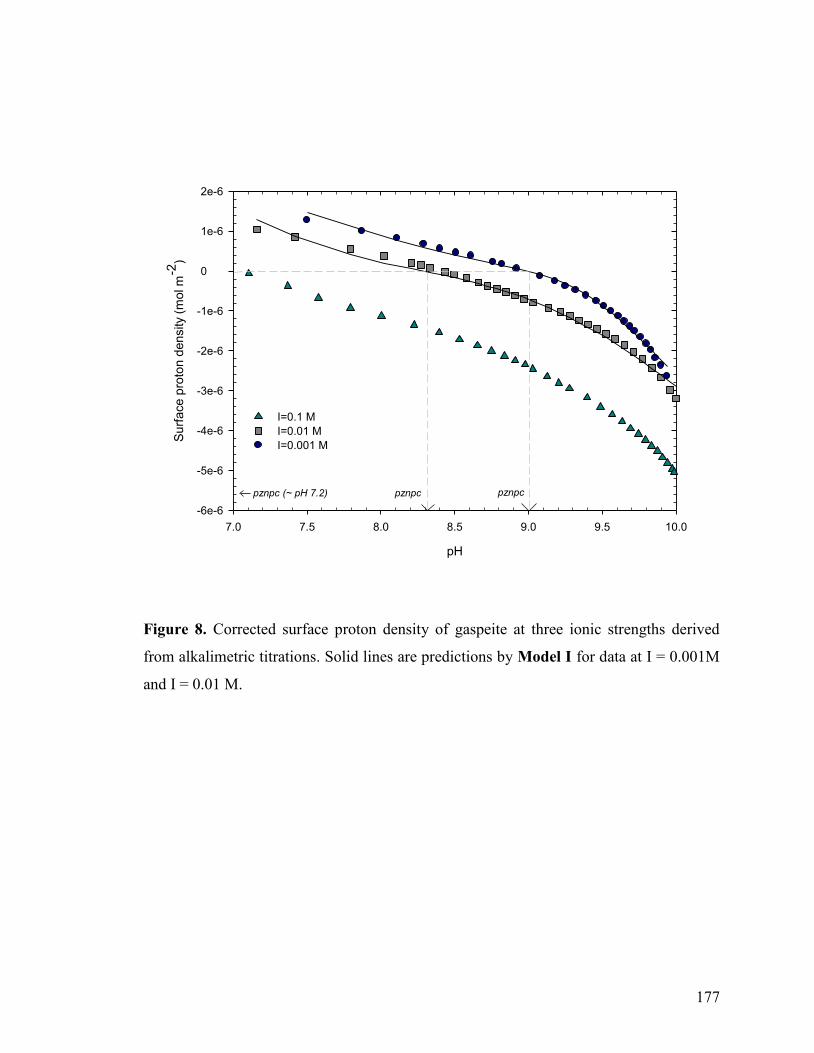
177
pH
7.0 7.5 8.0 8.5 9.0 9.5 10.0
Surf
ace p
roto
n d
ensity
(mol m
-2)
-6e-6
-5e-6
-4e-6
-3e-6
-2e-6
-1e-6
0
1e-6
2e-6
I=0.1 M
I=0.01 M
I=0.001 M
pznpc pznpcpznpc (~ pH 7.2)
Figure 8. Corrected surface proton density of gaspeite at three ionic strengths derived
from alkalimetric titrations. Solid lines are predictions by Model I for data at I = 0.001M
and I = 0.01 M.
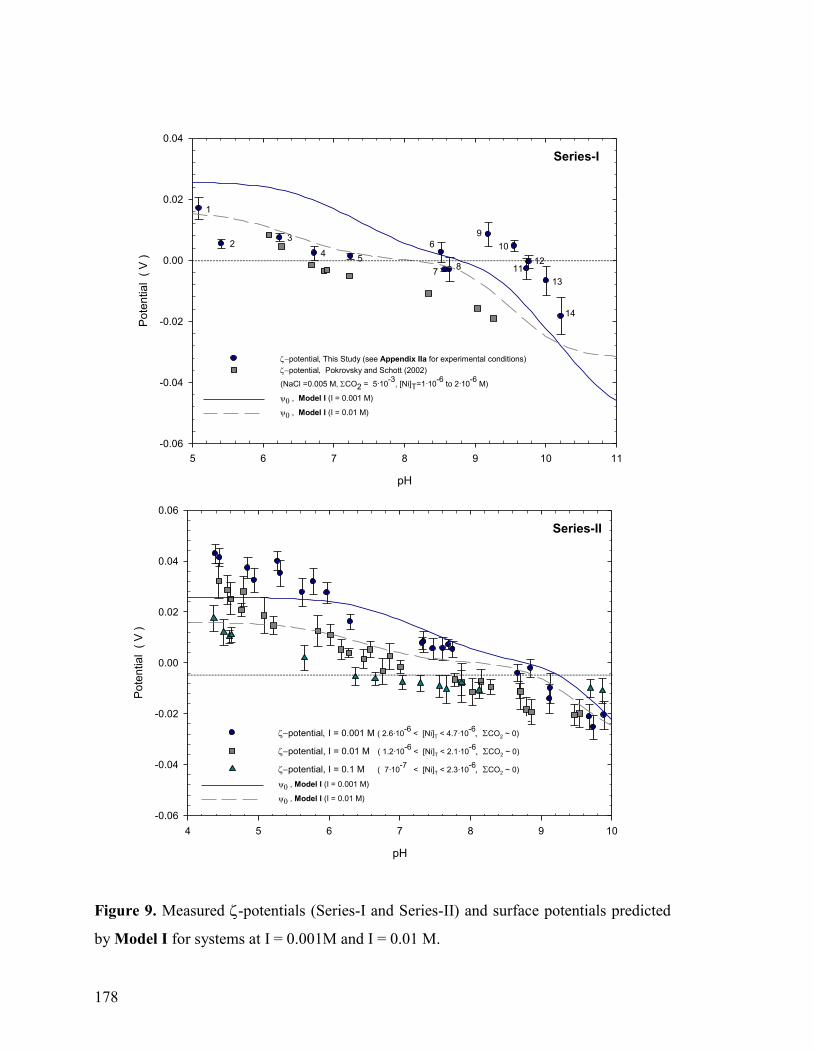
178
pH
5 6 7 8 9 10 11
Po
ten
tia
l (
V )
-0.06
-0.04
-0.02
0.00
0.02
0.04
potential This Study (see Appendix IIa for experimental conditions)
potential Pokrovsky and Schott (2002)
(NaCl =0.005 M, CO2 = 5·10-3
, [Ni]T=1·10-6
to 2·10-6
M)
, Model I (I = 0.001 M)
, Model I (I = 0.01 M)
1
23
45
6
78
9
10
1112
13
14
Series-I
pH
4 5 6 7 8 9 10
Po
ten
tia
l (
V )
-0.06
-0.04
-0.02
0.00
0.02
0.04
0.06
potential I = 0.001 M ( 2.6·10-6
< [Ni]T < 4.7·10-6CO2 ~ 0)
potential, I = 0.01 M ( 1.2·10-6
< [Ni]T < 2.1·10-6CO2 ~ 0)
potential, I = 0.1 M ( 7·10-7
< [Ni]T < 2.3·10-6CO2 ~ 0)
, Model I (I = 0.001 M)
, Model I (I = 0.01 M)
Series-II
Figure 9. Measured -potentials (Series-I and Series-II) and surface potentials predicted
by Model I for systems at I = 0.001M and I = 0.01 M.

179

180
PREFACE TO CHAPTER 5
In contrast to gaspeite, calcite is one of the most reactive naturally-occurring
rhombohedral carbonate minerals in aqueous solution and, by far, the most ubiquitous
in aquatic systems. Accordingly, numerous studies have focused on the
characterization of the calcite surface reactivity in aqueous solutions. Among these,
molecular modeling studies have pressed forward because of the experimental
difficulties inherent in the characterization of the surface reactivity of this mineral. In
most of these studies, atomistic and Molecular Dynamics force-field-based methods
and, to a lesser extent, Density Functional Theory (DFT) techniques were applied and
provided important insights on the structural registry of dry and wet calcite surfaces.
Nevertheless, despite all these efforts, no systematic ab initio molecular orbital study
on the ground-state structural and electronic properties of the first and second
hydration layers at the (10.4) calcite surface has been conducted. In the following
chapter, “Theoretical Insights into the Hydrated (10.4) Calcite Surface: Structure,
Energetics and Bonding Relationships” we exploit ab initio molecular orbital methods
using cluster models to represent the idealized (10.4) calcite surface, obtain a detailed
description of its 3D structural registry and elucidate the bonding relationships
governing the hydration process. Results of this study are in reasonable agreement
with earlier findings and further support the single-site scheme postulated for
rhombohedral carbonate mineral surfaces in Chapter 3 and subsequently used in
Chapter 4. This study revealed a significant Ca-O bonding reorganization at the
mineral surface leading to the weakening of the topmost atomic layer with respect to
the bulk.

181
CHAPTER 5
THEORETICAL INSIGHTS INTO THE HYDRATED (10.4) CALCITE SURFACE: STRUCTURE, ENERGETICS, AND BONDING RELATIONSHIPS
Adrián Villegas-Jiménez*1, Alfonso Mucci
1 and Michael Anthony Whitehead
2
1 Earth and Planetary Sciences, McGill University, 3450 University Street
Montréal, Qc H3A 2A7, Canada.
2 Chemistry, McGill University, 801 Sherbrooke Street W. Montréal, Qc Canada
*Corresponding Author
E-mail: [email protected]
“Reproduced with permission from the American Chemical Society:
Adrián Villegas-Jiménez, Alfonso Mucci, and Michael Anthony Whitehead (2009) Theoretical Insights into the
Hydrated (10.4) Calcite Surface: Structure, Energetics, and Bonding Relationships Langmuir 25(12) 6813-6824
Copyright 2009 American Chemical Society."

182
ABSTRACT
Roothaan-Hartree-Fock molecular orbital methods were applied to investigate the
ground-state structural, energetic properties, and bonding relationships of the hydrated
(10.4) calcite surface. The adsorption of water molecules was modelled at the 6-31G(d, p)
level of theory using Can(CO3)n slab cluster models (4 n 18), with a varying number
of H2O monomers (2 (H2O)n 6) interacting with the surface. Modelling results add
fresh insights into the detailed 3D structural registry of the 1st and 2
nd hydration layers
and the reconstructed (10.4) calcite surface, complementary to the information acquired
from earlier Atomistic, Density Functional, X-ray Scattering and Grazing Incidence X-
Ray Diffraction studies. Both the modelled energies and geometries agree best with
results of earlier Density Functional calculations, supporting the associative character of
adsorbed water molecules. Two adsorption configurations are postulated: i) H2O
molecules interacting with surface Ca through ionic bonding and by Hydrogen Bonding
to a surface O with their dipole slightly oblique above the surface (1st hydration layer)
and, ii) H2O molecules that Hydrogen Bond to surface O and to H2O molecules in the 1st
hydration layer with their dipole nearly parallel to the surface (2nd
hydration layer). These
interactions are consistent with the “chemisorption” and “physisorption” of H2O on
calcite surfaces, proposed on the basis of previous thermogravimetric and Fourier-
Transformed Infrared studies. Most significant is the distortion of the surface Ca-O
octahedra caused by the relaxation (and possibly rupture) of some Ca-O bonds upon
hydration, weakening the topmost atomic layer. These findings are consistent with
interpretations of X-ray Photoelectron Spectroscopy, Density Functional Theory and
Electrokinetic studies that suggest the preferential release of surface Ca atoms over

183
surface CO3 groups upon hydration of the cleavage surface. These insights will help to
elucidate mechanisms of carbonate mineral dissolution, the rearrangement of surface
layers, ion replacement, charge development and solute transport through subsurface
lattice layers.
Keywords: Molecular Orbital theory; surface hydration; calcite clusters; Ca-O bond
stretching, Ca-O octahedron distortion.

184
1. INTRODUCTION
Among CaCO3 polymorphs, calcite is the most abundant and ubiquitous form in natural
aquatic environments, where it plays a critical role on the regulation of pH, alkalinity, and
heavy metal transport/mobility through exchange and co-precipitation reactions (Morse
and Mackenzie, 1990). Calcite finds numerous industrial applications that range from the
production of paper, paints, plastics, pharmaceuticals and cosmetics to raw material in the
construction industry and agriculture (Vanerek et al., 2000; Usher et al., 2003).
Given its environmental significance and broad industrial applications, calcite has
been the subject of extensive experimental studies (Morimoto et al., 1980; Ahsan, 1992;
Davis et al., 1987; Zachara et al., 1991) which revealed the critical role that its surface
properties play on the macroscopic chemical behaviour of this mineral in aqueous
suspensions. These properties result from the interplay of intermolecular and surface
forces, such as hydrogen bonding, van der Waals interactions and solvation at the mineral
surface-water interface (Sposito, 1990; Israelachvili, 1992). Hydration is the most
fundamental phenomenon to which dry mineral surfaces are subjected when immersed in
aqueous solution. Surface forces modify the structure and properties of adsorbed and
interfacial water, relative to the bulk solution, by breaking down water clusters and
limiting the ability of H2O molecules to reorient their dipoles (Israelachvili, 1992) which
affects the properties of the mineral surface.
Surface-sensitive, non-invasive, X-ray, Electron Diffraction and spectroscopic
techniques as well as Atomic Force microscopic methods were used to investigate the
structural properties of the calcite mineral-water interface (Neagle and Rochester, 1990;
Stipp and Hochella, 1991; Chiarello et al, 1993; Stipp et al., 1994; Liang et al., 1996;
Stipp, 1999; Pokrovsky et al., 2000; Fenter et al., 2000; Geissbühler et al., 2004;

185
Magdans et al., 2006). They provided direct characterization of the molecular structure of
the hydrated calcite surface, revealing a degree of surface reconstruction upon hydration.
In addition, recent X-ray Scattering (Geissbühler et al., 2004) and Grazing Incidence X-
ray Diffraction (Magdans et al., 2006) studies unveiled, for the first time, 3D structural
details (interlayer spacing, inter-atomic distances and lateral registry) of the hydrated
(10.4) calcite surface.
Similarly, molecular modelling techniques are powerful tools to investigate the
energy and structure of hydrated CaCO3 surfaces. Several computer-assisted Atomistic
simulations (Force-Field-based) of the H2O interactions with calcite and magnesium-
bearing calcite surfaces were performed by numerous research groups (de Leeuw and
Parker, 1997; de Leeuw and Parker, 1998; de Leeuw et al., 1998; Stöckelmann and
Hentschke, 1999; de Leeuw and Parker, 2000; Kuriyavar et al., 2000; de Leeuw and
Parker, 2002; Hwang et al., 2001; Wright et al., 2001; Parker et al., 2003; Kerisit et al.,
2003; Kerisit and Parker, 2004; Kerisit et al., 2005a; Perry et al., 2007). Despite small
discrepancies in the estimated inter-atomic distances between surface atoms and H2O
molecules, results of most studies suggested the formation of a monolayer of
associatively adsorbed H2O in a nearly-flat arrangement, relative to the surface, adopting
a herringbone pattern (de Leeuw and Parker, 1997; de Leeuw and Parker, 1998; Parker et
al., 2003; Kerisit et al., 2003). A slight vertical displacement of surface Ca atoms and
rotation of the surface CO3 groups were also reported in these studies.
Electronic structure studies, based on Density Functional Theory (DFT),
investigated the hydration of the (10.4) calcite surface (Parker et al., 2003; Kerisit et al.,
2003; Archer, 2004; Kerisit et al., 2005b) and examined its surface composition upon
contact with a gaseous phase containing H2O and CO2(g) (Kerisit et al., 2005b). Whereas

186
some of these calculations (Parker et al., 2003; Kerisit et al., 2003) predict a similar
configuration of the associatively adsorbed H2O molecules to those of the Atomistic
studies (de Leeuw et al., 1997; de Leeuw and Parker, 1998; Kerisit et al., 2003), the latter
consistently overestimated the energies of dry and wet calcite surfaces because do not
contain explicit chemical information and lack the full electronic relaxation offered by
DFT techniques (Parker et al., 2003).
Ab initio Molecular Orbital methods have also been used to investigate the
ground-state properties of CaCO3 polymers and clusters. Roothaan-Hartree-Fock (RHF)
techniques were applied to i) investigate the bonding and charge distribution in CaCO3
monomers (Thackeray and Siders, 1998), ii) study protonation and H2O attachment to
CaCO3 monomers and dimers (Mao and Siders, 1997), and iii) evaluate the performance
of different protocols to stabilize Can(CO3)n clusters of different size (4 n 22)
expressing the (001) surface (Ruuska et al., 1999). It was concluded that H2O surrounding
the calcite clusters, simulating hydration, stabilize the clusters while decreasing the time
required to achieve the Self-Consistent-Field (SCF) convergence. Finally, partial
geometric optimisations of Can(CO3)n clusters (n 21), where only the geometries of the
adsorbates were optimised, were carried out to investigate the interactions of anionic
collectors, oleate and oleoyl sarcosine anions, with the calcite surface with RHF/3-21G
(Hirva and Tikka, 2002). This study confirmed that moderately large Can(CO3)n cluster
models (n ≥ 14) are adequate surrogate models to correctly describe the effect of
neighbouring surface atoms, model the infinite calcite surface and investigate adsorption
reactions.

187
Despite all these efforts, no systematic ab initio Molecular Orbital study of the
structure and energy of the hydration layers at the (10.4) calcite surface has been
conducted. The present investigation is an extension of earlier studies, based on the
application of RHF techniques, to exploit the power of Roothan-Hartree-Fock methods
using moderately large cluster models (n = 18), to properly represent the idealized
stoichiometric (10.4) calcite surface and accurately describe hydrogen bonding (Tossel
and Vaughan, 1992). The results from earlier Atomistic simulations and DFT calculations
were used to select reasonable geometric constraints on the clusters and assign an initial
configuration to the H2O molecules. A series of RHF/6-31G(d,p) partial geometric
optimisations, involving specific “reactive” atoms at the (10.4) calcite surface and a
varying number of H2O molecules, were performed to obtain information on the structure,
the energy and the bonding relationships governing the hydration process.
2. METHODS
2.1 Computational Methods and Cluster Models
Gaussian 03 software
(Frisch et al., 2003) was used to perform the geometric
optimisations of finite charge-neutral slabs (clusters) taken from the bulk calcite structure
(Graf, 1961). The ideal stoichiometric (10.4) cleavage plane was represented in the slab
and adopted as the molecular model of the calcite surface as in earlier studies (de Leeuw,
and Parker, 1997; Wright et al., 2001; Hirva and Tikka, 2002; Parker et al., 2003; Kerisit
et al., 2003; Kerisit and Parker, 2004). The validity of this representation was confirmed
using a standard protocol devised to determine the most stable surface atomic
configuration of oxide minerals according to residual charge and bond strength
minimization criteria (Koretsky et al., 1998).

188
Cluster models have been commonly used to study the electronic structure of
carbonate (Mao and Siders, 1997; Thackeray and Siders, 1998; Ruuska et al., 1999; Hirva
and Tikka, 2002) and metal oxide minerals (Xiao and Lasaga,, 1994; Kubicki and Bleam,
2003). Specifically, clusters models were found suitable for investigations of adsorption
reactions on CaCO3 surfaces (Mao and Siders, 1997; Thackeray and Siders, 1998; Ruuska
et al., 1999; Hirva and Tikka, 2002). Therefore, this approach was adopted throughout
this study. The absence of periodic boundary conditions in cluster models requires a
formalism to treat crystal structure terminations and prevent “edge effects”. Hydrogen
atoms are commonly used as terminators of covalent compounds (Xiao and Lasaga, 1994)
and oxide minerals (Tossel and Vaughan, 1992; Xiao and Lasaga, 1994; Kubicki and
Bleam, 2003) but this approach is not suitable for systems in which hydrogen bonding is
involved, as for H2O adsorption (Tossel and Vaughan, 1992). In addition, for semi-ionic
minerals such as CaCO3, the use of standard point embedding techniques can be
problematic because of possible polarization of cations near the borders of the cluster and
because they cannot prevent unrealistic delocalisation of the cluster wave function arising
from the neglect of Pauli’s exclusion effects (Stefanovich and Truong, 1997).
Furthermore, the embedded point charges depend on the geometry and charge relaxation
of the crystal surface and, therefore, the results must be subjected to validation against
multiple point embedding models, a process that is both tedious and time-consuming
(Ruuska et al., 1999). Alternatively, stabilisation of CaCO3 surfaces can be improved by
placing H2O molecules in the vicinity of the exposed surfaces (Ruuska et al., 1999).
Consequently, the use of sufficiently large charge-neutral cluster models, in
combination with appropriate geometric constraints, is considered an adequate alternative
to simulate semi-infinite calcium carbonate surfaces and minimize edge effects (Ruuska

189
et al., 1999; Hirva and Tikka, 2002). The selected cluster must realistically represent the
mineral surface and the underlying bulk crystal to accurately describe local and long-
range interactions while keeping the cluster size practical for ab initio calculations
(Rosso, 2001).
In this study, the Can(CO3)n cluster size ranged from 4 n 18 with a varying
number of H2O monomers, 0 (H2O)n 6. The Ca9(CO3)9 and Ca18(CO3)18 clusters were
used to represent one and two full surface unit cells, respectively. The surface atomic
layer of the Ca9(CO3)9 cluster was composed of 3 Ca atoms and 6 CO3 groups, whereas
the subatomic layer contained 3 CO3 groups and 6 Ca atoms (Fig. 1). In contrast, the
composition of the surface and subsurface atomic layers of the Ca18(CO3)18 cluster were
identical: 9 CaCO3 units in each layer (Fig. 2). Most of our calculations were performed
using these two clusters which will hereafter be referred to, respectively, as the small and
large cluster models. The selected density of H2O molecules per surface area unit
composing the 1st hydration layer (4.9 nm-
2) is consistent with the density of
exchangeable surface Ca atoms (5 nm-2
), measured experimentally (Möller and Sastri,
1974) and reflects a 1:1 H2O:Ca stoichiometry.
Preliminary all-atom RHF/6-31G(d,p) optimisations of smaller clusters,
Ca4(CO3)4 and Ca5(CO3)5, were carried out for comparison with the geometry of the bulk
crystal. These calculations revealed that, when all atom positions are allowed to relax, the
cluster geometry is significantly distorted, particularly at the edges, and unrealistic
interactions are obtained, such as several Ca atoms bonding directly to C atoms or to an
unreasonable number of O atoms. Consequently, before performing further optimisations,
criteria were developed to select the atoms whose positions could be restricted to those of

190
the bulk structure. This step is critical to prevent the generation of unrealistic interactions
between neighbouring atoms and minimize computational time. To impose some control
on the 3D symmetry of the selected cluster and mimic the bulk crystal, all the atoms
present in the second atomic layer, the subsurface atoms, were fixed and the computed
geometry was examined for modifications in the coordination environment of both the
surface and subsurface atoms. This condition was slightly modified later as explained
below.
In adsorption studies, it is common practice to fully optimise the internal
coordinates of the adsorbates while the surface atom positions are kept frozen (Hirva and
Tikka, 2002). However, this approach does not exploit the full potential of electronic
relaxation offered by ab initio methods. It is more realistic to unlock some surface atoms,
such as those that participate directly in the adsorption process, which will be referred to
as “reactive” surface atoms throughout this paper. A careful selection of these geometric
constraints will allow a more realistic description of the configuration of the hydrated
surface layer and account for the interactions between the surface and the adsorbate
(water) while controlling the 2D symmetry of the mineral surface and minimizing edge
effects. The number of surface atoms allowed to relax must be selected on the basis of the
adsorption reaction of interest, the cluster size and the available computational
capabilities.
Earlier theoretical studies (de Leeuw and Parker, 1997; Wright et al., 2001; Parker
et al., 2003; Kerisit et al., 2003; Archer, 2004; Perry et al., 2007) showed that each
adsorbed H2O, in the 1st hydration layer, interacts with one Ca and one or two O atoms at
the (10.4) calcite surface. These “reactive” atoms are well represented in our cluster
models (Figs. 1 and 2) and were allowed to relax during the cluster geometry

191
optimisations. For the Ca9(CO3)9/2H2O and Ca9(CO3)9/3H2O clusters, all other surface
atoms were frozen in their original crystallographic positions.
To optimise computational time and prevent edge effects, water molecules were
only allowed to interact with surface atoms within a full surface unit cell at the centre of
the large cluster surface. The initial position of the H2O molecules in the 1st hydration
layer were chosen to reflect results common to earlier studies: i) herringbone adsorption
pattern (de Leeuw and Parker, 1997; Parker et al., 2003), ii) flat alignment of H2O
molecules (de Leeuw and Parker, 1997; Parker et al., 2003; Kerisit et al., 2003) with
respect to the surface and, iii) H2O oxygen atoms located at a distance of approximately
2.37 Å with respect to the surface calcium atoms (de Leeuw and Parker, 1997; Kerisit et
al., 2003). Based on results of Molecular Dynamics (Kerisit and Parker, 2004) and X-ray
Scattering (Geissbühler., 2004), H2O molecules in the 2nd
hydration layer, formally
ascribed to the 1st hydration layer in earlier studies (Geissbühler., 2004; Kerisit and
Parker, 2004; Magdans et al., 2006) were initially placed at a greater distance from the
surface (~3.3 Å), with a larger x-y displacement from the surface Ca (~3.9 Å) and
according to a 2:1 Ca:H2O stoichiometry (Fig. 2).
3. RESULTS
3.1 Structural Details of the Hydrated Clusters
Preliminary cluster geometry optimisations of the small cluster models were performed
using the STO3G and 3-21G basis sets for a quick comparison against the highest level of
theory selected in this study, 6-31G(d,p). Very similar configurations of associatively
adsorbed H2O molecules were obtained with the 3-21G and the 6-31G(d,p) basis sets,
with the inter-atomic distances between H2O and the surface “reactive atoms” differing

192
by less than 16%. In contrast, the STO-3G basis set predicted the dissociation of H2O
upon adsorption whereas the position of H2O constituents differed by up to 31% with
respect to results generated by the 6-31G(d,p) basis set. Both the 3-21G and 6-31G(d,p)
basis set simulations revealed that H2O molecules do not lie flat on the mineral surface
but are tilted with one of their H atoms pointing towards one of the “reactive” surface O
atoms and the other pointing away from the surface. In other words, only one hydrogen
bond can form between each H2O monomer and the surface (see discussion below).
As in an earlier study (Kerisit et al., 2003), to further confirm the associative
adsorption character of H2O, the adsorption of the H2O constituents, H+ and OH
-, on the
calcite surface was simulated at the RFH/6-31G(d,p) level of theory using the small
cluster. H+ were initially bonded to surface O at 1 Å and OH
- were bonded to surface Ca
at the bulk crystal Ca-O bond length of 2.37 Å. The optimised structure revealed that the
H+
and OH- spontaneously associated to H2O with an identical configuration and SCF
energy as when undissociated H2O was considered as the initial configuration. That the 6-
31G(d,p) basis sets predicts associative adsorption of H2O from both initially dissociated
and undissociated H2O and yields identical Ca(surface)-O(water) inter-atomic distances further
supports its use in the optimisation of larger CaCO3 clusters.
The optimised inter-atomic distances of the Ca-O octahedra imply the substantial
relaxation of the Ca-O bonds between the surface Ca and the subsurface O. The surface
Ca atoms shift out from the surface, increasing their inter-atomic distances to subsurface
O to the extent that the bond is substantially weakened (average Ca(surface)-O(subsurface) bond
length of 2.5 Å). To confirm this observation, we performed: i) a geometric optimisation
of the Ca9(CO3)9/2H2O cluster for which the subsurface O bonded to the surface Ca was
unlocked and, ii) a geometric optimisation of a three atomic layer cluster, Ca12(CO3)12+

193
2H2O, for which the entire CO3 group in the second atomic layer associated with the
surface Ca was unlocked (Fig. 3) and the third layer was frozen to impose the bulk
symmetry of the cluster. Both calculations confirmed the substantial relaxation of at least
one Ca-O bond per surface Ca-O octahedron and yielded nearly identical structural results
to those of the first RHF/6-31G(d, p) calculation.
The optimised Ca12(CO3)12/2H2O cluster shows that the Ca atoms move out of the
surface whereas the subsurface O atoms move away from the Ca both horizontally (x-y
directions) and vertically (z-direction). As subsurface CO3 groups relax, they rotate
towards the subsurface plane (-1 layer in Fig. 3) resulting in the stretching of more
surface Ca-O bonds than in the small cluster without significantly affecting the average
structure of the reconstructed surface. This observation validates the criteria we selected
to impose geometric constraints and supports our premise that at least one Ca-O bond per
surface Ca-O octahedron is significantly weakened, and approaches rupture, upon
hydration. A thorough discussion on this issue is given below.
Having ascertained the self-consistency of our results using different cluster
thicknesses, we can confidently focus on the results of the large clusters,
Ca18(CO3)18/4H2O and Ca18(CO3)18/6H2O, which provide a reasonable representation of
the infinite calcite surface. Although the calcite surface undergoes some reconstruction
upon hydration, the average 2D dimensions of the (10.4) surface unit cell increase only by
approximately 1% along the x direction while they remain unchanged along the y
direction (Fig. 2). Table 1 summarizes the structural details of the reconstructed hydrated
calcite surface for the Ca18(CO3)18/6H2O cluster. They, respectively, reflect the average
Cartesian and internal coordinates of the relaxed surface atoms and the H2O. It is
noteworthy that the Ca and C atoms are displaced differently along the three Cartesian

194
coordinates. Their differential displacement along the z- direction, normal to the surface,
determines the height of the reconstructed surface layer, relative to the subsurface atomic
layer, and yields the extent of surface relaxation. Conventionally, as in previous studies
(Geissbühler, 2004), the averaged, relaxed positions of the Ca atoms were selected to
define the reconstructed surface atomic layer and reference the position of H2O in the
hydration layer.
Using the reconstructed lattice spacing, d12, the average surface relaxation, 12, can
be expressed as a percentage of the perfect lattice spacing, d, according to (Markmann et
al., 2006):
100δ
d
dd12
12 (1)
An average surface relaxation of 5.6% relative to surface Ca was observed. The
extent of surface corrugation is given by the surface rumpling parameter (z) which
represents the difference between the z-coordinates of surface anions (Zanion
) and cations
(Zcation
) with respect to the ideal lattice spacing (Markmann et al., 2006):
100d
zzz
cationanion (2)
At the ideal (10.4) unreconstructed calcite surface, one O of each surface CO3 lies
0.8 Å above the surface Ca and C atoms. Thus, the intrinsic rumpling (z) of the surface
is 26.4% which, upon reconstruction, decreases to 24%. If the z-displacement of the

195
central C of each CO3, rather than the surface O, is considered relative to the surface Ca,
the surface rumpling would go from zero for the unreconstructed surface, to 4.9% in the
reconstructed surface. On reconstruction, the glide symmetry at the surface is slightly
broken because of the differential displacement, along the z- direction, of the surface Ca
and the alternatively oriented CO3 groups (Config-1 and Config-2 in Fig. 4).
Upon hydration, surface Ca-O octahedra are distorted following changes in the
Ca-O bond lengths. The average surface Ca-O bond length is stretched by ~ 4 % (2.46 Å),
35% of the relaxed Ca-O bonds are stretched by less than 4.5% and one Ca-O bond per
surface Ca-O octahedra stretches by at least 10% whereas Ca-O bond contraction up to
5% is also observed in some Ca-O octahedra. Conversely, the CO3 groups are not
significantly distorted from their original trigonal planar geometry, although rigorously
speaking, their D3h symmetry is lost as a result of the differential displacement of the
three O atoms along the z-axis (Fig. 4). The average change in the CO3 dihedral angle (O-
C-O-O) is only 1.8º, which reflects the small out-of-plane distortion of the CO3 group.
The C-O bond lengths are only slightly shortened ( 0.8%) whereas the average O-C-O
angle remains unchanged. CO3 groups are tilted towards the plane of the relaxed surface
Ca atoms by an average of 4.1° but their atomic positions along the x-y directions are
significantly modified (Table 1). Because of the relative rigidity of the CO3 groups, their
average x-y displacement is expressed in terms of the optimised x-y coordinates of the
central C atom, the CO3 centre of mass, rather than relative to the surface O.
The associative character of adsorbed H2O in the 1st and 2
nd hydration layers is
confirmed in the large cluster, Ca18(CO3)18/6H2O. H2O molecules in the 1st hydration
layer (Mode-I) are slightly oblique to the surface with one H oriented towards one surface
O and the other towards H2O in the 2nd
hydration layer. H2O molecules in the 2nd

196
hydration layer align their dipole nearly parallel to the surface (Mode-II). The average O-
H-O angle of H2O in the 2nd
hydration layer is smaller than that of H2O in the 1st
hydration layer whereas the average O-H bond length of H2O in both layers is identical,
0.95 Å. Nevertheless, H2O in the 2nd
hydration layer display a short (0.94 Å) and a long
O-H bond (0.97 Å), the longer being oriented towards surface O following hydrogen
bonding (see below) and the shorter one pointing away from the surface. Structural details
of the reconstructed calcite surface, including the two hydration layers modelled in this
study, are illustrated in Figure 5.
3.2 Energies of Adsorption
The energy of interaction between H2O molecules and the calcite surface, Eads, can be
computed from (Cao and Chen, 2006):
Eads = Eslab/water n
– ( Eslab + n·Ewater ) (3)
where Eslab/watern represents the energy of the optimised cluster covered with n H2O
molecules (n = 2, 4 or 6, depending on the cluster model) at their adsorption
configurations, Eslab is the single-point energy of the cluster model with no H2O attached
(dry cluster model) and Ewater is the energy of a single H2O in the gas phase. The
adsorption energy of n H2O attached to the cluster is equal to –172.8 and –306 kJ mol-1
for the small (n=2) and large (n=4) clusters respectively. Upon normalization to the
number of attached H2O monomers, their respective energies become –86.4 and –76.5 kJ
mol-1, corresponding to the binding energy of a single H2O monomer at the cluster

197
surface. The adsorption energy of the 2nd
hydration layer, Eads-2nd, to the hydrated surface
is given by:
Eads-2nd = Eslab/water 6-(Eslab/1st+2·Ewater) (4)
where Eslab/1st is the total energy of the large cluster with the four 1st layer H2O monomers
attached. Once normalized to the total number of adsorbed H2O, Eads-2nd is –106.1 kJ mol-
1. To calculate the interaction energy between H2O and the dry surface with no other
adsorbates attached, the adsorption energy must be corrected by the average interaction
energy, Einter, among H2O in the 1st and 2
nd hydration layers (Cao and Chen, 2006):
Einter = Ewater/1st/2nd – (6·EH2O) (5)
where Ewater/1st/2nd is the single-point energy of the H2O of the 1st and 2
nd hydration layers
of the large hydrated cluster at their adsorbed configurations. To estimate Einter for H2O
molecules constituting the 1st hydration layer, we subtracted the gas phase single-point
energy of 4 H2O molecules from the single-point energy of the 4 H2O molecules in the 1st
hydration layer at their adsorbed configurations. In both cases, the estimated Einter values
per H2O molecule are nearly equal (–11.9 kJ mol-1
for n= 6 and –12 kJ mol-1
for n=4)
and are in excellent agreement with the Einter estimated by DFT calculations (–12.5 kJ
mol-1
after Basis Set Superposition Error, BSSE, correction; Archer, 2004). The
agreement between the two independent energy estimates strongly supports the suitability
of RHF methods, in combination with cluster models, to investigate hydration reactions at

198
CaCO3 surfaces, making computationally-expensive periodic boundary condition
calculations unnecessary.
Finally, the following energy decomposition:
interE - layer-adsE = slab-layerE (6)
provides the individual interaction energy of H2O in the 1st and 2
nd hydration layer with
the dry surface (Elayer-slab), where Eads-layer corresponds to the interaction energy of a
single H2O molecule of either the first (Eads-1st-layer) or second (Eads-2nd-layer) hydration layer
and Einter corresponds to the estimated interaction energy at each hydration layer.
Equation 6 yields energies of –64.5 kJ mol-1
and –94.2 kJ mol-1
per adsorbed H2O
molecule for the 1st and 2
nd hydration layers, respectively.
3.3 H2O Interlayer Penetration
The surface reconstruction and weakening of the surface atomic layer that result from the
relaxation of Ca-O bonds upon hydration may allow H2O to penetrate the subsurface
layers as it is the case in other minerals such as scheelite (CaWO4, de Leeuw and Cooper,
2003). To model this effect, two additional H2O were placed in the interlayer of the
RHF/6-31G(d,p) optimised Ca9(CO3)9/2H2O cluster and a geometric optimisation of the
Ca9(CO3)9/4H2O cluster, with identical geometric constraints (i.e., freezing of subsurface
lattice atoms and unlocking of “reactive” surface atoms and H2O molecules) to those used
for the small cluster, was performed. This optimisation revealed that one of the H2O
remains un-dissociated and lies at the centre of the subsurface interlayer with its dipole

199
paralel to the surface whereas the second H2O is repelled and migrates towards one end of
the cluster, dissociates and interacts with atoms at the cluster edge forming Ca-OH(water)
and O-H(water) bonds (see Supporting Information). The interlayer H2O significantly
stretches one Ca(surface)-O(surface) bond (3.06Å) whereas the average Ca-O bond stretching is
of 6 % (2.52 Å). In addition to the Ca-O octahedra distortion induced upon hydration of
the surface, following Ca(surface)-O(subsurface) bond stretching, the subsurface H2O further
weakens the topmost atomic layer. The energy of H2O incorporation from the bulk gas-
phase to the subsurface interlayer, Einc, is computed from:
Einc= Etot - Eads2 (7)
where, Eads2 is the energy of adsorption of two H2O to the surface calculated with
Equation 3, whereas ETot is the total energy of H2O adsorption and interlayer
incorporation as given for the Ca9(CO3)9/4H2O cluster model by:
Etot = Eslab/water4
- (Eslab +4·EH2O) (8)
Equation 7 yields a value of Einc equal to –4.8 kJ mol-1
. Because of the vastly
different configurations that H2O molecules adopt upon incorporation into the calcite
lattice, this value was not normalized to the number of H2O molecules and, hence, it
reflects the energy of incorporation of two H2O molecules.

200
4. DISCUSSION
4.1 Reliability of RHF/6-31G(d,p) Results
Proper evaluation of the accuracy of our theoretical predictions must be ultimately made
against reliable experimental data. The only available experimental data describing the
3D structure of the hydrated calcite surface was obtained via X-ray Scattering and
GIXRD techniques (Geissbühler et al., 2004; Magdans et al., 2006). Nevertheless, the
uniqueness of a structural model derived from these techniques largely depends on their
ability to resolve discrete, model-independent, structural features and to correct for
systematic errors in the raw data exceeding the expected statistical error, issues difficult
to address in practice (Fenter and Sturchio, 2004). More specifically, bond lengths derived
from these X-ray measurements might incur systematic errors and should be treated with
caution (Fenter and Sturchio, 2004). This is well illustrated by the differences between the
structural models constructed from X-ray Scattering and GIXRD data (e.g., equilibrium
positions of surface atoms, inter-atomic distances) which makes it difficult to adopt one
data set as “benchmark” for evaluating the accuracy of our theoretical results.
Alternatively, the accuracy of our RHF calculations, uncorrected by BSSE and
Zero Point Vibrational Energy (ZPVE) effects, can be estimated by comparing published
data acquired at different levels of theory ranging from uncorrelated RHF methods to
higher levels of theory, accounting for electron correlation, BSSE and ZPVE effects.
Numerous first- and second-row element-containing polyatomic models have been
studied at different levels of theory including RHF, DFT and Møller-Plesset perturbation
methods (MP2) (deFrees and McLean, 1985; Saebø et al., 1993; Scott and Radom, 1996;
Maheshwary et al., 2001; Zhou et al., 2004; Rozmanov et al., 2004). These studies
revealed that, for moderate basis sets coupled with suitable polarization functions, e.g. 6-

201
31G(d,p), MP2 approaches do not offer any substantial improvement over the less
demanding uncorrelated, BSSE- and ZPVE-uncorrected RHF methods for the calculation
of Zeroth-order (e.g., structure, association and stabilization energies; Saebø et al., 1993;
Maheshwary et al., 2001; Zhou et al., 2004; Rozmanov et al., 2004) or Second-order (e.g.,
harmonic frequencies, entropies and enthalpies; deFrees and McLean, 1985; Scott and
Radom, 1996; Rozmanov et al., 2004) chemical properties. In fact, in some cases (e.g.,
thermochemical quantities; Scott and Radom, 1996; Rozmanov et al., 2004), uncorrelated
RHF and correlated DFT methods performed better than MP2 suggesting an intrinsic
compensation of electron correlation, BSSE and ZPVE effects. Based on these
considerations and considering the excellent agreement of our results with those obtained
with BSSE-corrected correlated DFT techniques (see above), we estimated the
uncertainty of our RHF/6-31G(d,p) calculations to be 5%, what we believe to be an
excellent compromise between accuracy and computational cost for the investigation of
surface reactions at the ab initio level.
4.2 Three-D Structural Registry
Our results show that the hydrated calcite surface undergoes significant reconstruction
upon hydration, including: bond relaxation, differential displacement of surface atoms
along the x-, y- and z- directions and rupture of Ca-O bonds. This partly contrasts,
qualitatively and/or quantitatively, with the results of some previous theoretical (de
Leeuw and Parker, 1997; Hwang et al., 2001; Wright et al., 2001; Parker et al., 2003;
Kerisit et al., 2003) and experimental studies (Geissbühler et al., 2004; Magdans, 2006).
Associative adsorption of H2O is observed under various adsorption scenarios,
consistent with earlier results of Atomistic studies (de Leeuw and Parker, 1997; Hwang et

202
al., 2001; Wright et al., 2001; Parker et al., 2003; Kerisit et al., 2003), Molecular
Dynamic simulations (Stöckelmann and Hentschke, 1999; Kerisit and Parker, 2004; Perry
et al., 2007) and DFT (Kerisit et al., 2003; Archer, 2004) calculations. These common
findings challenge the traditional idea that water hydrolysis products are attached to
individual surface atoms upon dissociative H2O adsorption (Stipp and Hochella, 1991;
Van Cappellen et al., 1993). Within the context of adsorption and surface complexation
theory, the present results have fundamental implications to the definition of reactive
surface sites, including charge and mass assignment that reflect on the formulation of
mass action laws and the calibration of surface reactions as discussed in Chapter 3 of this
thesis (see Villegas-Jiménez et al., 2009a).
The configuration of adsorbed H2O computed in this study is not flat relative to
the surface (de Leeuw and Parker, 1997; Parker et al., 2003; Kerisit et al., 2003) nor does
it display a herringbone pattern (de Leeuw and Parker, 1997; Parker et al., 2003). The
H2O dipole in the 1st hydration layer lies slightly oblique to the surface. To minimize
electrostatic repulsion between neighbouring H2O molecules, one of the H of adsorbed
H2O in the 1st hydration layer is oriented towards an O of the adjacent H2O (H2Oadj) in the
2nd
hydration layer whereas the other H is oriented towards a surface O. This
configuration is intermediate between those predicted by Atomistic and Molecular
Dynamics studies: i) flat-orientation (de Leeuw and Parker, 1997; Parker et al., 2003;
Kerisit et al., 2003), ii) aligned near the surface with both H pointing to the surface
(Wright et al., 2001; Kerisit et al., 2005b; Perry et al., 2007), and iii) slightly angled
above the surface with the two H pointing away from the surface (Hwang et al., 2001).
The discrepancy reflects the chemical information contained in the RHF/6-31G(d,p)
technique which contrasts with the non-chemically informative Force Field-based

203
calculations mentioned above. As expected, based upon the superior performance of
electronic structure methods for the modelling of structural and energetic properties of
minerals (Parker et al., 2003), the RHF/6-31G(d,p) simulations agree best with results of
the BSSE-corrected DFT calculations (Archer, 2004) which also showed one H2O
hydrogen pointing to one surface O and the other H away from the surface.
The predicted average distance between surface Ca and O(water) atoms in the 1st
hydration layer (2.48 Å), is in excellent agreement with values of earlier studies:
Atomistic simulations (2.35 to 2.73 Å) and Density Functional Theory (2.37 to 2.42 Å),
but contrasts with X-ray specular and Non-specular Scattering and Grazing Incidence X-
ray Diffraction data that yield inter-atomic distances of 2.97 Å and 2.1 Å, respectively. In
the former case, the large Ca- O(water) distance was explained by a significant lateral
displacement of H2O relative to surface Ca along the x- and y- directions whereas, in the
latter, no explanation was provided to explain such a short Ca-O(water) distance.
Estimated distances between the H atoms of H2O in the 1st hydration layer and
surface O atoms differ from earlier theoretical investigations. Results of the RHF/6-
31G(d, p) simulations show that one of the water H points towards a surface O at a
distance of 2.01 Å, suggesting hydrogen bonding, whereas the other H points away from
the surface at an average distance of 3.3 Å from the nearest surface O, precluding
hydrogen bonding with the surface (see next section). These results agree with DFT
results (based upon the Generalized Gradient Approximation, GGA; Parker et al., 2003;
Kerisit et al., 2003) insomuch as the formation of one hydrogen bond per adsorbed H2O
(H(water)-O (surface) distance of 2.42 Å) but they differ with respect to the other H(water)-
O(surface) inter-atomic distance (1.66 Å) which was assumed to reflect a second H-Bond

204
in that study. A much better agreement is obtained with BSSE-corrected DFT calculations
that predict the formation of only one hydrogen bond and only slightly different H(water)-
O(surface) distances (1.81 Å and 3 Å; Archer, 2004). These discrepancies must reflect the
absence of Self Interaction Corrections in DFT (Suba and Whitehead, 1995) and improper
treatment by the DFT functionals of the van der Waals (dispersion) attractive forces,
arising from the long-range correlations of electronic density fluctuations (Ireta et al.,
2004; Santra et al., 2007; Santra et al., 2008), which makes this technique less accurate
than RHF/6-31G(d,p) in predicting hydrogen bonding and bridging.
Furthermore,
whereas DFT-GGA yields varying C–O bond lengths in CO3 (1.29 to 1.37 Å; Parker et
al., 2003), the RHF/6-31G(d,p) optimised C-O bond lengths agree with the bulk calcite
bond lengths to within 1%. The lack of explicit chemical information in Atomistic
calculations can explain why they predict the formation of two hydrogen bonds rather
than the single one found in the present study and in an earlier DFT investigation (Archer,
2004). Unfortunately, because of the weak X-ray Scattering power of the H atoms, the
structural models produced from least-squares fitting of X-ray Scattering (Geissbühler et
al., 2004) or GIXRD (Magdans, 2006) data are not sensitive enough to reliably determine
the orientation of adsorbed water (no rotational degrees of freedom), and therefore,
comparisons of the internal coordinates of H2O and H atoms positions against our results
are unwarranted.
According to our RHF calculations, the H2O molecules in the 1st hydration layer
lie 2.36 Å from the surface, along the z-plane, in agreement with results of Molecular
Dynamics (2.2 Å and 2.3 Å) and X-ray Scattering (2.3 Å) studies. The H2O molecules in
the 2nd
hydration layer sit 0.93 Å above the 1st hydration layer and 3.3 Å from the calcite

205
surface, also in good agreement with Molecular Dynamics (3.2 Å and 3.0 Å) and X-ray
Scattering (3.45 Å) results.
Upon hydration and relaxation of the calcite surface, Atomistic simulations
(Wright et al., 2001) and X-ray Scattering (Geissbühler, 2004) studies yield negative
displacements of the Ca and C along the z -direction. In contrast, results of this study
predict a positive outward displacement of 0.17 Å, in good agreement with a recent
Molecular Dynamics study (0.12 Å; Perry et al., 2007). Our simulations revealed a x-y
displacement of H2O molecules with respect to surface Ca atoms of 0.57 Å, much smaller
than the one derived from X-ray Scattering (1.9 Å) but is in excellent agreement with
results of Molecular Dynamics studies (0.6 Å; Kerisit et al., 2005a; Parker, private
communication in Geissbühler, 2004) which also consider the presence of multiple
hydration layers near the surface to better represent solvent effects. It is noteworthy that
these additional H2O (or solvent) layers have been shown to have little effect on the
bonded H2O (Whitehead et al., 2004). This further justifies our application of RHF
techniques on sufficiently large Can(CO3)n clusters (n 18) with a reduced number of
H2O (≥ 6) to adequately model the semi-infinite hydrated calcite surface, as usually
represented in periodic DFT studies.
Disruption of the glide symmetry observed in our study (by 0.11 Å) arises from
the differential displacement of Ca surface atoms along the z- direction and contrasts with
X-ray reflectivity (Geissbühler, 2004) and GIXRD
(Magdans, 2006) data that show no
evidence of such reconstruction and assume that the glide symmetry of the calcite surface
is passed on to the hydration layer. The observed loss of symmetry in our study is,
however, compatible with results of Atomic Force Microscopy (AFM) studies (Stipp et
al., 1994; Liang et al., 1996) that imply a vertical relaxation of approximately 0.35 Å

206
(Stipp et al., 1994) of the two alternatively oriented CO3 groups within the surface unit
cell (Fig. 4) and agrees well with Atomistic simulations of partially hydrated surfaces (de
Leeuw and Parker, 1997) that predict a differential displacement of the two CO3 groups
by 0.05 Å along the z-axis. Because AFM is believed to be plagued by technical artefacts
(surface deformation by interaction with the probe tip), which may significantly
overestimate the vertical relaxation of surface atoms (Stipp et al., 1994), a more subtle
loss of symmetry, such as that suggested by the present results, is thought to be more
realistic. Interestingly, in a more recent Atomistic study (Rohl et al., 2003), a (2x1)
reconstruction of the calcite surface (with rotation of half of the surface CO3 groups) was
predicted and it was concluded that the extent of reconstruction largely depends on the
experimental conditions which, in turn, may explain why surface reconstruction went
undetected in some of the above-mentioned studies.
4.3 Bonding Relationships: Geometric and Energetic Criteria
On thermodynamic grounds, the formation of the 1st and 2
nd hydration layers is
favourable, in agreement with earlier theoretical (de Leeuw and Parker, 1997; Wright et
al., 2001; Parker et al., 2003) and experimental findings (Liang et al., 1996) that indicate
an increasing stabilization of the (10.4) calcite surface following the adsorption of H2O
layers. At identical H2O adsorption densities (2 H2O molecules per unit cell in the 1st
hydration layer), the estimated adsorption or hydration energy per H2O molecule in the 1st
hydration layer, corrected for H2O-H2O interactions (-64.5 kJ mol-1
), lies within the range
of values reported in earlier Atomistic studies (de Leeuw and Parker, 1998; de Leeuw et
al., 1998; Wright et al., 2001; Kerisit et al., 2003), -53.9 to -93.9 kJ mol
-1, and is in
excellent agreement (3%) with the one estimated by BSSE-corrected DFT calculations

207
also corrected for H2O-H2O interactions (-62.7 kJ mol-1
, Archer, 2004). This suggests that
the BSSE associated to our RHF/6-31G(d,p) calculations is either very small or is largely
cancelled out by other effects (e.g., correlation effects, ZPVE) not accounted for in our
calculations (note that our Einter differs by less than 5% from the BSSE-corrected DFT
value (Archer, 2004, see above). Unfortunately, the energy of adsorption of H2O in the
2nd
hydration layer predicted in our study (–94.2 kJ mol-1
, corrected for H2O-H2O
interactions) cannot be evaluated against experimental or other theoretical approaches
since this information is not available in any previous study.
The relaxation of surface atoms following hydration results in a significant
weakening of some Ca-O bonds and of the topmost atomic layer of the calcite mineral
with respect to the bulk. Because of steric hindrance, H2O can only approach the mineral
surface to within approximately 2.37 Å, the ideal Ca-O bond length in the bulk crystal
structure. In response to their affinity for H2O, Ca surface atoms are vertically displaced,
increasing the inter-atomic distances of Ca to adjacent surface or subsurface CO3 groups
and relaxing and possibly breaking some Ca-O bonds. This can be analysed in terms of
Bond Valence Theory (Brown, 1981), an empirical approach based upon Pauling‟s
Valence Sum Principle (Pauling, 1929) and parameterised on the basis of bulk crystal
inter-atomic distances. Although the applicability of Bond Valence concepts to surface
structures subjected to external stresses is controversial (Bickmore et al., 2004), the
progressive convergence of this theory with molecular-orbital models of chemical
bonding (Burdett and Hawthorne, 1993) makes it a very promising approach to rationalise
bond orders at reconstructed surfaces in terms of inter-atomic distances.
Using the RHF/6-31G(d,p) optimised Ca-O inter-atomic distances, we computed
their respective Bond Valences from (Brown, 1981):

208
B
rrS
ijo
ij exp (9)
where Sij is the bond strength for a given cation-anion pair (i,j) in valence units, v.u., r0 is
an empirical parameter specific to that pair of atoms (r0 = 1.967 Å for Ca-O), rij represents
the experimental bond length and B is a fitted parameter equal to 0.37 (Brown, 1981). For
all bonds formed by a given central atom, the Valence Sum Principle must be satisfied
(Pauling, 1929; Brown, 1981):
j
iji Sv (10)
where vi is the formal valence of the central atom i and the right-hand term is the
calculated Bond Valence, vCalc
. Any deviations from vi are typically considered to
represent the unsatisfied or residual valence exhibited by atom i (Bickmore et al., 2004).
Nevertheless, it has been recently proposed that electronic and steric effects may generate
substantial deviations from integer stoichiometric valences, vStoich
, for some atoms and
specific crystal structures (Wang and Liebau, 2009). This might be the case of bulk
lattice Ca atoms which are shown to exhibit a net increase in charge of ~ 0.24 electrons,
per atom slightly decreasing the divalent stoichiometric valence (vStoich
=2) typically
ascribed to the Ca cation in calcite which, in turn, should give a structural valence
(vStruct
) of 1.76 (Skinner et al., 1994). Application of Eq. 9 and 10 using the average
optimised RHF/6-31G(d,p) Ca-O bond lengths (Table 2) yielded a vCalc
of 1.62 for the

209
surface Ca atoms, significantly smaller than vStoich
. In other words, in terms of Bond
Valence and vStoich
, it would seem that contraction of some Ca-O bonds do not fully
compensate for the stretching of others and, hence, a net positive unsatisfied valence of
approximately 0.4 v.u. per surface Ca atom is predicted. However, this unsatisfied
valence would reduce to 0.14 v.u. if the structural valence predicted for Ca atoms in the
bulk calcite lattice (vStruct
=1.76; Skinner et al., 1994) applies to the surface as well.
Alternatively, an hypothetical error of 4% in the r0 value (for Ca-O interactions) would
suffice to fulfil the Valence Sum Principle, vCalc
vStoich
. Such an error would be
compatible with the accuracy of the Bond Valence method for ionic compounds (5-7%;
Brown and Shannon, 1973) and would fall within the variability of r0 values designated
for H-O ( 25%; Yu et al., 2006), OH (15%; Yu et al., 2006) and lanthanide-O
interactions (1-4%; Zocchi, 2007) which show a dependency on the type or Coordination
Number of the specific compounds used in r0 calibration.
Regardless of the accuracy of the computed vCalc
values, the estimated Sij values
undoubtedly represent useful estimates of bond strengths which can be used as bond order
indexes. Hence, as a rule, Ca-O Bond Valences reduced to 50% (0.17 v.u.) of their
bulk calcite value were considered to reflect very weak Ca-O bonds approaching rupture
which are hereafter referred to as “significantly-weakened bonds” (SWBs). This
reduction in Bond Valence corresponds to a cut-off bond length 2.6 Å which is
equivalent to approximately 10% of bond stretching. Application of Emri‟s equation of
bond orders (Emri, 2003) shows that the selected cut-off bond length corresponds to a
bond order of 0.5 which is half of that in the bulk calcite lattice. This supports our
premise that at distances 2.6 Å, the Ca-O bond strength weakens to at least 50% of its

210
value in the bulk crystal and substantiates our hypothesis that some Ca-O bonds may
break upon hydration. Future Extended X-ray Absorption Fine Structure (EXAFS)
investigations designed to resolve the coordination number, CN, of surface Ca atoms at
hydrated cleavage calcite surfaces will be instrumental in ascertaining whether the SWBs
are part of the coordination shell of the central Ca atoms.
In Table 2, we show the average individual Ca-O bond lengths predicted in this
study for wet conditions (n= 6 H2O) as well as those obtained by an earlier GIXRD
investigation (Magdans, 2006). It is noteworthy that although the distortion of the Ca-O
octahedron is observed in the dry and wet scenarios, the Ca-O stretching is more subtle in
the former. For both scenarios, the ranges of Ca-O inter-atomic distances postulated by
GIXRD data are broader (dry: 1.9-2.5 Å; wet: 2.1-3 Å) than in our study (dry: 2.3-2.4 Å;
wet: 2.32-2.63 Å). Our predicted Ca-O bond lengths agree better with those estimated by
an earlier Atomistic study (dry: 2.3-2.7 Å; wet: unspecified bond lengths; Wright et al.,
2001). It follows that the disruption of the bulk crystal periodicity at the calcite surface
leads per se to a detectable distortion of Ca-O octahedra which increases upon interaction
of the water layer with the calcite surface. Although earlier X-ray Scattering data
(Geissbühler et al., 2004) also suggested a distortion of the surface Ca-O octahedra upon
contraction of the in-plane Ca–O bond lengths and expansion of the Ca(surface)-O(water) bond
length, the Ca-O bond lengths are not specified in such study and comparisons are not
possible.
The above considerations show that in addition to the stabilizing effect of H2O on
the calcite surface, H2O also plays an important role in the reorganization of Ca-O bonds.
The significant relaxation of at least one Ca-O bond per surface Ca atom reflects the
strong affinity of Ca for H2O and must therefore be a precursory step to the eventual

211
release of surface Ca atoms to the bulk solution which, in turn, may lead to a provisional
non-stoichiometric calcite dissolution regime. It is worth noting that earlier findings could
also be taken as indirect evidence of the preferential dissolution of Ca atoms over CO32-
ions by H2O following the stabilization of the hydrated mineral surface. For instance,
DFT calculations (Kerisit et al., 2005b), revealed that, at 100% relative humidity, a non-
stoichiometric, calcium-deficient surface may predominate over the ideal (10.4)
stoichiometric termination whereas XPS data showed a slight depletion in both O and Ca
relative to C atoms in the surface of dissolving calcite samples (Stipp and Hochella,
1991). Furthermore, interpretations of the electrokinetic behaviour of calcite in aqueous
suspensions suggest the greater tendency of Ca2+
than CO32-
ions to pass into solution
(Douglas and Walker, 1950). Finally, recent acid-base surface titrations of calcite
suspensions revealed that Ca2+
is released in exchange for H+ under circum-neutral and
alkaline conditions, reflecting the strong susceptibility of Ca atoms to leave the surface
(Villegas-Jiménez et al., 2009b).
It has also been hypothesized that Ca-O bonds may break upon attachment of
protons to the calcite surface (via bonding to surface O atoms) under acidic conditions
(Sjöberg, 1978), but the substantial weakening or rupture of Ca-O bonds following the
adsorption of H2O has not been postulated before. Furthermore, our results show that H2O
in the 1st hydration layer may diffuse to the subsurface interlayer further weakening the
topmost layer by rupture of additional Ca-O bonds. We did not thoroughly examine the
effect of H2O interlayer incorporation on the stability of the calcite surface but we believe
that this mechanism deserves further investigation as it possibly plays a key role on
mineral dissolution, rearrangement of surface layers, ion replacement and solute transport
through subsurface lattice layers in aqueous solutions.

212
The similarity in the bond lengths between surface Ca and H2O (2.48 Å) with Ca-
O bonds in the bulk lattice (2.36 Å) and the strong interaction between a single H2O and
the calcite surface (Eads-1st-layer= -64.5 kJ mol-1
) suggests similarities between the character
of binding of the Ca(surface)-O(water) and the Ca(surface)-O(calcite) bonds. Although the latter has
been traditionally considered ionic, there is some theoretical evidence that the 3p orbital
of Ca may hybridise slightly with the 2s and 2p orbitals of C and O and, therefore,
contribute to the electron density on the C-O bond in CO3, implying some covalent
character (Archer, 2004). In contrast, the Bond Valence scale only ascribes a covalent
character to bonds with Bond Valences 0.6 (Altermatt and Brown, 1985) which is not
the case of the Ca-O interactions observed in our study. Regardless of whether the
Ca(surface)-O(water) interaction is purely ionic or not, our findings are consistent with
experimental results confirming the chemisorption of H2O molecules in direct contact
with the calcite surface (Morimoto et al., 1980).
H2O-calcite surface interactions are illustrated by the Delocalised Molecular
Orbitals, DLMO-198 and DLMO-309, in Figure 6. DLMO-198 shows a local interaction
between H2O in the 1st hydration layer and a surface Ca, whereas in DLMO-309, the
interaction involves Ca, C and O surface atoms and H2O molecules in the 1st and 2
nd
hydration layers. They show that, in the 1st layer, H2O is directly bonded to the surface Ca
while interacting, through hydrogen bonding, with surface O and adjacent H2O in the 2nd
hydration layer.
The formation of hydrogen bonds between water and surface atoms and between
pairs of adjacent adsorbed water monomers can be discussed within the context of the
geometry of the hydrogen bond, as previously done for liquid water (Mezei and
Beverdige, 1981) and a large number of aqueous mixtures (Ferrario et al., 1990; Luzar

213
and Chandler, 1993; Chowdhuri and Chandra, 2002). Under this scheme, the hydrogen
bond is defined by geometric criteria and maximum internal coordinates (Luzar and
Chandler, 1993). These coordinates are illustrated in Fig. 7 for the calcite surface-H2O
and H2O-H2O interactions. Let RO represent either the oxygen in the CO3 group or in the
H2O molecule, then ROO represents the inter-atomic distance between the oxygen of the
bridging H2O molecule and RO, H is the angle between the H-O bond of H2O and RO, and
ROH is the length of the hydrogen bond.
The cut-off values are those specified earlier for H2O-H2O interactions
(Chowdhuri and Chandra, 2002): ROO ≤ 3.5 Å, ROH ≤ 2.45Å and H ≤ 30°. This geometric
approach is convenient since it applies to discretely H-bonded molecules and can be
generalized directly to systems other than water and water-like systems (Mezei and
Beverdige, 1981). On the basis of these criteria (Table 3), one hydrogen bond is formed
between H2O in the 1st hydration layer and a surface O (O
2-H
2 in Fig. 7) and, in
agreement with Atomistic studies (de Leeuw and Parker, 1997; Wright et al., 2001), there
is no hydrogen bond between adjacent H2O in the 1st hydration layer. In addition, H2O of
the 2nd
hydration layer are hydrogen-bonded to a surface O atom and to one adjacent H2O
in the 1st hydration layer (Fig. 5).
In agreement with earlier findings (de Leeuw and Parker, 1997), the hydrogen
bond network within the 1st hydration layer is disrupted by its strong interaction with the
surface. Ordering of the 2nd
hydration layer cannot be properly evaluated with this model
because only two H2O monomers are used to model this layer and the influence exerted
by adjacent multiple H2O layers is neglected. However, as emphasized earlier, solvent
effects are most likely negligible (Whitehead et al., 2004), and therefore, the present

214
results are considered as reliable first-order descriptors of the 2nd
hydration layer registry
obtained, for the first time, at the ab initio level.
The different nature of the interaction established by H2O in the 2nd
layer
(hydrogen bonding) and by H2O in the 1st layer (ionic bond and hidrogen bonding) with
the surface is consistent with thermogravimetric (Morimoto et al., 1980; Ahsan, 1992)
and Fourier-Transformed Infrared data (Ahsan, 1992) revealing the presence of strongly
adsorbed H2O, “chemisorbed”, and weakly adsorbed H2O, “physisorbed”, at hydrated
calcite surfaces.
5. CONCLUSIONS
The power of ab initio RHF Molecular Orbital methods, coupled to moderately large
cluster models and adequate basis sets, was exploited to investigate the ground-state
structural, energetic properties, and bonding relationships of the hydrated (10.4) calcite
surface. Fresh insights into the 3D structural registry and adsorption energetics of the 1st
and 2nd
hydration layers at the reconstructed (10.4) calcite surface complement the
information derived from earlier Atomistic and X-ray Scattering and GIXRD studies.
Whereas small discrepancies in the configuration of adsorbed H2O molecules and
their lateral registry are observed with respect to results of Atomistic and Molecular
Dynamic studies, in general, there is a good agreement with earlier DFT calculations,
especially with those corrected to the BSSE. The extent of surface reconstruction upon
hydration is more important than previously suggested. This includes bond stretching and
the differential 3D displacement of surface atoms which results in surface relaxation (5.6
%) and a decrease in the intrinsic surface rumpling (2.4 %) following the rotation of CO3
groups towards the surface.

215
The stabilizing effect of associatively adsorbed water on the (10.4) calcite surface,
previously postulated by Atomistic and DFT studies, was confirmed at the RHF/6-
31G(d,p) level of theory. The formation of two ordered hydration layers is
thermodynamically favourable where each H2O in the 1st hydration layer is bonded to a
single surface Ca by ionic bonding and to a surface O by a hydrogen bond. There is
therefore “chemisorption” of H2O to the calcite surface, as shown by experiment.
According to geometric criteria, the strong H2O-surface interaction disrupts the hydrogen
bond network of H2O in the bulk solution which prevents hydrogen bonding between
adjacent H2O in the 1st
layer. H2O in the 2nd
layer hydrogen bonds with a surface O and
with adjacent H2O in the 1st layer, reflecting a weaker interaction with the surface relative
to H2O in the 1st layer. This interaction is interpreted as H2O “physisorption”, in
agreement with earlier experimental data.
Most noteworthy is the role that H2O plays in the reorganization of Ca-O bonds at
the calcite surface. In agreement with X-ray Scattering and GIXRD results, surface Ca-O
octahedra undergo substantial distortion upon H2O binding to Ca, but in contrast to earlier
suggestions, the six-fold coordination shell of surface Ca atoms is probably not restored
because of the relaxation of surface atoms. This significantly weakens at least one Ca-O
bond per surface Ca atom and possibly leads to bond rupture. Alternate methods such as
EXAFS techniques must be applied in future studies to determine the CN of surface Ca
atoms and confidently ascertain whether the SWBs at the hydrated (10.4) calcite surface
reflect bond breaking or not. We conclude that, to stabilize the hydrated surface, H2O
may provisionally dissolve surface Ca preferentially over CO3 groups. This observation is
critical in the understanding of molecular mechanisms of mineral dissolution,
rearrangement of surface layers, ion replacement, charge development and solute
transport through subsurface lattice layers.

216
6. ACKNOWLEDGMENTS
A.V.-J. thanks Prof. Theo G. M. van de Ven for critical discussions at earlier stages of
this investigation as well as Dr. Nora de Leeuw, Dr. Kate Wright and Dr Paul Fenter
for providing additional information on their results. The constructive comments
provided by two anonymous reviewers have substantially improved the quality of our
work. This research was supported by a student grant to A.V.-J. from the Geological
Society of America (GSA) and by the Natural Sciences and Engineering Research
Council of Canada (NSERC) Discovery through Discovery grants to M.A.W. and
A.M. A.V.-J. also received post-graduate scholarships from Consejo Nacional de
Ciencia y Tecnología of Mexico and benefited from additional financial support from
Consorcio Mexicano Flotus-Nanuk, the Department of Earth and Planetary Sciences
of McGill University and the GEOTOP-McGill-UQAM Research Centre.

217
7. REFERENCES
Ahsan T. (1992) The surface properties of pure and modified precipitated calcium
carbonate by adsorption of nitrogen and water vapor. Colloids Surf. 64, 167.
Altermatt D. and Brown I.D. (1985) The automatic searching for chemical bonds in
inorganic crystal structures. Acta Cryst. B41, 240-244.
Archer T.D., Computer Simulations of Calcite. Ph. D. Thesis, University of Cambridge,
2004.
Bickmore B.R., Tadanier C.J., Rosso K.M., Monn W.D. and Egget D.L. (2004). Bond-
Valence methods for pKa prediction: critical reanalysis and a new approach.
Geochim. Cosmochim. Acta. 68(9), 2025-2042.
Brown I.D. and Shannon R.D. (1973) Empirical bond-strength-bond-length curves for
oxides. Acta Cryst. A29(3), 266-282.
Brown I.D. (1981) In Structure and Bonding in Crystals.; Vol. 2, O'Keeffe, M.,
Navrotsky, A. Eds.; Academic Press: New York USA,1981; pp. 1-30.
Burdett J.K. and Hawthorne F.C. (1993) An orbital approach to the theory of bond
valence. Am. Mineral. 78, 884-892.
Cao Y. and Chen Z.-X. (2006) Theoretical studies on the adsorption and decomposition
of H2O on Pd(111) surface. Surf. Sci. 600(19), 4572-4583.
Chiarello R.P., Wogelius R.A. and Sturchio N. (1993) In-situ synchrotron X-ray
reflectivity measurements at the calcite-water interface. Geochim. Cosmochim.
Acta 57(16), 4103-4110.

218
Chowdhuri S. and Chandra A. (2002) Hydrogen bonds in aqueous electrolyte solutions:
statistics and dynamics based on both geometric and energetic criteria. Physical
Rev. E. 66, 041203-1-7.
Davis J.A., Fuller C.C. and Cook A.D. (1987) A model for trace metal sorption processes
at the calcite surface: Adsorption of Cd2+
and subsequent solid solution formation.
Geochim. Cosmochim. Acta 51(6), 1477-1490.
deFrees D.J. and McLean A.D. (1985) Molecular orbital predictions of the vibrational
frequencies of some molecular ions J. Chem. Phys., 82(1), 333-341.
de Leeuw N.H. and Parker S.C. (1997) Atomistic simulation of the effect of molecular
adsorption of water on the surface structure and energies of calcite surfaces. J.
Chem. Soc.,Faraday Trans. 93(3), 467-475.
de Leeuw N.H. and Parker S.C. (1998) Surface structure and morphology of calcium
carbonate polymorphs calcite, aragonite, and vaterite: an atomistic approach. J.
Phys. Chem. B. 102, 2914-2922.
de Leeuw N.H. and Parker S.C. (2000) Modeling absorption and segregation of
magnesium and cadmium ions to calcite surfaces: Introducing MgCO3 and CdCO3
potential models. J. Chem. Phys. 112(9), 4326.
de Leeuw N.H. and Parker S.C. (2002) Surface structures, stabilities, and growth of
magnesian calcites: a computational investigation from the perspective of
dolomite formation. Am. Mineral. 87, 679-689.
de Leeuw N.H. and Cooper T.J. (2003) The layering effect of water on the structure of
scheelite. Phys. Chem. Chem. Phys., 5, 433-436.

219
de Leeuw N.H., Parker S.C., and Hanumantha Rao K. (1998) Modeling the competitive
adsorption of water and methanoic acid on calcite and fluorite surfaces. Langmuir
14, 5900-5906.
Douglas H.W. and Walker R.A. (1950) The electrokinetic behavior of iceland spar
against aqueous electrolyte solution. Trans. Faraday Soc. 46, 559-568.
Emri J. (2003) Using a novel relationship between bond length and bond order to
calculate accurate bond orders for carbon-carbon bonds. J. Mol. Struct.
(Theochem), 620, 283-290.
Fenter P., Geissbühler P., DiMasi E, Srajer G, Sorensen B and Sturchio N.C. (2000)
Surface speciation of calcite observed in situ by high-resolution X-ray reflectivity.
Geochim. Cosmochim. Acta 64(7), 1221-1228.
Fenter P. and Sturchio N.C. (2004) Mineral-water interfacial structures revealed by
synchrotron X-ray scattering . Progress Surf. Sci. 77, 171–258.
Ferrario M., Haughney M., McDonald I.R. and Klein M. (1990) Molecular-dynamics
simulation of aqueous mixtures: Methanol, acetone, and ammonia. J. Chem. Phys.
93(7), 5156-5166.
Frisch, M. J. ; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman,
J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, R. E.; Burant, J. C.;
Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas,
O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.;
Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.;
Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.;
Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko,
A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.;
Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.;

220
Gill, P. M. W.; Johnson, B. G.; Chen, W. M.; Wong, W.; Andres, J. L.; Head-
Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 03W, Revision B.02,
Gaussian, Inc.: Pittsburgh, PA, 2003.
Geissbühler P, Fenter P, DiMasi E., Sorensen, L.B. and Sturchio N.C. (2004) Three-
dimensional structure of the calcite–water interface by surface X-ray scattering.
Surf. Sci. 573, 191-203.
Graf D.L. (1961) Crystallographic tables for the rhombohedral carbonates. Am. Mineral.
46, 1283-1316.
Hirva P. and Tikka H.-K. (2002) Ab initio study on the interaction of anionic collectors
with calcite and dolomite surfaces. Langmuir 8, 5002-5006.
Hwang S., Blanco M. and Goddard W.A. III (2001) Atomistic simulations of corrosion
inhibitors adsorbed on calcite surfaces I. Force field parameters for calcite. J.
Phys. Chem. B. 105, 10746-10752.
Ireta J., Neugebauer J. and Scheffler M. (2004) On the accuracy of DFT for describing
hydrogen bonds: dependence on the bond directionality. J. Phys. Chem. A,
108(26), 5692-5698.
Israelachvili J. (1992) Intermolecular and Surface Forces, Academic Press, 2nd
Edition,
San Diego CA, 450 p.
Kerisit S. and Parker S.C. (2004) Free energy of adsorption of water and metal ions on
the (10.4) calcite surface. J. Am. Chem. Soc. 126, 10152-10161.
Kerisit S., Parker S.C. and Harding J. H. (2003) Atomistic simulation of the dissociative
adsorption of water on calcite surfaces J. Phys. Chem. B. 107, 7676-7682.

221
Kerisit S., Cooke D.J., Spagnoli D. and Parker S.C. (2005a) Molecular dynamics
simulations of the interactions between water and inorganic solids. J. Mater.
Chem. 15, 1454-1462.
Kerisit S., Marmier A., Parker S.C. (2005b) Ab initio surface phase diagram of the (10.4)
calcite surface. J. Phys. Chem. B 109:39, 18211-18213.
Koretsky C.M., Sverjensky D.A. and Sahai N. (1998) A model of surface sites types on
oxide and silicate minerals based on crystal chemistry: Implications for site types
and densities, multi-site adsorption, surface infrared spectroscopy and dissolution
kinetics. Am. J. Sci. 298, 349-438.
Kubicki J.D. and Bleam W.F. (2003) Molecular Modeling of Clays and Mineral Surfaces,
The Clay Mineral Society CMS Workshop Lectures, Vol 12, 2003, pp 229.
Kuriyavar S.I., Vetrivel R., Hegde S.G., Ramaswamy A.V., Chakrabarty D. and
Mahapatra S. (2000) Insights into the formation of hydroxyl ions in calcium
carbonate: temperature dependent FTIR and molecular modeling studies. J. Mater.
Chem. 10, 1835-1840.
Liang Y., Lea A.S., Baer D.R. and Engelhard M.H. (1996) Structure of the cleaved
CaCO3 (104) surface in an aqueous environment. Surface Sci. 351, 172-182.
Luzar A. and Chandler D. (1993) Structure and hydrogen bond dynamics of water–
dimethyl sulfoxide mixtures by computer simulations. J. Chem. Phys., 98(10),
8160-8173.
Magdans U., Gies H., Torrelles X. and Rius J. (2006) Investigation of the {104} surface
of calcite under dry and humid atmospheric conditions with grazing incidence X-
ray diffraction (GIXRD). Eur. J. Mineral. 18, 83-92.

222
Maheshwary S., Patel N., Sathyamurthy N., Kulkarni A.D. and Gadre S.R. (2001)
Structure and stability of water clusters (H2O)n, n=8-20: An ab initio investigation.
J. Phys. Chem. A, 105:46, 10525-10537.
Mao Y. and Siders P.D. (1997) Molecular Hartree–Fock model of calcium carbonate.
Journal of Mol. Struct. (Theochem) 419, 173-184.
Markmann A., Gavartin J.L. and Shluger A. L. (2006) Chemisorption of HCl to the
MgO(001) surface: A DFT study. Phys. Chem. Chem. Phys. 8, 4359.
Mezei M. and Beverdige D.L. (1981) Theoretical studies of hydrogen bonding in liquid
water and dilute aqueous solutions. J. Chem. Phys. 74(1), 622-632.
Möller P. and Sastri C. S. (1974) Estimation of the number of surface layers of calcite
involved in Ca45
Ca istotopic exchange with solution. Zeit. Physik. Chem. Neue.
Folg. 89, 80-87.
Morimoto T., Kishi J., Okada O. and Kadota T. (1980) Interaction of water with the
surface of calcite. Bull. Chem. Soc. Jpn. 53(7), 1918-1921.
Morse J.W. and Mackenzie F.T. (1990) Geochemistry of Sedimentary Carbonates,
Develop. in Sedimentol. 48. Elsevier Science Pubs., Amsterdam, pp 707.
Neagle W. and Rochester C.H. (1990) Infrared study of the adsorption of water and
ammonia on calcium carbonate. J. Chem. Soc., Faraday Trans. 86(1), 181-183.
Parker S.C., private communications in Geissbühler et al., 2004.
Parker S.C., Kerisit S., Marmier A., Grigoleit S. and Watson G.W. (2003) Modeling
inorganic solids and their interfaces: A combined approach of atomistic and
electronic structure simulation techniques. Faraday Discuss. 124, 155–170.

223
Pauling L.J. (1929) The principles determining the structures of complex ionic crystals. J.
Am. Chem. Soc. 51, 1010-1026.
Pokrovsky O.S., Mielczarski J.A., Barres O., and Schott J. (2000) Surface speciation
models of calcite and dolomite/aqueous solution interfaces and their spectroscopic
investigation. Langmuir 16, 2677-2688.
Rohl A.L., Wright K. and Gale J.D. (2003) Evidence for the (2x1) reconstruction of the
(104) surface of calcite from computer simulation. Am. Mineral. 88, 921-925.
Rosso K.M. (2001) in: Molecular Modeling Theory: Application in the Geosciences, (ed.
R. T. Cygan and J. D. Kubicki), Mineralogical Society of America, Rev. Mineral.
Geochem. Vol 42, Washington, DC., 2001, pp 199.
Rozmanov D.A., Sizova O.S., Burkov K.A. (2004) Ab initio studies of the beryllium
aquahydrocomplexes. J. Mol. Struct. Theochem. 712(1), 123-130.
Ruuska H., Hirva P. and Pakkanen T.A. (1999) Cluster models for calcite surfaces: Ab
initio quantum chemical studies. J. Phys. Chem. B 1999, 103(32), 6734-6740.
Saebø S., Tong W. and Pulay P. (1993) Efficient elimination of basis set superposition
errors by the local correlation method: Accurate ab initio studies of the water
dimer. J. Chem. Phys., 98:3, 2170-2175.
Santra B., Michaelides A., Fuchs M., Tkatchenko A., Filippi C. and Scheffler M. (2007)
On the accuracy of density-functional theory exchange-correlation functionals for
H bonds in small water clusters: Benchmarks approaching the complete basis set
limit. J. Phys. Chem. 127,184104.
Santra B., Michaelides A., Fuchs M., Tkatchenko A., Filippi C. and Scheffler M. (2008)
On the accuracy of density-functional theory exchange-correlation functionals for

224
H bonds in small water clusters. II. The water hexamer and van der Waals
interactions. J. Phys. Chem. 129, 194111.
Scott A.P. and Radom L. (1996) Harmonic vibrational frequencies – An evaluation of
Hartree-Fock, Moller-Plesset, Quadratic configuration interaction, Density
functional theory, and Semiempirical scale factors. J. Phys. Chem. 100, 16502-
16513.
Sjöberg E.L. (1978) Kinetics and mechanism of calcite dissolution in aqueous solutions at
low temperatures. Stockholm Contrib. Geol., 1978, 32(1), 1-92.
Skinner A.J., LaFemina J.P. and Jansen H.J.F. (1994) Structure and bonding of calcite: A
theoretical study. Am. Min. 1994, 79, 205-214.
Sposito G. (1990) in: Mineral-Water Interface Geochemistry, ed. M F. Hochella Jr. and
A. F. White, Mineralogical Society of America, Rev. Mineral. Vol 23, 261 p.
Stefanovich E.V. and Truong T.N. (1997) A theoretical approach for modeling reactivity
at solid–liquid interfaces. J.Chem. Phys. 106, 7700.
Stipp S.L. (1999) Toward a conceptual model the calcite surface: hydration, hydrolysis,
and surface potential. Geochim. Cosmochim. Acta 63(19/20), 3121-3131.
Stipp S.L. and Hochella M.F. Jr. (1991) Structure and bonding environments at the calcite
surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy
electron diffraction (LEED). Geochim. Cosmochim. Acta 55, 1723-1736.
Stipp S.L.S., Eggleston C.M. and Nielsen B.S. (1994) Calcite surface structure observed
at microtopographic and molecular scales with atomic force microscopy (AFM).
Geochim. Cosmochim. Acta 58(14), 3023-3033.

225
Stöckelmann, E.; Hentschke, R. (1999) Adsorption isotherms of water vapor on calcite :
A molecular dynamics-Monte Carlo hybrid simulation using a polarizable water
model. Langmuir, 15, 5141-5149.
Suba S. and Whitehead M.A. In Recent Advances in Computational Chemistry- Vol. 1,
Recent Advances in Density Functional Methods Part 1, Chong, D. P. Ed.; World
Scientific: New Jersey, USA, 1995; pp 53-78.
Thackeray D. and Siders P.D. (1998) Molecular-orbital and empirical-potential
descriptions of CaCO3. J. Chem. Soc., Faraday Trans. 94, 2653-2661.
Tossel J.A. and Vaughan D.J. (1992) Theoretical Geochemistry: Applications of Quantum
Mechanics in the Earth and Mineral Sciences, Oxford University Press, New
York, pp 514.
Usher C.R., Michel A.E. and Grassian V. H. (2003) Reactions on mineral dust. Chem.
Rev. 103, 4883-4939.
Van Cappellen P., Charlet L., Stumm W. and Wersin P. (1993). A surface complexation
model of the carbonate mineral-aqueous solution interface. Geochim. Cosmochim.
Acta 57, 3505-3518.
Vanerek A., Alince B. and van de Ven T.G.M. (2000). Interaction of calcium carbonate
fillers with pulp fibres: Effect of surface charge and cationic polyelectrolytes. J.
Pulp Paper Sci. 26:9, 317-322.
Villegas-Jiménez A., Mucci A., Pokrovsky O.S. and Schott J. (2009a) Defining reactive
sites at hydrated mineral surfaces: Rhombohedral carbonate minerals. Geochim.
Cosmochim. Acta 73(15), 4326-4345.

226
Villegas-Jiménez A., Mucci, A., Paquette, J. (2009b) Proton/calcium ion exchange
behavior of calcite. Phys. Chem. Chem. Phys 39(11), 8895-8912.
Wang X. and Liebau F. (2009) On the optimization of bond-valence parameters: Artifacts
conceal chemical effects. Acta Cryst. Struct. Sci. B65, 96-98.
Whitehead M.A., van de Ven T.G.M. and Malardier-Jugroot C. (2004) Study of the water
conformation around hydrophilic and hydrophobic parts of stryene-maleic
anhydride J. Mol. Struct. (Theochem) 679, 171-177.
Wright K., Cygan R.T. and Slater B. (2001) Structure of the (10.4) surfaces of calcite,
dolomite and magnesite under wet and dry conditions. Phys. Chem. Chem. Phys.,
3, 839-844.
Xiao Y. and Lasaga A.C. (1994) Ab initio quantum mechanical studies of the kinetics and
mechanism of silicate dissolution: H+(H3O
+) catalysis. Geochim. Cosmochim.
Acta, 58(24), 5379-5400.
Yu D., Xue D. and Ratajczak H. (2006) Bond-valence parameters for characterizing O–
H O hydrogen bonds in hydrated borates. J. Mol. Struct. 792-793, 280-285.
Zachara J.M., Cowan C.E. and Resch C.T. (1991) Sorption of divalent metals on calcite.
Geochim. Cosmochim. Acta, 55, 1549-1562.
Zhou Z., Shi Y. and Zhou X. (2004) Theoretical studies on the hydrogen bonding
interaction of complexes of formic acid with water. J. Phys. Chem. A, 108(5),
813-822.
Zocchi F. (2007) Accurate bond valence parameters for lanthanide-oxygen bonds. J. Mol.
Struct. (Theochem) 805, 73-78.
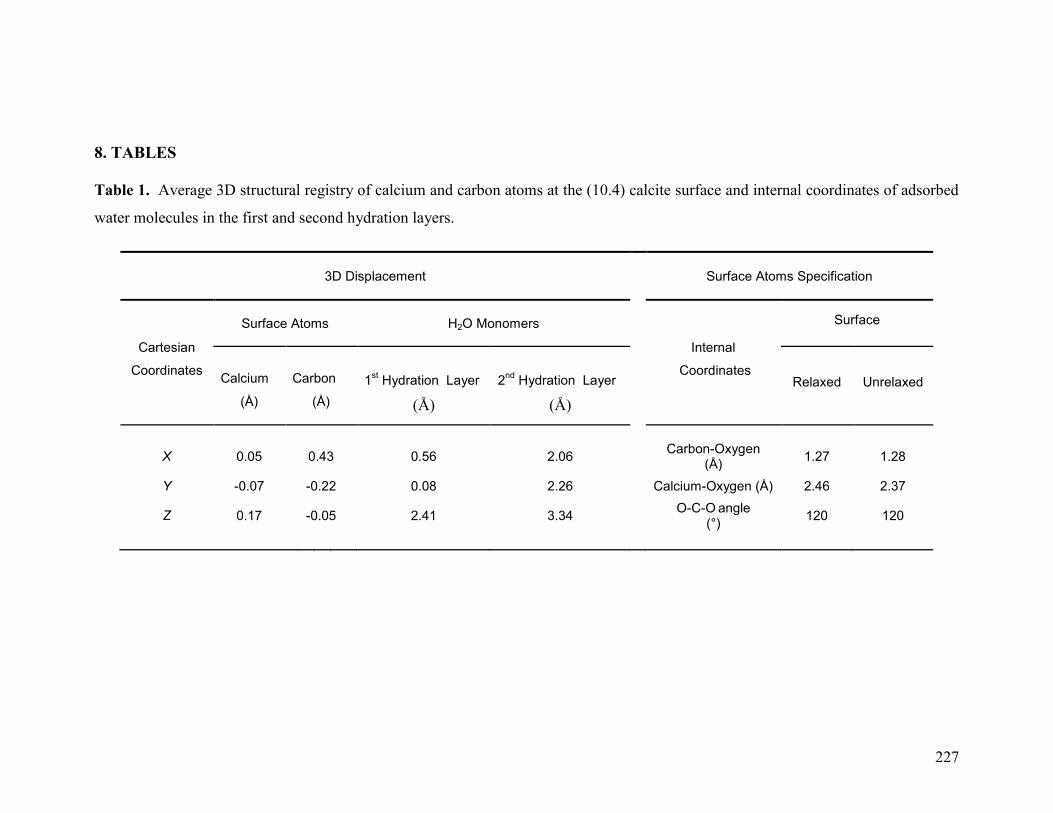
227
8. TABLES
Table 1. Average 3D structural registry of calcium and carbon atoms at the (10.4) calcite surface and internal coordinates of adsorbed
water molecules in the first and second hydration layers.
3D Displacement
Surface Atoms Specification
Cartesian
Coordinates
Surface Atoms H2O Monomers
Internal
Coordinates
Surface
Calcium
(Å)
Carbon
(Å)
1st Hydration Layer
(Å)
2nd
Hydration Layer
(Å)
Relaxed Unrelaxed
X 0.05 0.43 0.56 2.06 Carbon-Oxygen
(Å) 1.27 1.28
Y -0.07 -0.22 0.08 2.26 Calcium-Oxygen (Å) 2.46 2.37
Z 0.17 -0.05 2.41 3.34 O-C-O angle
(°) 120 120

228
Table 2. Average Relaxed Ca-O Bond Lengths per Hydrated Surface Ca-O Octahedron
Bond ID
GIXRD*
This Study
Ca-O(1) 2.1 2.32
Ca-O(2) 2.2 2.36
Ca-O(3) 2.55 2.46
Ca-O(4) 2.6 2.53
Ca-O(5) 3 2.63
Because Ca-O bond interactions are not explicitly identified by earlier authors (Magdans et al.,
2006) Ca-O bond lengths of both studies are tabulated in ascending order. Sub-indexes are
arbitrary identification labels.

229
Table 3. Geometric coordinates confirming the formation of Hydrogen Bonds
between H2O molecules and calcite surface atoms as well as among adjacent H2O
molecules.
Type of interaction
Internal Coordinatesa
ROH ( Å )
ROO
( Å )
H (°)
H2O(1st-Layer) - Surface Oxygen 2.01 2.82 26.4
H2O(2nd-Layer) - Surface Oxygen 1.78 2.75 3.9
H2O(1st-Layer) - H2O(2nd-Layer) 2.04 2.9 18

230
9. FIGURES
Figure 1. Plan view of the constituents of the (CaCO3)9/2H2O cluster. Ovals highlight
“reactive” surface atoms that were allowed to relax in the optimizations in addition to
H2O monomers. Subsurface atoms are represented by shaded areas. Dashed line defines
the surface unit cell. x-y axes are arbitrary and are consistent with those selected in earlier
studies (Geissbühler, 2004).
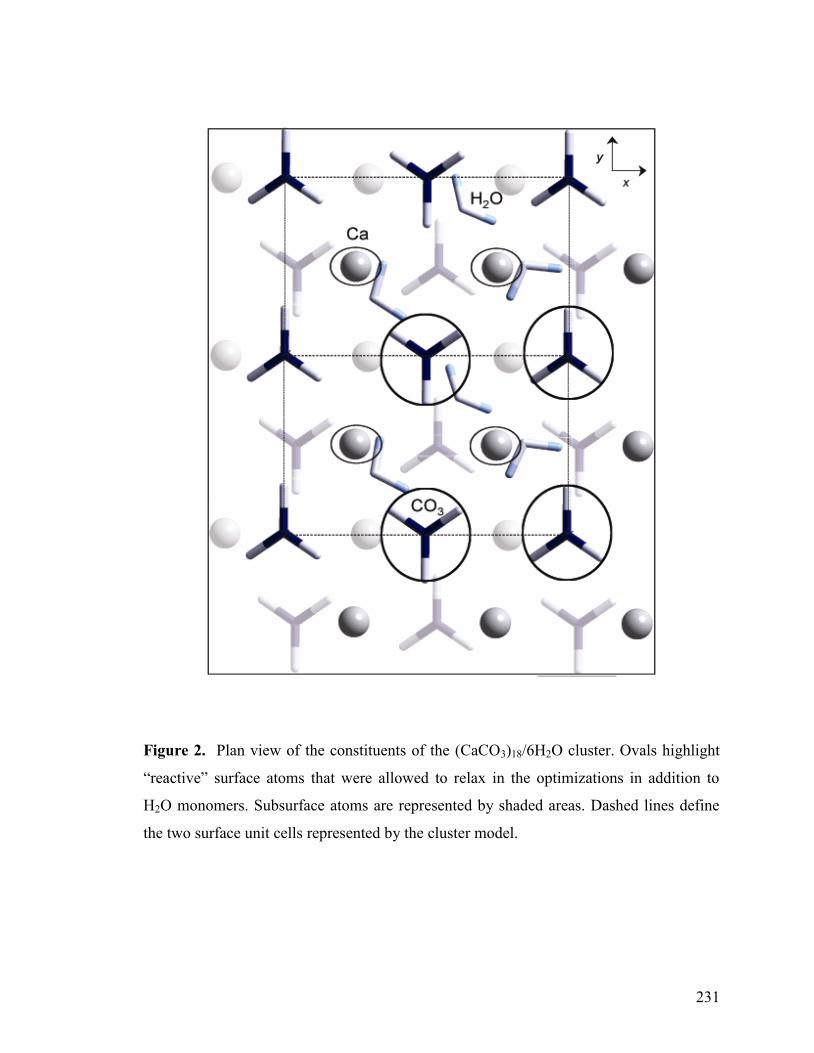
231
Figure 2. Plan view of the constituents of the (CaCO3)18/6H2O cluster. Ovals highlight
“reactive” surface atoms that were allowed to relax in the optimizations in addition to
H2O monomers. Subsurface atoms are represented by shaded areas. Dashed lines define
the two surface unit cells represented by the cluster model.

232
Figure 3. Snapshot of the Ca12(CO3)12/2H2O cluster model. Ovals highlight reactive
surface and subsurface atoms that were not frozen in the optimizations in addition to H2O
monomers. Two subatomic layers below the (10.4) surface are displayed. The
approximate rhombohedral morphology of the cluster is defined by the dashed lines.
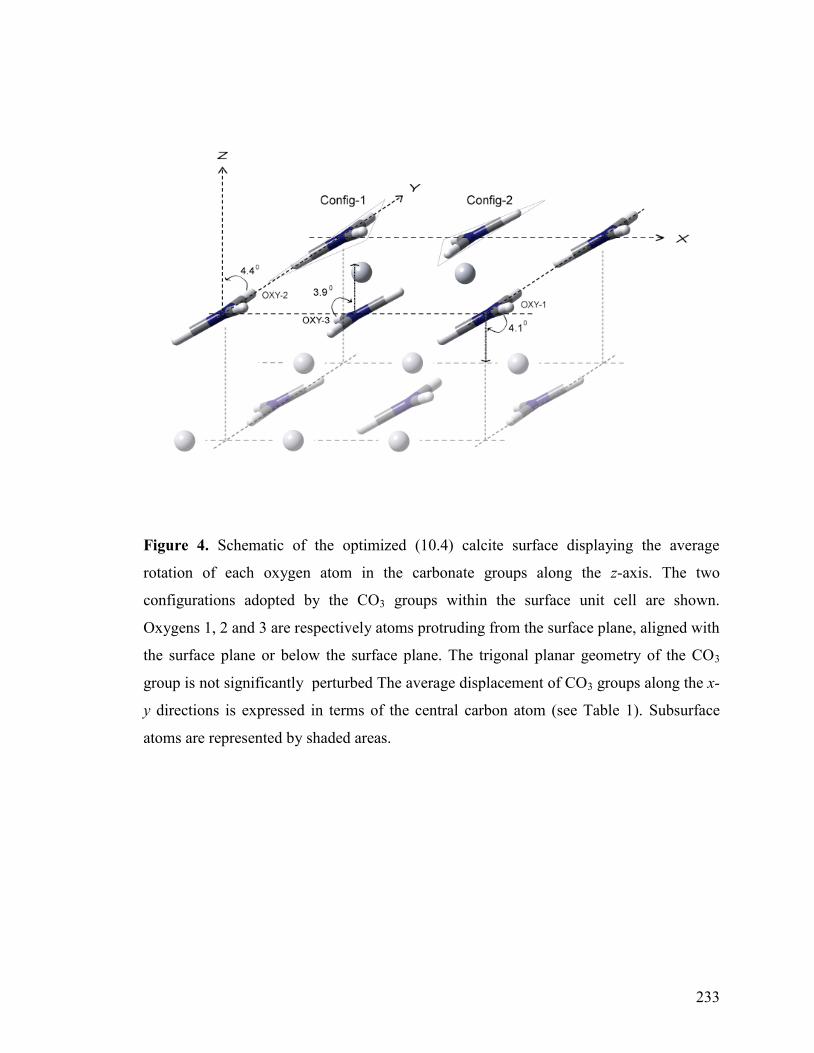
233
Figure 4. Schematic of the optimized (10.4) calcite surface displaying the average
rotation of each oxygen atom in the carbonate groups along the z-axis. The two
configurations adopted by the CO3 groups within the surface unit cell are shown.
Oxygens 1, 2 and 3 are respectively atoms protruding from the surface plane, aligned with
the surface plane or below the surface plane. The trigonal planar geometry of the CO3
group is not significantly perturbed The average displacement of CO3 groups along the x-
y directions is expressed in terms of the central carbon atom (see Table 1). Subsurface
atoms are represented by shaded areas.
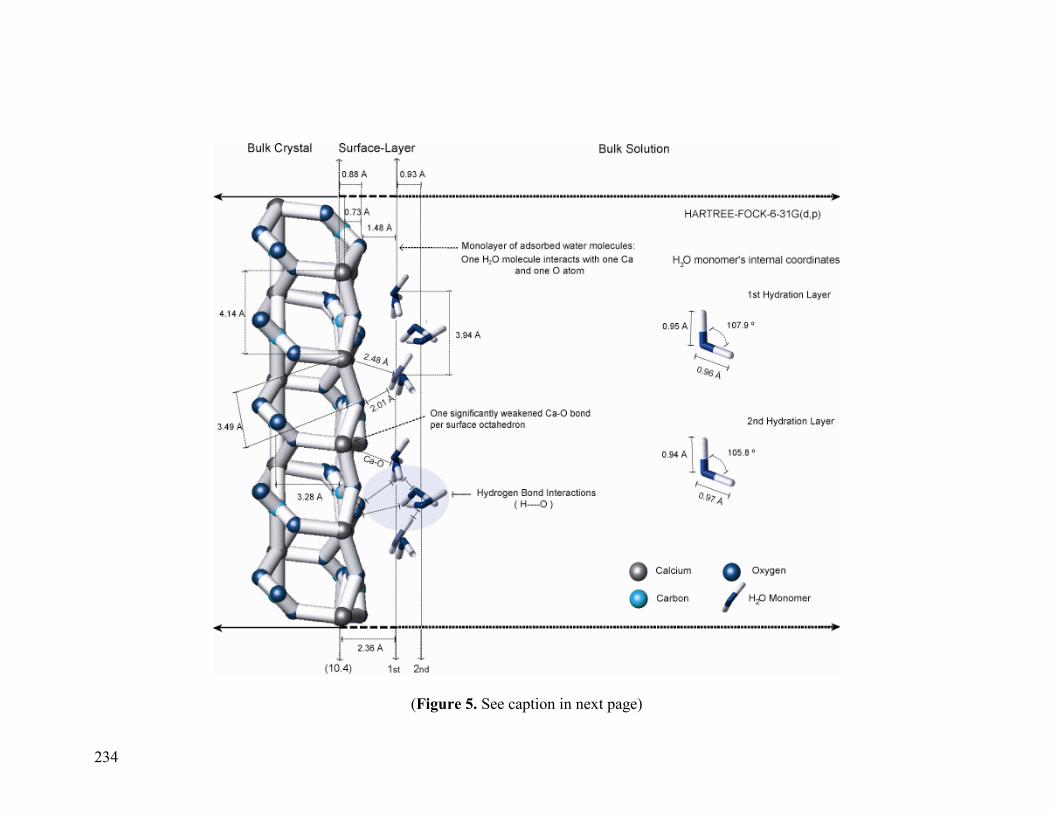
234
(Figure 5. See caption in next page)

235
Figure 5. Lateral view of the (10.4) reconstructed calcite surface as predicted at the HF-6-31G(d.p) level of theory. Structural details
of the first and second hydration layers are given. The average internal coordinates of the adsorbed water monomers are specified. Ca-
O and Hydrogen-bond interactions are shown.
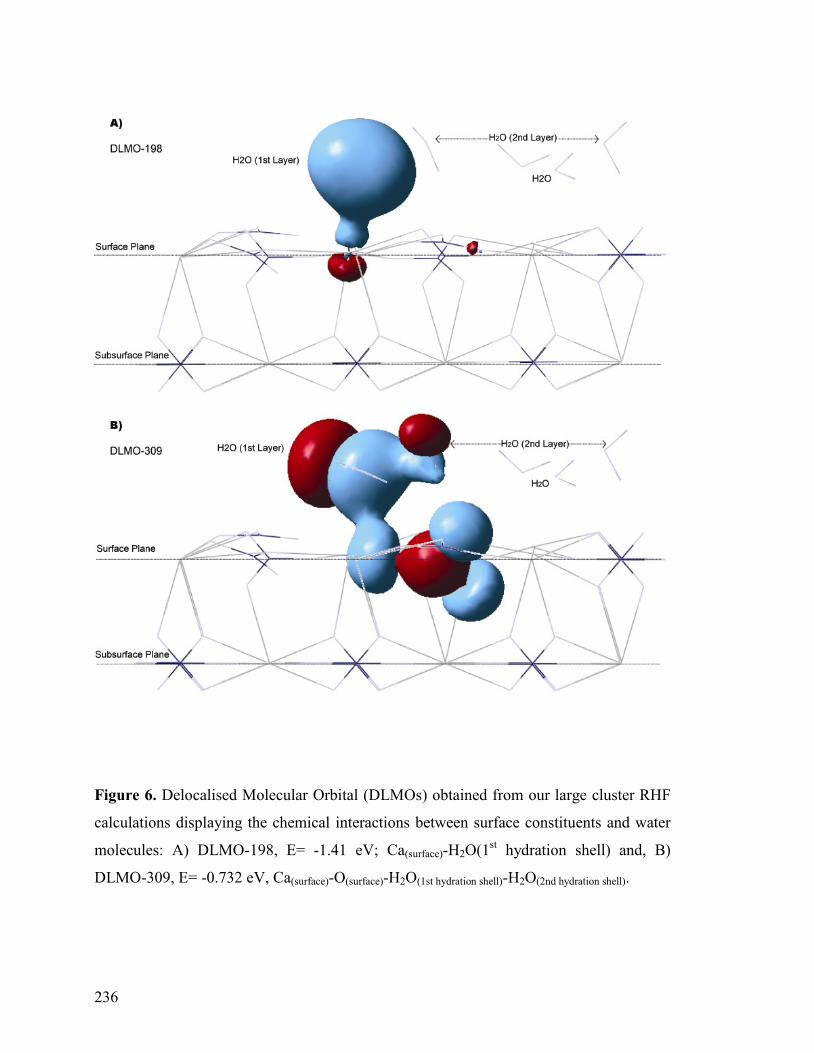
236
Figure 6. Delocalised Molecular Orbital (DLMOs) obtained from our large cluster RHF
calculations displaying the chemical interactions between surface constituents and water
molecules: A) DLMO-198, E= -1.41 eV; Ca(surface)-H2O(1st hydration shell) and, B)
DLMO-309, E= -0.732 eV, Ca(surface)-O(surface)-H2O(1st hydration shell)-H2O(2nd hydration shell).

237
Figure 7. Geometric definition of the hydrogen bond established between H2O molecules
and surface atoms as well as among adjacent H2O molecules. The optimised orientations
of H2O molecules in the 1st and 2nd layer are slightly modified for clarity. Subsurface
atoms.

238
PREFACE TO CHAPTER 6
Despite the usefulness of molecular modeling techniques in the investigation of the
structural and energetic properties of the calcite surface, experimental data are required to
validate these results and further improve our understanding of the surface properties of
this mineral in aqueous solutions. Given the lack of suitable experimental protocols, the
quantitative characterization of some of the more fundamental sorptive properties of
calcite (e.g., proton, calcium) are still poorly known because sorption reactions at
carbonate surfaces are typically: i) characterized over relatively narrow ranges of
chemical composition (e.g., batch adsorption experiments, thermogravimetry,
chromatography), ii) qualitatively evaluated with surface sensitive instrumental
techniques (e.g., X-ray, Electron Diffraction, Spectroscopy), or iii) semi-quantitatively
inferred from electrokinetic studies. It follows that the acquisition of adsorption data over
expanded compositional ranges is a critical step for probing of sorption phenomena at the
calcite surface. In the following chapter, “Proton/Calcium Ion Exchange Behavior of
Calcite”, we address this issue by introducing a novel surface titration protocol that
allows, for the first time, the rigorous quantitative characterization of the proton sorptive
properties of calcite in aqueous solutions over a relatively wide range of chemical
conditions. In contrast to other rhombohedral carbonate minerals (magnesite, dolomite,
gaspeite) whose proton sorptive properties could be rationalized in terms of surface
complexation reactions, our calcite data led us to postulate on the existence of a
proton/calcium ion exchange reaction involving subsurface calcite layers. This reaction,
never postulated before, significantly impacts the aqueous speciation of closed carbonate-
rock systems and open environments with poor CO2 ventilation, via pH and calcite
dissolution buffering and CO2(g) sequestration upon ion exchange-induced calcite
precipitation.

239
CHAPTER 6
PROTON/CALCIUM ION EXCHANGE BEHAVIOR
OF CALCITE
Adrián Villegas-Jiménez*1, Alfonso Mucci
1 and Jeanne Paquette
1
1 Earth and Planetary Sciences, McGill University, 3450 University Street
Montréal, Qc H3A 2A7, Canada.
*Corresponding Author
E-mail: [email protected]
“Reproduced by permission of the PCCP Owner Societies: Phys. Chem. Chem. Phys. 39(11) 8895-8912”
http://dx.doi.org./10.1039/B815198A

240
ABSTRACT
The characterization of the proton sorptive properties of calcite in aqueous solutions at 25
± 1ºC over a relatively wide range of chemical conditions (7.16 ≤ pH ≤ 9.7; 410-5
M ≤
Ca ≤ 5.210-3
M; 1.310-4
M ≤ CO2 ≤ 1.810-2
M) and solid:solution ratios (0.4 to 12.3 g
L-1
) was performed using a novel surface titration technique. A large net proton uptake,
coupled with a significant release of Ca2+
ions is consistently observed, greatly exceeding
the theoretical number of reactive surface sites. These observations are interpreted as a
fast proton/calcium exchange equilibrium between the solution and “exchangeable cation
sites” (e.g., lattice positions) at and/or beneath the calcite surface:
(CaCO3)2(exc) + 2 H+
Ca(HCO3)2(exc) + Ca2+
that leads to a transient, “apparent” incongruent dissolution regime and the formation of a
stable calcium-deficient proton-enriched calcite layer under circum-neutral and alkaline
conditions. The 2H+/Ca
2+ ion exchange is quantitatively described by the Langmuir-
power exchange function under the Vanselow convention:
2
2
)CaCO(
)HCO(Ca
)(
)(
X
X
2(exc)3
(exc)23
aH
aCaK
n
ex
where n=1 and log10 Kex = 13.0 ± 0.3. This calcite behavior, never reported before, masks
surface equilibria and directly impacts the aqueous speciation of carbonate-rock systems
with poor CO2 ventilation (e.g., aquifers, pore and deep sea waters, industrial reactors) via

241
the buffering of pH and calcite dissolution. In contrast, at fixed pCO2 conditions, aqueous
speciation remains unaffected upon CO2(g) sequestration resulting from ion exchange-
induced calcite precipitation:
(CaCO3)2(exc) + CO2(g)+ H2O Ca(HCO3)2(exc) + CaCO3(s)
Accordingly, reliable predictions of aqueous speciation in natural or engineered calcite-
containing systems at variable CO2(g) conditions must consider this exchange reaction and
associated Kex. The postulated proton/calcium exchange may have far-reaching
implications on the interpretation of kinetic and equilibrium data, and can partly explain
the anomalous solution chemistry observed in some field and laboratory carbonate
studies.
Keywords: Calcite titrations, proton sorptive properties, bicarbonate lattice species,
“apparent” incongruent calcite dissolution, calcium-deficient proton-enriched leached
layer.

242
1. INTRODUCTION
The most stable calcium carbonate polymorphs (calcite and aragonite) are highly reactive
minerals and ubiquitous in the environment. They are found: in aquatic systems as
suspended particles and carbonate-rich sediments whose occurrence range from tropical
environments (Morse and MacKenzie, 1990) to glacial settings (McGillen and Fairchild,
2005), in biological systems as the building blocks of shells and skeletons (MacKenzie et
al., 1990), and in the Earth‟s troposphere as mineral aerosols (Usher et al., 2003). They
also have numerous engineered applications, from fillers for paints, plastics, rubbers,
pharmaceuticals, cosmetics, optical devices, and paper (Vanerek et al., 2000) to raw
material in the construction industry, agriculture, as well as in the production of
biomedical scaffolds (Tas, 2007). In aqueous systems, they largely impact the solution
chemistry by regulating pH and alkalinity through their dissolution and precipitation, and
govern the mobility and bioavailability of trace and major elements via ion exchange
(Zachara et al., 1991) and sorption reactions (Martin-Garin, 2003). In the atmosphere,
these minerals regulate the CO2 exchange (Robbins and Fabry, 1994) and influence the
chemistry of volatile inorganic and organic acids (Usher et al., 2003; Al-Hosney and
Grassian, 2005).
Despite their environmental significance and broad industrial applications, critical
aspects of the reactivity of these minerals in aqueous solutions are still not fully
understood. Bulk phase CaCO3(s)-H2O equilibria are fairly well characterized
thermodynamically (Morse and MacKenzie, 1990) but surface properties, including the
pH of the isoelectric point (pHIEP) of calcite (Douglas and Walker, 1950; Prédali and
Cases, 1973; Mishra, 1978; Foxall et al., 1979; Siffert and Fimbel, 1984; Thompson and
Pownall, 1989; Huang et al., 1991; Cicerone et al., 1992; Moulin and Roques, 2003) and

243
the sorption behavior of potential-determining ions such as hydrolysis products, H+, OH
-
(Prédali and Cases, 1973; Mishra, 1978; Foxall et al., 1979), and lattice-derived
(constituent) ions, Ca2+
, CO32-
and HCO3- (Douglas and Walker, 1950; Siffert and Fimbel,
1984; Thompson and Pownall, 1989; Huang et al., 1991; Cicerone et al., 1992; Moulin
and Roques, 2003), that reflect on the macroscopic behavior of these minerals (e.g.,
coagulation, dissolution, etc.), remain poorly described. This is because proton and
constituent ion sorption equilibria are often difficult to resolve experimentally from the
dissolution/precipitation reactions that rapidly respond to minute variations in the solution
chemistry. This is particularly problematic when the experimental solution can exchange
CO2 with a gas phase, impacting the proton and carbon balance in solution to an unknown
extent, and hence, prohibiting the use of conventional titration techniques commonly
applied to the characterization of the ion sorptive properties of less reactive minerals such
as metal oxides (Huang, 1981) and sparingly reactive carbonates (Charlet et al., 1990).
The first attempt to quantitatively describe the surface reactivity of calcite in
aqueous solutions in terms of acid-base and constituent ion adsorption reactions was
provided by Van Cappellen and coworkers (1993) in the form of a surface complexation
model (SCM). In that study, formation constants of surface species were adjusted
manually until the predicted surface speciation closely reproduced the pHIEP (8.2)
recorded by electrokinetic measurements under specific solution conditions (Mishra,
1978). This procedure is useful but not rigorous because of the limited amount of
experimental data used in model calibration (a single data point: the pHIEP) which renders
the system an undetermined one: the number of adjustable model parameters (model
degrees of freedom) largely exceeds the number of data points used in parameter
calibration. Furthermore, in contrast to adsorption data, electrokinetically-derived data

244
(i.e., zeta potentials) are often obtained at conditions far from thermodynamic equilibrium
and do not allow direct probing of sorption reactions (an arbitrarily-selected electrostatic
model is required to relate zeta potentials to charge data which can, in turn, be used for
the calibration of sorption reactions, Westall and Hohl, 1980). Consequently, the
contribution of individual surface reactions to the development of surface charge cannot
be resolved nor can the formation constants of surface species be estimated accurately by
the above procedure. Recently, a multi-site ion complexation approach was applied to the
calibration of proton and constitutent ion surface complexation reactions for calcite using
selected electrokinetic data from the literature (Wolthers et al., 2008). It was concluded
that a straightforward validation of the postulated multi-site SCM was not possible
because of the uncertainties associated with the nature and magnitude of potential
artifacts inherent to the electrokinetic data. Clearly, reliable proton and constituent ion
adsorption data, acquired over a wide range of chemical conditions, are required for the
proper calibration of acid-base and constituent ion adsorption reactions at the calcite
surface.
Fast surface acid-base titration techniques were successfully applied to
determine ion sorption and/or surface charge development on sparingly reactive
carbonate minerals such as siderite and rhodochrosite (Charlet et al., 1990; Van
Cappellen et al., 1993), magnesite (Pokrovsky et al., 1999a) and dolomite (Pokrovsky
et al., 1999b; Brady et al., 1996; Brady et al., 1999). However, application of these
and more conventional
titration techniques (Huang, 1981) is unsuitable to the
characterization of the acid-base properties of highly reactive carbonate minerals such
as calcite and its polymorphs because their fast dissolution and precipitation kinetics
largely impacts the sorbate molar balance in solution, and hence, affects the

245
computation of surface charge (Van Cappellen et al., 1993; Wolthers et al., 2008).
Despite these considerations, conventional acidimetric surface titration techniques
were recently used in an attempt to estimate proton adsorption on calcite in aqueous
suspensions at different ionic strengths (Eriksson et al., 2007, 2008). Unfortunately,
the experimental protocol adopted in these studies suffers from serious deficiencies
among which: i) calcite dissolution was neglected in the computation of proton
adsorption data; ii) CO2(g) exchange with the atmosphere was neither prevented nor
monitored during the titrations thus affecting the CO2 and proton mass balance in
solution; and iii) the time intervals selected for the acquisition of serial experimental
data throughout the titrations were ill-defined (presumably very short) and reflect, at
best, partial restoration of bulk CaCO3(s)-CO2(g)-H2O thermodynamic equilibrium.
Consequently, the acquired proton sorption data are unsuitable for the calibration of
proton sorption reactions, as previously suggested (Wolthers et al., 2008).
To date, no reliable proton or carbonate adsorption data for the calcite surface
are available. To our knowledge, only one systematic constituent cation adsorption
study (Huang et al., 1991), conducted under a narrow range of solution conditions (9.3
≤ pH ≤ 9.9; 210-5
M ≤ CO2 ≤ 1.210-4
M; 10-5
M ≤ Ca ≤ 10
-2 M; solid:solution ratio:
250 g L-1
, equivalent to a surface area:solution ratio of 2160 m2 L
-1),
provides
quantitative calcium adsorption data suitable for the calibration of calcium adsorption
reaction(s) on the calcite surface.
It follows that the design of appropriate
experimental protocols for the acquisition of quantitative proton and constituent ion
adsorption data over a wide compositional range is a critical step for the quantitative
characterization of sorption equilibria of CaCO3(s) polymorphs (e.g., surface

246
complexation, ion exchange, etc.) and its interpretation within a self-consistent
theoretical framework.
In this paper, we address this issue by introducing a novel titration technique
that allows us, for the first time, to perform acidimetric and calcium ion titrations in
calcite suspensions, resolve sorption processes from bulk calcite
dissolution/precipitation reactions and, obtain reliable proton sorption and calcium
desorption data over a wide range of solution compositions. These are then interpreted
quantitatively on the basis of binary ion exchange equilibria established between the
solution and “exchangeable cation sites” (e.g., lattice positions).
2. MATERIALS AND METHODS
2.1 Principle of Calcite Titrations
In contrast to previous surface titration protocols where dissolution/precipitation
reactions are either circumvented or minimized (e.g., Huang, 1981; Charlet et al.,
1990) the high reactivity of calcite is exploited in the new technique. The
characterization of ion sorption is based on the following premises: i) an accurate
mass balance registry of all chemical components is maintained throughout the
titrations and ii) bulk-phase thermodynamic equilibrium is fully re-established upon
each titrant addition. The latter aspect contrasts with fast titration techniques (Charlet
et al., 1990; Van Cappellen, et al., 1993; Pokrovsky et al., 1999a; Pokrovsky et al.,
1999a; Brady et al., 1996; Brady et al., 1999) that are premised on the establishment
of surface rather than bulk equilibrium. Briefly, the titration is conducted over a
suitable range of chemical conditions in a closed-system in the absence of a gas phase
(i.e., headspace, dead volume) where known amounts of titrant are added

247
incrementally to a pre-equilibrated CaCO3(s) suspension. Under this scenario, no
CO2(g) is transferred from or to the suspension throughout the experiment (total
inorganic carbon, CO2, and proton mass conservation conditions). The solution
chemistry of the suspension is fully characterized before and after each titrant addition
(upon restoration of bulk equilibrium) without perturbing the experimental system
which permits the computation of sorption densities after consideration of
dissolution/precipitation reactions via thermodynamic speciation calculations. The
faster the system reacts to restore equilibrium, the shorter the duration of the titration.
Detailed explanations on the computation of sorption data are given below.
2.2 Description of the Reaction Vessel
A gas-tight glass reaction vessel was constructed specifically for this study (Fig. 1).
The reaction vessel is composed of two pieces joined by an O-ring and firmly held
together with a Plexiglas®
clamp. The top part of the reaction vessel is equipped with
threaded glass ports through which three ion selective electrodes (ISEs) and one
titrant dispenser tube are inserted. The ISEs and the titrant dispenser were secured to
the vessel with gas-tight, plastic threaded stoppers. The ISEs were used to monitor H+,
Ca2+
and CO32-
ion activities at each titration point and allow the full chemical
characterization of the system via the aqueous equilibra given in Table 1 and the
Davies Equation (Morel and Hering, 1993). Note that whereas the three ISEs were
used in most preliminary titration experiments, only the pCa and pH ISEs were used
in subsequent acidimetric and calcium titration experiments (see appendices). To
exclude the presence of a gas phase (i.e., headspace), the reaction vessel was

248
completely filled with the calcite suspension whereas, to accommodate the added
titrant, a 5 or 10 mL high density polyethylene (HDPE) syringe was fitted on a
protruding glass inlet at the top of the reaction vessel and carefully sealed with
Teflon®
tape and Parafilm®
. A gas-tight, two-way polyethylene stopcock was mounted
on the top of the reaction vessel to evacuate excess solution and/or adventitious air
bubbles before initiating the titration. To minimize grinding of the calcite powder, the
suspension was stirred with a Teflon-coated, suspended stir bar positioned at the
bottom of the reaction vessel. The total dead volume of the fully-assembled reaction
vessel, including all components displayed in Figure 1, was determined
gravimetrically using Milli-Q®
water at 25C. A suitable correction was made to
account for the volume of the calcite powder present in each titration experiment.
Details on chemical analyses, preparation and standardization of titrant solutions,
characterization and pre-treatment of the calcite powder, calibration of ISEs, and
preliminary titrations are given in the appendices to this thesis.
2.3 Surface Titration Conditions
Preliminary thermodynamic calculations revealed that highly concentrated calcite
suspensions are required to properly resolve the contribution of potential sorption
reactions from dissolution/precipitation equilibria. However, exceedingly high
solid:solution ratios must be avoided to ensure that crystals remain in suspension,
prevent possible interferences of CaCO3 particles with the ISEs, and circumvent
electrolyte-induced coagulation. Accordingly, titrations were performed at
solid:solution ratios ranging from 0.4 to 12.3 g L-1
(surface area:solution ratios of 0.2

249
to 5.7 m2/L) which proved to be effective for the acquisition of sorption data. The
ionic strength of the CaCO3 suspensions was adjusted to ~ 0.02 M with a 3 M KCl
solution. Two types of experiments were conducted at 1 atm and 25 1C:
acidimetric and calcium surface titrations. These were performed with a Radiometer
Titralab 865 titrator to which the pH electrode and the titrant dispenser tube (Fig. 1)
were connected. An Elite Ion Analyzer multichannel potentiometer, interfaced to a PC
computer, was used to monitor the Ca2+
and CO32-
ISE responses, although, as
mentioned earlier, the latter was used only in the preliminary titrations (see
appendices to this thesis). Because of the differential response times of the ISEs, the
titrations could not readily be pre-programmed to perform automatic incremental
titrant additions through the selection of a universal electrode stability criterion (e.g.,
mV/time). Alternatively, discrete titrant additions were performed manually at pre-
determined time intervals. The selection of these time intervals for each type of
titration is critical in the acquisition of reliable sorption data because the length of the
titration must be kept to a minimum to limit electrode drift or titrant leaks by diffusion
through the dispenser tube, while maximizing the number of titration points that
reflect complete restoration of bulk equilibrium. In addition, detectable changes in the
solution chemistry must be generated throughout the titration. To this end, preliminary
equilibrium speciation calculations were performed to select suitable initial chemical
conditions for our titration experiments (Table 2) and titrant concentrations.
Prior to titrations, a few mg of calcite were pre-equilibrated for at least 14 days
in the solutions described in Table 2 using stoppered 1 L Pyrex®
glass bottles (with
minimum headspace) before use (first equilibration). After standardization of the

250
titrant solutions and calibration of the ISEs, the supernatant of the pre-equilibrated
and decanted calcite suspension (accompanied by a few calcite particles to preserve
saturation) was transferred to the reaction vessel (containing known amounts of dry,
un-reacted, non-titrated, calcite powder) which was immediately assembled, closed,
and sealed to minimize CO2 exchange with the atmosphere. After full assembly of the
titration system (Fig. 1), the suspension was allowed to re-equilibrate for a minimum
of 24 hours under vigorous stirring before starting the titrations. Throughout this
second equilibration period, pH and pCa were continuously monitored to verify the
re-establishment of equilibrium conditions. The presence of adventitious air bubbles
inside the reaction vessel was monitored before and during the surface titrations. As a
rule, if bubbles were detected at the end of the second equilibration period, these
were evacuated, the reaction vessel was replenished with the supernatant remaining
from the first equilibration and the reaction vessel closed, sealed, and allowed to re-
equilibrate for another 24 hrs before initiating the titrations (third equilibration). If
recalcitrant bubbles (e.g., infiltrated air, evolved CO2(g)) were detected after the third
equilibration period or during the titration, the experiment was interrupted and all
data discarded. Details on the validation of our titration system and of our titration
experiments are given in the appendices to this thesis. Unless otherwise specified,
MINEQL+ v.4.6 software was used in all equilibrium calculations in this paper.
2.4 Computation of Sorption Data
All relevant definitions, mass and mole balance equations as well as associated
nomenclature for the Tableau-based aqueous phase definition of the CaCO3(s)-KCl-
H2O chemical equilibrium (Morel and Hering, 1993)1
and the computation of sorption

251
data are provided in Table 3. Apparent proton and calcium sorption densities (Happ
and Caapp
, respectively) are computed from:
Happ
= (1/A·S)·(TOTH*Theo - TOTH*Exp) (1)
Caapp
=(1/A·S)·(Ca*Theo – Ca*Exp) (2)
where A is the specific surface area (m2
g-1
) and S is the solid:solution ratio (g L-1
).
In contrast to proton- and OH-bearing minerals such as metal oxides (goethite,
lepidocrocite etc.), pure calcite powders do not display a net proton imbalance at their dry
terminal surfaces (no protons or hydroxyl groups within their lattices or at their surfaces),
and hence, no excess or deficit in protons is introduced to the mineral-H2O system (in
addition to CA-CB, see Table 3) upon calcite immersion in water. Thus, assuming CO2(g) is
not exchanged with a gas-phase (as in our experiment), net proton densities, Hnet
, of the
calcite surface, at the beginning and throughout the titration, can be obtained directly
from pH and pCa measurements, the amounts of CA and CB added, and application of Eq.
1. This yields the condition: Happ
=Hnet
. This contrasts with proton- and OH-bearing
minerals that require a pre-determination of the Point of Zero Net Proton Charge
(PZNPC) before Hnet
can be computed (Sposito, 1998). Similarly, potential calcium
imbalances (with respect to a stoichiometric number of CO3 groups) at the calcite surface,
possibly arising from differential constituent ion re-adsorption or transient non-
stoichiometric calcite dissolution before titration (Douglas and Walker, 1950; Stipp and
Hochella, 1991; Villegas-Jiménez et al., 2009), can be estimated from pH and pCa

252
measurements, the initial amounts of Ca and CO2 in the system, and Eq. 2. This yields
the condition: Caapp
= Canet
where the latter term represents net calcium densities.
The simplest way to verify that the above conditions are met is to compute Happ
and Caapp
from the solution chemistry measured in the simplest possible CaCO3(s)-H2O
scenario: an equilibrated suspension of calcite (≥ 99% purity) in pure water (Milli-Q®, ~
18 Mohm cm, without CO2(g) exchange with a gas phase) which should correspond to
the solution condition: TOTH0=TOTCa
0=0. Under this condition, if no significant proton
or calcium adsorption/desorption occurs (i.e., Happ
= Caapp
=0), the pH and pCa
measured at 25 ± 1ºC must be nearly identical to those predicted thermodynamically. The
excellent agreement between the measured and theoretical pH and pCa values (>99% and
>97%, respectively) under these chemical conditions confirmed the validity of the
assumption that sorbateapp
=sorbatenet
=0 where the subscript “sorbate” represents proton or
calcium ions This chemical scenario will be referred to as the zero net sorption reference
condition (ZNSRC) This is a common assumption in carbonate mineral surface studies
(Charlet et al., 1990; Van Cappellen, et al., 1993; Pokrovsky et al., 1999a, b), even though
it is rarely defined explicitly (Charlet et al., 1990) or verified experimentally.
Whereas the ZNSRC prevails at the beginning of Experiments TH-I, TH-III, TH-
IV and TH-VI, it does not necessarily hold for titrations conducted under the following
initial chemical conditions: TOTH0 and/or TOTCa
0 0 (Experiments: TH-II, TH-V, TCa-
I-TCa-IV), because the proton and/or constituent ion imbalance in solution may induce
ion sorption and modify the chemistry of the solution (TOTH0
Exp TOTH0
Theo and/or
Ca0Exp
Ca
0Theo) and the calcite surface (H
0 and/or Ca
0 0) during the first and
second equilibration periods and prior to titration. Consequently, to correctly compute

253
the net sorption densities, sorbatenet
, in these experiments, new TOTH0
Theo and Ca0
Theo
values must be “experimentally” determined from the pH and pCa measured in the
supernatant at the end of the first equilibration period and before exposure to additional
calcite (i.e., the powder subjected to titration). Once transferred to the reaction vessel, a
new sorption equilibrium with the un-reacted calcite powder is established, and thus, Hnet
and Canet
can be estimated using the new TOTH0
Theo and Ca0
Theo values, pH and pCa
measurements, and Eqs. 1 and 2. Alternatively, once the pre-equilibrated supernatant
(first equilibration) is transferred to the reaction vessel and after the second
equilibration period, one can determine TOTH0
and Ca0 (total calcium concentration
before titration) from the measured pH and pCa values and the definitions given in Table
3, substitute these in Eqs. 1 and 2 respectively (as the TOTH*
Theo and Ca*Theo values
prior to titration: TOTH0
Theo and Ca0
Theo, and compute the initial extent of proton
occupancy of the calcite sample (subsequently subjected to acidimetric or calcium
titrations) via a suitable mathematical relationship. This relationship, derived from
experiments initiated at the ZNSRC, is described in the “sorption modeling section”,
whereas detailed explanations on the computation of sorbatenet
values for titrations
initiated away from the ZNSRC are given in the appendices to this thesis.
3. RESULTS AND DISCUSSION
3.1 Qualitative Interpretation of Data
Before proceeding to the quantitative interpretation of the sorption data, some
preliminary discussion is required. Firstly, the Happ
values computed via Eq. 1 in all
our acidimetric and calcium titrations (Hnet
>> 0), largely exceed the theoretical

254
number of reactive surface sites at the (10.4) calcite surface (8.2 moles m-2
) which is a
striking result, particularly in the case of calcium titrations considering that no HCl
was added in these experiments. Secondly, the Caapp
values computed in all titration
experiments by Eq. 2 reflect a substantial calcium release (Canet
<< 0). In other
words, Ca*Exp values were consistently much larger than Ca*Theo reflecting a large
excess of Ca over CO2 in solution (TOTCa*
Exp >> 0). Because stoichiometric
calcite dissolution should not affect the Ca:CO2 ratios, the registered TOTCa*
Exp
values are unexpected in all cases (including experiment TH-V initiated at TOTCa*
Exp
> 0), but most particularly, in experiments initiated at TOTCa0
Exp = 0 (TH-I, TH-III,
TH-IV, TH-VI, in which no calcium addition was made) and at TOTCa0
<< 0 (TCa-I-
TCa-IV). These results are counterintuitive since, under the conditions of our calcium
titration experiments (stepwise additions of CaCl2 rather than HCl), a net calcium
removal from solution via calcite precipitation and calcium adsorption (Huang et al.,
1991) was expected, possibly coupled with a net proton release (rather than a net
proton uptake) induced by calcium/proton exchange surface reactions as those
postulated earlier (Van Cappellen et al., 1993; Wolthers et al., 2008). Keeping in mind
that precipitation/dissolution reactions are accounted for in the determination of the
Ca0
Theo values, it is clear that other mechanism(s) must be called upon to explain the
“anomalous” TOTH*
Exp, Ca0
Exp, and TOTCa*
Exp values registered in our acidimetric
and calcium titrations.
Whereas contamination of our suspensions by impurities carried-over from
previous experiments can be dismissed (see precautions described in the appendices to
this thesis), neither can the observed Ca*Exp and TOTCa*Exp values be explained by

255
the desorption of Ca2+
ions (potentially pre-adsorbed on the calcite surface) because
the release of several hypothetically “Ca-enriched” atomic layers (up to 16 layers,
according to the maximum proton uptake and calcium release observed in our
experiments at a pH of 7.2, see below) would be required to account for the high
Ca values measured at the end of the titrations. This is clearly an unrealistic
scenario. Similarly, the “non-stoichiometric” release of Ca2+
over CO32-
ions from the
calcite outmost surface layer (Douglas and Walker, 1950; Stipp and Hochella, 1991;
Villegas-Jimenez et al., 2009)
and/or the preferential detachment of Ca2+
from
subsurface atomic layers are either insufficient to account for our observations or
would generate an unreasonable number of ion vacancies and negative charges within
the lattice. Likewise, the extent of carbonate adsorption required to increase
TOTCa*Exp to the observed levels largely exceeds the theoretical number of reactive
surface sites available in our experimental systems (7.5·10-7
-2.4·10-5
moles, as
computed from the theoretical site density, the specific surface area, the mass:volume
ratio, and the total volume of the suspensions). Finally, incongruent calcite dissolution
upon the formation of a secondary solid phase (e.g., calcium hydroxide) is
thermodynamically unfavorable and could not account for either proton uptake or
calcium release. Consequently, we propose that a proton/calcium ion exchange takes
place between calcite and the solution according to the following reaction:
(CaCO3)2(exc) + 2 H+ Ca(HCO3)2(exc) + Ca
2+ (3)

256
where species identified with the subscript “exc” correspond to reactive units at and/or
beneath the calcite surface which we will refer to hereafter as “exchangeable cation
sites”. Reaction 3 describes the non-stoichiometric release of one Ca2+
ion, with
respect to CO32-
ions (“apparent” incongruent dissolution), upon substitution by two
protons at exchangeable cation sites, which preserves charge-neutrality within the
mineral lattice. Alternatively, reaction 3 could be equally formulated as:
CaCO3(exc) + 2 H+ H2CO3(exc) + Ca
2+ (4)
which only contrasts with reaction 3 in the nature of the resultant exchangeable cation
species.
A representative example of the mirror-image displayed by the net proton and
calcium densities is shown in Figure 2. Although the computed average Hnet
:Canet
ratio (~1.5) differs from the ideal stoichiometry (2.0) of reactions 3 and 4, the
discrepancy is ascribed to cumulative and systematic errors associated to the
computation of TOTH*Exp and TOTCa*Exp. For instance, calculations using our
sorption data, the Ca2+
ISE calibration curves, and the chemical definitions given in
Table 3 reveal that a systematic decrease in the Ca2+
ISE signal by 1-3%, consistent
with the estimated electrode drift (see appendices to this thesis), would lead to a net
increase in the TOTH*Exp and a concomitant net decrease in Ca*Exp by 2-6%
depending on the pH range. Whereas errors of this magnitude would not impact
significantly on the quantitative interpretation of the sorption data (see below), they
would account for most of the discrepancy between the observed and ideal Hnet
:Canet

257
ratios (R) suggested by reactions 3 and 4. Furthermore, propagation of error analysis
based on the experimental uncertainties of our experimental protocol (ISE readings,
titrant concentrations, volumetric titrant additions, etc.) adds up to a 10% random
error in the estimated Happ
and Caapp
values which, in turn, yields a random error of
14% in the computed R values. This translates into R values ranging from 1.3 to 1.7,
in close agreement with the variability of the experimental R values (1.25-1.8). In
brief, a combination of random and systematic errors inherent to our experimental
protocol likely accounts for the discrepancy between the observed and “ideal” R
values.
Alternatively, a Hnet
:Canet
ratio of 1, dictated by the stoichiometry of the
proton/calcium ion exchange reaction involving the background electrolyte cation in
our experimental system (K+):
CaCO3 (exc) + H+
+ K+ KHCO3 (exc) + Ca
2+ (5)
could only be obtained if the pCa values derived from Ca2+
ISE readings carried much
larger systematic errors (~ 12-18%, depending on the pH) than those required to achieve a
Hnet
:Canet ratio of 2, an unlikely scenario given the maximum Ca
2+ ISE drift (3 %, see
appendices to this thesis). In addition, experimental studies reveal that potassium defects
in natural calcite samples (incorporated during early mineral crystallization) are highly
unstable under Earth‟s surface conditions and potassium ions tend to migrate
spontaneously out of the lattice and accumulate in crystallites at the calcite surface (Stipp
et al., 1997; Stipp et al., 1998). These observations strongly argue against the stability of

258
the KHCO3 (exc) species and the viability of reaction 5, a premise that was further
confirmed by our sorption modeling work described below. Because no other potentially
exchangeable cation is present in the system, the last possibility to the observed R values
is that the proton/calcium exchange is not governed by a reaction with an integer 2:1
stoichiometry (reactions 3 and 4) but rather, by a reaction involving a fractional
stoichiometry which would lead to a large net negative charge buildup within the calcite
lattice (because more positive charges are leaving the calcite lattice than are incorporated
via proton uptake), a largely speculative hypothesis that would be difficult to explain
thermodynamically. It follows that cation exchange reactions 3 and 4 are the most viable
mechanisms for the quantitative interpretation of our sorption data.
If we consider that, upon HCl additions to the suspensions, calcite will dissolve
according to:
CaCO3(s) + 2 H+
Ca2+
+ H2CO3* (6)
combining reactions 3 and 6 yields:
(CaCO3)2(exc) + CaCO3(s) + 4 H+
Ca(HCO3)2(exc) + 2 Ca2+
+ H2CO3* (7)
Estimated values of TOTCa*Exp as a function of the deficit in CO2 with respect to the
amount expected from calcite dissolution (i.e., CO2* = CO2*Theo - CO2*Exp; see
Table 3), obtained in all the acidimetric titrations, are compiled in Figure 3. Note that

259
to properly account for TOTCa*Exp arising solely from reactions between the bulk
solution and calcite at conditions: TOTCa0 0 (Experiments TH-II and TH-V),
TOTCa0 was subtracted from the corresponding TOTCa*Exp values and the corrected
values reported in Figure 3. A linear regression of these data yields a slope of 2.1, in
excellent agreement with the Ca:C stoichiometry dictated by reaction 7. The CO2*
values are explained by the net decrease in calcite dissolution following proton uptake
and Ca2+
release to the solution. In other words, upon HCl additions, proton/calcium
ion buffers the pH, increases the Ca2+
ion activity (yielding higher TOTCa*Exp) in
solution which, in turn, buffers the calcite saturation state, limits calcite dissolution,
and progressively decreases CO2*
Exp (i.e. increases CO2
*). This is consistent with
the non-stoichiometric release of Ca2+
over CO32-
ions postulated by reaction 3 upon
strong acid additions.
On the other hand, upon incremental additions of CaCl2 to the experimental
suspensions containing large concentrations of HCO3- ions (Experiments TCa-I to
TCa-IV), calcite precipitation occurs and generates increasing levels of H2CO3*
according to:
Ca2+
+ 2 HCO3- CaCO3(s) + H2CO3
* (8)
Hence, for this scenario, we can illustrate the 2H+/Ca
2+ exchange mechanism by
combining reactions 3, 8 and the following equilibrium:
2 H2CO3* 2 H
+ + 2 HCO3
- (9)

260
to yield:
(CaCO3)2(exc) + H2CO3*
Ca(HCO3)2(exc) + CaCO3(s) (10)
Reaction 10 suggests that carbonic acid, generated by CaCO3(s) precipitation upon
CaCl2 additions, can be removed from solution upon 2H+/Ca
2+ ion exchange-induced
calcite precipitation.
To summarize, under all chemical scenarios examined in our study (see Table
2) we observe that: i) proton removal from solution (i.e., TOTH*Exp << TOTH*
Theo)
largely exceeds the theoretical number of reactive surface sites (by up to ~32-fold,
according to the maximum proton uptake and calcium release observed in our
experiments at a pH of 7.2 and assuming a 1:1 sorbate:surface site stoichiometry,
see below) and ii) a significant TOTCa*Exp increase is generated throughout the
titrations that cannot be explained by the desorption of potentially pre-adsorbed Ca2+
ions on the calcite surface. Both observations (eventually confirmed by Hnet
and
Canet
data) can only be explained by invoking the presence of reactive sites other than
those conventionally defined at the calcite surface (e.g., exchangeable cation sites at
and/or beneath the calcite surface). This is the basis of the postulated 2H+/Ca
2+ ion
exchange mechanism proposed by reactions 3 and 4.
3.2 Possible Mechanisms of “Proton Uptake/Calcium Release” and “Apparent”
Incongruent Calcite Dissolution
Several mechanisms could equally account for the stoichiometry of reactions 3 and 4,
including mechanisms that might involve possible OH-bearing defects (e.g.,

261
CaCO3nH2O(s) and/or Ca(OH)2(s)) homogeneously embedded within the calcite
lattice (i.e., subsurface layers) prior to titration, and subsequently released to the
solution via co-dissolution, ion migration along micro-fractures and/or rapid physical
rearrangement of the near-surface layers (see Figure 4). In mechanism A, n H2O
molecules are presumably embedded in the calcite lattice through interactions with
CaCO3 lattice units whereas in mechanism B, 2n hydroxyl groups replace nCO32-
lattice units preserving charge neutrality in the bulk crystal. Under these scenarios,
lattice OH- groups and Ca
2+ ions are released either by: i) co-dissolution of n
Ca(OH)2(s) / n CaCO3(s) units, leading to a hypothetical substitution of
(CaCO3H2O)x “hypothetical” defects by CaCO3(x-n)H2Ox-2n and (CO3H2)n lattice
units, or ii) coupled Ca2+
/OH- ion migration outwards from the crystal. Both
mechanisms would lead to a net decrease of TOTH*Exp (upon proton neutralization by
releasing OH- ions) and a net increase of TOTCa*Exp in solution. Nevertheless, these
mechanisms are unlikely under our experimental conditions. Firstly, although H2O
and/or OH-bearing defects have been observed within the lattice of Mg-rich biogenic
calcites (Gaffey, 1995), precursor metastable amorphous CaCO3 phases (Elfil and
Roques, 2001), and aragonite (B. Phillips, personal communication); available NMR
evidence is still inconclusive to confirm their presence in synthetic Mg-free calcite
samples as the one used in this study (B. Phillips, personal communication; Feng et
al., 2006). Furthermore, the aging pre-treatment, to which our calcite powder was
subjected (see calcite specimen section in the appendices to this thesis), would have
removed most of these “hypothetical” OH- and/or H2O-bearing defects. In turn, this
would have affected the solution chemistry recorded in individual titration

262
experiments, yielding inconsistent results between experiments. Secondly, mechanism
A would be expected to occur only under a dissolution regime (acidimetric tirations),
and hence, would not explain results obtained in the calcium titration experiments
where precipitation takes place. Conversely, mechanism B would be independent of
dissolution/precipitation processes but would generate numerous lattice vacancies that
would destabilize the crystal lattice, an aspect difficult to explain thermodynamically.
Finally, because solid-state ion diffusion is unlikely within the time-scale of our
experiments (Fisler and Cygan, 1999),
an unrealistic number of micro-fractures
serving as ion conduits would have to pervade the calcite crystals used in all titrations.
Consequently, we believe that the most plausible mechanism involves a fast
2H+/Ca
2+ ion exchange (mechanism C in Fig. 4) between the solution and the solid,
following the dynamic rearrangement of the near-surface calcite layers (i.e.,
spontaneous recrystallisation, Hoffmann and Stipp, 2001). The latter would lead to a
renewal of the adsorption sites, a net increase in proton uptake capacity, and the
generation of a proton-enriched, calcium-deficient layer, analogous to the chemically
and structurally altered “leached” layers developed by some chain-silicate minerals
(e.g., wollastonite, diopside, albite, labradorite) following their incongruent
dissolution under acidic regimes (Casey et al., 1993; Green and Lüttge, 2006). In fact,
rearrangement of the calcite surface has been shown to extend over at least 10 atomic
layers upon exposure to air at 30% relative humidity or more (Stipp et al., 1997) and
is consistent with mechanisms of fast ion incorporation into the calcite lattice (i.e.,
lattice penetration or surface recrystallization) postulated by other researchers
(Zachara et al., 1988; Zachara et al., 1991; Stipp et al., 1992; Stipp et al., 1996;
Hoffmann and Stipp, 2001; Curti et al., 2005). These observations have led to the

263
suggestion of the presence of an “interfacial region” conceptualized either as a
hydrated CaCO3(s) “gel-like” phase (Somasundaran and Agar, 1967), a porous
membrane composed of adsorbed and surface layers (Mucci et al., 1985), or as a
hydrated “multi-atomic disordered CaCO3(s) layer” (Davis et al., 1987) through which
ions presumably diffuse, segregate, and dehydrate before incorporation in the bulk
lattice. These conceptualizations of the hydrated calcite interface would provide a
suitable environment for the fast proton incorporation and calcium release postulated
by mechanism C. Unlike mechanisms A and B, mechanism C does not require an
homogeneous distribution (most likely unrealistic) of hydroxyl-bearing defects
(CaCO3nH2O(s) and/or Ca(OH)2(s)) over the calcite lattice prior to titration nor does
it lead to the generation of lattice vacancies that could destabilize the crystal.
Results of a recent Nuclear Magnetic Resonance (NMR) spectroscopy study
(Feng et al., 2006) reveal the presence of stable bicarbonate species within the bulk
lattice of organic defect-free synthetic calcite samples whereas Knudsen flow reactor-
based adsorption experiments suggest the presence of bicarbonate-bearing species at
hydrated calcite surfaces after their exposure to CO2(g) (Santschi and Rossi, 2006),
consistent with the formation of the bicarbonate species postulated by reaction 3.
Similarly, recent theoretical (Tossel, 2006) and experimental studies (Usher et al.,
2003, Al-Hosney and Grassian, 2005) suggest that H2CO3 might be stable in solid
phase either in some oligomeric form (Tossel, 2006) or as an adsorbed species at acid-
treated carbonate mineral surfaces (Usher et al., 2003, Al-Hosney and Grassian,
2005), supporting the formation of H2CO3–like species such as produced by reaction
4. Whether HCO3- or H2CO3-bearing species form or not within the calcite lattice is

264
beyond the scope of the present study but we believe that the formation of
≡Ca(HCO3)2(exc) is more likely to explain proton stabilization within the calcite lattice
than the presence of ≡H2CO3(exc) species. Accordingly, the following discussion will
focus on reaction 3. Nevertheless, as explained below, the quantitative treatment
performed on our sorption data is equivalent whether based on reactions 3 or 4.
3.3 Sorption Modeling
As discussed above, the chemical conditions selected in this study induce
proton/calcium exchange and are thus suitable for the quantitative description of
reaction 3. Given the inter-dependency of Ca2+
and H+ activities under equilibrium
conditions, sorption isotherms calibrated to specific pH values are not useful for the
interpretation of sorption data. Instead, the pH-dependency of this reaction allows the
simultaneous quantitative interpretation of sorption data acquired at different pH
values, as is commonly done in surface complexation modeling studies.
The mass action law describing the binary exchange (reaction 3) can be
represented by the selectivity constant (Kex) defined by the classical Langmuir-power
exchange function based upon the Vanselow convention:
2
2
)CaCO(
)HCO(Ca
)(
)(
X
X
2(exc)3
(exc)23
aH
aCaK
n
ex (11)
which is defined in terms of the mole fractions (X) of the
exchangeable cation sites, the activities (a) of the aqueous species and an empirical

265
exponent (n) that accounts for deviations from ideal cation mixing (n1) arising from
steric and electrostatic effects (i.e., local electrostatic balance) within the crystal
lattice. The activity of solid component i, ai, is given by:
ai = i Xi (12)
where i is the corresponding solid activity coefficient and depends on the
composition of the system (Stoessell, 1998). Hence, if mole fractions realistically
reflect the activities of exchange sites (=1) and local electrostatic balance is fully
achieved (n=1), then Kex corresponds to the conditional equilibrium constant
describing the 2H+/Ca
2+ exchange,
cKex (Stoessell, 1998).
We assume that only a small fraction of the calcite lattice participates to
proton/calcium exchange which, hereafter, we will refer to as the “exchangeable
cation site density” (ECSD). According to reactions 3 and 4, the ECSD represents half
of the proton uptake capacity (PUC) of calcite. Both parameters are expressed in mol
m-2
units for consistency with adsorption data normalized to the experimental BET
specific surface area. Nevertheless, it must be emphasized that because proton uptake
appears to extend beneath the topmost surface atomic layer, the PUC is unlikely a
surface property, and instead, presumably reflects the intrinsic ability of calcite to
develop a partial solid solution of the type: Ca(1-x)H2xCO3 under specific chemical
conditions. In addition, one must note that, although the specific BET-surface area of
the titrated calcite sample was not significantly different from that of the non-titrated
material, BET measurements on reacted, rinsed, and dried powders would not reveal

266
the presence of features (e.g., gels, channels, crystallographic re-arrangements) that
may have developed upon proton/calcium exchange (and possibly affected the
available reactive surface area) since they may only be persistent in solution.
The concentration of the so-called “cation-specific surface sites”, presumably
corresponding to the concentration of exchangeable Ca at the topmost surface layer(s)
of calcite crystals, was previously estimated, using 45
Ca isotopic exchange
experiments, at 3.6·10-6
moles g-1
(or 0.72·10-6
moles m-2
; Zachara et al., 1991). This
value, however, is too low to successfully model our sorption data using equation 11.
Attempts to model our data, using the experimental total CaCO3(s) molar concentration
(0.004-0.12 M) to estimate molar fractions, without optimization of the ECSD, were
also unsuccessful. Consequently, Kex and ECSD were estimated via numerical
optimization using FITEQL v.3.2 (Herbelin and Westall, 1996), in analogy to the
procedures commonly adopted to estimate intrinsic formation constants and the total
surface site density within the framework of surface complexation theory (Dzombak
and Morel, 1990). Preliminary estimates of Kex and ECSD were obtained using net
proton sorption data from experiments initiated at the ZNSRC (experiments TH-I, TH-
III, TH-IV and TH-VI). Since successful convergence was achieved with n=1 (ideal
cation mixing) for all selected data sets, this value was used in subsequent
optimization runs. An average log10 Kex of 13.1 0.4 and an average ECSD of 12
(3.7) ·10-5
moles m-2
, corresponding to ~ 0.7 % of the total CaCO3 in the system and
equivalent to 32 calcite monolayers (according to reaction 3), were obtained. Since
most solid solutions behave ideally when the mole fraction of the solvent crystal
approaches unity, we can confidently assume that the solid solution Ca (1-x)H2xCO3

267
(where x < 0.01) satisfies the same criterion over the compositional range of study,
and hence, the activities of exchange species can be equated to their mole fractions in
equation 11. This shows that the constant returned by FITEQL optimization of
reaction 3 represents indeed cKex, and thus, we will refer as such hereafter.
As mentioned earlier, apparent proton sorption densities derived from
experiments TH-II and TH-V were referenced to the ZNSRC to account for the extent
of proton occupancy at the beginning of these titrations and compute the
corresponding net sorption densities. The latter were used in subsequent optimizations
of cKex and ECSD (see appendices to this thesis). An average log10
cKex of 13.1 0.4
and an average ECSD of 13 ( 3.1) ·10-5
moles m-2
were obtained upon optimization
of these data, values statistically identical to those obtained in our previous
optimizations.
Net proton sorption densities computed from all our acidimetric titrations are
displayed in Figure 5. The good reproducibility of the data obtained under different
chemical conditions confirms that the experimental protocol can quantitatively resolve
sorption or ion exchange reactions from bulk dissolution equilibria. It is noteworthy
that, although the initial conditions of Experiments TH-II and TH-V differ
significantly from others (including a lower initial pH), the net proton densities
derived from these are consistent with results of other titrations and extend the
investigational pH range to circum-neutral pH values. In all cases, proton uptake
increases smoothly from pH 10 to about 8.5 and displays a significant increase at pH
< 8.5. The solid curve in Figure 5 shows the net proton densities predicted by
speciation calculations including reaction 3, the optimized cKex, and ECSD values for

268
a solid:solution ratio of 9.61 g L-1
(surface area: solution ratio of 4.42 m2 L
-1). Clearly,
reaction 3 successfully simulates data over a wide range of chemical conditions with
small deviations at pH > 8.5 where proton sorption values are relatively low.
Similarly, because all calcium titrations were performed at TOTH0 > 0 and
TOTCa0
< 0, proton sorption densities computed from these experiments using Eqs. 1
and 2 were corrected and referenced to the ZNSRC (see appendices to this thesis). As
calcium was added incrementally to the solution, the calcite saturation state increased,
triggering its precipitation and the concomitant formation of H2CO3* (reaction 8).
Thus, intuitively, either positive or null calcium sorption densities (Canet
≥ 0) are
expected but, as stated earlier, the opposite behavior (i.e., Ca releaseCanet
<< 0,) was
observed accompanied by a substantial proton uptake. These results can be explained
by the net removal of H2CO3* from solution upon proton/calcium ion exchange,
following calcite precipitation, resulting in a net proton uptake and net Ca release
(reaction 10). Consequently, the computed amounts of calcium released to the solution
(i.e., “calcium desorption data”) were used to fit Kex and ECSD. To this end, and
according to the stoichiometry of reaction 3, the amount of Ca released to the solution
(in absolute molar units: |Canet
|·S·A) was subtracted from the total molar
concentration of exchangeable cation sites, (CaCO3)2(exc), available in each
experiment (Experiments TCa-I to TCa-III: 6.0·10-4
mol L-1
, Experiment TCa-IV:
2.4·10-5
mol L-1
). The latter was computed from the product of the solid:solution ratio
in each experiment, the specific surface area, and the average ECSD value previously
optimized with acidimetric titration data (13·10-5
moles m-2
). This subtraction yields
the apparent molar concentration of unreacted exchangeable cation sites,

269
(CaCO3)2(exc)*, at each titration point. The molar concentration of exchangeable
cation sites initially occupied by protons, Ca(HCO3)2(exc)o, after the second
equilibration and prior to titration (see appendices to this thesis), was then subtracted
from (CaCO3)2(exc)* to normalize data to the ZNRSC and obtain the net unreacted
(available) exchangeable cation site concentration, (CaCO3)2(exc)*net
, which was
subsequently used in the FITEQL optimizations. The net molar densities of unreacted
exchangeable cation sites (i.e., ECSD-Hnet
) and the model predictions are shown in
Figure 6. The data from experiment TCa-III could not be fitted with FITEQL and
show significant deviations from the model predictions. The inability of our model to
fit these data is explained by the high CO2 in this experiment ( 0.023 M) promoting
calcite precipitation (i.e., reaction 8 was favored), largely masking calcium
desorption, and affecting the accuracy of the estimated (CaCO3)2(exc)* used in
FITEQL optimizations. In Figure 6, the experimental H2CO3(aq)* values are also
displayed for each titration experiment. Note that, for a given H2CO3(aq)* value
(dictated by the pH and the pCa), an identical fraction of unreacted exchangeable
cation sites is generated, regardless of the TOTH0
Theo and TOTCa0
Theo values, as
observed in experiments TCa-I, TCa-II and TCa-IV, in conformity with reaction 10.
The average conditional equilibrium constant derived from the calcium
titration data, log10 cKex = 12.9 ± 0.2 (with n=1 as well), is in excellent agreement
with the value computed from the acidimetric titrations (13.1 0.3). Averaging all the
optimized cKex values yields a log10
cKex of 13.0 ± 0.3, the conditional equilibrium
constant for reaction 3 over the compositional range of this study. Combining this
value, the calcite solubility product (Kºsp), the product of the dissociation constants of

270
carbonic acid (K°H2CO3* K°HCO3, Table 1), and their associated uncertainties (Plummer
and Busenberg, 1982), we obtain a log10 cKex2 of 4.8 ± 0.4 for the following mass
action law describing reaction 10:
n
)*COaH((
(
exKc
322
2(exc)3
(exc)23
)CaCOX
)HCOCaX
(13)
With the exception of data from experiment TCa-III, the model predictions fit
experimental data reasonably well and confirm the validity of reaction 3 beyond the
chemical conditions under which the acidimetric titrations were conducted. The
uncertainty of our sorption data (~ 10%) and of the optimized log10 cKex value (± 0.3)
describing reaction 3 confirm that the maximum errors potentially associated to the
computation of TOTCa*Exp and TOTH*Exp (see above) do not affect significantly the
optimization of cKex. Note that modification of the solid:solution ratios resulting from
calcite dissolution (acidimetric titrations, Eq. 7) or precipitation (calcium titrations,
Eq. 10) do not affect the cKex value because the concentration units of the
exchangeable species, (CaCO3)2(exc) and Ca(HCO3)2(exc), in reaction 3 cancel each
other, and thus, eliminate their mass and surface area dependency. All attempts to fit
sorption data to reaction 5 with FITEQL were unsuccessful, further dismissing the
viability of this ion exchange mechanism as explained above.
In view of the successful model results provided by reaction 3, we believe that
the estimated cKex and ECSD values are reliable predictors to model proton/calcium
exchange over a circum-neutral and alkaline pH regime (preponderant in calcite-

271
containing systems) but we have yet to establish if these values vary significantly with
the properties of the calcite substrate (e.g., purity, pre-treatment, specific surface area)
or the solution chemistry, particularly under acidic conditions where the proton-
enriched, calcium-deficient leached layer might be destabilized upon enhanced calcite
dissolution. Because of the formalism adopted in reactions 3 and 4 to define the
exchangeable lattice species, Ca(HCO3)2(exc) and H2CO3(exc), the calibrated cKex and
ECSD values apply to both reactions. The only distinction is that, because lattice
species in reaction 3 involve full unit cells (i.e., 2 CaCO3 units), the corresponding
ECSD represents 32 calcite monolayers, whereas for reaction 4 only half this value
is involved. Application of Nuclear Magentic Resonance techniques (Feng et al.,
2006), Raman Spectroscopy (Casey et al.,1993), Ion Beam Analyses (Casey et al.,
1993; Petit et al., 1987; Bureau et al., 2009) to acid-reacted single calcite crystals
should shed light on the identity of the lattice species and the extent of proton
penetration within the calcite lattice. This information will be key in ascertaining the
mechanisms accounting for the 2H+/Ca
2+ ion exchange behavior of calcite and in the
characterization of the proton-enriched, calcium-deficient leached layer postulated in
this study.
As suggested by our model predictions in Figure 5, under circum-neutral and
slightly acidic conditions, exchangeable cation sites may become fully saturated by
protons, and thus, provided no calcium is added to the system, proton/calcium ion
exchange will likely be interrupted; unless additional exchangeable cation sites (i.e.,
higher PUC), not revealed in our study, are available in calcite. Under the former
scenario, a progressive decrease of pH, following incremental additions of a strong

272
acid (HA), should lead to the dissolution of the calcium-deficient, proton-enriched
layer (presumably located near the topmost surface layer) as well as of some of the
underlying bulk CaCO3(s) layers. This may allow the system to restore the TOTH*Theo,
TOTCa*Theo and Ca/CO2 values predicted by thermodynamic calculations without
consideration of proton/calcium exchange.
The predicted behavior of TOTH*Theo, TOTCa*Theo and Ca/CO2 upon HA
additions with and without consideration of proton/calcium exchange at different
solid:solution ratios is displayed in Figure 7. As a result of 2H+/Ca
2+ exchange,
TOTH*Theo decreases with increasing solid concentration whereas TOTCa*Theo and
Ca/CO2 increase upon the non-stoichiometric Ca2+
release over CO32-
ions. The
salient feature in Figure 7 is that, once the exchangeable cation sites are fully titrated,
the slopes (m) of TOTH*Theo and TOTCa*Theo become equal to those predicted in the
absence of ion exchange (m=1), reflecting the prevalent role of calcite dissolution
(over ion exchange) in dictating the solution chemistry behavior under these chemical
conditions. The curves do not overlap at this point because the solution chemistry is
affected differentially (depending on the amount of calcite) by the chemical
composition of the calcium-deficient, proton-enriched leached layer developed in the
ion exchange scenario. Nevertheless, all curves will overlap if the leached layer
constituents are fully released to the solution, following a destabilization of the
calcium-deficient, proton-enriched layer, a phenomenon not observed in our
experiments and neglected in our modeling but potentially important in studies
conducted under acidic conditions or at other PVT conditions not investigated in our
study.

273
3.4 Ion Exchange vs Surface Equilibria
The postulated proton/calcium ion exchange mechanism is consistent with the fast
incorporation of some divalent metals within the calcite lattice (Stipp et al., 1992;
Stipp et al., 1997; Hoffmann and Stipp, 2001), possibly coupled with proton
incorporation (Curti et al., 2005), and reveals that ion sorption likely extends over
several subsurface calcite layers within the so-called “gel-like” interfacial region
(Somasundaran and Agar, 1967).
Results of numerous sorption (Davis et al., 1987; Huang et al., 1991; Stipp et
al., 1992; Zhong an Mucci, 1995; Hoffmann and Stipp, 2001; Eriksson et al., 2007,
2008) ion exchange (Zachara et al., 1988, 1991; Curti et al., 2005) and surface
complexation (Comans and Middelburg 1987; Van Cappellen et al., 1993; Pokrovsky
et al., 2000; Martin-Garin et al., 2003; Wolthers et al., 2008) studies of metal ions at
the calcite surface can be found in the literature but none was designed for the
quantitative evaluation of the proton/cation exchange equilibria. For example, Huang
et al (1991) obtained calcium adsorption data that were interpreted in terms of a
Langmuir-type isotherm over a very limited pH range (9.3-9.9) but the authors could
not observe 2H+/Ca
2+ ion exchange because their experimental conditions (i.e.,
TOTHTheo=0 and TOTCaTheo 0) did not promote proton/calcium exchange. In other
words, the driving force (TOTHTheo > 0 and/or TOTCaTheo< 0) necessary to induce
proton/calcium ion exchange in pure calcite samples was absent. Similarly, earlier
proton adsorption data, obtained from acidimetric titrations of calcite suspensions
conducted in open reaction vessels (Eriksson et al., 2007, 2008) are not useful for the
evaluation of 2H+/Ca
2+ exchange because an accurate mass balance registry of H (or

274
TOTH, see Table 3), CO2 and Ca cannot be obtained by the employed methods.
Consequently, neither adsorption nor ion exchange reactions can be properly
evaluated with these data.
In fact, it is possible that, under most experimental conditions, proton surface
equilibria and proton/calcium exchange are intrinsically coupled in calcite, the former
dictating the surface speciation and the development of surface charge while the latter
affects the composition of the solution and the speciation of exchangeable cation sites.
If so, the experimental decoupling of these two processes is not trivial (if possible at
all) since ion exchange may affect proton adsorption by modifying the chemical
composition of the near-surface calcite layers which, in turn, should reflect on its
surface properties (e.g., ion affinity). Similarly, it is possible that additional
adsorption reactions involving other metals (e.g. Mg2+
) or large adsorbates (e.g.,
phosphate, sulfate, organics) interfere with ion exchange equilibria by blocking
reactive surface sites and/or inhibiting the rearrangement of the calcite surface which,
in turn, may affect ion transport to or from the exchangeable cation sites. The complex
interplay of these equilibria must exert a key role on determining the surface
properties of calcite and its macroscopic behavior in aqueous solutions (e.g.,
coagulation, dissolution, pHIEP). Consequently, quantitative interpretations of its
surface reactivity, including its surface charge, electrokinetic behavior (Huang et al.,
1991; Cicerone et al., 1992; Van Cappellen et al., 1993; Wolthers et al., 2008) and
dissolution kinetics (Sjöberg and Rickard, 1984; Van Cappellen et al., 1993; Arakaki
and Mucci, 1995) based upon surface reactions calibrated against data acquired under

275
proton/calcium exchange inducing conditions
will likely be more intricate than
previously considered.
3.5 Implications of Proton/Calcium Ion Exchange
Proton/calcium ion exchange has numerous and important implications with respect to
the interpretation of experimental and field data. The best way to illustrate this is by
comparing speciation calculations performed with and without consideration of
proton/calcium exchange. Because MINEQL+ v.4.6, as well as many other
equilibrium speciation computer codes, does not allow the user to decouple the mass
action law and the mass balance matrices necessary to properly define the ion
exchange equilibrium problem, these calculations were performed using an in-house
Matlab subroutine specifically adapted for this purpose. Nevertheless, identical
calculations can be performed within any equilibrium speciation code in which
suitable modifications can be implemented to decouple the mass action law and the
mass balance matrices. Detailed explanations on these calculations are provided in the
appendices to this thesis.
Figure 8 shows the speciation predicted in a closed CaCO3(s)-H2O system upon
additions of a strong acid (HA) without (Scenario I) and with (Scenario II)
consideration of ion exchange at a high solid:solution ratio (100 g L-1
or 46 m2 L
-1) to
magnify the impact of ion exchange on the solution chemistry. For equivalent strong
acid additions, the values of Ca, H2CO3* and Total Alkalinity (TA) predicted in
Scenario I are consistently higher whereas pH displays the opposite behavior. As a
result of ion exchange, solution pH is significantly buffered, decreasing the extent of

276
calcite dissolution which, in turn, shifts the equilibrium towards lower Ca,
H2CO3*, and TA values. Note that, although reactions 6 (calcite dissolution
scenario, acidimetric titrations) and 8 (calcite precipitation scenario, calcium
titrations) predict increasing levels of H2CO3*, proton/calcium exchange results in a
lowered H2CO3*. This is because in the dissolution scenario, less H2CO3* is
produced upon ion exchange since more protons are required for H2CO3* generation
than when ion exchange is neglected (see the differential H+:H2CO3* stoichiometries
of reactions 6 and 7). Similarly, a net H2CO3* consumption takes place in the
precipitation scenario upon ion exchange (reaction 10), in contrast with what is
predicted when ion exchange is not considered (reaction 8). For a given pH, Ca
values are higher and H2CO3* and TA are lower in Scenario II than in Scenario I
because more strong acid equivalents (CA) are needed in Scenario II to reach an
identical pH because of the pH buffering induced by ion exchange. Additions of CA
favor 2H+/Ca
2+ exchange and increase Ca but decrease the amount of carbonate
equivalents that must be dissolved to restore saturation, as manifested by the lower
H2CO3* and TA values. The difference in CA equivalents required to achieve a given
pH increases at lower pH, reflecting the enhanced pH-buffering capacity of calcite
induced by proton/calcium ion exchange under these solution conditions.
In contrast to closed-systems, in open carbonate systems where the pCO2 is
fixed or at steady-state, the aqueous speciation is identical for Scenarios I and II
whereas the relative concentrations of exchangeable cation species, (Ca(HCO3)2(exc)
and (CaCO3(exc))2), are solely dictated by the pCO2 and cKex, and thus, remain fixed
throughout the entire pH scale in Scenario II (see reaction 10). Hence, in open

277
systems, provided enough exchangeable cation sites are available for proton uptake,
net non-stoichiometric Ca2+
ion release (over CO3-2
) will be observed upon addition of
CO2(g) to aquatic carbonate-rock systems, as defined by (reaction 10 minus reaction
8):
(CaCO3)2(exc) + 2 H2CO3* Ca(HCO3)2(exc) + Ca2+
+ 2 HCO3- (14)
It follows that model predictions of the response of carbonate-rich (sediments,
suspended particles) aquatic environments to rising atmospheric pCO2 (i.e., enhanced
[H2CO3]*) must consider the role of proton/calcium exchange on the carbonate
mineral equilibria as the reaction will buffer pH and the calcite saturation state of the
waters, partly mitigating the postulated negative effects (e.g., decalcification of
calcifying organisms, deleterious development of coral reefs; Andersson et al., 2003,
2006) of anthropogenic CO2(g) invasion in marine shelf waters. Interestingly, this
geochemically-driven ion exchange mechanism is similar to the physiologically-
driven 2H+/Ca
2+ ion exchange exhibited by some calcifying marine species
(McConnaughey, 1991; McConnaughey and Falk, 1991; Al-Horani et al., 2003). The
calcification of some marine algae and coral species is believed to be promoted by
extra-cellular transport of Ca2+
ions to calcification sites and removal of protons
through ATP-driven 2H+/Ca
2+ ion exchange which induces aragonite supersaturation
within the calcifying environment (McConnaughey, 1991; McConnaughey and Falk,
1991; Al-Horani et al., 2003). It follows that the quantitative decoupling of these two,

278
possibly overlapping proton/calcium exchange mechanisms, represents a new
scientific challenge to carbonate marine geochemists.
Clearly, quantitative interpretations of field or laboratory data that rely on
speciation calculations of the CaCO3(s)-H2O system should consider reaction 3 when
experiments are conducted under conditions favorable for proton/calcium exchange
such as: i) when pCO2 is variable, ii) when strong acid is added to a calcite suspension
and/or iii) when calcite powder is subjected to chemical pre-treatments (such as dilute
HCl leaching) before use in order to remove potential impurities or reduce surface
roughness (Zachara et al., 1991; Elzinga et al., 2006; Ahmed et al., 2008). For instance,
some researchers have reported anomalously high Ca equilibrium concentrations
upon HCl additions to highly concentrated calcite suspensions (Eriksson et al., 2007).
Similarly, field data reveal large Ca and TA anomalies in water column samples in
equilibrium with CaCO3(s) collected from the deep ocean that reflect an apparent
excess of calcium (or TA deficit) over that predicted from stoichiometric calcite
dissolution (Brewer et al., 1975). These findings could be, a priori, interpreted as the
incongruent dissolution of calcite whereas they more likely reflect the non-
stoichiometric release of Ca2+
over CO32-
ions induced by proton/calcium exchange.
The observed “apparent” incongruent dissolution regime is presumably a transient
stage of the overall dissolution process that remains active until saturation of available
exchangeable cation sites and might be eventually masked by a possible
destabilization of the calcium-deficient, proton-enriched leached layer under acidic
conditions (or at other PVT conditions) and the re-establishment of the congruent
dissolution regime. A detailed quantitative discussion of the role of proton/calcium

279
ion exchange in dictating aqueous speciation in carbonate-rock aquatic systems under
different chemical scenarios and its implications for the in-situ, long-term geological
storage of atmospheric CO2(g) and on the responses of marine carbonate-rich shelf
sediments to rising atmospheric pCO2 will be the subject of future work.
4. CONCLUSIONS
A novel experimental protocol was developed to quantitatively characterize the proton
sorptive properties of calcite in aqueous suspensions. Sorption data were acquired via
acidimetric and CaCl2 titrations conducted over a relatively wide range of chemical
conditions (pH, Ca, CO2 and solid:solution ratios). In contrast to expectations, a
large net proton uptake, coupled with a significant release of Ca2+
ions is consistently
observed. Because proton uptake greatly exceeds the theoretical number of available
reactive surface sites at the calcite surface, sorption data cannot be quantitatively
interpreted by adsorption or surface complexation reactions. Alternatively, these data
were interpreted on the basis of a proton/calcium ion exchange reaction, a possible
analogue of the physiologically-driven 2H+/Ca
2+ ion exchange behavior exhibited by
some calcifying marine organisms. The postulated 2H+/Ca
2+ ion exchange would
occur by a fast, chemically-driven equilibrium mechanism between the solution and
“exchangeable cation sites” (e.g., lattice positions) at and/or beneath the calcite
surface following the dynamic rearrangement of the near-surface calcite layers. The
latter would lead to a renewal of the adsorption sites, a net increase in proton uptake
capacity and the generation of a calcium-deficient, proton-enriched layer, under
circum-neutral and alkaline conditions, analogous to the leached layer developed by

280
chain-silicate minerals under acidic conditions. The application of NMR, Ion Beam
Analysis, and Raman Spectroscopy techniques is required for a detailed chemical
characterization of this layer and for determining the depth of proton penetration
within the calcite lattice.
This newly postulated proton/calcium exchange behavior of calcite largely
masks surface equilibria and directly impacts the solution chemistry of carbonate-rock
systems isolated from the atmosphere (closed-system) or where CO2 ventilation is
restricted (e.g., aquifers, pore and deep sea waters, industrial reactors) via pH and
calcite dissolution buffering. In contrast, in systems exposed to fixed pCO2 conditions
(e.g., open-systems), aqueous speciation remains unaffected because of CO2(g)
sequestration arising from 2H+/Ca
2+ ion exchange-induced calcite precipitation. The
postulated mechanism may partly explain the anomalous solution chemistry observed
in some field and laboratory studies. Accordingly, quantitative interpretations of
experimental data acquired at proton/calcium exchange inducing conditions (i.e.,
TOTH0 > 0 and/or TOTCa
0 < 0) require consideration of this reaction via the
cKex and
the ECSD values calibrated in this study. For instance, dissolution kinetics data
acquired following the incremental addition of a strong (e.g., HCl) or a weak acid
(i.e., CO2(g)) without full characterization of the solution chemistry may require a re-
evaluation since these may partly reflect the transient, non-stochiometric release of
Ca2+
and CO32-
ions to the solution upon ion exchange (i.e., “apparent” incongruent
dissolution), with the concomitant formation of a leached layer, rather than the
progressive destruction of the 3D crystallographic framework (i.e., congruent calcite
dissolution). Similarly, accurate predictions of the response of carbonate-rich shelf
sediments to rising atmospheric pCO2 must consider the pH and calcite saturation state

281
buffering capacity imparted by the proton/calcium exchange behavior of calcite and
possibly other carbonate minerals that make up the sediment assemblage.
Finally, the titration protocol introduced in this study is recommended for the
quantitative characterization of ion exchange and/or sorption reactions between calcite
(or its polymorphs: aragonite and vaterite) and other potential sorbates whose
activities can be measured with available Ion Selective Electrodes.
5. ACKNOWLEDGMENTS
A.V.-J. is grateful to Mrs. Rosy J.-R. for offering a stimulating environment
throughout the preparation of this paper. Special thanks go to Dr. Brian Phillips and
Dr Michel Rossi for providing additional information on their results. The insightful
comments of two anonymous reviewers are greatly appreciated. This research was
supported by a graduate student grant to A.V.-J. from the Geological Society of
America (GSA) and Natural Sciences and Engineering Research Council of Canada
(NSERC) Discovery grants to A.M. and J.P. A.V.-J. also benefited from post-graduate
scholarships from the Consejo Nacional de Ciencia y Tecnología (CONACyT) of
Mexico as well as financial support from the Department of Earth and Planetary
Sciences, McGill University and Consorcio Mexicano Flotus-Nanuk.

282
6. REFERENCES
Ahmed I.A.M., Crout N.M.J. and Young S.D. (2008) Kinetics of Cd sorption, desorption
and fixation by calcite: A long-term radiotracer study. Geochim. Cosmochim. Acta
72, 1498-1512.
Al-Horani F.A., Al-Moghrabi S.M., de Beer D. (2003) The mechanism of calcification
and its relation to photosynthesis and respiration in the scleractinian coral Galaxea
fascicularis. Mar. Biol. 142, 419-426.
Al-Hosney H.A. and Grassian V.H. (2005) Water, sulfur dioxide and nitric acid
adsorption on calcium carbonate: A transmission and ATR-FTIR study. Phys.
Chem. Chem. Phys. 7, 1266-1276.
Andersson A.J., Mackenzie F.T., Ver L.M. (2003) Solution of shallow-water carbonates:
An insignificant buffer against rising atmospheric. CO2. Geology 31(6), 513-516.
Andersson A.J., Mackenzie F.T., Lerman A. (2006) Coastal ocean CO2–carbonic acid–
carbonate sediment system of the Anthropocene. Global Biogeochem. Cycles
20:GB1S92, 1-13.
Arakaki T. and Mucci A. (1995) A continouous and mechanistic representation of calcite
reaction-controlled kinetics in dilute solutions at 25° and 1 atm total pressure.
Aquatic Geochem. 1, 105-130.
Brady P.V., Krumhans J.L. and Papenguth, H.W. (1996) Surface complexation clues to
dolomite growth. Geochim. Cosmochim. Acta 60(4), 727-731.
Brady P.V., Papenguth, H.W. and Kelly J.W. (1999) Metal sorption to dolomite surfaces.
Applied Geochem. 14(5), 569-579.

283
Brewer P.G., Wong G.T.F., Bacon M.P. and Spencer D.W. (1975) An oceanic calcium
problem? Earth Planet. Sci. Lett. 26, 81-87.
Bureau H., Raepsaet C., Khodja H., Carraro A. and Aubaud C. (2009) Determination of
hydrogen content in geological samples using elastic recoil detection analysis
(ERDA). Geochim. Cosmochim. Acta 73(11), 3311-3322.
Casey W.H., Westrich H.R., Banfield J.F., Ferruzi G. and Arnold G.W. (1993) Leaching
and reconstruction at surfaces of dissolving chain-silicate minerals. Nature 366,
253-256.
Charlet L., Wersin P. and Stumm W. (1990) Surface charge of MnCO3 and FeCO3.
Geochim. Cosmochim. Acta 54, 2329-2336.
Cicerone D.S., Regazzoni A.E. and Blesa M.A. (1992) Electrokinetic properties of the
calcite/water interface in the presence of magnesium and organic matter. J.
Colloid Interface Sci. 154, 423-433.
Comans R.N.J. and Middelburg J.J. (1987) Sorption of trace metals on calcite:
Applicability of the surface precipitation model. Geochem. Cosmochim. Acta 51,
2587-2591.
Curti E., Kulik D.A. and Tits J. (2005) Solid solutions of trace Eu(III) in calcite:
Thermodynamic evaluation of experimental data over a wide range of pH and
pCO2. Geochim. Cosmochim. Acta 69(7), 1721-1737.
Davis J.A., Fuller C.C. and Cook A.D. (1987) A model for trace metal sorption processes
at the calcite surface: Adsorption of Cd2+
and subsequent solid solution formation.
Geochim. Cosmochim. Acta 51(6), 1477-1490.

284
Douglas H.W. and Walker R.A. (1950) The electrokinetic behavior of iceland spar
against aqueous electrolyte solution. Trans. Faraday Soc. 46, 559-568.
Dzombak D.A. and Morel F.M.M. (1990) Surface Complexation Modeling. John Wiley
and Sons Inc.: New York, NY, Chapters 1-3.
Elfil H. and Roques H. (2001) Role of hydrate phases of calcium carbonate on the scaling
phenomenon. Desalination, 137, 177-186.
Elzinga E.J., Rouff A.A. and Reeder R.J. (2006) The long-term fate of Cu2+
, Zn2+
, and
Pb2+
adsorption complexes at the calcite surface: An X-ray absorption
spectroscopic study. Geochim. Cosmochim. Acta, 70, 2715-2725.
Eriksson R., Merta J. and Rosenholm J.B. (2007) The calcite/water interface I. Surface
charge in indifferent electrolyte media and the influence of low-molecular-weight
polyelectrolyte. J. Colloid Interface Sci. 313, 184-193.
Eriksson R., Merta J. and Rosenholm J.B. (2008) The calcite/water interface II. Effect of
added lattice ions on the charge properties and adsorption of sodium polyacrylate.
J. Colloid Interface Sci., 2008, 326, 396-402.
Felmy A.R., Dixon D.A., Rustad J.R., Mason M.J. and Onishi L.M. (1998) The
hydrolysis and carbonate complexation of strontium and calcium in aqueous
solution. Use of molecular modeling calculations in the development of aqueous
thermodynamic models. J. Chem. Thermodynamics 30, 1103-1120.
Feng J., Lee Y.J., Reeder R.J. and Phillips B.L. (2006) Observation of bicarbonate in
calcite by NMR spectroscopy. Am. Mineral. 91, 957-960.
Fisler D.K. and Cygan R.T. (1999) Diffusion of Ca and Mg in calcite. Am. Mineral. 84,
1392-1399.

285
Foxall T., Peterson G.C., Rendall H.M. and Smith A. L. (1979) Charge determination at
calcium salt/aqueous solution interface. J. Chem. Soc. Farad. Trans. 175, 1034-
1039.
Gaffey S.J. (1995) H2O and OH in echinoid calcite: A spectroscopic study. Am. Mineral.
80, 947-959.
Green E. and Lüttge A. (2006) Incongruent dissolution of wollastonite measured with
vertical scanning interferometry. Am. Mineral. 91, 430-434.
Herbelin A. and Westall, J. (1996) FITEQL- A computer program for determination of
chemical equilibrium constants from experimental data; version 3.2: user‟s
manual. Department of Chemistry. Oregon State University, Corvallis, OR,
Report 96-01.
Hoffmann U. and Stipp S.L.S. (2001). The behavior of Ni2+
on calcite surfaces. Geochim.
Cosmochim. Acta 65(22), 4131-4139.
Huang C.P. (1981). The surface acidity of hydrous solids in: Adsorption of Inorganics at
Solid-Liquid Interfaces. M.A. Anderson and A.J. Rubin (eds.) Ann Arbor Science,
Ann Arbor, Mich., pp. 183-217.
Huang, Y.C., Fowkes F.M., Lloyd, T.B. and Sanders, N.D. (1991) Adsorption of calcium
ions from calcium chloride solutions onto calcium carbonate particles. Langmuir
7, 1742-1748.
Mackenzie F.T., Bischoff W. D., Bishop F. C., Loijens M., Schoonmaker J. and Wollast,
R. In Carbonates: Mineralogy and chemistry; Reeder, R.J., Eds.; Rev. Mineral.
Mineralogical Society of America: Michigan, 1990; Vol 11, pp 97-144.

286
Martin-Garin A; Van Cappellen P. and Charlet, L. (2003) Aqueous cadmium uptake by
calcite: A stirred flow-through reactor study. Geochim. Cosmochim. Acta 67(15),
2763-2774.
McConnaughey T.A. (1991) Calcification in Chara corallina: CO2, hydroxylation
generates protons for bicarbonate assimilation. Limnol. Oceanogr. 36(4), 619-628.
McConnaughey T.A. and Falk R.H., (1991) Calcium-proton exchange during algal
calcification. Biol. Bull. 180, 185-195.
McGillen M.R. and Fairchild, I.J. (2005) An experimental study of incongruent
dissolution of CaCO3 under analogue glacial conditions. J. Glaciol. 51(174), 383-
390.
Mishra S.K. (1978) The electrokinetics of apatite and calcite in inorganic electrolyte
environment. Int. J. Miner. Process. 5, 69-83.
Morel F.M.M. and Hering, J. G. Principles and Applications of Aquatic Chemistry; John
Wiley and Sons Inc.: New York, NY, 1993; Chapters 1-6..
Morse J.W. and Mackenzie F.T. (1990) Geochemistry of Sedimentary Carbonates;
Develop. Sedimentol., 48. Elsevier: Amsterdam, Chapter 1.
Moulin P. and Roques H. (2003) Zeta potential measurement of calcium carbonate. J.
Colloid. Inter. Sci. 261, 115-126.
Mucci A., Morse J.W. and Kaminsky M.S. (1985) Auger spectroscopy analysis of
magnesian calcite overgrowths precipitated from seawater and solutions of similar
composition Am. J. Sci. 285, 289-305.

287
NIST (1998) Critically Selected Stability Constants of Metal Complexes, Standard
Reference Database 46, Version 5, National Institute of Standards and
Technology, US Department of Commerce, Gaithersburg, MD, USA.
Petit J.-C., Delia Mea G., Drand J.-C., Schott J. and Berner R.A. (1987) Mechanism of
diopside dissolution from hydrogen depth profiling. Nature, 325, 705-707.
Plummer L.N. and Busenberg E. (1982) The solubilities of calcite, aragonite and vaterite
in CO2-H2O solutions between 0 and 90°C, and an evaluation of the aqueous
model for the system CaCO3-CO2-H2O. Geochim. Cosmochim. Acta 46(6), 1011-
1040.
Pokrovsky O.S., Schott J. and Thomas F. (1999a) Processes at the magnesium-bearing
carbonates/solution interface. I. A surface speciation model for magnesite.
Geochim. Cosmochim. Acta 63(6), 863-880.
Pokrovsky O.S., Schott J. and Thomas F. (1999b) Dolomite surface speciation and
reactivity in aquatic systems. Geochim. Cosmochim. Acta 63(19/20), 3133-3143.
Pokrovsky O.S., Mielczarski J.A., Barres O., and Schott J. (2000) Surface speciation
models of calcite and dolomite/aqueous solution interfaces and their spectroscopic
investigation. Langmuir 16, 2677-2688.
Robbins L. L. and Fabry V. J. (1994) In Carbon Dioxide Chemistry: Environmental
Issues; Paul, J., Pradier, C., Eds.; The Royal Society of Chemistry: Cambridge,
U.K., pp 301-304.
Santschi Ch. and Rossi M.J. (2006) Uptake of CO2, SO2, HNO3 and HCl on calcite
(CaCO3) at 300 K: Mechanism and the role of adsorbed water. J. Phys. Chem. A
110, 6789-6802.

288
Siffert B. and Fimbel, P. (1984) Parameters affecting the sign and the magnitude of the
electrokinetic potential of calcite. Colloids Surf. 11, 377-389.
Somasundaran P. and Agar G.E. (1967) The zero point of charge of calcite. J. Colloid
Interface Sci. 24, 433-440.
Sposito G. (1998) On points of zero charge. Environ. Sci. Technol. 32(19), 2815-2819.
Stipp S.L. and Hochella M.F. Jr. (1991) Structure and bonding environments at the calcite
surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy
electron diffraction (LEED). Geochim. Cosmochim. Acta 55, 1723-1736.
Stipp S.L.S., Hochella, F., Parks, G.A. and Leckie J.O. (1992) Cd2+
uptake by calcite,
solid-state diffusion, and the formation of solid-solution: Interface processes
observed with near-surface sensitive techniques (XPS, LEED, and AES).
Geochim. Cosmochim. Acta 56, 1941-1954.
Stipp S.L.S., Gutmannsbauer W. and Lehmann, T. (1996) The dynamic nature of calcite
surfaces in air. Am. Mineral. 81,1-8.
Stipp, S.L.S., Kulik A.J., Franzreb K., Benoit W. and Mathieu H.J. A (1997) Combination
of SFM and TOF-SIMS imaging for observing local inhomogenieties in
morphology and composition: Aged calcite surfaces. Surf. Interface Analysis 25,
959-965.
Stipp S.L.S., Konnerup-Madsen K., Franzreb K., Kulik A. and Mathieu H.J. (1998)
Spontaneous movement of ions through calcite at standard temperature and
pressure. Nature 396, 356-359.
Stoessell R.K. (1998) Binary cation exchange reactions. Clays Clay Miner. 46(2), 215-
218.

289
Tas C.A. (2007) Porous, biphasic CaCO3-calcium phosphate biomedical cement scaffolds
from calcite (caco3) powder Int. J. Appl. Ceram. Technol, 4(2), 152-163.
Thompson D.W. and Pownall P.G. (1989) Surface electrical properties of calcite. J.
Colloid. Inter. Sci. 131, 74-83.
Tossel J.A. (2006) H2CO3 and its oligomers: Structures, stabilities, vibrational and NMR
spectra, and acidities. Inorg. Chem., 45, 5961-5970.
Usher C.R., Michel A.E. and Grassian V.H. (2003) Reactions on mineral dust. Chem.
Rev. 103, 4883-4939.
Van Cappellen P., Charlet L., Stumm W. and Wersin P. (1993) A surface complexation
model of the carbonate mineral-aqueous solution interface. Geochim. Cosmochim.
Acta 57, 3505-3518.
Vanerek A., Alince B. and van de Ven T. G. M. (2000) Interaction of calcium carbonate
fillers with pulp fibres: Effect of surface charge and cationic polyelectrolytes. J.
Pulp Paper Sci. 26:9, 317-322
Villegas-Jiménez A., Mucci A. and Whitehead M.A. (2009) Theoretical insights into the
hydrated (10.4) calcite surface: structure, energetic, and bonding relationships.
Langmuir 25(12), 6813-6824.
Westall J. and Hohl H. (1980) A comparison of electrostatic models for the oxide/solution
interface. Adv. Colloid Int. Sci. 12, 265-294.
Wolthers M., Charlet L., and Van Cappellen P.V. (2008) The surface chemistry of
divalent metal carbonate minerals; A critical assessment of surface charge and
potential data using the charge distribution multi-site ion complexation model.
Am. J. Sci. 308, 905-941.

290
Zachara J.M., Kittrick, J.A., Harsh J.B. (1988) The mechanism of Zn2+
adsorption on
calcite. Geochim. Cosmochim. Acta 52, 2281-2291.
Zachara J.M., Cowan C.E. and Resch C.T. (1991) Sorption of divalent metals on calcite.
Geochim. Cosmochim. Acta 55, 1549-1562.
Zhong S. and Mucci A. (1995) Partitioning of rare earth elements (REEs) between calcite
and seawater solutions at 25°C and 1 atm and high dissolved REE concentrations.
Geochem. Cosmochim. Acta 59, 443-453.
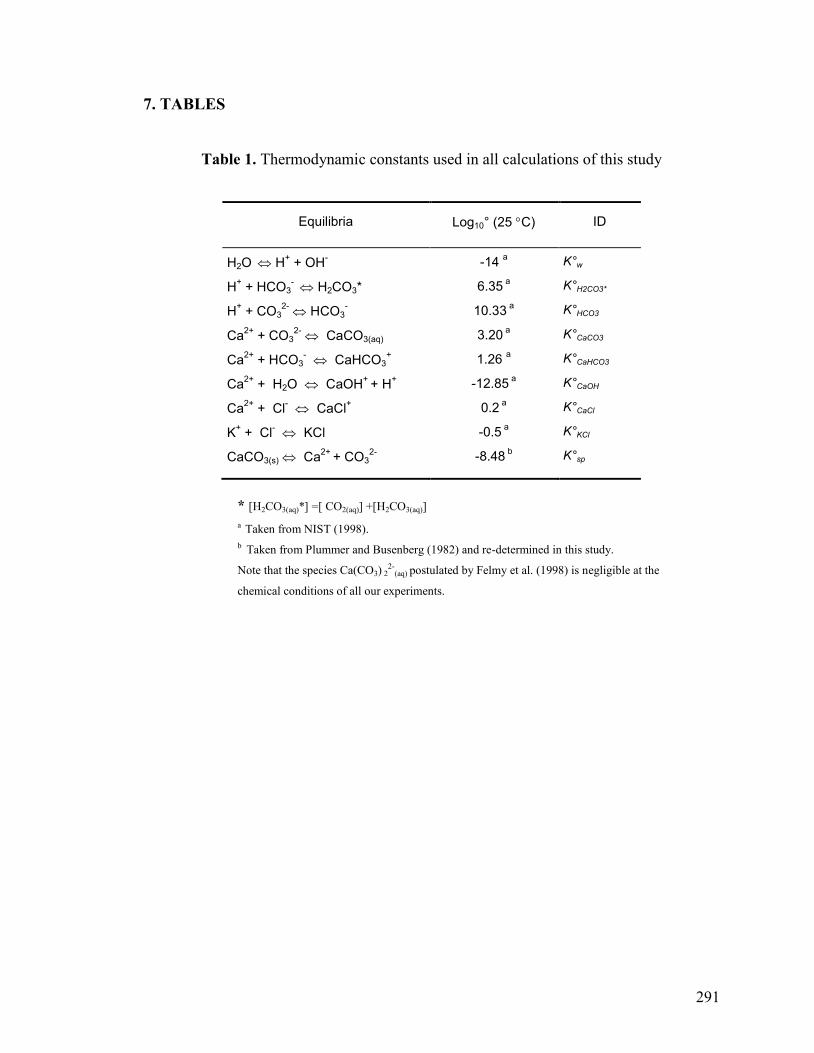
291
7. TABLES
Table 1. Thermodynamic constants used in all calculations of this study
Equilibria Log10° (25 C) ID
H2O
H
+ + OH
- -14
a K°w
H+ + HCO3
- H2CO3* 6.35
a K°H2CO3*
H+ + CO3
2- HCO3
- 10.33
a K°HCO3
Ca2+
+ CO32-
CaCO3(aq) 3.20 a K°CaCO3
Ca2+
+ HCO3- CaHCO3
+ 1.26
a K°CaHCO3
Ca2+
+ H2O CaOH+
+ H+ -12.85
a K°CaOH
Ca2+
+ Cl- CaCl
+ 0.2
a K°CaCl
K+ + Cl
- KCl -0.5
a K°KCl
CaCO3(s) Ca2+
+ CO32-
-8.48 b K°sp
* [H2CO3(aq)*] =[ CO2(aq)] +[H2CO3(aq)]
a Taken from NIST (1998).
b Taken from Plummer and Busenberg (1982) and re-determined in this study.
Note that the species Ca(CO3) 22-
(aq) postulated by Felmy et al. (1998) is negligible at the
chemical conditions of all our experiments.
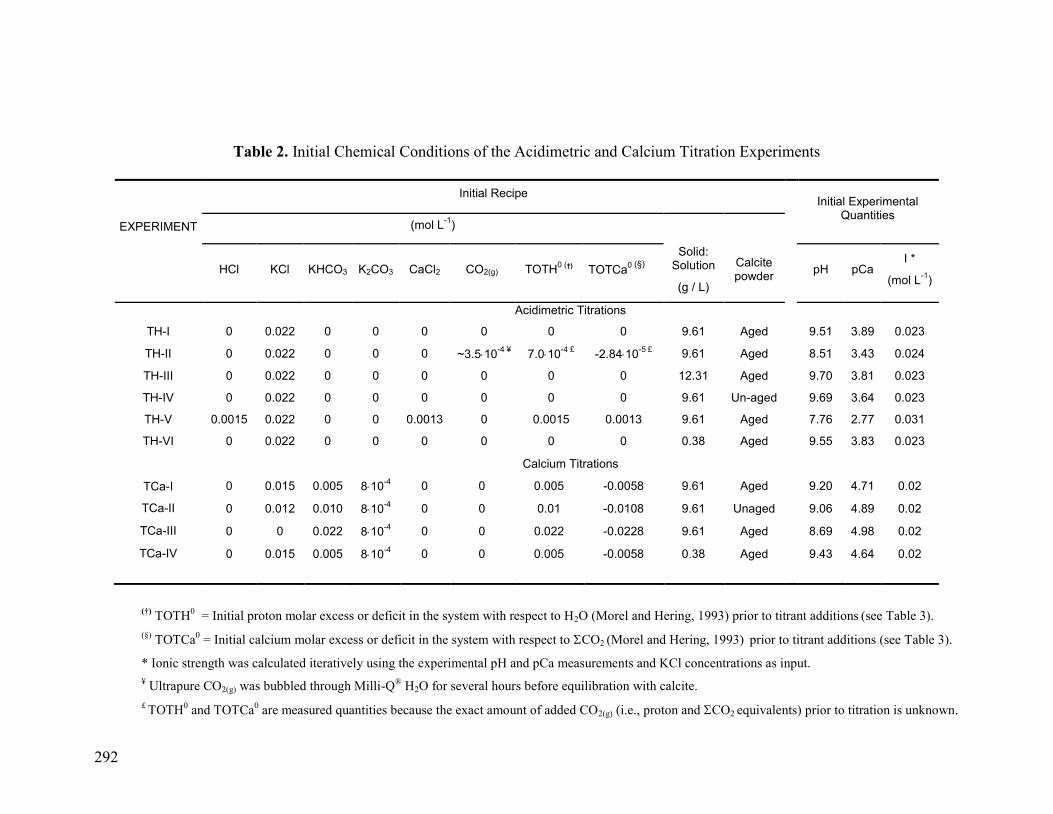
292
Table 2. Initial Chemical Conditions of the Acidimetric and Calcium Titration Experiments
EXPERIMENT
Initial Recipe Initial Experimental
Quantities (mol L
-1)
HCl KCl KHCO3 K2CO3 CaCl2 CO2(g) TOTH0 (†) TOTCa
0 (§)
Solid: Solution
(g / L)
Calcite powder
pH pCa I *
(mol L-1
)
Acidimetric Titrations
TH-I 0 0.022 0 0 0 0 0 0 9.61 Aged 9.51 3.89 0.023
TH-II 0 0.022 0 0 0 ~3.510-4 ¥
7.010-4 £
-2.8410-5 £
9.61 Aged 8.51 3.43 0.024
TH-III 0 0.022 0 0 0 0 0 0 12.31 Aged 9.70 3.81 0.023
TH-IV 0 0.022 0 0 0 0 0 0 9.61 Un-aged 9.69 3.64 0.023
TH-V 0.0015 0.022 0 0 0.0013 0 0.0015 0.0013 9.61 Aged 7.76 2.77 0.031
TH-VI 0 0.022 0 0 0 0 0 0 0.38 Aged 9.55 3.83 0.023
Calcium Titrations
TCa-I 0 0.015 0.005 810-4
0 0 0.005 -0.0058 9.61 Aged 9.20 4.71 0.02
TCa-II 0 0.012 0.010 810-4
0 0 0.01 -0.0108 9.61 Unaged 9.06 4.89 0.02
TCa-III 0 0 0.022 810-4
0 0 0.022 -0.0228 9.61 Aged 8.69 4.98 0.02
TCa-IV 0 0.015 0.005 810-4
0 0 0.005 -0.0058 0.38 Aged 9.43 4.64 0.02
(†) TOTH
0 = Initial proton molar excess or deficit in the system with respect to H2O (Morel and Hering, 1993) prior to titrant additions
(see Table 3).
(§)
TOTCa0 = Initial calcium molar excess or deficit in the system with respect to CO2 (Morel and Hering, 1993)
prior to titrant additions (see Table 3).
* Ionic strength was calculated iteratively using the experimental pH and pCa measurements and KCl concentrations as input.
¥ Ultrapure CO2(g) was bubbled through Milli-Q
® H2O for several hours before equilibration with calcite.
£ TOTH
0 and TOTCa
0 are measured quantities because the exact amount of added CO2(g) (i.e., proton and CO2 equivalents) prior to titration is unknown.

293
Table 3. Tableau-based aqueous phase definitions, mass and mole balance equations and
associated nomenclature relevant to the computation of sorption data for the experimental
CaCO3(s)-KCl-H2O chemical system
Mass Balance Equations
H= [H+] + [HCO3-] + 2 [H2CO3]* + [CaHCO3
+] – [CaOH+] – [OH-]
Ca = [Ca2+] + [CaOH+] + [CaCl+] + [CaHCO3+] + [CaCO3(aq)]
CO2 = [H2CO3]* + [HCO3-] + [CO3
2-] + [CaHCO3+] + [CaCO3(aq)]
K = [K+] + [KCl]
Cl = [Cl-] + [CaCl+] + [KCl]
Mole Balance Equations
TOTH = H = CA - CB: Proton molar excess or deficit in the system with respect to H2O
CA: Total molar concentration of acid in the system
CB: Total molar concentration of base in the system
TOTCa = Ca - CO2: Calcium molar excess or deficit in the system with respect to CO2
TOTCO2 = CO2 - Ca: Carbon molar excess or deficit in the system with respect to Ca
TOTK = K, TOTCl = Cl
Associated Nomenclature
PROTON
TOTH0 = CA - CB at initial conditions prior to titrant additions = [HCl] + [KHCO3] + 2 [CO2(g)] (see Table 2)
TOTH*Theo= Theoretical† TOTH values upon cumulative titrant additions
TOTH*Exp = Experimental£ TOTH values upon cumulative titrant additions
CALCIUM
TOTCa0 = Ca - CO2 at initial conditions prior to titrant additions = [CaCl2] - [K2CO3] - [KHCO3] - [CO2(g)] (see Table 2)
TOTCa*Theo= Theoretical¥ TOTCa values upon cumulative titrant additions
TOTCa*Exp = Experimental£ TOTCa values upon cumulative titrant additions
Ca*Theo = Theoretical§ Ca
Ca*Exp = Experimental cumulative £Ca
CARBON
CO2*Theo = Theoretical§ CO2 upon cumulative titrant additions
CO2*Exp = Experimental£ CO2 upon cumulative titrant additions
CO2* = CO2*Theo - CO2*Exp : CO2 molar excess or deficit relative to the theoretical CO2 arising from
the stoichiometric calcite dissolution
TOTAL ALKALINITY (TA) = -[H+] + [OH-]+ [HCO3
-] + 2 [CO32-] + [CaHCO3
+] + 2 [CaCO3o] + [CaOH+]
Symbology
† = TOTH0 + [HCl]stepwise-additions
£ = Computed from ISEs readings, aqueous equilibria in Table 1 and the Davies Equation41
¥ = TOTCa0 + [CaCl2]stepwise-additions
§ = Computed from speciation calculations in the CaCO3(s)-KCl-H2O system with MINEQL v4.3 using the corresponding
values of TOTCa*Theo, TOTH*Theo, K, and Cl, at each titration point, as input
All quantities are given in molar concentrations. Titrant: HCl or CaCl2. The superscripts specify the state of the titration (0:
prior to titration; *: cumulative) whereas the subscripts specify the nature of the parameter of interest (Theo: Theoretical; Exp:
Experimental). The adopted nomenclature is consistent with the Tableau method.
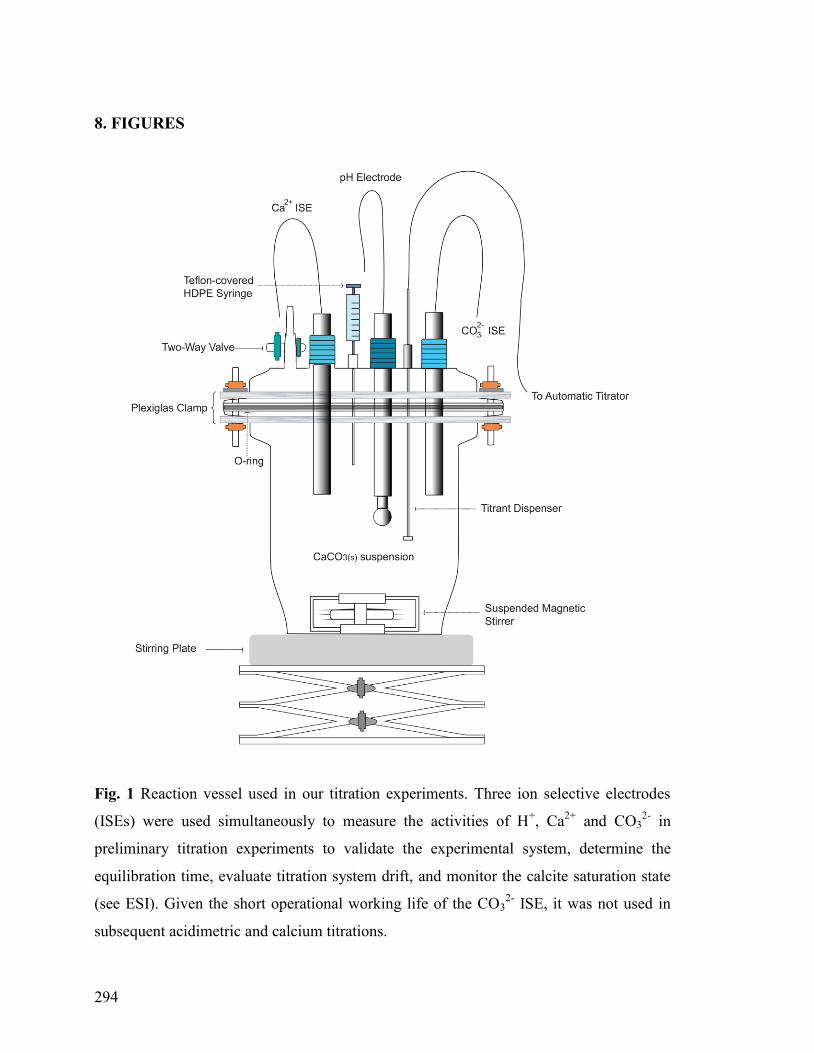
294
8. FIGURES
Fig. 1 Reaction vessel used in our titration experiments. Three ion selective electrodes
(ISEs) were used simultaneously to measure the activities of H+, Ca
2+ and CO3
2- in
preliminary titration experiments to validate the experimental system, determine the
equilibration time, evaluate titration system drift, and monitor the calcite saturation state
(see ESI). Given the short operational working life of the CO32-
ISE, it was not used in
subsequent acidimetric and calcium titrations.
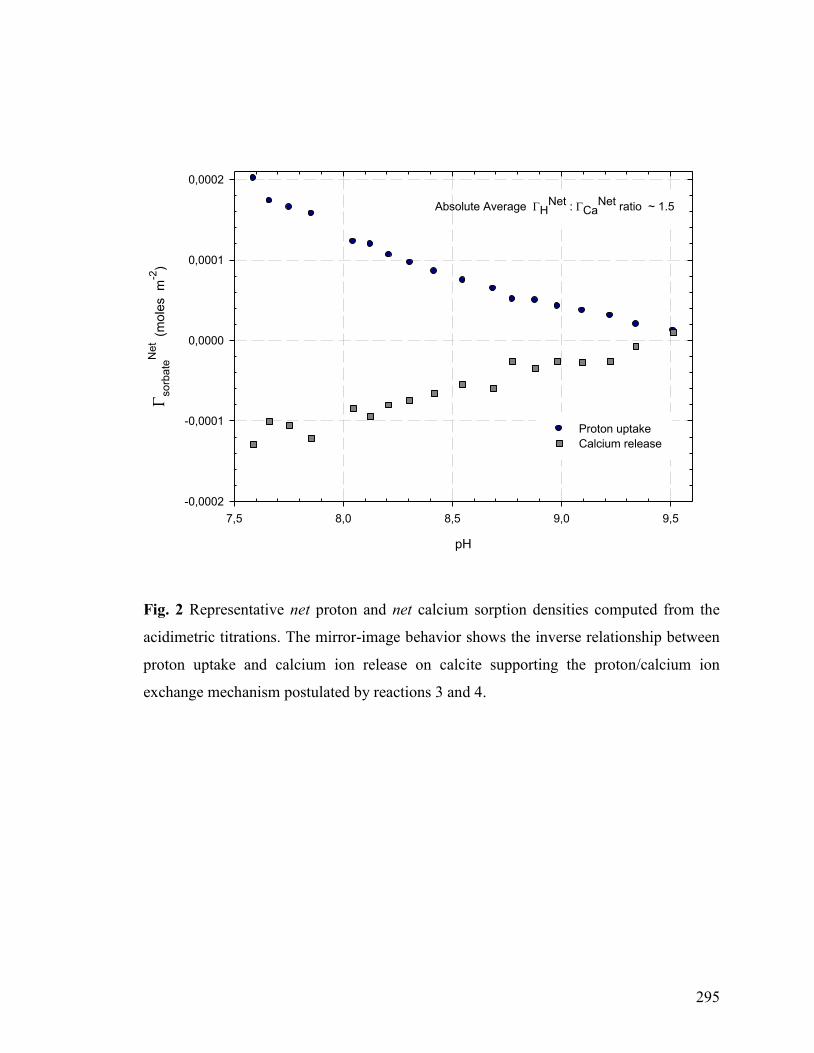
295
pH
7,5 8,0 8,5 9,0 9,5
sorb
ate
Net (m
ole
s m
-2)
-0,0002
-0,0001
0,0000
0,0001
0,0002
Proton uptake
Calcium release
Absolute Average HNet : Ca
Net ratio ~ 1.5
Fig. 2 Representative net proton and net calcium sorption densities computed from the
acidimetric titrations. The mirror-image behavior shows the inverse relationship between
proton uptake and calcium ion release on calcite supporting the proton/calcium ion
exchange mechanism postulated by reactions 3 and 4.
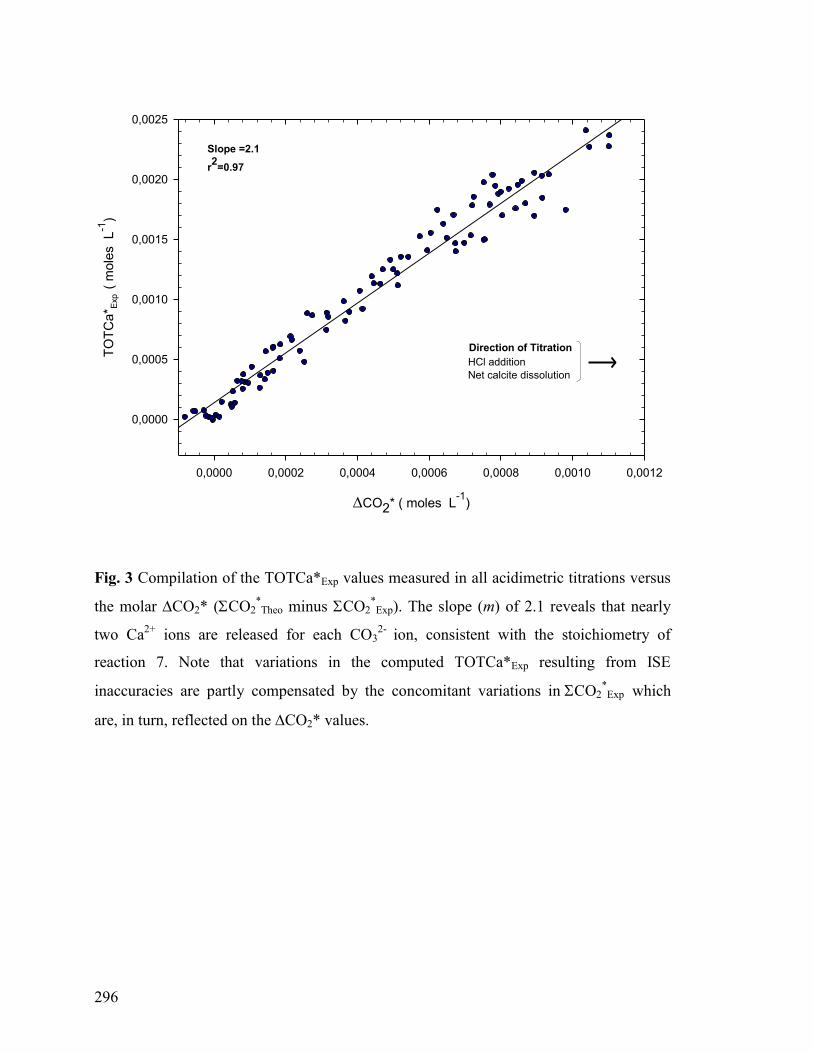
296
Slope =2.1
r2
=0.97
CO2* ( moles L-1
)
0,0000 0,0002 0,0004 0,0006 0,0008 0,0010 0,0012
TO
TC
a* E
xp
(
mole
s L
-1)
0,0000
0,0005
0,0010
0,0015
0,0020
0,0025
Direction of Titration
HCl addition
Net calcite dissolution
Fig. 3 Compilation of the TOTCa*Exp values measured in all acidimetric titrations versus
the molar CO2* (CO2*Theo minus CO2
*Exp). The slope (m) of 2.1 reveals that nearly
two Ca2+
ions are released for each CO32-
ion, consistent with the stoichiometry of
reaction 7. Note that variations in the computed TOTCa*Exp resulting from ISE
inaccuracies are partly compensated by the concomitant variations inCO2*Expwhich
are, in turn, reflected on the CO2* values.
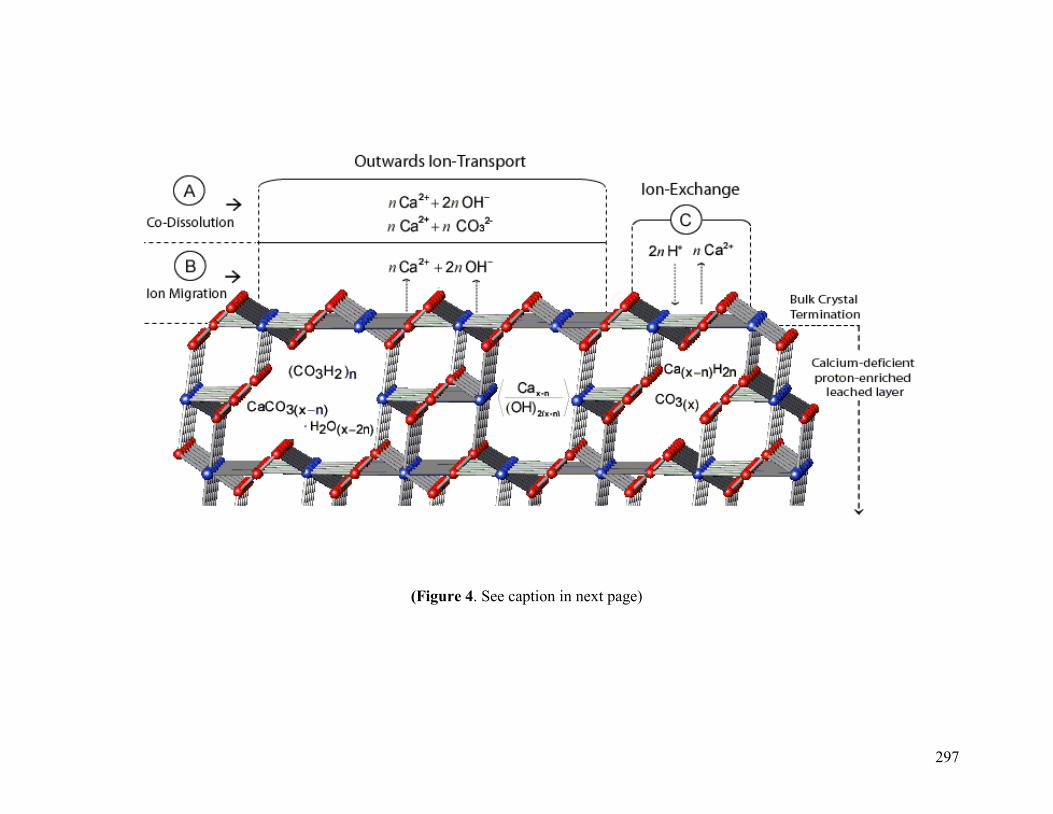
297
(Figure 4. See caption in next page)

298
Fig. 4 Schematic representation of three equivalent mechanisms accounting for the pH and pCa behavior observed in the acidimetric
and calcium titrations. Panels A and B consider the hypothetical presence of hydroxyl-bearing species (CaCO3nH2O(s) and/or
Ca(OH)2(s)) embedded within the calcite lattice prior to titrations. If co-dissolution of Ca(OH)2(s) and CaCO3(s) species occurs
(Panel A), no lattice vacancy is generated. In contrast, if Ca2+
and OH- ions are removed from their lattice positions by ion migration
along surface features such as micro-fractures, lattice vacancies might be generated (Panel B). Panel C schematizes the substitution of
Ca2+
lattice ions by protons via a chemically-driven 2H+/Ca
2+ ion exchange mechanism between the bulk solution and a “labile”,
possibly hydrated, interfacial region following the dynamic rearrangement of the calcite topmost atomic layers. In scenario C, no
lattice vacancies are generated because protons occupy lattice sites formerly occupied by Ca2+
ions at exchangeable cation sites and
yield Ca(HCO3)2(exc) and/or H2CO3(exc) lattice species as defined respectively by reactions 3 and 4.
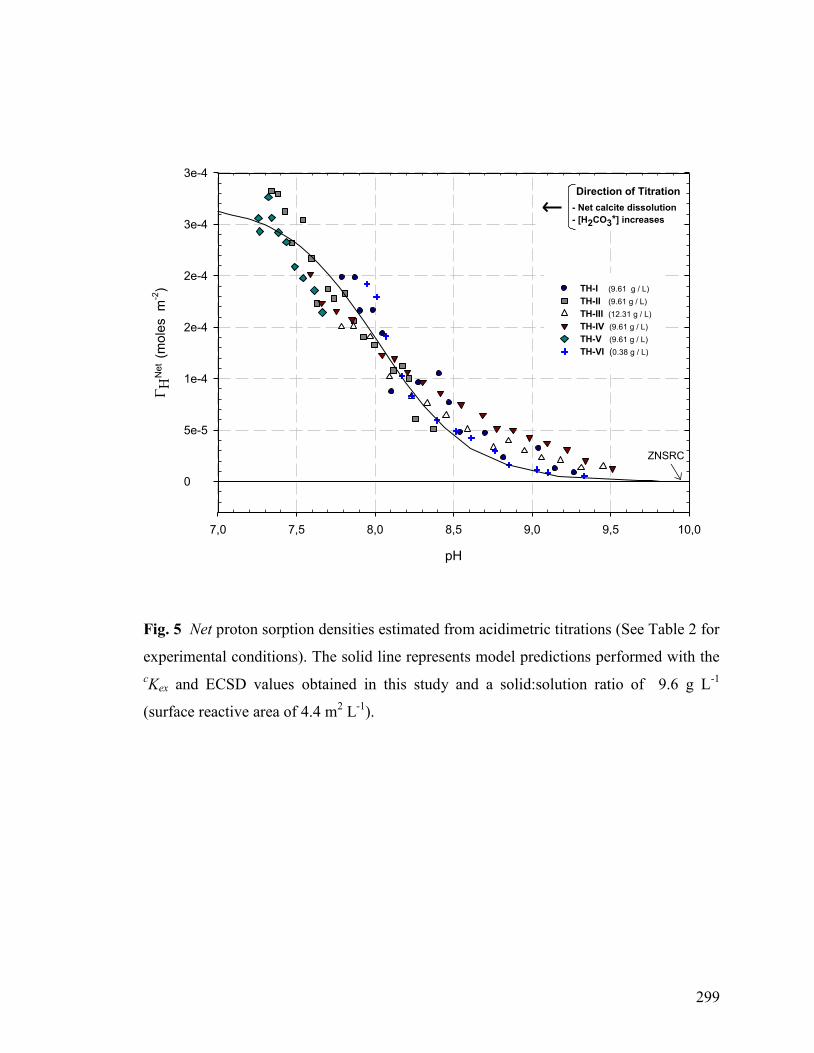
299
pH
7,0 7,5 8,0 8,5 9,0 9,5 10,0
Net (m
ole
s m
-2)
0
5e-5
1e-4
2e-4
2e-4
3e-4
3e-4
TH-I (9.61 g / L)
TH-II (9.61 g / L)
TH-III (12.31 g / L)
TH-IV (9.61 g / L)
TH-V (9.61 g / L)
TH-VI (0.38 g / L)
Direction of Titration
- Net calcite dissolution
- [H2CO3*] increases
ZNSRC
Fig. 5 Net proton sorption densities estimated from acidimetric titrations (See Table 2 for
experimental conditions). The solid line represents model predictions performed with the
cKex and ECSD values obtained in this study and a solid:solution ratio of 9.6 g L
-1
(surface reactive area of 4.4 m2 L
-1).
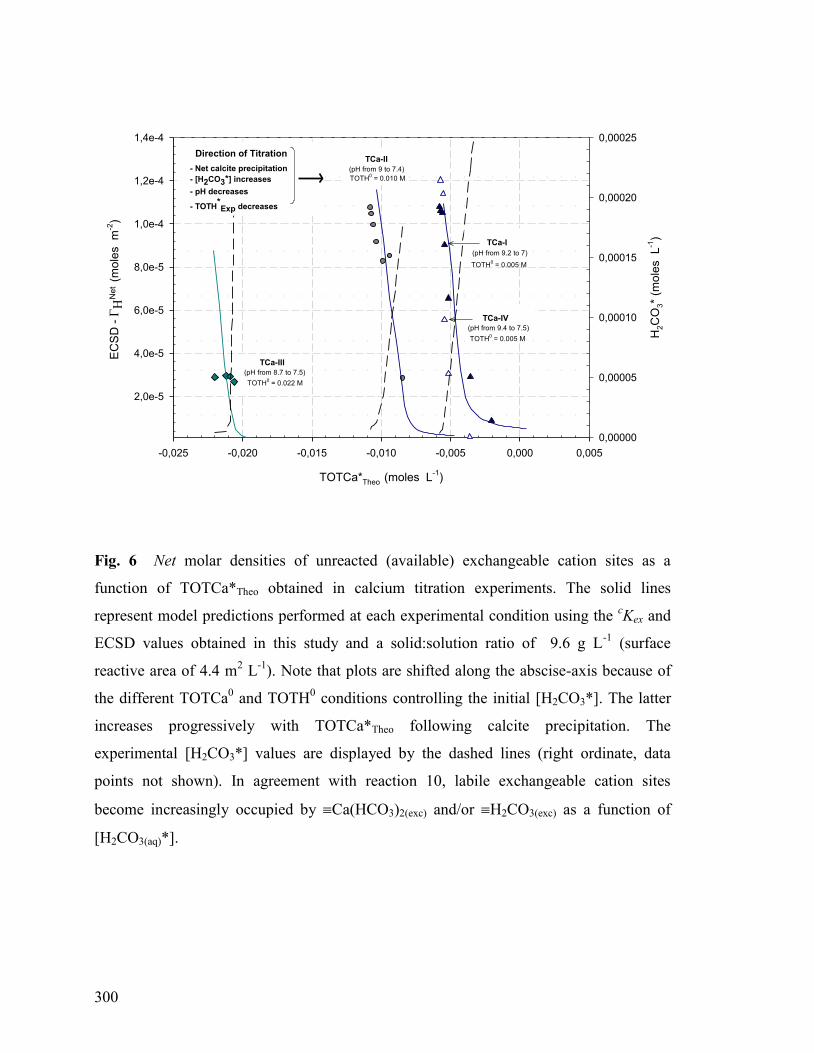
300
TOTCa*Theo (moles L-1
)
-0,025 -0,020 -0,015 -0,010 -0,005 0,000 0,005
EC
SD
-
Net (m
ole
s m
-2)
2,0e-5
4,0e-5
6,0e-5
8,0e-5
1,0e-4
1,2e-4
1,4e-4
H2C
O3*
(mo
les L
-1)
0,00000
0,00005
0,00010
0,00015
0,00020
0,00025
Direction of Titration
- Net calcite precipitation
- [H2CO3*] increases
- pH decreases
- TOTH*Exp decreases
TCa-I
(pH from 9.2 to 7)
TOTH0 = 0.005 M
TCa-II
(pH from 9 to 7.4)
TOTH0 = 0.010 M
TCa-IV(pH from 9.4 to 7.5)
TOTH0 = 0.005 M
TCa-III (pH from 8.7 to 7.5)
TOTH0 = 0.022 M
Fig. 6 Net molar densities of unreacted (available) exchangeable cation sites as a
function of TOTCa*Theo obtained in calcium titration experiments. The solid lines
represent model predictions performed at each experimental condition using the cKex and
ECSD values obtained in this study and a solid:solution ratio of 9.6 g L-1
(surface
reactive area of 4.4 m2 L
-1). Note that plots are shifted along the abscise-axis because of
the different TOTCa0 and TOTH
0 conditions controlling the initial [H2CO3*]. The latter
increases progressively with TOTCa*Theo following calcite precipitation. The
experimental [H2CO3*] values are displayed by the dashed lines (right ordinate, data
points not shown). In agreement with reaction 10, labile exchangeable cation sites
become increasingly occupied by Ca(HCO3)2(exc) and/or H2CO3(exc) as a function of
[H2CO3(aq)*].
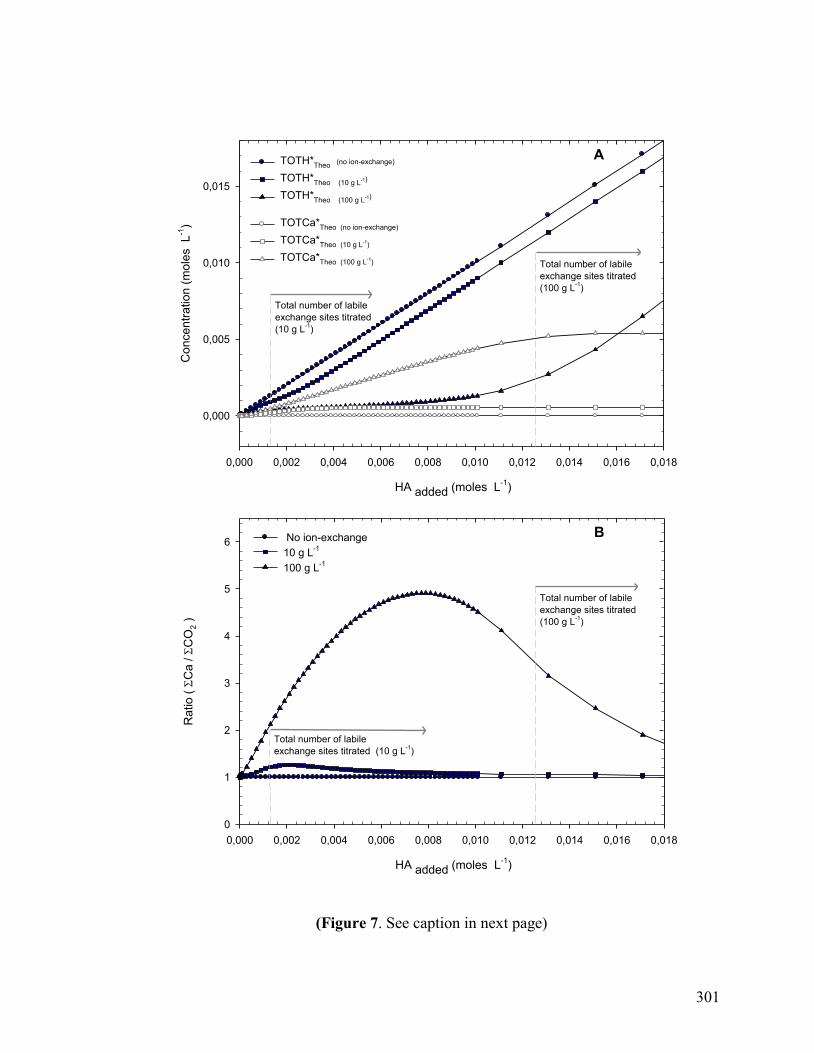
301
HA added (moles L-1
)
0,000 0,002 0,004 0,006 0,008 0,010 0,012 0,014 0,016 0,018
Concentr
ation (
mole
s L
-1)
0,000
0,005
0,010
0,015
TOTH*Theo
(no ion-exchange)
TOTH*Theo (10 g L
-1)
TOTH*Theo (100 g L
-1)
TOTCa*Theo (no ion-exchange)
TOTCa*Theo (10 g L
-1)
TOTCa*Theo (100 g L
-1)
A
Total number of labile
exchange sites titrated
(10 g L-1)
Total number of labile
exchange sites titrated
(100 g L-1)
HA added (moles L-1
)
0,000 0,002 0,004 0,006 0,008 0,010 0,012 0,014 0,016 0,018
Ratio (
C
a /
CO
2 )
0
1
2
3
4
5
6 No ion-exchange
10 g L-1
100 g L-1
B
Total number of labile
exchange sites titrated (10 g L-1)
Total number of labile
exchange sites titrated
(100 g L-1)
(Figure 7. See caption in next page)

302
Fig. 7 Predicted TOTH*Theo and TOTCa*Theo values (Panel A) and Ca:CO2 ratios
(Panel B) upon a hypothetical acidimetric titration (HA: strong acid) conducted in a
closed CaCO3(s)-H2O system (at TOTH0 and TOTCa
0=0), with and without consideration
of proton/calcium ion exchange and at different solid:solution ratios using log10 cKex = 13
and the average ECSD value obtained in this study (13.5·10-5
moles m-2
or 6.2·10-5
moles
g-1
).
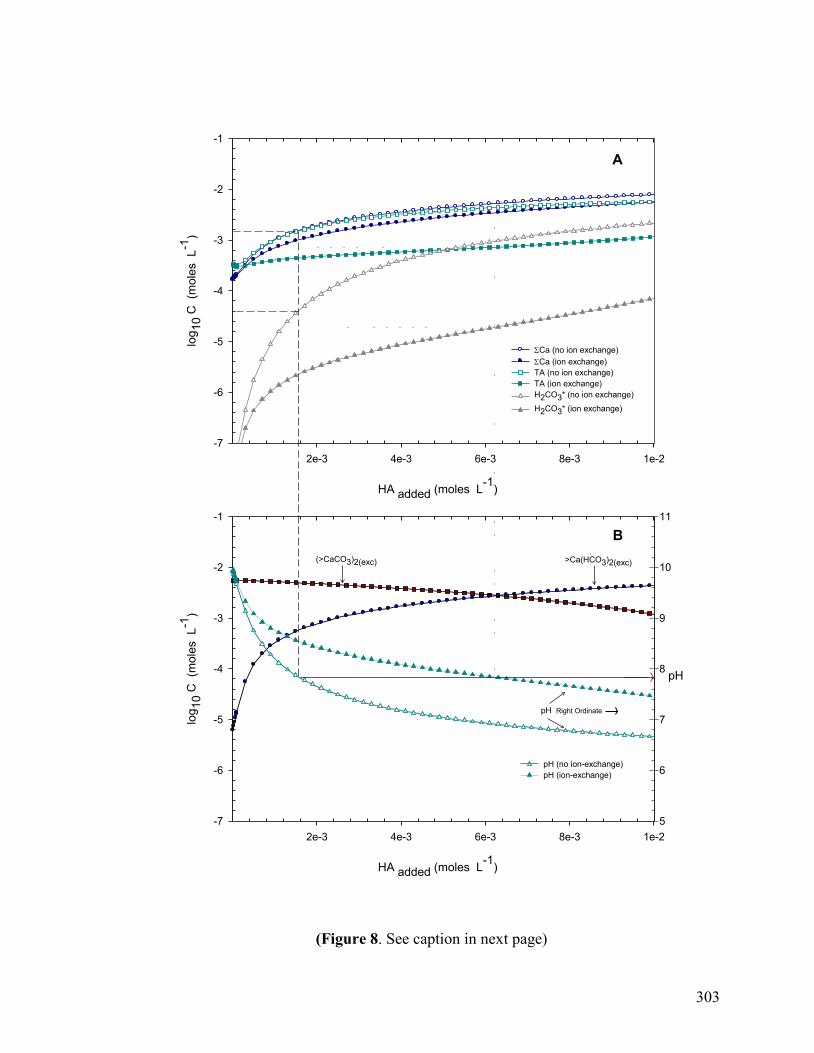
303
HA added (moles L-1
)
2e-3 4e-3 6e-3 8e-3 1e-2
log10
C (m
ole
s L
-1)
-7
-6
-5
-4
-3
-2
-1
HA added (moles L-1
)
2e-3 4e-3 6e-3 8e-3 1e-2
pH
log10
C (m
ole
s L
-1)
-7
-6
-5
-4
-3
-2
-1
5
6
7
8
9
10
11
>Ca(HCO3)2(exc)(>CaCO3)2(exc)
pH Right Ordinate
Ca (no ion exchange)
Ca (ion exchange)
TA (no ion exchange)
TA (ion exchange)
H2CO3* (no ion exchange)
H2CO3* (ion exchange)
pH (no ion-exchange)
pH (ion-exchange)
A
B
(Figure 8. See caption in next page)

304
Fig. 8 Predicted Ca, Total Alkalinity (TA), [H2CO3*] (Panel A), pH and speciation of
labile exchangeable cation sites (Panel B) upon a hypothetical acidimetric titration (HA:
strong acid) in a closed CaCO3(s)-H2O system with (solid symbols) and without (open
symbols) consideration of proton/calcium ion exchange. Ion exchange speciation
predictions were performed at a fixed solid:solution ratio of 100 g L-1
(reactive surface
area of 0.46 m2 L
-1) using log10
cKex = 13 and the average ECSD value obtained in this
study (13.5·10-5
moles m-2
or 6.2·10-5
moles g-1
). Under these conditions, equimolar
concentrations of protonated and non-protonated (“unreacted”) labile exchangeable sites
are registered at a pH of 7.8. Drop dotted lines intersecting panels A and B are
references to facilitate comparison of Ca, TA and [H2CO3*] values predicted with and
without consideration of ion proton/calcium exchange at pH=7.8.

305
CHAPTER 7
GENERAL CONCLUSIONS
CONTRIBUTIONS TO KNOWLEDGE
Despite significant scientific efforts focused on carbonate minerals, some fundamental
aspects of the surface reactivity of these minerals in aqueous solutions, such as ion
sorption processes, remain poorly characterized. Because of the higher reactivities of
carbonates relative to other minerals such as metal oxides, silicates, and clays, and the
occurrence of stepwise and/or parallel reactions, the experimental characterization of
these processes represents a sizable challenge. This explains why the systematic
acquisition of experimental data and the elaboration, calibration, and validation of
empirical, semi-empirical, and/or theoretical models describing sorption phenomena on
carbonate minerals have lagged behind those of less reactive minerals. Consequently, the
combination of multiple investigative approaches represents a suitable strategy to
improve our understanding of the sorptive properties of these minerals. In addition, the
development of novel experimental protocols extending the range of experimental
conditions under which the sorptive properties of carbonate minerals can be investigated
would be desirable. In this dissertation, we combined theoretical, experimental, computer-
assisted molecular modeling, and numerical simulation approaches to investigate sorption
phenomena at carbonate surfaces in aqueous solutions under ambient conditions. In doing
so, we introduced several elements of novelty that allowed new interpretations and fresh
insights into the reactivity of these surfaces.
We implemented a powerful evolutionary programming technique, the Genetic
Algorithm (GA) that allowed, for the first time, the rigorous calibration of SCMs for

306
magnesite and dolomite via stochastic numerical optimization. The GA is simple,
flexible, and robust and possesses important advantages over conventional numerical
techniques used in commercially-available programs with inverse modeling applications
such as FITEQL. The GA is a powerful tool to estimate intrinsic formation constants of
mineral surface species, a critical step in the generation of a reliable thermodynamic
database. Given the ability of our GA-based optimization code to simultaneous fit
intrinsic ionization and affinity constants, capacitance(s), and/or site densities, we
strongly recommend its use for the calibration of sophisticated Surface Complexation
Models (SCMs), particularly those that invoke numerous adjustable parameters or when
extensive data sets are not available for model calibration. A future user-friendly version
will help generalize its use among SCM practitioners and modelers.
Despite the success of earlier SCMs to simulate the surface charge behavior of
rhombohedral carbonate mineral surfaces, a critical analysis of the definition of surface
sites revealed that the postulated reactions may not necessarily reflect realistic processes
at the carbonate/water interface and may yield questionable predictions of surface
speciation. Consequently, we formulated, calibrated, and tested chemically-sound and
simplified SCMs for two representative rhombohedral carbonate minerals: MgCO3(s)
(magnesite) and CaMg(CO3)2(s) (dolomite). The models include proton/bicarbonate ion
co-adsorption as one of the fundamental mechanisms to explain the surface charge-
buffering behavior and the relatively wide range of pH values of isoelectric point (pHiep)
displayed by these minerals in aqueous solutions. In analogy to the one-site 2-pKa
ionization model frequently adopted for metal oxides, our SCMs are formalized in terms
of a single-site that renders them more mathematically-tractable than earlier multi-site
complexation models that require numerous reactions to reproduce experimental data.

307
Although, we recognize that, under specific chemical conditions, multi-site complexation
may occur at carbonate surfaces, its relevance for the quantitative description of available
experimental data is debatable because it is well known that surface irregularities (e.g.,
steps, kinks and dislocations), chemical micro-heterogeneities, and the existence of multi-
domain crystal surfaces, presumably allowing for the presence of multiple reactive sites,
are properties that are difficult to assess quantitatively. Accordingly, the applicability of
the multi-site approach is limited to well-characterized surfaces and to high quality
experimental data suitable for the proper calibration of the multi-site-based surface
reaction(s) of interest. Because the simplified SCMs are a fair compromise between the
quality of the experimental data available for model calibration, the compatibility of the
models with physical/chemical constraints, and the viability of the SCM predictions, it is
a convenient and realistic approach to use in the construct of SCMs for other
rhombohedral carbonate minerals and represents an important advance in the
rationalization/interpretation of available experimental surface charge data for this type of
minerals.
The applicability of the one-site scheme was extended to other rhombohedral
carbonate minerals using the least reactive of known naturally-occurring rhombohedral
carbonate minerals in aqueous solutions, gaspeite (NiCO3(s)), as a surrogate of calcite-type
minerals. Conventional surface titrations applied to gaspeite allowed us to obtain
quantitative proton adsorption data for the calibration of a SCM for this mineral. To this
end, we employed conventional titration techniques, never applied before to carbonate
minerals, and generated abundant electrokinetic data to qualitatively evaluate the
predictive power of the derived SCM. The most important insights obtained in this study
is that the background electrolyte (NaCl) affects the properties of the gaspeite surface

308
(surface protonation and the development of surface charge) possibly through
modification of the structure of the electrified interfacial layer, perturbation of the solvent
structure dynamics and the affinity of water molecules and adsorbing ions towards the
mineral surface. These observations challenge earlier conceptions on carbonate mineral
surfaces that traditionally considered these minerals as chemically inert to background
electrolyte ions. Clearly, carbonate surfaces behave differently than most metal oxides,
silicates, and clay surfaces in aqueous solutions and, hence, electrolyte effects should be
carefully examined in future studies through alternative experimental approaches and/or
using different background electrolytes. This should help fine tune the postulated SCMs
and extend their applicability to solution conditions beyond those of model calibration.
Regardless of the physical significance of the calibrated SCM parameters, those obtained
at low and intermediate ionic strengths (I=0.001 and 0.01 M) can be considered as useful
operational quantities that can yield reasonable predictions of the surface protonation and
the electrokinetic behavior of gaspeite over compositional ranges similar to those
investigated in our study.
Our other line of investigation involved the study of the interactions between
one of the most reactive naturally-occurring rhombohedral carbonate minerals in
aqueous solution, calcite, and H2O and/or H2O constituents using molecular modeling
and experimental techniques. Quantitative insights into the energetics of H2O
adsorption on the (10.4) calcite surface and on the 3D structural registry of the 1st and
2nd
hydration layers were obtained using ab initio molecular orbital methods and
cluster models. This theoretical approach had never been applied before to
characterize the structure of the hydrated calcite surface and elucidate the bonding
relationships governing the hydration process. The results are in reasonable agreement

309
with earlier findings of force-field-based and Density Functional Theory studies and
show that H2O molecules in the 1st hydration layer adsorb associatively to the surface
through ionic bonding with calcium atoms and hydrogen bonding with one surface
oxygen atom. This scenario is consistent with the generalized single primary surface
site scheme postulated in this study for rhombohedral carbonate mineral surfaces. In
addition, our ab initio study revealed a significant reorganization of the mineral
surface, specifically Ca-O bond relaxation and possible bond rupture leading to the
weakening of the topmost atomic layer with respect to the bulk. This new insight may
have important implications with respect to the elucidation of mechanisms of mineral
dissolution, rearrangement of surface layers and, possibly, solute transport through
subsurface lattice layers.
Finally, we developed a novel titration protocol that allowed, for the first time,
the rigorous quantitative characterization of the proton sorptive (or hydroxyl ion
desorptive) properties of calcite in aqueous solutions over a relatively wide range of
chemical conditions. Using this approach we generated data that lead us to postulate
the existence of a dynamic proton/calcium ion exchange reaction that extends beneath
the topmost surface calcite layer. This process may significantly impact the aqueous
speciation of closed (e.g., aquifers, pore waters) and open aquatic environments with
poor CO2 ventilation (e.g., deep sea waters, industrial reactions), via pH buffering and
CO2(g) sequestration upon ion exchange-induced calcite precipitation. Although the
observed fast proton/calcium ion exchange cannot be ascribed to any specific
transport mechanism such as ion physical entrapment, “solid-state” ion diffusion, or
ion migration through micro-fractures, it certainly reflects the highly dynamic nature
of the topmost surface calcite layers and lends support to earlier ideas that

310
conceptualize the interface between the bulk calcite crystal and the bulk solution as a
porous membrane of adsorbed or surface layers (or as a hydrated “gel-like” region)
through which processes such as site competition, dehydration, segregation, and/or ion
diffusion occur. In turn, these ideas are compatible conceptually with the weakening
of the topmost atomic layer as suggested by our ab initio molecular orbital study. The
observed proton/calcium ion exchange reaction could be directly involved in the
physiologically-driven 2H+/Ca
2+ exchange behavior exhibited by some calcifying
marine organisms, may have far-reaching implications on the interpretation of field
and laboratory data as well as on on the predicted responses of carbonate-rich shelf
sediments to rising atmospheric pCO2 and ocean acidification. Finally, although our
results cannot support or challenge earlier ideas on the origin of surface charge, the
electrokinetic behavior of calcite and/or potential mechanisms of calcite dissolution, it
raises serious questions on the validity of quantitative interpretations of adsorption,
electrokinetic, and dissolution kinetic data based upon surface speciation concepts and
suggest that these may require revision.
RECOMMENDATIONS FOR FUTURE RESEARCH
Despite the valuable scientific insights obtained in this thesis, numerous issues about the
reactivity of carbonate surfaces in aqueous solutions remain to be tackled. The best way
to address these is through a combination of multiple investigative approaches and a
careful acquisition and interpretation of novel data.
Because of their high reactivity, carbonate surfaces are not as “investigator-
friendly” as most metal oxides, a serious limitation to experimentalists wishing to apply

311
conventional experimental techniques. The corollary to this is straightforward. Either
novel experimental protocols must be developed, alternate investigative approaches (e.g.,
theoretical) must be used, or a combination of both must be adopted to obtain further
insight into processes at these mineral surfaces.
In doing so, it is critical to remember that, within specific time-scales, not all
carbonate minerals will react similarly as it was clearly demonstrated by our acidimetric
titration experiments conducted on: (i) the least reactive of known naturally-occurring
rhombohedral carbonate minerals in aqueous solutions: gaspeite and, (ii) one of the most
reactive ones: calcite. It follows that, whereas some fundamental concepts may be
common to all carbonate surfaces (e.g., hydration), others may not (e.g., proton uptake)
and therein lies the importance of compiling self-consistent and well-characterised data
sets (in terms of time scale, solution chemistry, nature, and history of mineral specimen,
etc.) for a specific carbonate mineral. Statistically- and geochemically-sound data
treatment is critical for the reliable description of the surface reactivity of these minerals.
Another important consideration is that the high reactivity of carbonate minerals
can be sometimes exploited to the experimentalist‟s advantage (as was demonstrated in
our calcite titration experiments) as it may allow the characterization of surface and near-
surface processes (e.g., ion exchange, surface rearrangement, solute transport through
lattice layers), a task that would be difficult or prohibitive for other minerals displaying
slow dissolution/equilibrium kinetics.
It is critical to carefully test and validate the experimental protocols and avoid
using these beyond the operational conditions at which they were validated. For instance,
in this study, by simulating a strict closed carbonate mineral system, we were able to
investigate the proton sorption behavior of calcite over an alkaline pH regime of interest

312
for many environmental applications. Because the nature of the carbonate system
(substantial evolution of CO2(g)) precludes extension of this protocol to acidic regimes (to
further examine the sorptive properties of calcite), the implementation of a novel
experimental protocol would be required. A feasible alternative would be to implement a
closed-system titration protocol, involving the presence of a confined gas phase in contact
with a calcite suspension, where the solution chemistry (pH and pCa) and the pCO2(g) in
the headspace are constantly monitored. Such a protocol would be analogous to that
implemented by Villalobos and Leckie (2000) for the evaluation of carbonate adsorption
at the goethite surface.
Clearly, a multitude of research projects could be designed to gain further insight
into the surface reactivity of carbonate minerals but they cannot be all enumerated here. It
suffices to say that, regardless of the specific carbonate surface of interest, emphasis must
be placed on the following aspects:
Implementing novel experimental protocols for the characterization of the sorptive
properties of carbonate minerals.
Generating well-constrained, well-characterised, self-consistent experimental data
sets for a specific carbonate mineral by multiple approaches (such as batch and
automatised titrations, calorimetric, radiometric and electrokinetic techniques). A
critical evaluation of the suitability of these composite data sets to describe
specific surface processes is warranted.
Examining the role of different background electrolytes on the sorptive properties
and reactivity of the carbonate mineral of interest (particularly important for
environmental applications such as saline brines and oceans).

313
Studying the proton sorptive properties of aragonite and natural magnesian
calcites (predominant in marine environments) using either the titration protocol
described in this thesis or analogous titration protocols.
Investigating the role of protons and water molecules on carbonate dissolution by
ab initio methods and/or molecular dynamics techniques.
Describing the nature of the postulated “proton-enriched, calcium-deficient
leached calcite layer” by spectroscopic and ab initio molecular modeling
techniques.
Developing novel semi-empirical or theoretical schemes to investigate and/or
interpret ion sorption at carbonate surfaces.
Finally, it would also be useful to fine tune the GA technique presented in this
study for the optimization of intrinsic constants (e.g., test other GA operators). A
combination of the GA with other powerful global search algorithms (e.g., Particle
Swarm or Differential Evolution) may render it more efficient for this type of application.
Clearly, a user-friendly version of the Matlab
-based GA optimization subroutines
(preferably written in Fortran or C++ languages) would be of interest to surface
complexation modelers and practitioners.

314
REFERENCES
Ahmed I.A.M., Crout N.M.J. and Young S.D. (2008) Kinetics of Cd sorption, desorption
and fixation by calcite: A long-term radiotracer study. Geochim. Cosmochim. Acta
72, 1498-1512.
Al-Horani F.A., Al-Moghrabi S.M., de Beer D. (2003) The mechanism of calcification
and its relation to photosynthesis and respiration in the scleractinian coral Galaxea
fascicularis. Mar. Biol. 142, 419-426.
Al-Hosney H.A. and Grassian V.H. (2005) Water, sulfur dioxide and nitric acid
adsorption on calcium carbonate: A transmission and ATR-FTIR study. Phys.
Chem. Chem. Phys. 7, 1266-1276.
Allison J. D., Brown D.S. and Novo-Gradac, K.J. (1991) MINTEQA2/PRODEFA2, a
geochemical assessment model for environmental systems: version 3.0. User‟s
manual, Environmental Research Laboratory, Office of Research and
Development, U.S. Environmental Protection Agency, Athens, GA, 106 pp.
Andersson A.J., Mackenzie F.T., Ver L.M. (2003) Solution of shallow-water carbonates:
An insignificant buffer against rising atmospheric CO2. Geology 31(6), 513-516.
Andersson A.J., Mackenzie F.T., Lerman A. (2006) Coastal ocean CO2–carbonic acid–
carbonate sediment system of the Anthropocene. Global Biogeochem. Cycles
20:GB1S92, 1-13.
APHA-AWWA-WPCF (1998). Standard Methods for the Examination of Water and
Wastewater Sewage and Industrial Wastes. Washington, D.C.
Arakaki T. and Mucci A. (1995) A continouous and mechanistic representation of calcite
reaction-controlled kinetics in dilute solutions at 25° and 1 atm total pressure.
Aquatic Geochem 1, 105-130.

315
Archer T. D. (2004) Computer Simulations of Calcite. Ph.D. Thesis, University of
Cambridge. UK.
Austen K. T., Wright K., Slater B and Gale J.D. (2005) The interaction of dolomite
surfaces with metal impurities: a computer simulation study. Phys. Chem. Chem.
Phys. 7, 4150-4156.
Ball J. W., Jenne E. A. and Norstrom D. K. (1981) WATEQ2- A computerized chemical
model for trace and major element speciation and mineral equilibrium of natural
waters. In: E. A. Jenne (Ed.) Chemical Modeling in Aqueous Systems, Symposium
series 93, American Chemical Society, Washington, D.C. p. 815-836.
Bandstra J.Z. and Brantley S.L. (2008) Surface evolution of dissolving minerals
investigated with a kinetic ising model. Geochim. Cosmochim. Acta 72:11, 2587-
2600.
Barrow N. J., Brümer G. W. and Strauss R. g (1993) Effects of surface heterogeneity on
ion adsorption by metal oxides and by soils. Langmuir. 9, 2606-2611.
Benjamin M. (2002) Modeling the mass-action expression for bidentate adsorption.
Environ. Sci. Technol. 36, 307-313.
Bermanec V., Sijarić, Kniewald G. and Mandarino J.A. (2000). Gaspeite and associated
Ni-rich minerals from veins in altered ultrabasic rocks from Duboštica, Bosnia
and Herzegovina. Can. Mineral. 38, 1371-1376.
Berner, R. A. and Morse, J. W. (1974) Dissolution kinetics of calcium carbonate in
seawater; IV, Theory of calcite dissolution. Am. J. Sci. 274, 108-134.

316
Bureau H., Raepsaet C., Khodja H., Carraro A. and Aubaud C. (2009) Determination of
hydrogen content in geological samples using elastic recoil detection analysis
(ERDA). Geochim. Cosmochim. Acta 73(11), 3311-3322.
Bickmore B. R., Rosso K. M., Tadanier C. J., Bylaska E. J. and Doud D. (2006). Bond-
Valence methods for pKa prediction: Bond-valence, electrostatic, molecular
geometry and solvation effects. Geochim. Cosmochim. Acta. 70, 4057-4071.
Bleam W. F. (1993) On the modeling proton affinity at the oxide/water interface. J.
Colloid Interface Sci. 159, 312-318.
Blesa M.A. and Kallay N. (1988) The metal oxide-electrolyte solution interface revisited.
Adv. Colloid Interface Sci. 28, 111-134.
Brady P.V., Krumhans J.L. and Papenguth, H.W. (1996) Surface complexation clues to
dolomite growth. Geochim. Cosmochim. Acta 60(4), 727-731.
Brady P.V., Papenguth H.W., Kelly J.W. (1999). Metal sorption to dolomite surfaces.
Applied Geochem. 14, 569-579.
Brewer P.G., Wong G.T.F., Bacon M.P. and Spencer D. W. (1975) An oceanic calcium
problem? Earth Planet. Sci. Lett. 26, 81-87.
Borkovec M. (1997). Origin of 1-pK and 2-pK models for ionizable water-solid
interfaces. Langmuir. 13, 2608-2613.
Borkovec M. and Koper G. J. M. (1994). Ising models of polyprotic acids and bases. J.
Phys. Chem. 98, 6038-6045.
Bruanuer S., Emmet P.H. and Teller E. (1938) Adsorption of gases in multimolecular
layers. J. Phys. Chem. 60, 309-316.

317
Casey W.H., Westrich H.R., Banfield J.F., Ferruzi G. and Arnold G.W. (1993) Leaching
and reconstruction at surfaces of dissolving chain-silicate minerals. Nature 366,
253-256.
Ĉerník M., Borkovec M. and Westall J. C. (1995). Regularized least-squares methods for
the calculation of discrete and continuous affinity distributions for heterogeneous
sorbents. Environ. Sci. Technol. 29, 413-425.
Chandler D. (1987) Introduction to Modern Statistical Mechanics. Oxford University
Press, New York, 288 p.
Chandra S.W. and Kollman P. (1984) An approach to computing electrostatic charges for
molecules. J. Comput. Chem. 5(2), 129-145.
Charlet L., Wersin P. and Stumm W. (1990). Surface charge of MnCO3 and FeCO3.
Geochim Cosmochim. Acta. 54, 2329-2336.
Charmas R. (1999) Four-layer complexation model for ion adsorption at energetically
heterogeneous metal oxide/electrolyte interfaces. Langmuir 15(17), 5635-5648.
Chiarello R.P., Wogelius R.A. and Sturchio N. (1993) In-situ synchrotron X-ray
reflectivity measurements at the calcite-water interface. Geochim. Cosmochim.
Acta. 57(16), 4103-4110.
Chou L., Garrels R.M. and Wollast R. (1989) Comparative study of the kinetics and
mechanisms of dissolution of carbonate minerals. Chem. Geol. 78, 269–282.
Cicerone D.S., Regazzoni A.E. and Blesa M. A. (1992) Electrokinetic properties of the
calcite/water interface in the presence of magnesium and organic matter. J.
Colloid Interface Sci. 154, 423-433.

318
Comans R.N.J. and Middelburg J.J. (1987) Sorption of trace metals on calcite:
Applicability of the surface precipitation model. Geochem. Cosmochim. Acta 51,
2587-2591.
Compton R.G. and Unwin P.R. (1990) The dissolution of calcite in aqueous solution at
pH less 4: kinetics and mechanism. R. Phil. Trans. R. Soc. Lond. A. 330, 1-45.
Compton R.G. and Pritchard K.L. (1990) The dissolution of calcite at pH > 7: kinetics
and mechanism. R. Phil. Trans. R. Soc. Lond. A. 330, 47-70.
Culberson C. and Pytkowicz R.M. (1968) Effect of pressure on carbonic acid, boric acid
and the pH in seawater. Limnol. Oceanogr. 13(3), 403-417.
Curti E., Kulik D.A. and Tits J. (2005) Solid solutions of trace Eu(III) in calcite:
Thermodynamic evaluation of experimental data over a wide range of pH and
pCO2. Geochim. Cosmochim. Acta 69(7), 1721-1737.
Davis J.A. and Kent D.B. (1990). Surface complexation modeling in aqueous
geochemistry. In Mineral-Water Interface Geochemistry. (ed. M.F. Hochella and
A.F. White). Rev. Mineral. 23. Mineral. Soc. Washington, DC. p. 177-260.
Davis J.A., Fuller C.C. and Cook A.D. (1987) A model for trace metal sorption processes
at the calcite surface: Adsorption of Cd2+
and subsequent solid solution formation.
Geochim. Cosmochim. Acta 51(6), 1477-1490.
Douglas, H.W. and Walker, R. A. (1950) The electrokinetic behavior of iceland spar
against aqueous electrolyte solution. Trans. Faraday Soc. 46, 559-568.
Duckworth O.W. and Martin S.T. (2003). Connections between surface complexation and
geometric models of mineral dissolution investigated for rhodochrosite. Geochim.
Cosmochim. Acta 67, 1787-1801

319
Dzombak D.A. and Morel F.M.M. (1990) Surface Complexation Modeling. John Wiley
and Sons Inc.: New York, NY, Chapters 1-3.
de Leeuw N. H. and Parker S. C. (1997). Atomistic simulation of the effect of molecular
adsorption of water on the surface structure and energies of calcite surfaces. J.
Chem. Soc., Faraday Trans., 93(3), 467-475.
de Leeuw N. H. and Parker S. C. (1998). Surface structure and morphology of calcium
carbonate polymorphs calcite, aragonite, and vaterite: an atomistic approach. J.
Phys. Chem. B., 102, 2914-2922.
de Leeuw N. H. and Parker S.C. (2001). Surface-water interactions in the dolomite
problem. Phys. Chem. Chem. Phys., 3, 3217-3221.
de Leeuw N. H. and Parker S.C. (2002). Surface structures, stabilities, and growth of
magnesian calcites: a computational investigation from the perspective of
dolomite formation. Am. Mineral., 87, 679-689.
de Leeuw N. H., Parker S.C., and Hanumantha Rao K. (1998). Modeling the competitive
adsorption of water and methanoic acid on calcite and fluorite surfaces. Langmuir,
14, 5900-5906.
Elfil H. and Roques H. (2001) Role of hydrate phases of calcium carbonate on the scaling
phenomenon. Desalination, 137, 177-186.
Elzinga E.J., Rouff A.A. and Reeder R.J. (2006) The long-term fate of Cu2+
, Zn2+
, and
Pb2+
adsorption complexes at the calcite surface: An X-ray absorption
spectroscopic study. Geochim. Cosmochim. Acta, 70, 2715-2725.
Epperson J. F. (2002) An Introduction to Numerical Methods and Analysis. John Wiley
and Sons, New York, NY, 556 pp.

320
Eriksson R., Merta J. and Rosenholm J.B. (2007) The calcite/water interface I. Surface
charge in indifferent electrolyte media and the influence of low-molecular-weight
polyelectrolyte. J. Colloid Interface Sci. 313, 184-193.
Eriksson R., Merta J. and Rosenholm J.B. (2008) The calcite/water interface II. Effect of
added lattice ions on the charge properties and adsorption of sodium polyacrylate.
J. Colloid Interface Sci., 2008, 326, 396-402.
Essaid H.I., Cozzarelli I.M., Eganhouse R.P., Herkelrath W.N., Bekins B.A. and Delin
G.N. (2003) Inverse modeling of BTEX dissolution and biodegradation at the
Bemidji, MN crude-oil spill site. J. Cont. Hydr. 67(1), 269-299.
Felmy A.R., Dixon D.A., Rustad J.R., Mason M.J. and Onishi L.M. (1998) The
hydrolysis and carbonate complexation of strontium and calcium in aqueous
solution. Use of molecular modeling calculations in the development of aqueous
thermodynamic models. J. Chem. Thermodynamics 30, 1103-1120.
Feng J., Lee Y.J., Reeder R.J. and Phillips B.L. (2006) Observation of bicarbonate in
calcite by NMR spectroscopy. Am. Mineral. 91, 957-960.
Fenter P., Geissbühler P., DiMasi E, Srajer G, Sorensen B and Sturchio N.C. (2000)
Surface speciation of calcite observed in situ by high-resolution X-ray reflectivity.
Geochim. Cosmochim. Acta 64(7), 1221-1228.
Fernández Alvarez J.P., Fernández Martínez J.L. and Menéndez Pérez C.O. (2008)
Feasibility analysis of the use of binary genetic algorithms as importance samplers
Application to 1-D DC resistivity inverse problem. Math. Geosci. 40, 375-408.
Foxall T., Peterson G.C., Rendall H.M. and Smith A. L. (1979) Charge determination at
calcium salt/aqueous solution interface. J. Chem. Soc. Farad. Trans. 175, 1034-
1039.

321
Franklin M.L. and Morse J.W. (1983) The interaction of manganese(II) with the surface
of calcite in dilute solutions and seawater. Mar. Chem., 12(4), 241-254.
Gaffey S.J. (1995) H2O and OH in echinoid calcite: A spectroscopic study. Am. Mineral.
80, 947-959.
Gans P. (1976) Numerical methods for data-fitting problems. Coord. Chem. Rev. 19, 99-
124.
Gao Y. and Mucci A. (2001) Acid base reactions, phosphate and arsenate complexation,
and their competitive adsorption at the surface of goethite in 0.7 M NaCl solution.
Geochim. Cosmochim. Acta. 65, 2361-2378.
Geissbühler P, Fenter P, DiMasi E., Sorensen, L.B. and Sturchio N.C. (2004). Three-
dimensional structure of the calcite–water interface by surface X-ray scattering .
Surf. Sci. 573, 191-203.
Gen M. and Cheng R. (1997) Genetic Algorithms and Engineering Design. John Wiley
and Sons, New York, NY, 411 pp.
Gen M. and Cheng R. (2000) Genetic Algorithms and Engineering Optimization. John
Wiley and Sons, New York, NY, 495 pp.Goldberg D. (1989) Genetic Algorithms
in Search, Optimization and Machine Learning. Addison-Wesley, Reading, MA,
412 pp.
Gence N. and Ozbay N. (2006) pH dependence of electrokinetic behavior of dolomite and
magnesite in aqueous electrolyte solutions. Appl. Surface Sci. 252, 8057-8061.
Gieskes J. M. Alkalinity, pH, Mg, Ca, Si, PO4 and NH4: Interstitial water studies, Leg
15: Initial Repts. Deep-Sea Drilling Proj., v. 15, 1974, p. 63-79.

322
Golberg D., Korb K. and Deb K. (1989) Messy genetic algorithms: motivation, analysis
and first results. Complex Systems 3, 493-530.
Goldberg S. (1991) Sensitivity of surface complexation modeling to the surface site
density parameter. J. Colloid Interface Sci. 145:1, 1-9.
Goldberg S. (1993) Constant capacitance model: Chemical surface complexation model
for describing adsorption of toxic trace elements on soil minerals. Am. Chem. Soc.
Symp. Ser. 518: 278-307.
Golderg S. (1995) Adsorption models incorporated into chemical equilibrium models. In:
Chemical Equilibrium and Reaction Models. (ed. R. Loeppert, A.P. Schwab and
S. Goldberg), Soil. Sci. Soc. Am. Special Publication 42, 75-95.
Green E. and Luttge A. (2006) Incongruent dissolution of wollastonite measured with
vertical scanning interferometry. Am. Mineral. 91, 430-434.
Hayes K.F., Redden G., ELA W. and Leckie J.O. (1991). Surface complexation models:
An evaluation of model parameter estimation using FITEQL and oxide mineral
titration. J. Colloid Interface Sci. 142(2), 448-469.
Healy T. W. and White L. R. (1978) Ionizable surface groups models of aqueous
interfaces. Adv. Colloid Interface Sci. 9, 303-345.
Herbelin A. and Westall J. (1996) FITEQL-. A computer program for determination of
chemical equilibrium constants from experimental data version 3.2: user‟s
manual. Department of Chemistry. Oregon State University, Corvallis, OR,
Report 96-01.
Hiemstra T. and van Riemsdijk W. H. (1991). Physical chemical interpretation of primary
charging behavior of metal (hydr)oxides. Colloids and Surfaces 59, 7-25.

323
Hiemstra T. and van Riemsdijk W. H. (1996). A surface structural approach to ion
adsorption: The charge distribution (CD) model. J. Colloid Interface Sci. 179,
488-508.
Hiemstra T. Yong, H. and van Riemsdijk W.H. (1999) Interfacial charging phenomena of
aluminum (hydr)oxides. Langmuir 15(18), 5942-5955.
Hiemstra T., van Riemsdijk W. H. and Bolt G. H. (1989). Multi-site proton adsorption
modeling at the solid/solution interface of (hydr)oxides: A new approach. Journal
J. Colloid Interface Sci. 133(1), 91-104.
Hiemstra T., Venema P. and van Riemsdijk W. H. (1996). Intrinsic proton affinity of
reactive surface groups of metal (hydr)oxides: The bond valence principle. J.
Colloid Interface Sci. 184, 680-692.
Hoffmann U. and Stipp S.L.S. (2001). The behavior of Ni2+
on calcite surfaces. Geochim.
Cosmochim. Acta 65(22), 4131-4139.
Hohl H. and Stumm W. (1976) Interaction of Pb2+
with hydrous -Al2O3. J. Colloid
Interface Sci. 55, 281-288
Holland J. (1975) Adaptation in Natural and Artificial Systems. University of Michigan
Press, Ann Arbor MI, 183 pp.
Huang C.P. (1981). The surface acidity of hydrous solids in: Adsorption of Inorganics at
Solid-Liquid Interfaces. M.A. Anderson and A.J. Rubin (eds.) Ann Arbor Science,
Ann Arbor, Mich., pp. 183-217.
Huang, Y.C., Fowkes F.M., Lloyd, T.B. and Sanders, N.D. (1991) Adsorption of calcium
ions from calcium chloride solutions onto calcium carbonate particles. Langmuir
7, 1742-1748.

324
Hunter R.J. (2001). Foundations of Colloid Science. Oxford University, Oxford, 806 p.
Hwang S., Blanco M. and Goddard W.A. III (2001). Atomistic simulations of corrosion
inhibitors adsorbed on calcite surfaces I. Force field parameters for calcite. J.
Phys. Chem. B. 105, 10746-10752.
Jäger I. (1991). Adsorption of pairwise interacting atoms on the derivation of the
interaction parameters between first and second neighbors from experimental data.
Surf. Sci. 254, 300-308.
Jakeman A.J., Letcher R.A. and Norton J.P. (2006) Ten iterative steps in development
and evaluation of environmental models. Environ. Model. Software 21, 602-614.
Jordan, G., Higgins, S.R., Eggleston, C.M., Knauss, K.G. and Schmahl, W.W., 2001.
Dissolution kinetics of magnesite in acidic aqueous solution, a hydrothermal
atomic force microscopy (HAFM) study: Step orientation and kink dynamics.
Geochim. Cosmochim. Acta. 65, 4257-4266
Kallay N. and Žalac S. (2000). Charged surfaces and interfacial ions. J. Colloid Interface
Sci. 230(1), 1-11.
Keizer M. G. and van Riemsdijk W. H. (1999) ECOSAT, Technical Report of the
departments of Soil Science and Plant Nutrition. Wageningen University,
Wageningen, The Netherlands.
Keller B and R. Lutz (1997) A new crossover operator for rapid function optimization
using a genetic algorithm, Proceedings of the Eighth Ireland Conference on
Artificial Intelligence (AI-97), Vol. 2, p. 48-57.
Kendall T.A. and Martin S.T. (2005) Mobile ions on carbonate surfaces. Geochim.
Cosmochim. Acta. 69, 3257-3263.

325
Kerisit S. and Parker S. C. (2004). Free energy of adsorption of water and metal ions on
the (10.4) calcite surface. J. Am. Chem. Soc. 126, 10152-10161.
Kerisit S., Parker S. C. and Harding J. H. (2003). Atomistic simulation of the dissociative
adsorption of water on calcite surfaces J. Phys. Chem. B. 107, 7676-7682.
Kerisit S., Cooke D.J., Spagnoli D. and Parker S.C. (2005a). Molecular dynamics
simulations of the interactions between water and inorganic solids. J. Mater.
Chem. 15, 1454-1462.
Kerisit S., Marmier A., Parker S.C. (2005b) Ab initio surface phase diagram of the (10.4)
calcite surface. J. Phys. Chem. B, 109:39, 18211-18213.
Kohls D.W. and Rodda J.L. (1966). Gaspeite, (Ni, Mg, Fe) (CO3), A new carbonate from
the Gaspé peninsula, Québec. Am. Mineral. 51, 677-684.
Koretsky C. M. Sverjensky D. A. and Sahai N. (1998) A model of surface sites types on
oxide and silicate minerals based on crystal chemistry: Implications for site types
and densities, multi-site adsorption, surface infrared spectroscopy and dissolution
kinetics. Am. J. Sci. 298, 349-438.
Kosmulski M., Maczka E. and Rosenholm J. B. (2002). Isoelectric points of metal oxides
at high ionic strengths. J. Phys. Chem. B 106, 2918-2921.
Kovačević D., Kobal I. and Kallay N. (1998) Adsorption of organic acids on metal
oxides. The umbrella effect. Croat. Chim. Acta 71(4), 1139-1153.
Kulik D.A. (2002) Gibbs energy minimization approach to modeling sorption equilibria
at the mineral-water interface: Thermodynamic relations for multi-site-surface
complexation. Am. J. Sci. 302, 227-279.

326
Kulik D. A., Aja S. U., Sinitsyn V. A. and Wood S. A. (2000) Acid–base surface
chemistry and sorption of some lanthanides on K1-saturated Marblehead illite: II.
A multisite–surface complexation modeling. Geochim. Cosmochim. Acta. 64:2,
195-213.
Kuriyavar S. I., Vetrivel R., Hegde S. G., Ramaswamy A.V., Chakrabarty D. and
Mahapatra S. (2000). Insights into the formation of hydroxyl ions in calcium
carbonate: temperature dependent FTIR and molecular modeling studies. J. Mater.
Chem. 10, 1835-1840.
Langmuir D. (1997) Aqueous Environmental Geochemistry. Prentice Hall, Upper Saddle
River, N.J., 600.
Liang Y., Lea A.S., Baer D.R. and Engelhard M.H. (1996). Structure of the cleaved
CaCO3 (104) surface in an aqueous environment. Surface Sci. 351, 172-182.
Lorens R. B. (1981) Sr, Cd, Mn and Co distribution coefficients in calcite as a function of
calcite precipitation rate. Geochim. Cosmochim. Acta 45, 553–561.
Ludwig C. and Schindler P. W. (1995). Surface complexation on TiO2. I Adsorption of
H+and Cu
2+ ions onto TiO2 (anatase). J. Interface Colloid Sci. 169, 284-290.
Lützenkirchen J. (1997). Ionic strength effects on cation sorption to oxides: macroscopic
observations and their significance in microscopic interpretation. J. Colloid
Interface Sci. 195, 149-155.
Lützenkirchen J. Surface Complexation Models of Adsorption: A Critical Survey in the
Context of Experimental Data In: Tóth J. (2002). Adsorption. Theory, Modeling
and Analysis, Marcel Dekker Inc., New York. pp 631-710.

327
Lützenkirchen J. (2003) Surface complexation models for adsorption: A critical survey in
the context of experimental data. In: Tóth J. (Ed) Adsorption: Theory, Modeling,
and Analysis, Surfactant Science Series 107, New York, NY, Marcel Dekker Inc.,
p. 631-710.
Lützenkirchen J. (2005). On derivatives of surface charge of carbonate minerals. J.
Colloid Interface Sci. 2(15), 489-497.
Mackenzie, F.T., Bischoff, W. D., Bishop, F. C., Loijens, M., Schoonmaker, J. and
Wollast, R. In Carbonates: Mineralogy and Chemistry; Reeder, R.J., Eds.; Rev.
Mineral. Mineralogical Society of America: Michigan, 1990; Vol 11, pp 97-144.
Martin-Garin A; Van Cappellen P. and Charlet, L. (2003) Aqueous cadmium uptake by
calcite: A stirred flow-through reactor study. Geochim. Cosmochim. Acta 67(15),
2763-2774.
Matott L.S. and Rabideau A.J. (2008) ISOFIT - A program for fitting sorption isotherms
to experimental data. Environ Model Software 23: 670-676.
McConnaughey T.A. (1991) Calcification in Chara coralline: CO2, hydroxylation
generates protons for bicarbonate assimilation. Limnol. Oceanogr. 36(4), 619-628.
McConnaughey T.A. and Falk R.H., (1991) Calcium-proton exchange during algal
calcification. Biol. Bull. 180, 185-195.
McGillen, M.R. and Fairchild, I.J. (2005) An experimental study of incongruent
dissolution of CaCO3 under analogue glacial conditions. J. Glaciol. 51(174), 383-
390.

328
Mestres J. and Scuseira G.E. (1995) Genetic algorithms, A robust scheme for geometry
optimizations and global minimum structure problems. J. Comput. Chem. 16(6),
729-742.
Michalewicz Z. (1996) Genetic Algorithms + Data Structure = Evolution Programs, 2nd
ed. Springer-Verlag, New York, NY, 387 pp.
Mishra S.K. (1978) The electrokinetics of apatite and calcite in inorganic electrolyte
environment. Int. J. Miner. Process. 5, 69-83.
Möller P. and Sastri C.S. (1974) Estimation of the number of surface layers of calcite
involved in Ca45
Ca istotopic exchange with solution. Zeit. Physik. Chem. Neue.
Folg. 89, 80-87.
Morel F. and Morgan J. (1972) A numerical method for computing equilibria in aqueous
chemical systems. Environ. Sci. Technol. 6, 58-87.
Morel F.M.M. and Hering, J. G. (1993) Principles and Applications of Aquatic
Chemistry; John Wiley and Sons Inc.: New York, NY, Chapters 1, 2 and 5.
Morse J.W. (1986) The surface chemistry of calcium carbonate minerals in natural
waters: An overview. Mar. Chem. 20, 91-112.
Morse J.W. and Berner R.A. (1972) Dissolution kinetics of calcium carbonate in sea
water; I, A kinetic origin for the lysocline. Am. J. Sci. 272, 840–851.
Morse J.W. and Mackenzie F.T. (1990). Geochemistry of Sedimentary Carbonates.
Develop. in Sedimentol. 48. Elsevier Science Pubs. Amsterdam, 707 p.
Morse J.W., Arvidson R.S. and Lüttge A. (2007) Calcium carbonate formation and
dissolution. Chem. Rev. 2007, 107, 342-381.

329
Mosegaard K. (1998) Resolution analysis of general inverse problems through inverse
Monte Carlo sampling. Inverse Probl. 14, 405-426.
Mosegaard K. and Sambridge M. (2002) Monte Carlo Analysis of inverse problems.
Inverse Probl. 18, 29-54.
Moulin P. and Roques H. (2003) Zeta potential measurement of calcium carbonate. J.
Colloid. Inter. Sci. 261, 115-126.
Mucci A., Morse J.W. and Kaminsky M.S. (1985) Auger spectroscopy analysis of
magnesian calcite overgrowths precipitated from seawater and solutions of similar
composition. Am. J. Sci. 285, 289-305.
Neagle W. and Rochester C.H. (1990) Infrared study of the adsorption of water and
ammonia on calcium carbonate. J. Chem. Soc., Faraday Trans. 86(1), 181-183.
Nicholson K. (1995) Linear Algebra with Applications. PWS Pub. Co., Boston, MD, 540
pp.
NIST (1998) Critically Selected Stability Constants of Metal Complexes, Standard
Reference Database 46, Version 5, National Institute of Standards and
Technology, US Department of Commerce, Gaithersburg, MD, USA.
Papelis C., Hayes K. F. and Leckie J.O. (1988) HYDRAQL: A program for the
computation of chemical equilibrium, composition of aqueous batch systems
including surface-complexation modeling of ion adsorption at the oxide/solution
interface. Report No. 306. Stanford University, Stanford, CA, 130 pp.
Parkhurst D. L. (1995) User‟s guide to PHREEQC. U.S. Geological Survey, Water
Resources Investigations Report 95-4277, 212 pp.

330
Pehrsson L, Ingman F and Johanssson A. (1976). Acid-Base titrations by stepwise
additions of equal volumes of titrant with special reference to automatic titrations-
I. Theory, discussion of the Gran functions, the Hofstee method and two proposed
methods for calculating equivalence volumes. Talanta. 23, 769-780.
Pingitore N.E. Jr., Eastman M.P., Sandidge M., Oden K. and Freiha B. (1988) The
coprecipitation of manganese(II) with calcite: an experimental study. Mar. Chem.
25(2), 107-120.
Plummer L.N. and Busenberg E. (1982) The solubilities of calcite, aragonite and vaterite
in CO2-H2O solutions between 0 and 90°C, and an evaluation of the aqueous
model for the system CaC03-CO2-H20. Geochim. Cosmochim. Acta 46(6), 1011-
1040.
Pokrovsky O.S. and Schott J. (2002). Surface chemistry and dissolution of divalent metal
carbonates. Environ. Sci. Technol. 36(3), 426-432.
Pokrovsky O.S., Schott J. and Thomas F. (1999a) Processes at the magnesium-bearing
carbonates/solution interface. I. A surface speciation model for magnesite.
Geochim. Cosmochim. Acta 63(6), 863-880.
Pokrovsky O.S., Schott J. and Thomas F. (1999b) Dolomite surface speciation and
reactivity in aquatic systems. Geochim. Cosmochim. Acta. 63(19/20), 3133-3143.
Pokrovsky O.S., Mielczarski J.A., Barres O., and Schott J. (2000) Surface speciation
models of calcite and dolomite/aqueous solution interfaces and their spectroscopic
investigation. Langmuir 16, 2677-2688.
Prédali J.-J. and Cases J.-M.J. (1973) Zeta potential of magnesian carbonates in inorganic
electrolytes. J. Colloid Interface Sci. 45(3), 449-458.

331
Robbins L. L. and Fabry V. J. (1994) In Carbon Dioxide Chemistry: Environmental
Issues; Paul, J., Pradier, C., Eds.; The Royal Society of Chemistry: Cambridge,
U.K., pp 301-304.
Saebø S., Tong W. and Pulay P. (1993) Efficient elimination of basis set superposition
errors by the local correlation method: Accurate ab initio studies of the water
dimer. J. Chem. Phys., 98:3, 2170-2175.
Sahai N. and Sverjensky D.A. (1997). Evaluation of internally consistent parameters for
the triple-layer model by the systematic analysis of oxide surface titration data.
Geochim. Cosmochim. Acta. 61(14), 2801-2826.
Sahai N. and Sverjensky D. A. (1998) GEOSURF: A computer program for modeling
adsorption on mineral surfaces from aqueous solution. Comp. Geosc. 24(9), 853-
873.
Sait S. M. and Youssef H. (1999) Iterative Computer algorithms with Applications in
Engineering Solving Combinatorial Optimization Problems. IEEE Computer
Society Los Alamitos, CA, 387 pp.
Santra B., Michaelides A., Fuchs M., Tkatchenko A., Filippi C. and Scheffler M. (2007)
On the accuracy of density-functional theory exchange-correlation functionals for
H bonds in small water clusters: Benchmarks approaching the complete basis set
limit. J. Phys. Chem. 127,184104.
Santra B., Michaelides A., Fuchs M., Tkatchenko A., Filippi C. and Scheffler M. (2008)
On the accuracy of density-functional theory exchange-correlation functionals for
H bonds in small water clusters. II. The water hexamer and van der Waals
interactions. J. Phys. Chem. 129, 194111.

332
Santschi Ch. and Rossi M.J. (2006) Uptake of CO2, SO2, HNO3 and HCl on calcite
(CaCO3) at 300 K: Mechanism and the role of adsorbed water. J. Phys. Chem. A
110, 6789-6802.
Sarmiento J.L. and Sundquist E.T. (1992) Revised budget for the oceanic uptake for
anthropogenic carbon dioxide. Nature 356, 589-593.
Schecher W.D. and McAvoy D.C. (1992) MINEQL+: A software environment for
chemical equilibrium modeling. Computers, Environment and Urban Systems 16,
65-76.
Scheidegger A. M., and Sparks D. L. (1996). A critical assessment of sorption-desorption
mechanisms at the soil mineral/water interface. Soil Sci. 161, 813-831.
Schindler P.W. and Kamber H.R. (1968) Die acidität von silanolgruppen. Helv. Chim.
Acta 51, 1781-1786.
Scott A.P. and Radom L. (1996) Harmonic vibrational frequencies – An evaluation of
Hartree-Fock, Moller-Plesset, Quadratic configuration interaction, Density
functional theory, and Semiempirical scale factors. J. Phys. Chem. 100, 16502-
16513.
Siffert B. and Fimbel, P. (1984) Parameters affecting the sign and the magnitude of the
electrokinetic potential of calcite. Colloids Surf. 11, 377-389.
Sjöberg E.L. and Rickard D.T. (1984) Calcite dissolution kinetics: surface speciation and
the origin of the variable pH dependence. Chem. Geol. 42, 119-136.
Skinner A.J., LaFemina J.P. and Jansen H.J.F. (1994) Structure and bonding of calcite: A
theoretical study. Am. Min. 1994, 79, 205-214.

333
Somasundaran P. and Agar G.E. (1967) The zero point of charge of calcite. J. Colloid
Interface Sci. 24, 433-440.
Sposito G. (1983). On the surface complexation model of the oxide-aqueous solution
interface. J. Colloid Interface Sci. 91(2), 329-340.
Sposito G. (1984). The Surface Chemistry of Soils. Oxford University Press, New York,
234 p.
Sposito G. (1989). Surface reactions in natural aqueous colloidal systems. Chimia 43,
169-176.
Sposito G. (1990) in: Mineral-Water Interface Geochemistry, ed. M F. Hochella Jr. and
A. F. White, Mineralogical Society of America, Rev. Mineral. Vol 23, 261 p.
Sposito G. (1998) On points of zero charge. Environ. Sci. Technol. 32(19), 2815-2819.
Sposito G. and Coves J. (1988) SOILCHEM: A computer program for the calculation of
chemical speciation in soils. Keamey Found. Soil Sci., Univ. California,
Riverside.
Stefanovich E.V. and Truong T.N. (1997) A theoretical approach for modeling reactivity
at solid–liquid interfaces. J.Chem. Phys. 106, 7700.
Stipp S.L.S. (1999) Toward a conceptual model the calcite surface: hydration, hydrolysis,
and surface potential. Geochim. Cosmochim. Acta 63(19/20), 3121-3131.
Stipp S.L. and Hochella M.F. Jr. (1991) Structure and bonding environments at the calcite
surface as observed with X-ray photoelectron spectroscopy (XPS) and low energy
electron diffraction (LEED). Geochim. Cosmochim. Acta 55, 1723-1736.

334
Stipp S.L.S., Hochella, F., Parks, G.A. and Leckie J.O. (1992) Cd2+
uptake by calcite,
solid-state diffusion, and the formation of solid-solution: Interface processes
observed with near-surface sensitive techniques (XPS, LEED, and AES).
Geochim. Cosmochim. Acta 56, 1941-1954.
Stipp S.L.S., Eggleston C.M. and Nielsen B.S. (1994) Calcite surface structure observed
at microtopographic and molecular scales with atomic force microscopy (AFM).
Geochim. Cosmochim. Acta 58(14), 3023-3033.
Stipp S.L.S., Gutmannsbauer W. and Lehmann, T. (1996) The dynamic nature of calcite
surfaces in air. Am. Mineral. 81,1-8.
Stipp, S.L.S., Kulik A.J., Franzreb K., Benoit W. and Mathieu H.J. A (1997) Combination
of SFM and TOF-SIMS imaging for observing local inhomogenieties in
morphology and composition: Aged calcite surfaces. Surf. Interface Analysis 25,
959-965.
Stipp S.L.S., Konnerup-Madsen K., Franzreb K., Kulik A. and Mathieu H.J. (1998)
Spontaneous movement of ions through calcite at standard temperature and
pressure. Nature 396, 356-359.
Stoessell R.K. (1998) Binary cation exchange reactions. Clays Clay Miner. 46(2), 215-
218.
Stumm W. and Morgan J. (1996). Aquatic Chemistry: Chemical Equilibria and Rates in
Natural Waters. John Wiley and Sons Inc., 3rd
Edition New York, 1022 p.
Sverjensky D.A. (2003) Standard states for the activities of mineral surface sites and
species. Geochim. Cosmochim. Acta 67, 17–28.

335
Tas C.A. (2007) Porous, biphasic caco3-calcium phosphate biomedical cement scaffolds
from calcite (CaCO3) powder. Int. J. Appl. Ceram. Technol, 4(2), 152-163.
Teng H.H. and Dove P.M. (1997) Surface site-specific interactions of aspartate with
calcite during dissolution: Implications for biomineralization. Am. Mineral. 82,
878-887.
Tesoriero A. and Pankow J. (1996) Solid solution partitioning of Sr2+
, Ba2+
, and Cd2+
to
calcite. Geochim. Cosmochim. Acta. 60(6), 1053-1063.
Thompson D.W. and Pownall P.G. (1989) Surface electrical properties of calcite. J.
Colloid. Inter. Sci. 131, 74-83.
Tipping E. (1994) WHAM- A chemical equilibrium model and computer code for water,
sediments and soils incorporating a discrete site/electrostatic model of ion-binding
by humic substances. Comp. Geosci. 20, 973-1023.
Tossel J.A. (2006) H2CO3 and its oligomers: Structures, stabilities, vibrational and NMR
spectra, and acidities. Inorg. Chem., 45, 5961-5970.
Tossel J.A. and Vaughan D.J. (1992) Theoretical Geochemistry: Applications of Quantum
Mechanics in the Earth and Mineral Sciences, Oxford University Press, New
York, pp 514.
Turner B.F. and Fein J.B. (2006) Protofit: A program for determining surface protonation
constants from titration data. Comp. Geosci. 32, 1344–1356.
Usher C. R., Michel A. E. and Grassian V. H. (2003) Reactions on mineral dust. Chem.
Rev. 103, 4883-4939.

336
Vanerek A., Alince B. and van de Ven T. G. M. (2000) Interaction of calcium carbonate
fillers with pulp fibres: Effect of surface charge and cationic polyelectrolytes. J.
Pulp Paper Sci. 26:9, 317-322
Van Cappellen P., Charlet L., Stumm W. and Wersin P. (1993). A surface complexation
model of the carbonate mineral-aqueous solution interface. Geochim. Cosmochim.
Acta. 57, 3505-3518.
van der Lee J. and de Windt L. (1999) CHESS Tutorial and cookbook. Technical report
No. LHM/RD/99/05. École des Mines de Paris. Fontainebleu, France. 77.
Van Riemsdijk W. H., De Wit J. C.M., Koopal L. K and Bolt G.H. (1987). Metal ion
adsorption on heterogeneous surfaces: Adsorption models. J. Colloid Interface
Sci. 116(2), 511-522.
Villalobos M. and Leckie J. O. (2000) Carbonate adsorption on goethite under closed and
open CO2 conditions. Geochim. Cosmochim. Acta 64(22), 3787–3802.
Villalobos M. and Leckie J. O. (2001). Surface complexation modeling and FTIR study
of carbonate adsorption to goethite. Geochim. Cosmochim. Acta. 235, 15-32.
Villegas-Jiménez A., Mucci A. and Whitehead M.A. (2005). Ab initio molecular orbital
investigation of the chemical interactions of water with the (10.4) calcite surface.
Proceeding of the First Applied Pulp and Paper Molecular Modeling Symposium.
Montréal, Canada. pp. 227-244.
Villegas-Jiménez A. and Mucci A. (2009) Estimating intrinsic formation constants of
mineral surface species using a genetic algorithm. Math. Geosci. (Accepted).

337
Villegas-Jiménez A., Mucci A. and Whitehead M.A. (2009) Theoretical insights into the
hydrated (10.4) calcite surface: structure, energetic, and bonding relationships.
Langmuir 25(12), 6813-6824.
Villegas-Jiménez A., Mucci A., Paquette J. (2009) Proton/calcium ion exchange behavior
of calcite. Phys. Chem. Chem. Phys. 39(11), 8895-8912.
Villegas-Jiménez A., Mucci A., Pokrovsky O.S. and Schott J. (2009) Defining reactive
sites at hydrated mineral surfaces: rhombohedral carbonate minerals. Geochim.
Cosmochim. Acta 73(15), 4326-4345.
Wasserman E., Rustad J. R., Felmy A. R. (1999) Molecular modeling of surface charging
of hematite: I. The calculation of proton affinities and acidities on a surface. Surf.
Sci. 424, 19-27.
Westall J.C. (1979) MICROQL II: Computation of adsorption equilibria in BASIC.
Technical Report. Swiss Federal Institute of Technology, EAWAG, 8600
Dübendorf, Switzerland.
Westall J.C. (1982) FITEQL: A computer program for determination of chemical
equilibrium constants from experimental data. Version 1.2 Report 82-01,
Department of Chemistry, Oregon State University, Corvallis, OR, USA.
Westall J. and Hohl H. (1980) A comparison of electrostatic models for the
oxide/solution interface. Adv. Colloid Int. Sci. 12, 265-294.
Wolery T.J. (1992) EQ3NR: A computer program for geochemical aqueous speciation-
solubility calculations: theoretical manual, user‟s guide and related documentation
(Version 7.0), Report No. UCRL-MA-110662-PT-III, Lawrence Livermore
National Laboratory, Livermore, CA.

338
Wiesner A.D, Katz L.E. and Chen C. (2006) The impact of ionic strength and background
electrolyte on pH measurements in metal ion adsorption experiments. J. Colloid
Interface Sci. 301, 329-332.
Wright K., Cygan R.T. and Slater B. (2001) Structure of the (10.4) surfaces of calcite,
dolomite and magnesite under wet and dry conditions. Phys. Chem. Chem. Phys.
3, 839-844.
Xiao Y. and Lasaga A.C. (1994) Ab initio quantum mechanical studies of the kinetics and
mechanism of silicate dissolution: H+(H3O
+) catalysis. Geochim. Cosmochim.
Acta, 58(24), 5379-5400.
Xie Z. and Walther J. (1994) Dissolution stoichiometry of alkali and alkaline earth
elements to the acid-reacted wollastonite surface at 25ºC. Geochim. Cosmochim.
Acta. 58, 2587-2598.
Yoon R. H., Salman T. and Donnay G. (1979) Predicting points of zero charge of oxides
and hydroxides. J. Colloid Interface Sci. 70(3), 483-493.
Yu D., Xue D. and Ratajczak H. (2006) Bond-valence parameters for characterizing O–
H O hydrogen bonds in hydrated borates. J. Mol. Struct. 792-793, 280-285.
Zachara, J. M. and Westall, J. C. (1999) Chemical modeling of ion adsorption in soils, In
Soil Physical Chemistry, 2nd ed. (ed. D. L. Sparks), CRC Press LLC, Boca Raton,
FL, 47–95.
Zachara J.M., Kittrick, J.A., Harsh J.B. (1988) The mechanism of Zn2+
adsorption on
calcite. Geochim. Cosmochim. Acta 52, 2281-2291.
Zachara J.M., Cowan C.E. and Resch C.T. (1991) Sorption of divalent metals on calcite.
Geochim. Cosmochim. Acta 55, 1549-1562.

339
Zeleznik F.J. and Gordon S. (1968) Calculation of complex chemical equilibria. Ind. Eng.
Chem. 60, 27-57.
Zhong S. and Mucci A. (1995) Partitioning of rare earth elements (REEs) between calcite
and seawater solutions at 25°C and 1 atm and high dissolved REE concentrations
Geochem. Cosmochim. Acta 59, 443-453.
Zhou Z., Shi Y. and Zhou X. (2004) Theoretical studies on the hydrogen bonding
interaction of complexes of formic acid with water. J. Phys. Chem. A, 108(5),
813-822.
Zocchi F. (2007) Accurate bond valence parameters for lanthanide-oxygen bonds. J. Mol.
Struct. (Theochem) 805, 73-78.
Zuyi T., Taiwei C. and Weijuan L. (2000) On the application of surface complexation
models to ionic adsorption. J. Colloid Interface Sci. 232(1), 174-177.

340
APPENDICES
XVI. Chapter 1: Gedanken Experiment Data
XVII. Chapter 4: Gaspeite: Acidimetric Titration Data
XVIII. Chapter 4: Gaspeite: Alkalimetric Data
XIX. Chapter 4: Gaspeite: Electrokinetic Data
XX. Chapter 5: Optimised Small Calcite Cluster
XXI. Chapter 5: Optimised Large Calcite Cluster
XXII. Chapter 5: Geometrically-optimized (CaCO3)9/4H2O cluster
XXIII. Chapter 6: CaCO3(s) Solubility Product Data
XXIV. Chapter 6: CaCO3(s) Acidimetric Titration Data
XXV. Chapter 6: CaCO3(s) Calcium Titration Data
XXVI. Chapter 6: Methods and Calculations
XXVII. Chapter 6: Referencing of data to the ZNSRC
XXVIII. Chapter 6: Equilibrium Speciation Calculations involving Ion Exchange
XXIX. Chapter 6: Tableau-based Formulation : CaCO3(s)-KCl-H2O System
XXX. Chapters 2, 3, 4 and 6: Matlab© Subroutines
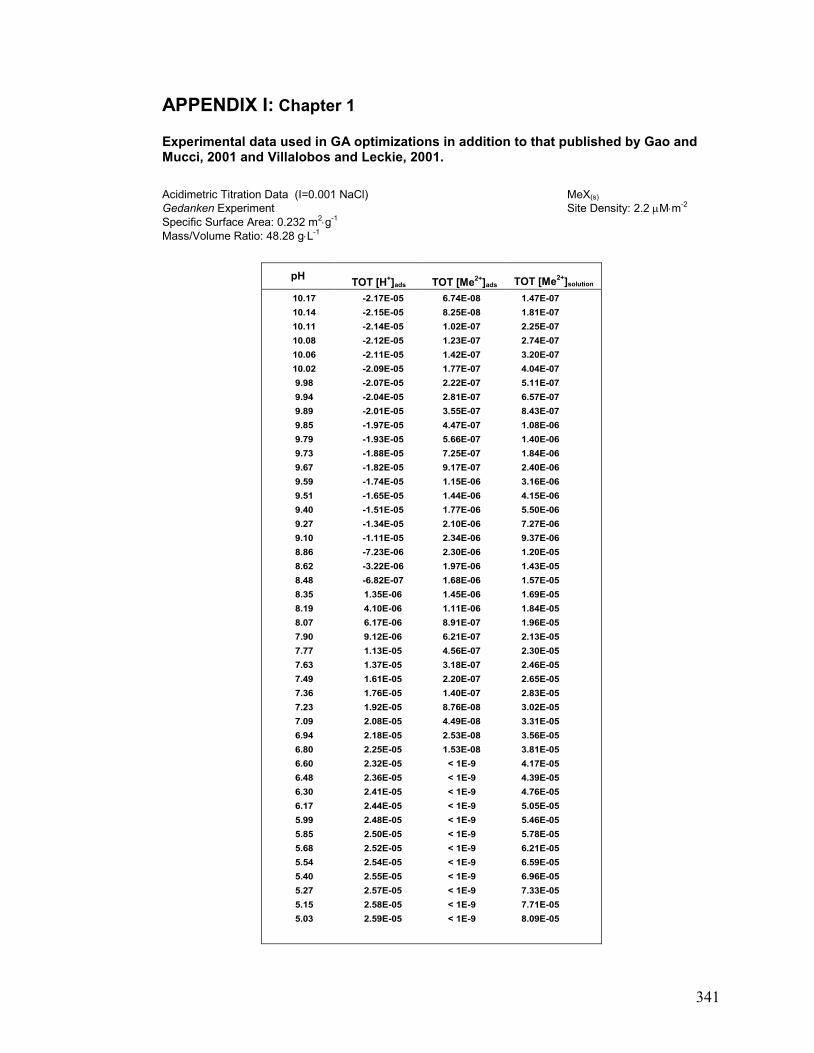
341
APPENDIX I: Chapter 1
Experimental data used in GA optimizations in addition to that published by Gao and Mucci, 2001 and Villalobos and Leckie, 2001.
Acidimetric Titration Data (I=0.001 NaCl) MeX(s)
Gedanken Experiment Site Density: 2.2 Mm-2
Specific Surface Area: 0.232 m2g-1
Mass/Volume Ratio: 48.28 gL-1
pH
TOT [H+]ads
(mol · L
-1)
TOT [Me
2+]ads
(mol · L
-1)
TOT [Me
2+]solution
(mol · L
-1)
10.17 -2.17E-05 6.74E-08 1.47E-07
10.14 -2.15E-05 8.25E-08 1.81E-07
10.11 -2.14E-05 1.02E-07 2.25E-07
10.08 -2.12E-05 1.23E-07 2.74E-07
10.06 -2.11E-05 1.42E-07 3.20E-07
10.02 -2.09E-05 1.77E-07 4.04E-07
9.98 -2.07E-05 2.22E-07 5.11E-07
9.94 -2.04E-05 2.81E-07 6.57E-07
9.89 -2.01E-05 3.55E-07 8.43E-07
9.85 -1.97E-05 4.47E-07 1.08E-06
9.79 -1.93E-05 5.66E-07 1.40E-06
9.73 -1.88E-05 7.25E-07 1.84E-06
9.67 -1.82E-05 9.17E-07 2.40E-06
9.59 -1.74E-05 1.15E-06 3.16E-06
9.51 -1.65E-05 1.44E-06 4.15E-06
9.40 -1.51E-05 1.77E-06 5.50E-06
9.27 -1.34E-05 2.10E-06 7.27E-06
9.10 -1.11E-05 2.34E-06 9.37E-06
8.86 -7.23E-06 2.30E-06 1.20E-05
8.62 -3.22E-06 1.97E-06 1.43E-05
8.48 -6.82E-07 1.68E-06 1.57E-05
8.35 1.35E-06 1.45E-06 1.69E-05
8.19 4.10E-06 1.11E-06 1.84E-05
8.07 6.17E-06 8.91E-07 1.96E-05
7.90 9.12E-06 6.21E-07 2.13E-05
7.77 1.13E-05 4.56E-07 2.30E-05
7.63 1.37E-05 3.18E-07 2.46E-05
7.49 1.61E-05 2.20E-07 2.65E-05
7.36 1.76E-05 1.40E-07 2.83E-05
7.23 1.92E-05 8.76E-08 3.02E-05
7.09 2.08E-05 4.49E-08 3.31E-05
6.94 2.18E-05 2.53E-08 3.56E-05
6.80 2.25E-05 1.53E-08 3.81E-05
6.60 2.32E-05 < 1E-9 4.17E-05
6.48 2.36E-05 < 1E-9 4.39E-05
6.30 2.41E-05 < 1E-9 4.76E-05
6.17 2.44E-05 < 1E-9 5.05E-05
5.99 2.48E-05 < 1E-9 5.46E-05
5.85 2.50E-05 < 1E-9 5.78E-05
5.68 2.52E-05 < 1E-9 6.21E-05
5.54 2.54E-05 < 1E-9 6.59E-05
5.40 2.55E-05 < 1E-9 6.96E-05
5.27 2.57E-05 < 1E-9 7.33E-05
5.15 2.58E-05 < 1E-9 7.71E-05
5.03 2.59E-05 < 1E-9 8.09E-05
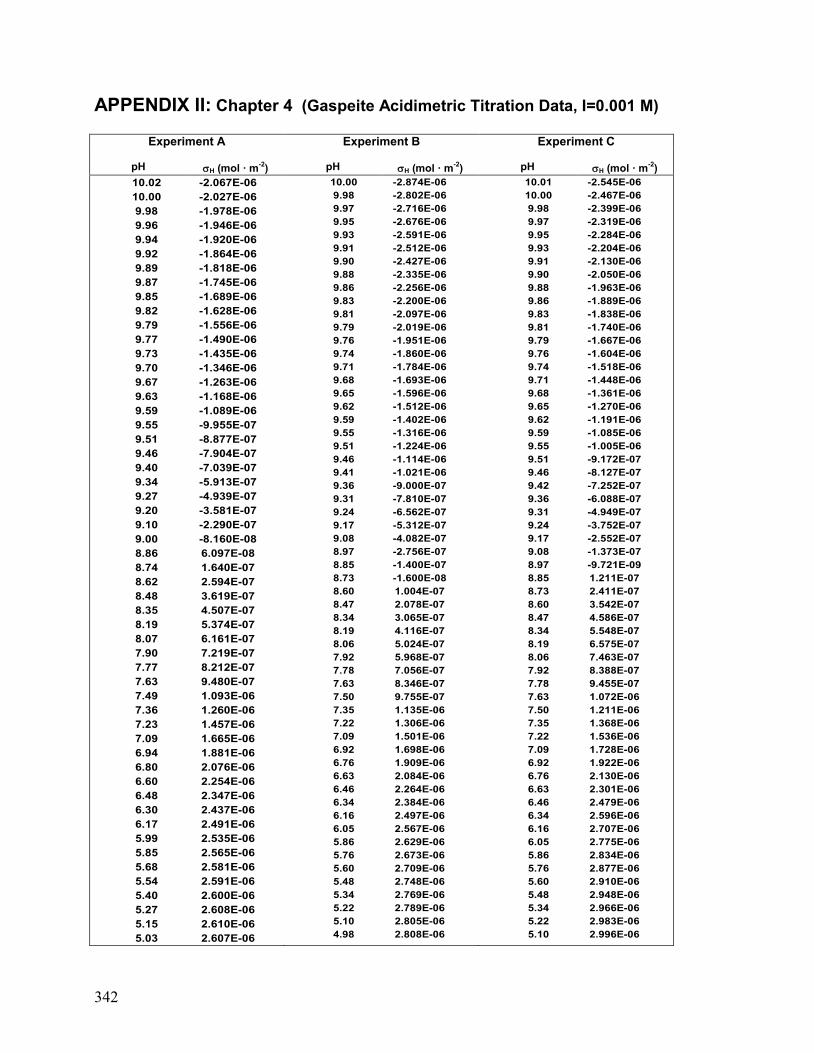
342
APPENDIX II: Chapter 4 (Gaspeite Acidimetric Titration Data, I=0.001 M)
Experiment A Experiment B Experiment C
pH
H (mol · m-2)
pH
H (mol · m-2)
pH
H (mol · m-2)
10.02 -2.067E-06
10.00 -2.027E-06
9.98 -1.978E-06
9.96 -1.946E-06
9.94 -1.920E-06
9.92 -1.864E-06
9.89 -1.818E-06
9.87 -1.745E-06
9.85 -1.689E-06
9.82 -1.628E-06
9.79 -1.556E-06
9.77 -1.490E-06
9.73 -1.435E-06
9.70 -1.346E-06
9.67 -1.263E-06
9.63 -1.168E-06
9.59 -1.089E-06
9.55 -9.955E-07
9.51 -8.877E-07
9.46 -7.904E-07
9.40 -7.039E-07
9.34 -5.913E-07
9.27 -4.939E-07
9.20 -3.581E-07
9.10 -2.290E-07
9.00 -8.160E-08
8.86 6.097E-08
8.74 1.640E-07
8.62 2.594E-07
8.48 3.619E-07
8.35 4.507E-07
8.19 5.374E-07
8.07 6.161E-07
7.90 7.219E-07
7.77 8.212E-07
7.63 9.480E-07
7.49 1.093E-06
7.36 1.260E-06
7.23 1.457E-06
7.09 1.665E-06
6.94 1.881E-06
6.80 2.076E-06
6.60 2.254E-06
6.48 2.347E-06
6.30 2.437E-06
6.17 2.491E-06
5.99 2.535E-06
5.85 2.565E-06
5.68 2.581E-06
5.54 2.591E-06
5.40 2.600E-06
5.27 2.608E-06
5.15 2.610E-06
5.03 2.607E-06
10.00 -2.874E-06
9.98 -2.802E-06
9.97 -2.716E-06
9.95 -2.676E-06
9.93 -2.591E-06
9.91 -2.512E-06
9.90 -2.427E-06
9.88 -2.335E-06
9.86 -2.256E-06
9.83 -2.200E-06
9.81 -2.097E-06
9.79 -2.019E-06
9.76 -1.951E-06
9.74 -1.860E-06
9.71 -1.784E-06
9.68 -1.693E-06
9.65 -1.596E-06
9.62 -1.512E-06
9.59 -1.402E-06
9.55 -1.316E-06
9.51 -1.224E-06
9.46 -1.114E-06
9.41 -1.021E-06
9.36 -9.000E-07
9.31 -7.810E-07
9.24 -6.562E-07
9.17 -5.312E-07
9.08 -4.082E-07
8.97 -2.756E-07
8.85 -1.400E-07
8.73 -1.600E-08
8.60 1.004E-07
8.47 2.078E-07
8.34 3.065E-07
8.19 4.116E-07
8.06 5.024E-07
7.92 5.968E-07
7.78 7.056E-07
7.63 8.346E-07
7.50 9.755E-07
7.35 1.135E-06
7.22 1.306E-06
7.09 1.501E-06
6.92 1.698E-06
6.76 1.909E-06
6.63 2.084E-06
6.46 2.264E-06
6.34 2.384E-06
6.16 2.497E-06
6.05 2.567E-06
5.86 2.629E-06
5.76 2.673E-06
5.60 2.709E-06
5.48 2.748E-06
5.34 2.769E-06
5.22 2.789E-06
5.10 2.805E-06
4.98 2.808E-06
10.01 -2.545E-06
10.00 -2.467E-06
9.98 -2.399E-06
9.97 -2.319E-06
9.95 -2.284E-06
9.93 -2.204E-06
9.91 -2.130E-06
9.90 -2.050E-06
9.88 -1.963E-06
9.86 -1.889E-06
9.83 -1.838E-06
9.81 -1.740E-06
9.79 -1.667E-06
9.76 -1.604E-06
9.74 -1.518E-06
9.71 -1.448E-06
9.68 -1.361E-06
9.65 -1.270E-06
9.62 -1.191E-06
9.59 -1.085E-06
9.55 -1.005E-06
9.51 -9.172E-07
9.46 -8.127E-07
9.42 -7.252E-07
9.36 -6.088E-07
9.31 -4.949E-07
9.24 -3.752E-07
9.17 -2.552E-07
9.08 -1.373E-07
8.97 -9.721E-09
8.85 1.211E-07
8.73 2.411E-07
8.60 3.542E-07
8.47 4.586E-07
8.34 5.548E-07
8.19 6.575E-07
8.06 7.463E-07
7.92 8.388E-07
7.78 9.455E-07
7.63 1.072E-06
7.50 1.211E-06
7.35 1.368E-06
7.22 1.536E-06
7.09 1.728E-06
6.92 1.922E-06
6.76 2.130E-06
6.63 2.301E-06
6.46 2.479E-06
6.34 2.596E-06
6.16 2.707E-06
6.05 2.775E-06
5.86 2.834E-06
5.76 2.877E-06
5.60 2.910E-06
5.48 2.948E-06
5.34 2.966E-06
5.22 2.983E-06
5.10 2.996E-06
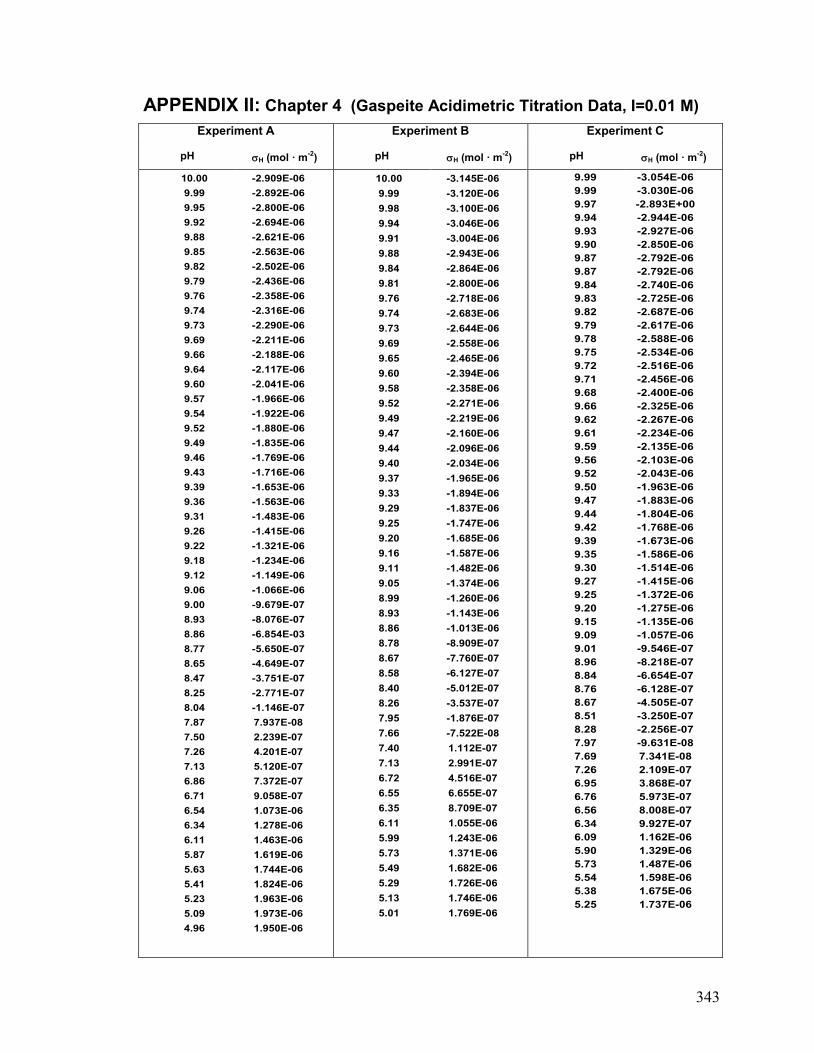
343
APPENDIX II: Chapter 4 (Gaspeite Acidimetric Titration Data, I=0.01 M)
Experiment A Experiment B Experiment C
pH
H (mol · m-2)
pH
H (mol · m-2)
pH
H (mol · m-2)
10.00 -2.909E-06
9.99 -2.892E-06
9.95 -2.800E-06
9.92 -2.694E-06
9.88 -2.621E-06
9.85 -2.563E-06
9.82 -2.502E-06
9.79 -2.436E-06
9.76 -2.358E-06
9.74 -2.316E-06
9.73 -2.290E-06
9.69 -2.211E-06
9.66 -2.188E-06
9.64 -2.117E-06
9.60 -2.041E-06
9.57 -1.966E-06
9.54 -1.922E-06
9.52 -1.880E-06
9.49 -1.835E-06
9.46 -1.769E-06
9.43 -1.716E-06
9.39 -1.653E-06
9.36 -1.563E-06
9.31 -1.483E-06
9.26 -1.415E-06
9.22 -1.321E-06
9.18 -1.234E-06
9.12 -1.149E-06
9.06 -1.066E-06
9.00 -9.679E-07
8.93 -8.076E-07
8.86 -6.854E-03
8.77 -5.650E-07
8.65 -4.649E-07
8.47 -3.751E-07
8.25 -2.771E-07
8.04 -1.146E-07
7.87 7.937E-08
7.50 2.239E-07
7.26 4.201E-07
7.13 5.120E-07
6.86 7.372E-07
6.71 9.058E-07
6.54 1.073E-06
6.34 1.278E-06
6.11 1.463E-06
5.87 1.619E-06
5.63 1.744E-06
5.41 1.824E-06
5.23 1.963E-06
5.09 1.973E-06
4.96 1.950E-06
10.00 -3.145E-06
9.99 -3.120E-06
9.98 -3.100E-06
9.94 -3.046E-06
9.91 -3.004E-06
9.88 -2.943E-06
9.84 -2.864E-06
9.81 -2.800E-06
9.76 -2.718E-06
9.74 -2.683E-06
9.73 -2.644E-06
9.69 -2.558E-06
9.65 -2.465E-06
9.60 -2.394E-06
9.58 -2.358E-06
9.52 -2.271E-06
9.49 -2.219E-06
9.47 -2.160E-06
9.44 -2.096E-06
9.40 -2.034E-06
9.37 -1.965E-06
9.33 -1.894E-06
9.29 -1.837E-06
9.25 -1.747E-06
9.20 -1.685E-06
9.16 -1.587E-06
9.11 -1.482E-06
9.05 -1.374E-06
8.99 -1.260E-06
8.93 -1.143E-06
8.86 -1.013E-06
8.78 -8.909E-07
8.67 -7.760E-07
8.58 -6.127E-07
8.40 -5.012E-07
8.26 -3.537E-07
7.95 -1.876E-07
7.66 -7.522E-08
7.40 1.112E-07
7.13 2.991E-07
6.72 4.516E-07
6.55 6.655E-07
6.35 8.709E-07
6.11 1.055E-06
5.99 1.243E-06
5.73 1.371E-06
5.49 1.682E-06
5.29 1.726E-06
5.13 1.746E-06
5.01 1.769E-06
9.99 -3.054E-06
9.99 -3.030E-06
9.97 -2.893E+00
9.94 -2.944E-06
9.93 -2.927E-06
9.90 -2.850E-06
9.87 -2.792E-06
9.87 -2.792E-06
9.84 -2.740E-06
9.83 -2.725E-06
9.82 -2.687E-06
9.79 -2.617E-06
9.78 -2.588E-06
9.75 -2.534E-06
9.72 -2.516E-06
9.71 -2.456E-06
9.68 -2.400E-06
9.66 -2.325E-06
9.62 -2.267E-06
9.61 -2.234E-06
9.59 -2.135E-06
9.56 -2.103E-06
9.52 -2.043E-06
9.50 -1.963E-06
9.47 -1.883E-06
9.44 -1.804E-06
9.42 -1.768E-06
9.39 -1.673E-06
9.35 -1.586E-06
9.30 -1.514E-06
9.27 -1.415E-06
9.25 -1.372E-06
9.20 -1.275E-06
9.15 -1.135E-06
9.09 -1.057E-06
9.01 -9.546E-07
8.96 -8.218E-07
8.84 -6.654E-07
8.76 -6.128E-07
8.67 -4.505E-07
8.51 -3.250E-07
8.28 -2.256E-07
7.97 -9.631E-08
7.69 7.341E-08
7.26 2.109E-07
6.95 3.868E-07
6.76 5.973E-07
6.56 8.008E-07
6.34 9.927E-07
6.09 1.162E-06
5.90 1.329E-06
5.73 1.487E-06
5.54 1.598E-06
5.38 1.675E-06
5.25 1.737E-06
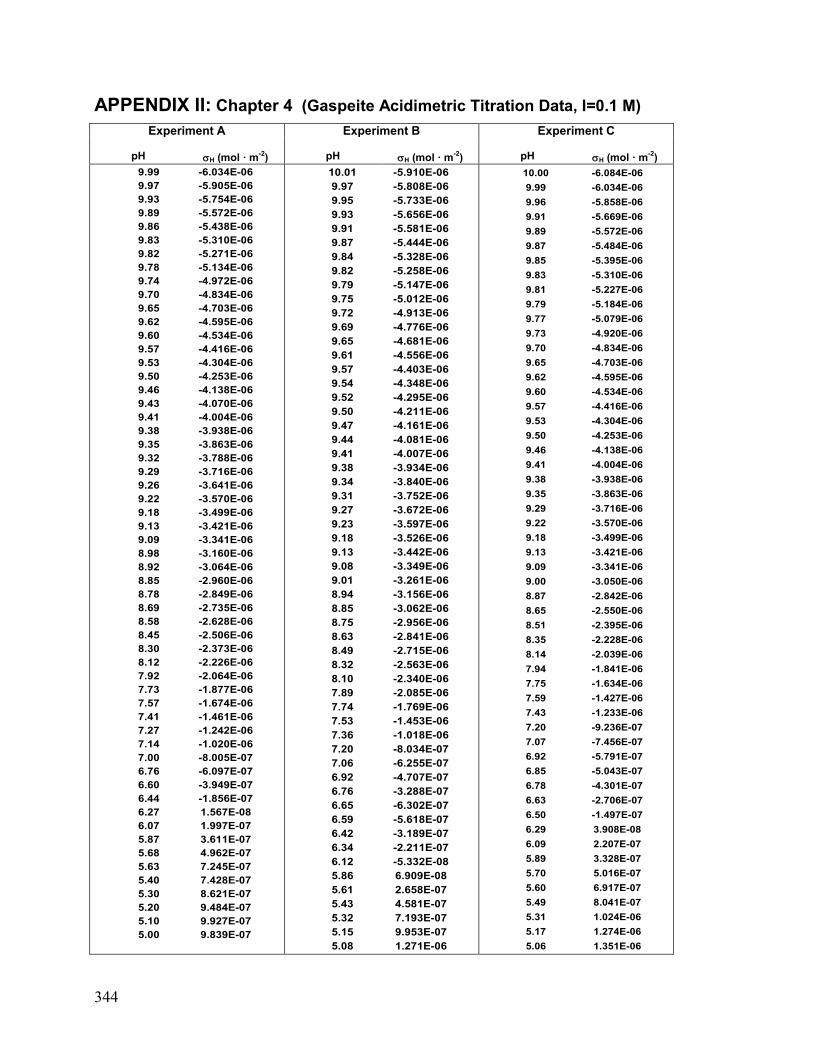
344
APPENDIX II: Chapter 4 (Gaspeite Acidimetric Titration Data, I=0.1 M)
Experiment A Experiment B Experiment C
pH
H (mol · m-2)
pH
H (mol · m-2)
pH
H (mol · m-2)
9.99 -6.034E-06
9.97 -5.905E-06
9.93 -5.754E-06
9.89 -5.572E-06
9.86 -5.438E-06
9.83 -5.310E-06
9.82 -5.271E-06
9.78 -5.134E-06
9.74 -4.972E-06
9.70 -4.834E-06
9.65 -4.703E-06
9.62 -4.595E-06
9.60 -4.534E-06
9.57 -4.416E-06
9.53 -4.304E-06
9.50 -4.253E-06
9.46 -4.138E-06
9.43 -4.070E-06
9.41 -4.004E-06
9.38 -3.938E-06
9.35 -3.863E-06
9.32 -3.788E-06
9.29 -3.716E-06
9.26 -3.641E-06
9.22 -3.570E-06
9.18 -3.499E-06
9.13 -3.421E-06
9.09 -3.341E-06
8.98 -3.160E-06
8.92 -3.064E-06
8.85 -2.960E-06
8.78 -2.849E-06
8.69 -2.735E-06
8.58 -2.628E-06
8.45 -2.506E-06
8.30 -2.373E-06
8.12 -2.226E-06
7.92 -2.064E-06
7.73 -1.877E-06
7.57 -1.674E-06
7.41 -1.461E-06
7.27 -1.242E-06
7.14 -1.020E-06
7.00 -8.005E-07
6.76 -6.097E-07
6.60 -3.949E-07
6.44 -1.856E-07
6.27 1.567E-08
6.07 1.997E-07
5.87 3.611E-07
5.68 4.962E-07
5.63 7.245E-07
5.40 7.428E-07
5.30 8.621E-07
5.20 9.484E-07
5.10 9.927E-07
5.00 9.839E-07
10.01 -5.910E-06
9.97 -5.808E-06
9.95 -5.733E-06
9.93 -5.656E-06
9.91 -5.581E-06
9.87 -5.444E-06
9.84 -5.328E-06
9.82 -5.258E-06
9.79 -5.147E-06
9.75 -5.012E-06
9.72 -4.913E-06
9.69 -4.776E-06
9.65 -4.681E-06
9.61 -4.556E-06
9.57 -4.403E-06
9.54 -4.348E-06
9.52 -4.295E-06
9.50 -4.211E-06
9.47 -4.161E-06
9.44 -4.081E-06
9.41 -4.007E-06
9.38 -3.934E-06
9.34 -3.840E-06
9.31 -3.752E-06
9.27 -3.672E-06
9.23 -3.597E-06
9.18 -3.526E-06
9.13 -3.442E-06
9.08 -3.349E-06
9.01 -3.261E-06
8.94 -3.156E-06
8.85 -3.062E-06
8.75 -2.956E-06
8.63 -2.841E-06
8.49 -2.715E-06
8.32 -2.563E-06
8.10 -2.340E-06
7.89 -2.085E-06
7.74 -1.769E-06
7.53 -1.453E-06
7.36 -1.018E-06
7.20 -8.034E-07
7.06 -6.255E-07
6.92 -4.707E-07
6.76 -3.288E-07
6.65 -6.302E-07
6.59 -5.618E-07
6.42 -3.189E-07
6.34 -2.211E-07
6.12 -5.332E-08
5.86 6.909E-08
5.61 2.658E-07
5.43 4.581E-07
5.32 7.193E-07
5.15 9.953E-07
5.08 1.271E-06
10.00 -6.084E-06
9.99 -6.034E-06
9.96 -5.858E-06
9.91 -5.669E-06
9.89 -5.572E-06
9.87 -5.484E-06
9.85 -5.395E-06
9.83 -5.310E-06
9.81 -5.227E-06
9.79 -5.184E-06
9.77 -5.079E-06
9.73 -4.920E-06
9.70 -4.834E-06
9.65 -4.703E-06
9.62 -4.595E-06
9.60 -4.534E-06
9.57 -4.416E-06
9.53 -4.304E-06
9.50 -4.253E-06
9.46 -4.138E-06
9.41 -4.004E-06
9.38 -3.938E-06
9.35 -3.863E-06
9.29 -3.716E-06
9.22 -3.570E-06
9.18 -3.499E-06
9.13 -3.421E-06
9.09 -3.341E-06
9.00 -3.050E-06
8.87 -2.842E-06
8.65 -2.550E-06
8.51 -2.395E-06
8.35 -2.228E-06
8.14 -2.039E-06
7.94 -1.841E-06
7.75 -1.634E-06
7.59 -1.427E-06
7.43 -1.233E-06
7.20 -9.236E-07
7.07 -7.456E-07
6.92 -5.791E-07
6.85 -5.043E-07
6.78 -4.301E-07
6.63 -2.706E-07
6.50 -1.497E-07
6.29 3.908E-08
6.09 2.207E-07
5.89 3.328E-07
5.70 5.016E-07
5.60 6.917E-07
5.49 8.041E-07
5.31 1.024E-06
5.17 1.274E-06
5.06 1.351E-06
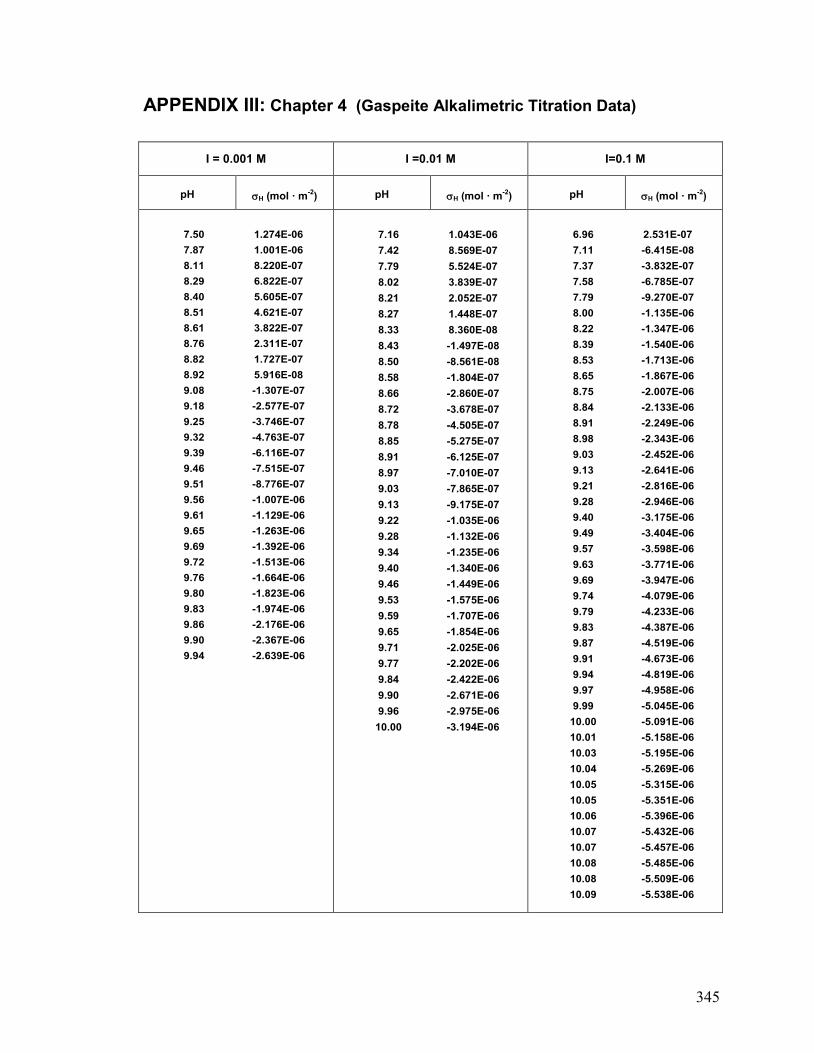
345
APPENDIX III: Chapter 4 (Gaspeite Alkalimetric Titration Data)
I = 0.001 M I =0.01 M I=0.1 M
pH
H (mol · m-2)
pH
H (mol · m-2)
pH
H (mol · m-2)
7.50 1.274E-06
7.87 1.001E-06
8.11 8.220E-07
8.29 6.822E-07
8.40 5.605E-07
8.51 4.621E-07
8.61 3.822E-07
8.76 2.311E-07
8.82 1.727E-07
8.92 5.916E-08
9.08 -1.307E-07
9.18 -2.577E-07
9.25 -3.746E-07
9.32 -4.763E-07
9.39 -6.116E-07
9.46 -7.515E-07
9.51 -8.776E-07
9.56 -1.007E-06
9.61 -1.129E-06
9.65 -1.263E-06
9.69 -1.392E-06
9.72 -1.513E-06
9.76 -1.664E-06
9.80 -1.823E-06
9.83 -1.974E-06
9.86 -2.176E-06
9.90 -2.367E-06
9.94 -2.639E-06
7.16 1.043E-06
7.42 8.569E-07
7.79 5.524E-07
8.02 3.839E-07
8.21 2.052E-07
8.27 1.448E-07
8.33 8.360E-08
8.43 -1.497E-08
8.50 -8.561E-08
8.58 -1.804E-07
8.66 -2.860E-07
8.72 -3.678E-07
8.78 -4.505E-07
8.85 -5.275E-07
8.91 -6.125E-07
8.97 -7.010E-07
9.03 -7.865E-07
9.13 -9.175E-07
9.22 -1.035E-06
9.28 -1.132E-06
9.34 -1.235E-06
9.40 -1.340E-06
9.46 -1.449E-06
9.53 -1.575E-06
9.59 -1.707E-06
9.65 -1.854E-06
9.71 -2.025E-06
9.77 -2.202E-06
9.84 -2.422E-06
9.90 -2.671E-06
9.96 -2.975E-06
10.00 -3.194E-06
6.96 2.531E-07
7.11 -6.415E-08
7.37 -3.832E-07
7.58 -6.785E-07
7.79 -9.270E-07
8.00 -1.135E-06
8.22 -1.347E-06
8.39 -1.540E-06
8.53 -1.713E-06
8.65 -1.867E-06
8.75 -2.007E-06
8.84 -2.133E-06
8.91 -2.249E-06
8.98 -2.343E-06
9.03 -2.452E-06
9.13 -2.641E-06
9.21 -2.816E-06
9.28 -2.946E-06
9.40 -3.175E-06
9.49 -3.404E-06
9.57 -3.598E-06
9.63 -3.771E-06
9.69 -3.947E-06
9.74 -4.079E-06
9.79 -4.233E-06
9.83 -4.387E-06
9.87 -4.519E-06
9.91 -4.673E-06
9.94 -4.819E-06
9.97 -4.958E-06
9.99 -5.045E-06
10.00 -5.091E-06
10.01 -5.158E-06
10.03 -5.195E-06
10.04 -5.269E-06
10.05 -5.315E-06
10.05 -5.351E-06
10.06 -5.396E-06
10.07 -5.432E-06
10.07 -5.457E-06
10.08 -5.485E-06
10.08 -5.509E-06
10.09 -5.538E-06
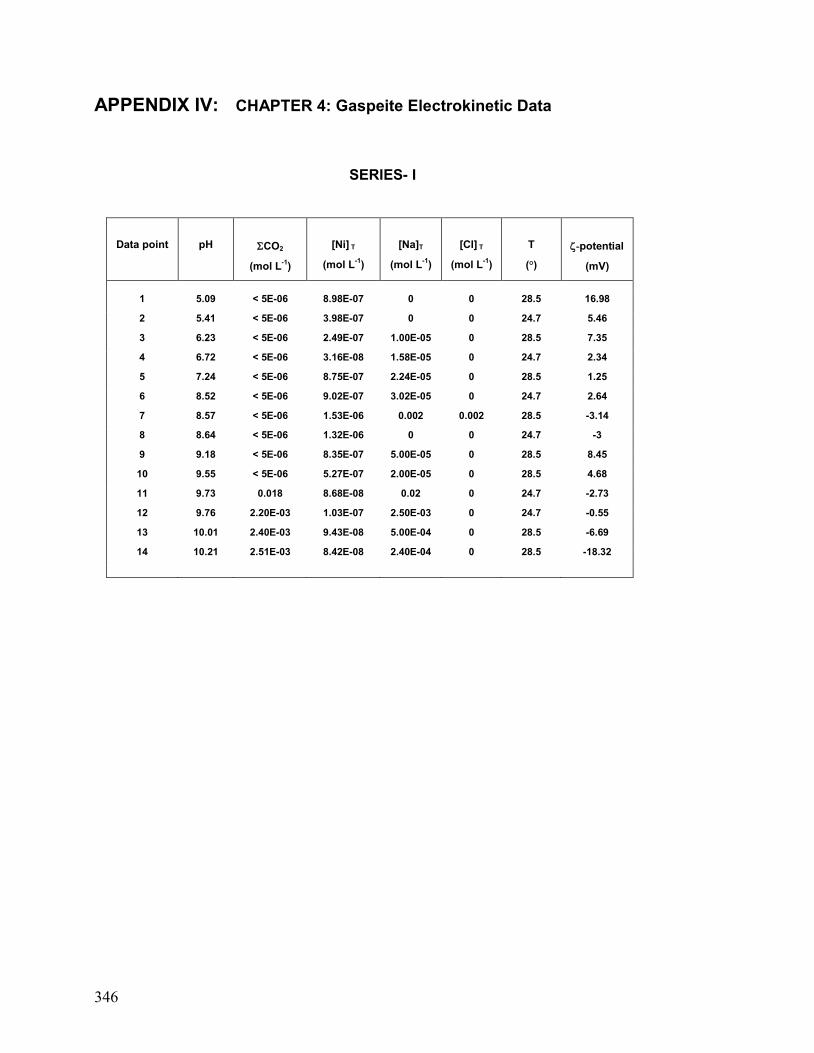
346
APPENDIX IV: CHAPTER 4: Gaspeite Electrokinetic Data
SERIES- I
Data point
pH
CO2
(mol L-1)
[Ni] T
(mol L-1)
[Na]T
(mol L-1)
[Cl] T
(mol L-1)
T
(°)
-potential
(mV)
1 5.09 < 5E-06 8.98E-07 0 0 28.5 16.98
2 5.41 < 5E-06 3.98E-07 0 0 24.7 5.46
3 6.23 < 5E-06 2.49E-07 1.00E-05 0 28.5 7.35
4 6.72 < 5E-06 3.16E-08 1.58E-05 0 24.7 2.34
5 7.24 < 5E-06 8.75E-07 2.24E-05 0 28.5 1.25
6 8.52 < 5E-06 9.02E-07 3.02E-05 0 24.7 2.64
7 8.57 < 5E-06 1.53E-06 0.002 0.002 28.5 -3.14
8 8.64 < 5E-06 1.32E-06 0 0 24.7 -3
9 9.18 < 5E-06 8.35E-07 5.00E-05 0 28.5 8.45
10 9.55 < 5E-06 5.27E-07 2.00E-05 0 28.5 4.68
11 9.73 0.018 8.68E-08 0.02 0 24.7 -2.73
12 9.76 2.20E-03 1.03E-07 2.50E-03 0 24.7 -0.55
13 10.01 2.40E-03 9.43E-08 5.00E-04 0 28.5 -6.69
14 10.21 2.51E-03 8.42E-08 2.40E-04 0 28.5 -18.32
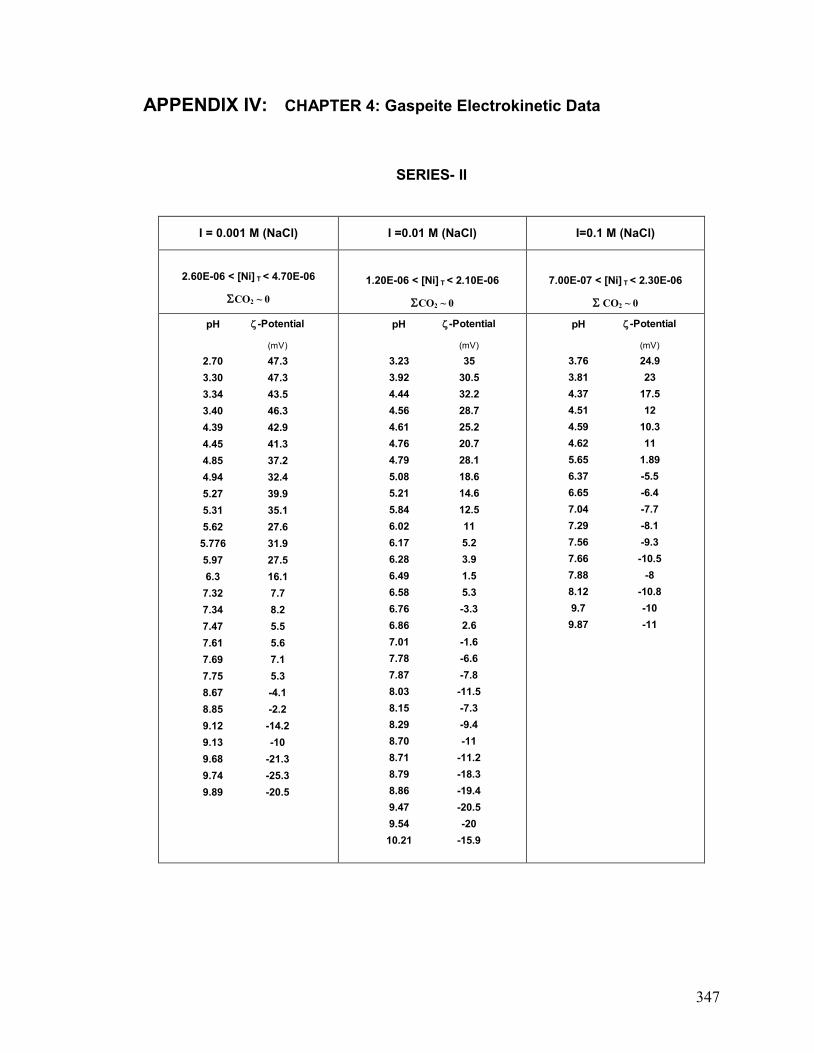
347
APPENDIX IV: CHAPTER 4: Gaspeite Electrokinetic Data
SERIES- II
I = 0.001 M (NaCl) I =0.01 M (NaCl) I=0.1 M (NaCl)
2.60E-06 < [Ni] T < 4.70E-06
CO2 ~ 0
1.20E-06 < [Ni] T < 2.10E-06
CO2 ~ 0
7.00E-07 < [Ni] T < 2.30E-06
CO2 ~ 0
pH -Potential
(mV)
2.70 47.3
3.30 47.3
3.34 43.5
3.40 46.3
4.39 42.9
4.45 41.3
4.85 37.2
4.94 32.4
5.27 39.9
5.31 35.1
5.62 27.6
5.776 31.9
5.97 27.5
6.3 16.1
7.32 7.7
7.34 8.2
7.47 5.5
7.61 5.6
7.69 7.1
7.75 5.3
8.67 -4.1
8.85 -2.2
9.12 -14.2
9.13 -10
9.68 -21.3
9.74 -25.3
9.89 -20.5
pH -Potential
(mV)
3.23 35
3.92 30.5
4.44 32.2
4.56 28.7
4.61 25.2
4.76 20.7
4.79 28.1
5.08 18.6
5.21 14.6
5.84 12.5
6.02 11
6.17 5.2
6.28 3.9
6.49 1.5
6.58 5.3
6.76 -3.3
6.86 2.6
7.01 -1.6
7.78 -6.6
7.87 -7.8
8.03 -11.5
8.15 -7.3
8.29 -9.4
8.70 -11
8.71 -11.2
8.79 -18.3
8.86 -19.4
9.47 -20.5
9.54 -20
10.21 -15.9
pH -Potential
(mV)
3.76 24.9
3.81 23
4.37 17.5
4.51 12
4.59 10.3
4.62 11
5.65 1.89
6.37 -5.5
6.65 -6.4
7.04 -7.7
7.29 -8.1
7.56 -9.3
7.66 -10.5
7.88 -8
8.12 -10.8
9.7 -10
9.87 -11
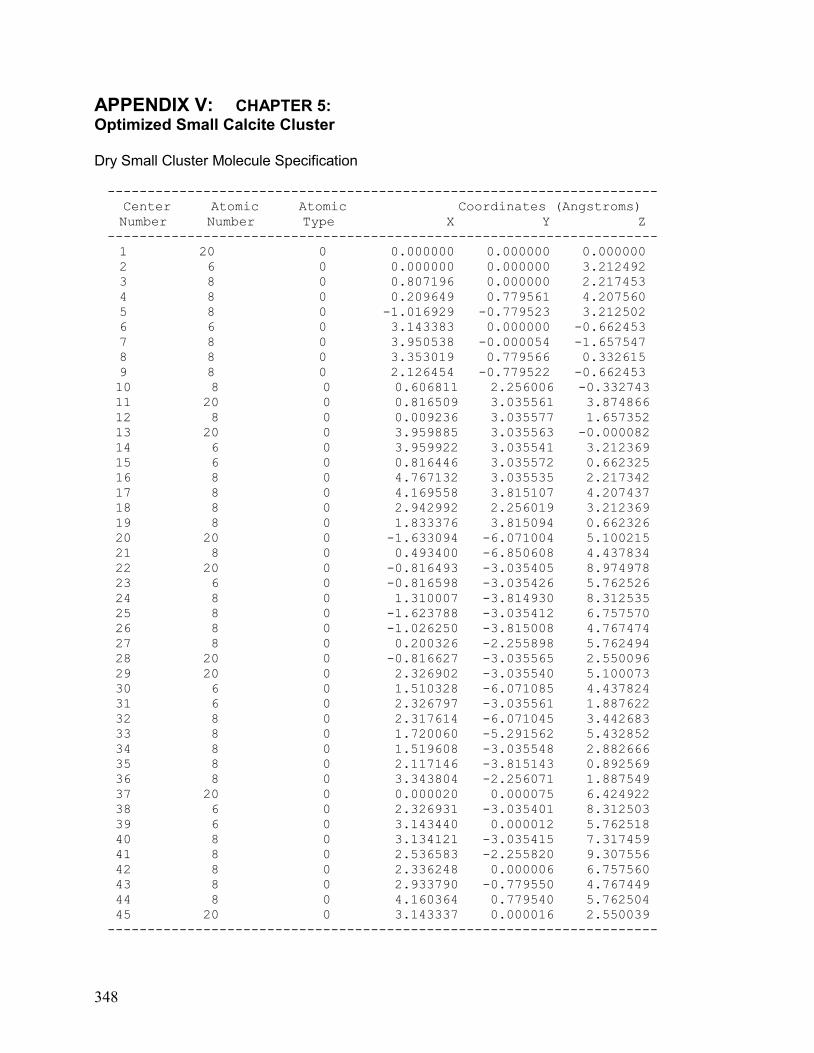
348
APPENDIX V: CHAPTER 5:
Optimized Small Calcite Cluster Dry Small Cluster Molecule Specification
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 20 0 0.000000 0.000000 0.000000
2 6 0 0.000000 0.000000 3.212492
3 8 0 0.807196 0.000000 2.217453
4 8 0 0.209649 0.779561 4.207560
5 8 0 -1.016929 -0.779523 3.212502
6 6 0 3.143383 0.000000 -0.662453
7 8 0 3.950538 -0.000054 -1.657547
8 8 0 3.353019 0.779566 0.332615
9 8 0 2.126454 -0.779522 -0.662453
10 8 0 0.606811 2.256006 -0.332743
11 20 0 0.816509 3.035561 3.874866
12 8 0 0.009236 3.035577 1.657352
13 20 0 3.959885 3.035563 -0.000082
14 6 0 3.959922 3.035541 3.212369
15 6 0 0.816446 3.035572 0.662325
16 8 0 4.767132 3.035535 2.217342
17 8 0 4.169558 3.815107 4.207437
18 8 0 2.942992 2.256019 3.212369
19 8 0 1.833376 3.815094 0.662326
20 20 0 -1.633094 -6.071004 5.100215
21 8 0 0.493400 -6.850608 4.437834
22 20 0 -0.816493 -3.035405 8.974978
23 6 0 -0.816598 -3.035426 5.762526
24 8 0 1.310007 -3.814930 8.312535
25 8 0 -1.623788 -3.035412 6.757570
26 8 0 -1.026250 -3.815008 4.767474
27 8 0 0.200326 -2.255898 5.762494
28 20 0 -0.816627 -3.035565 2.550096
29 20 0 2.326902 -3.035540 5.100073
30 6 0 1.510328 -6.071085 4.437824
31 6 0 2.326797 -3.035561 1.887622
32 8 0 2.317614 -6.071045 3.442683
33 8 0 1.720060 -5.291562 5.432852
34 8 0 1.519608 -3.035548 2.882666
35 8 0 2.117146 -3.815143 0.892569
36 8 0 3.343804 -2.256071 1.887549
37 20 0 0.000020 0.000075 6.424922
38 6 0 2.326931 -3.035401 8.312503
39 6 0 3.143440 0.000012 5.762518
40 8 0 3.134121 -3.035415 7.317459
41 8 0 2.536583 -2.255820 9.307556
42 8 0 2.336248 0.000006 6.757560
43 8 0 2.933790 -0.779550 4.767449
44 8 0 4.160364 0.779540 5.762504
45 20 0 3.143337 0.000016 2.550039
---------------------------------------------------------------------
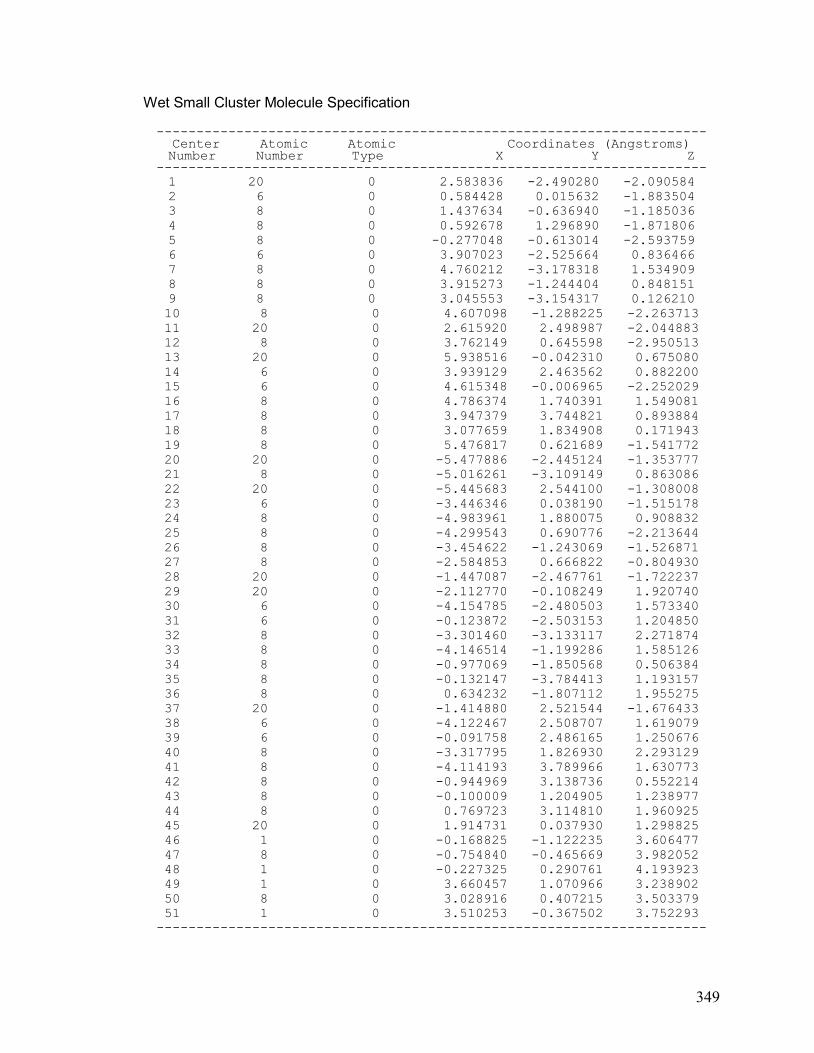
349
Wet Small Cluster Molecule Specification
--------------------------------------------------------------------- Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z
---------------------------------------------------------------------
1 20 0 2.583836 -2.490280 -2.090584
2 6 0 0.584428 0.015632 -1.883504
3 8 0 1.437634 -0.636940 -1.185036
4 8 0 0.592678 1.296890 -1.871806
5 8 0 -0.277048 -0.613014 -2.593759
6 6 0 3.907023 -2.525664 0.836466
7 8 0 4.760212 -3.178318 1.534909
8 8 0 3.915273 -1.244404 0.848151
9 8 0 3.045553 -3.154317 0.126210
10 8 0 4.607098 -1.288225 -2.263713
11 20 0 2.615920 2.498987 -2.044883
12 8 0 3.762149 0.645598 -2.950513
13 20 0 5.938516 -0.042310 0.675080
14 6 0 3.939129 2.463562 0.882200
15 6 0 4.615348 -0.006965 -2.252029
16 8 0 4.786374 1.740391 1.549081
17 8 0 3.947379 3.744821 0.893884
18 8 0 3.077659 1.834908 0.171943
19 8 0 5.476817 0.621689 -1.541772
20 20 0 -5.477886 -2.445124 -1.353777
21 8 0 -5.016261 -3.109149 0.863086
22 20 0 -5.445683 2.544100 -1.308008
23 6 0 -3.446346 0.038190 -1.515178
24 8 0 -4.983961 1.880075 0.908832
25 8 0 -4.299543 0.690776 -2.213644
26 8 0 -3.454622 -1.243069 -1.526871
27 8 0 -2.584853 0.666822 -0.804930
28 20 0 -1.447087 -2.467761 -1.722237
29 20 0 -2.112770 -0.108249 1.920740
30 6 0 -4.154785 -2.480503 1.573340
31 6 0 -0.123872 -2.503153 1.204850
32 8 0 -3.301460 -3.133117 2.271874
33 8 0 -4.146514 -1.199286 1.585126
34 8 0 -0.977069 -1.850568 0.506384
35 8 0 -0.132147 -3.784413 1.193157
36 8 0 0.634232 -1.807112 1.955275
37 20 0 -1.414880 2.521544 -1.676433
38 6 0 -4.122467 2.508707 1.619079
39 6 0 -0.091758 2.486165 1.250676
40 8 0 -3.317795 1.826930 2.293129
41 8 0 -4.114193 3.789966 1.630773
42 8 0 -0.944969 3.138736 0.552214
43 8 0 -0.100009 1.204905 1.238977
44 8 0 0.769723 3.114810 1.960925
45 20 0 1.914731 0.037930 1.298825
46 1 0 -0.168825 -1.122235 3.606477
47 8 0 -0.754840 -0.465669 3.982052
48 1 0 -0.227325 0.290761 4.193923
49 1 0 3.660457 1.070966 3.238902
50 8 0 3.028916 0.407215 3.503379
51 1 0 3.510253 -0.367502 3.752293
---------------------------------------------------------------------
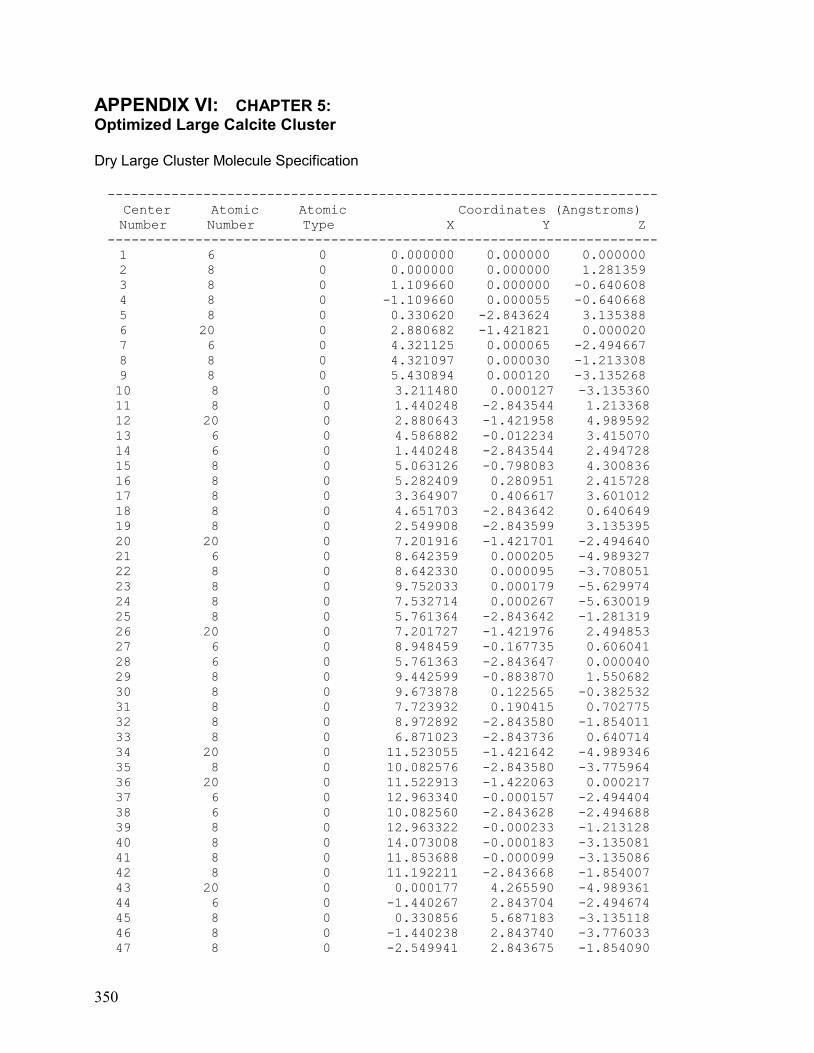
350
APPENDIX VI: CHAPTER 5:
Optimized Large Calcite Cluster Dry Large Cluster Molecule Specification
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 0.000000 0.000000 0.000000
2 8 0 0.000000 0.000000 1.281359
3 8 0 1.109660 0.000000 -0.640608
4 8 0 -1.109660 0.000055 -0.640668
5 8 0 0.330620 -2.843624 3.135388
6 20 0 2.880682 -1.421821 0.000020
7 6 0 4.321125 0.000065 -2.494667
8 8 0 4.321097 0.000030 -1.213308
9 8 0 5.430894 0.000120 -3.135268
10 8 0 3.211480 0.000127 -3.135360
11 8 0 1.440248 -2.843544 1.213368
12 20 0 2.880643 -1.421958 4.989592
13 6 0 4.586882 -0.012234 3.415070
14 6 0 1.440248 -2.843544 2.494728
15 8 0 5.063126 -0.798083 4.300836
16 8 0 5.282409 0.280951 2.415728
17 8 0 3.364907 0.406617 3.601012
18 8 0 4.651703 -2.843642 0.640649
19 8 0 2.549908 -2.843599 3.135395
20 20 0 7.201916 -1.421701 -2.494640
21 6 0 8.642359 0.000205 -4.989327
22 8 0 8.642330 0.000095 -3.708051
23 8 0 9.752033 0.000179 -5.629974
24 8 0 7.532714 0.000267 -5.630019
25 8 0 5.761364 -2.843642 -1.281319
26 20 0 7.201727 -1.421976 2.494853
27 6 0 8.948459 -0.167735 0.606041
28 6 0 5.761363 -2.843647 0.000040
29 8 0 9.442599 -0.883870 1.550682
30 8 0 9.673878 0.122565 -0.382532
31 8 0 7.723932 0.190415 0.702775
32 8 0 8.972892 -2.843580 -1.854011
33 8 0 6.871023 -2.843736 0.640714
34 20 0 11.523055 -1.421642 -4.989346
35 8 0 10.082576 -2.843580 -3.775964
36 20 0 11.522913 -1.422063 0.000217
37 6 0 12.963340 -0.000157 -2.494404
38 6 0 10.082560 -2.843628 -2.494688
39 8 0 12.963322 -0.000233 -1.213128
40 8 0 14.073008 -0.000183 -3.135081
41 8 0 11.853688 -0.000099 -3.135086
42 8 0 11.192211 -2.843668 -1.854007
43 20 0 0.000177 4.265590 -4.989361
44 6 0 -1.440267 2.843704 -2.494674
45 8 0 0.330856 5.687183 -3.135118
46 8 0 -1.440238 2.843740 -3.776033
47 8 0 -2.549941 2.843675 -1.854090
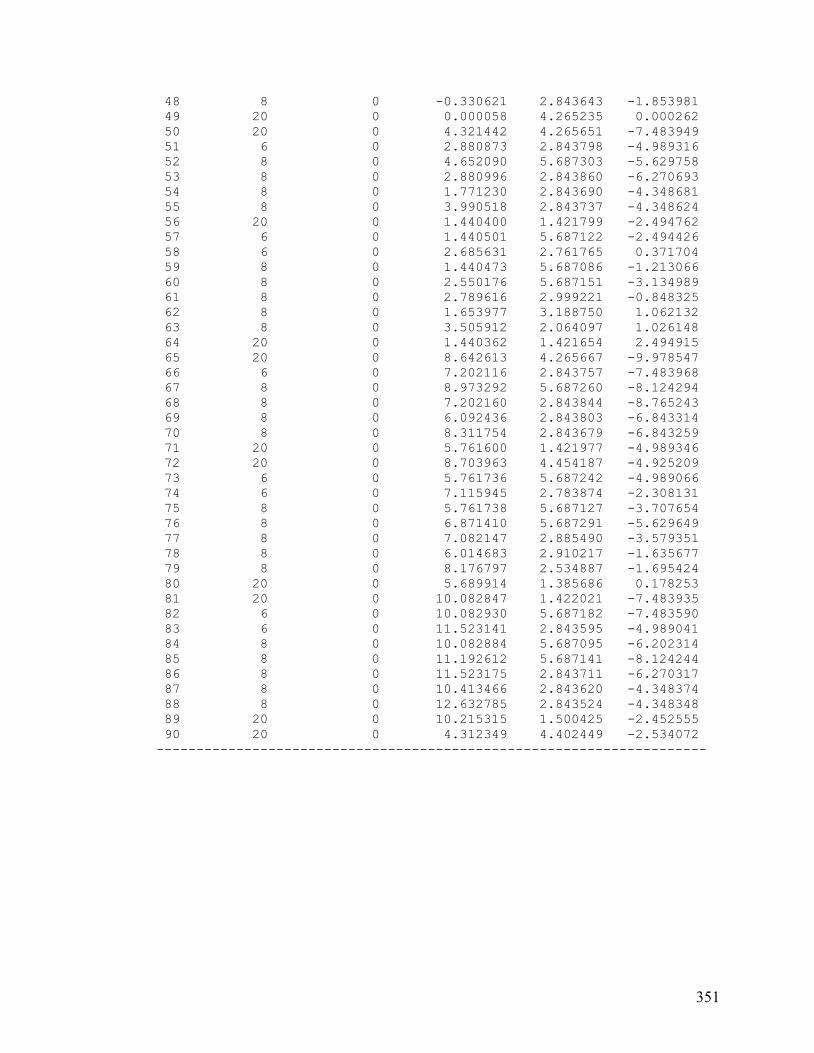
351
48 8 0 -0.330621 2.843643 -1.853981
49 20 0 0.000058 4.265235 0.000262
50 20 0 4.321442 4.265651 -7.483949
51 6 0 2.880873 2.843798 -4.989316
52 8 0 4.652090 5.687303 -5.629758
53 8 0 2.880996 2.843860 -6.270693
54 8 0 1.771230 2.843690 -4.348681
55 8 0 3.990518 2.843737 -4.348624
56 20 0 1.440400 1.421799 -2.494762
57 6 0 1.440501 5.687122 -2.494426
58 6 0 2.685631 2.761765 0.371704
59 8 0 1.440473 5.687086 -1.213066
60 8 0 2.550176 5.687151 -3.134989
61 8 0 2.789616 2.999221 -0.848325
62 8 0 1.653977 3.188750 1.062132
63 8 0 3.505912 2.064097 1.026148
64 20 0 1.440362 1.421654 2.494915
65 20 0 8.642613 4.265667 -9.978547
66 6 0 7.202116 2.843757 -7.483968
67 8 0 8.973292 5.687260 -8.124294
68 8 0 7.202160 2.843844 -8.765243
69 8 0 6.092436 2.843803 -6.843314
70 8 0 8.311754 2.843679 -6.843259
71 20 0 5.761600 1.421977 -4.989346
72 20 0 8.703963 4.454187 -4.925209
73 6 0 5.761736 5.687242 -4.989066
74 6 0 7.115945 2.783874 -2.308131
75 8 0 5.761738 5.687127 -3.707654
76 8 0 6.871410 5.687291 -5.629649
77 8 0 7.082147 2.885490 -3.579351
78 8 0 6.014683 2.910217 -1.635677
79 8 0 8.176797 2.534887 -1.695424
80 20 0 5.689914 1.385686 0.178253
81 20 0 10.082847 1.422021 -7.483935
82 6 0 10.082930 5.687182 -7.483590
83 6 0 11.523141 2.843595 -4.989041
84 8 0 10.082884 5.687095 -6.202314
85 8 0 11.192612 5.687141 -8.124244
86 8 0 11.523175 2.843711 -6.270317
87 8 0 10.413466 2.843620 -4.348374
88 8 0 12.632785 2.843524 -4.348348
89 20 0 10.215315 1.500425 -2.452555
90 20 0 4.312349 4.402449 -2.534072
---------------------------------------------------------------------
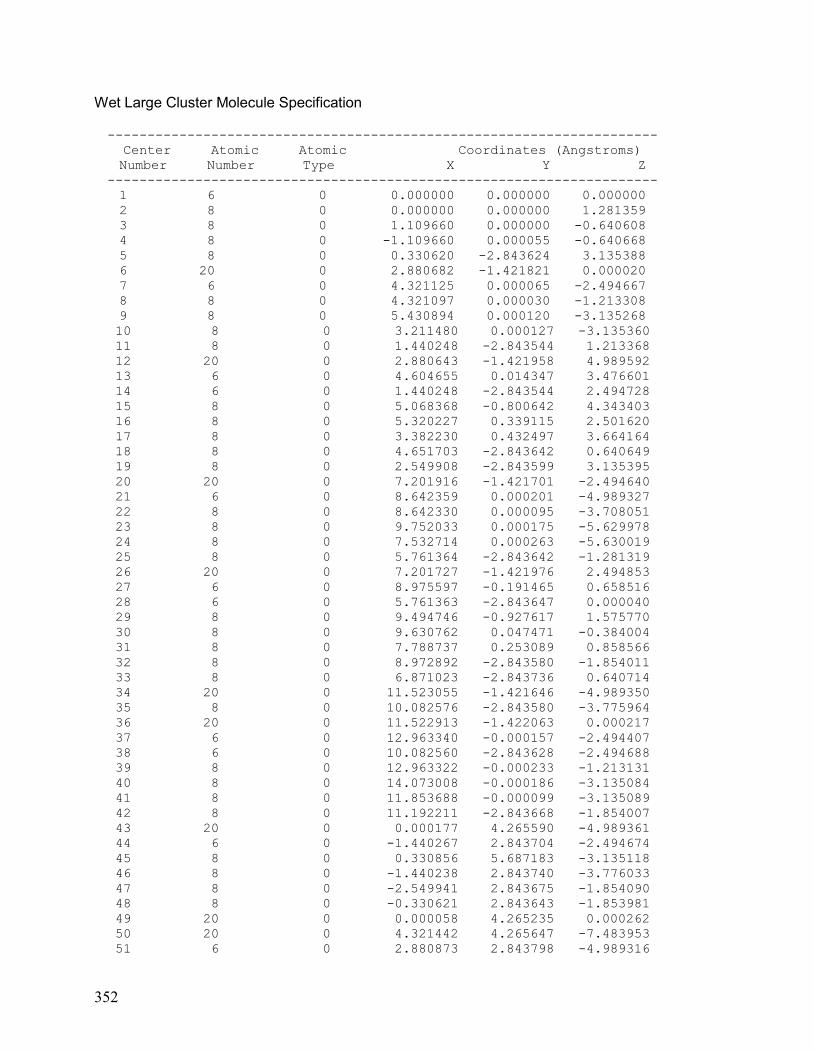
352
Wet Large Cluster Molecule Specification ---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 0.000000 0.000000 0.000000
2 8 0 0.000000 0.000000 1.281359
3 8 0 1.109660 0.000000 -0.640608
4 8 0 -1.109660 0.000055 -0.640668
5 8 0 0.330620 -2.843624 3.135388
6 20 0 2.880682 -1.421821 0.000020
7 6 0 4.321125 0.000065 -2.494667
8 8 0 4.321097 0.000030 -1.213308
9 8 0 5.430894 0.000120 -3.135268
10 8 0 3.211480 0.000127 -3.135360
11 8 0 1.440248 -2.843544 1.213368
12 20 0 2.880643 -1.421958 4.989592
13 6 0 4.604655 0.014347 3.476601
14 6 0 1.440248 -2.843544 2.494728
15 8 0 5.068368 -0.800642 4.343403
16 8 0 5.320227 0.339115 2.501620
17 8 0 3.382230 0.432497 3.664164
18 8 0 4.651703 -2.843642 0.640649
19 8 0 2.549908 -2.843599 3.135395
20 20 0 7.201916 -1.421701 -2.494640
21 6 0 8.642359 0.000201 -4.989327
22 8 0 8.642330 0.000095 -3.708051
23 8 0 9.752033 0.000175 -5.629978
24 8 0 7.532714 0.000263 -5.630019
25 8 0 5.761364 -2.843642 -1.281319
26 20 0 7.201727 -1.421976 2.494853
27 6 0 8.975597 -0.191465 0.658516
28 6 0 5.761363 -2.843647 0.000040
29 8 0 9.494746 -0.927617 1.575770
30 8 0 9.630762 0.047471 -0.384004
31 8 0 7.788737 0.253089 0.858566
32 8 0 8.972892 -2.843580 -1.854011
33 8 0 6.871023 -2.843736 0.640714
34 20 0 11.523055 -1.421646 -4.989350
35 8 0 10.082576 -2.843580 -3.775964
36 20 0 11.522913 -1.422063 0.000217
37 6 0 12.963340 -0.000157 -2.494407
38 6 0 10.082560 -2.843628 -2.494688
39 8 0 12.963322 -0.000233 -1.213131
40 8 0 14.073008 -0.000186 -3.135084
41 8 0 11.853688 -0.000099 -3.135089
42 8 0 11.192211 -2.843668 -1.854007
43 20 0 0.000177 4.265590 -4.989361
44 6 0 -1.440267 2.843704 -2.494674
45 8 0 0.330856 5.687183 -3.135118
46 8 0 -1.440238 2.843740 -3.776033
47 8 0 -2.549941 2.843675 -1.854090
48 8 0 -0.330621 2.843643 -1.853981
49 20 0 0.000058 4.265235 0.000262
50 20 0 4.321442 4.265647 -7.483953
51 6 0 2.880873 2.843798 -4.989316

353
52 8 0 4.652090 5.687303 -5.629762
53 8 0 2.880996 2.843860 -6.270693
54 8 0 1.771230 2.843690 -4.348681
55 8 0 3.990518 2.843737 -4.348624
56 20 0 1.440400 1.421799 -2.494762
57 6 0 1.440501 5.687122 -2.494426
58 6 0 2.624743 2.734907 0.395448
59 8 0 1.440473 5.687086 -1.213066
60 8 0 2.550176 5.687151 -3.134993
61 8 0 2.714486 2.967435 -0.821638
62 8 0 1.636567 3.221194 1.112711
63 8 0 3.405815 1.972883 1.033582
64 20 0 1.440362 1.421654 2.494915
65 20 0 8.642613 4.265663 -9.978549
66 6 0 7.202116 2.843753 -7.483968
67 8 0 8.973292 5.687256 -8.124298
68 8 0 7.202160 2.843840 -8.765243
69 8 0 6.092436 2.843799 -6.843314
70 8 0 8.311754 2.843675 -6.843259
71 20 0 5.761597 1.421973 -4.989346
72 20 0 8.715971 4.547224 -4.798165
73 6 0 5.761736 5.687242 -4.989070
74 6 0 7.060296 2.684957 -2.344034
75 8 0 5.761738 5.687127 -3.707658
76 8 0 6.871410 5.687287 -5.629653
77 8 0 7.189152 2.782233 -3.600004
78 8 0 5.901690 2.776652 -1.801008
79 8 0 8.049922 2.470265 -1.588306
80 20 0 5.632710 1.483148 0.172931
81 20 0 10.082845 1.422017 -7.483939
82 6 0 10.082930 5.687178 -7.483594
83 6 0 11.523140 2.843595 -4.989045
84 8 0 10.082884 5.687091 -6.202318
85 8 0 11.192612 5.687136 -8.124248
86 8 0 11.523173 2.843707 -6.270321
87 8 0 10.413466 2.843620 -4.348378
88 8 0 12.632785 2.843524 -4.348352
89 20 0 10.137314 1.546532 -2.401857
90 8 0 5.133493 5.442819 -0.321180
91 1 0 6.089056 5.450648 -0.302737
92 1 0 4.828074 6.326240 -0.185179
93 1 0 6.680104 2.638194 2.679882
94 8 0 7.053362 2.796457 1.826888
95 1 0 7.741044 2.150173 1.703085
96 1 0 9.301588 5.734964 -1.886283
97 8 0 9.647984 5.296011 -2.647326
98 1 0 10.599582 5.229323 -2.569447
99 1 0 12.177248 3.319319 -1.162930
100 8 0 11.782595 2.521463 -0.818036
101 1 0 12.449912 1.834806 -0.825848
102 1 0 8.091095 4.191689 -0.636802
103 8 0 7.968290 5.007258 -0.154820
104 1 0 8.057777 4.765604 0.753669
105 1 0 13.084608 5.363889 -2.650458
106 8 0 12.433860 4.700834 -2.485661
107 1 0 12.550864 4.032377 -3.195608
108 20 0 4.309359 4.446754 -2.457821
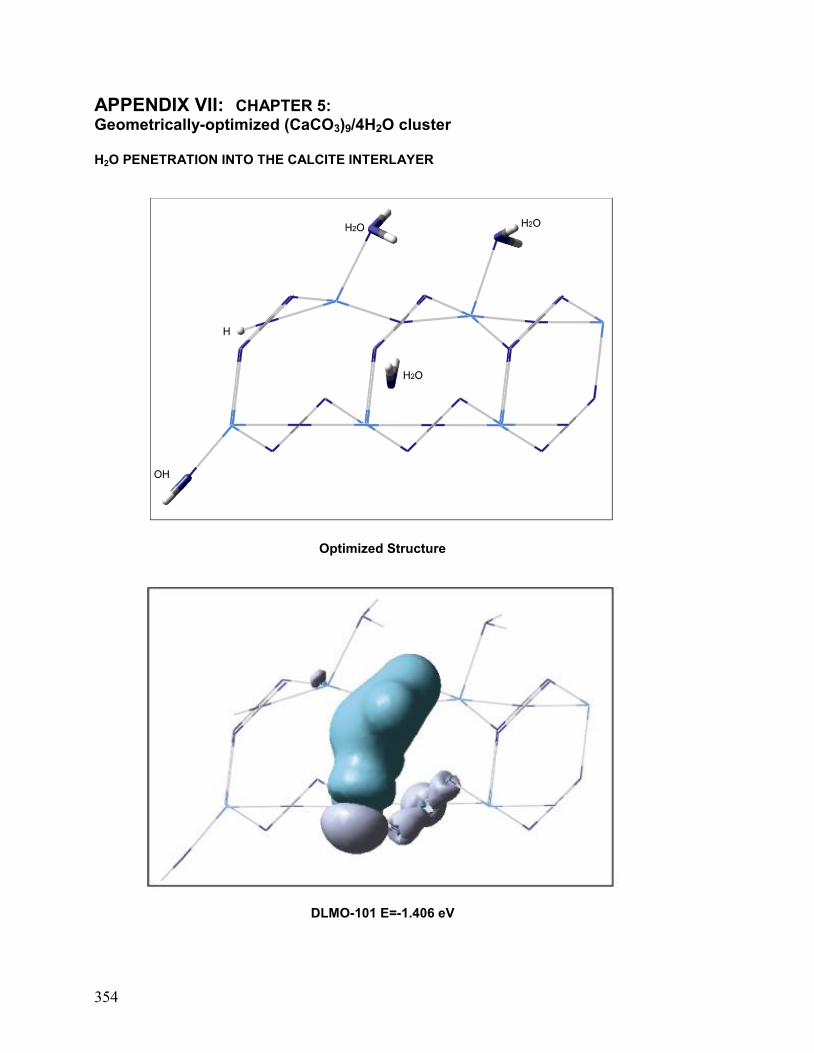
354
APPENDIX VII: CHAPTER 5:
Geometrically-optimized (CaCO3)9/4H2O cluster H2O PENETRATION INTO THE CALCITE INTERLAYER
Optimized Structure
DLMO-101 E=-1.406 eV
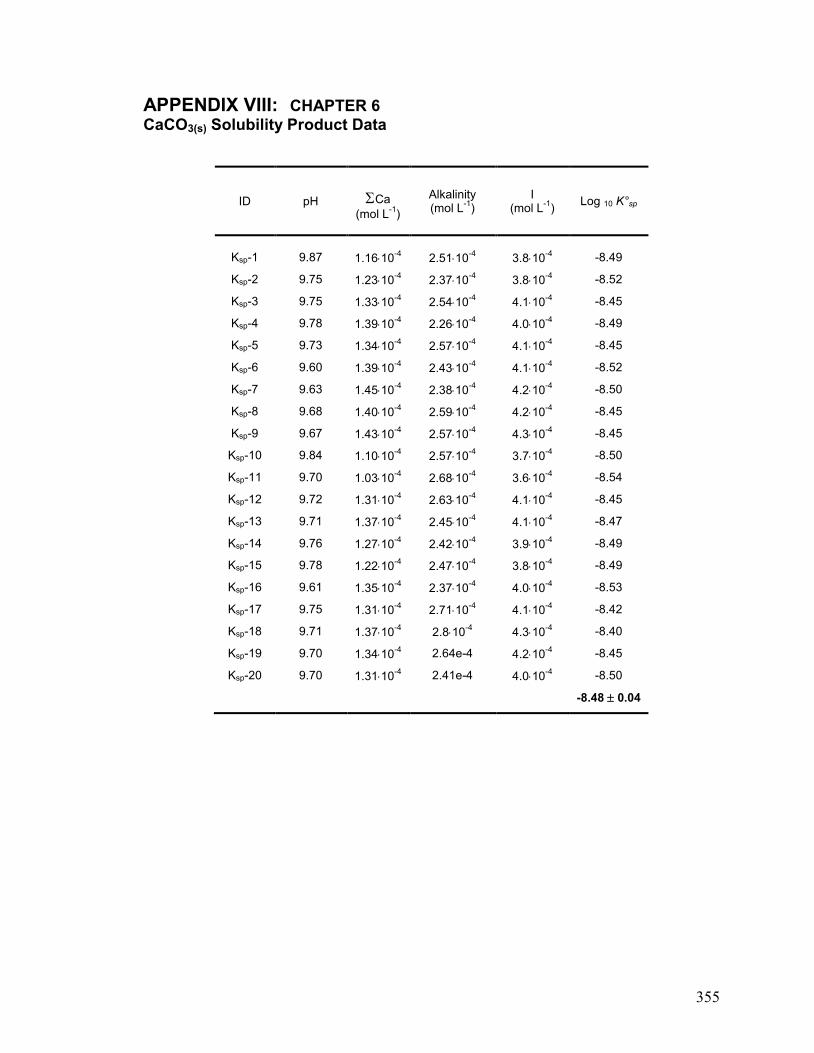
355
APPENDIX VIII: CHAPTER 6
CaCO3(s) Solubility Product Data
ID pH
Ca
(mol L-1
)
Alkalinity (mol L
-1)
I
(mol L-1
)
Log 10 K°sp
Ksp-1 9.87 1.1610
-4 2.5110
-4 3.810
-4 -8.49
Ksp-2 9.75 1.2310-4
2.3710-4
3.810-4
-8.52
Ksp-3 9.75 1.3310-4
2.5410-4
4.110-4
-8.45
Ksp-4 9.78 1.3910-4
2.2610-4
4.010-4
-8.49
Ksp-5 9.73 1.3410-4
2.5710-4
4.110-4
-8.45
Ksp-6 9.60 1.3910-4
2.4310-4
4.110-4
-8.52
Ksp-7 9.63 1.4510-4
2.3810-4
4.210-4
-8.50
Ksp-8 9.68 1.4010-4
2.5910-4
4.210-4
-8.45
Ksp-9 9.67 1.4310-4
2.5710-4
4.310-4
-8.45
Ksp-10 9.84 1.1010-4
2.5710-4
3.710-4
-8.50
Ksp-11 9.70 1.0310-4
2.6810-4
3.610-4
-8.54
Ksp-12 9.72 1.3110-4
2.6310-4
4.110-4
-8.45
Ksp-13 9.71 1.3710-4
2.4510-4
4.110-4
-8.47
Ksp-14 9.76 1.2710-4
2.4210-4
3.910-4
-8.49
Ksp-15 9.78 1.2210-4
2.4710-4
3.810-4
-8.49
Ksp-16 9.61 1.3510-4
2.3710-4
4.010-4
-8.53
Ksp-17 9.75 1.3110-4
2.7110-4
4.110-4
-8.42
Ksp-18 9.71 1.3710-4
2.810-4
4.310-4
-8.40
Ksp-19 9.70 1.3410-4
2.64e-4 4.210-4
-8.45
Ksp-20 9.70 1.3110-4
2.41e-4 4.010-4
-8.50
-8.48 0.04
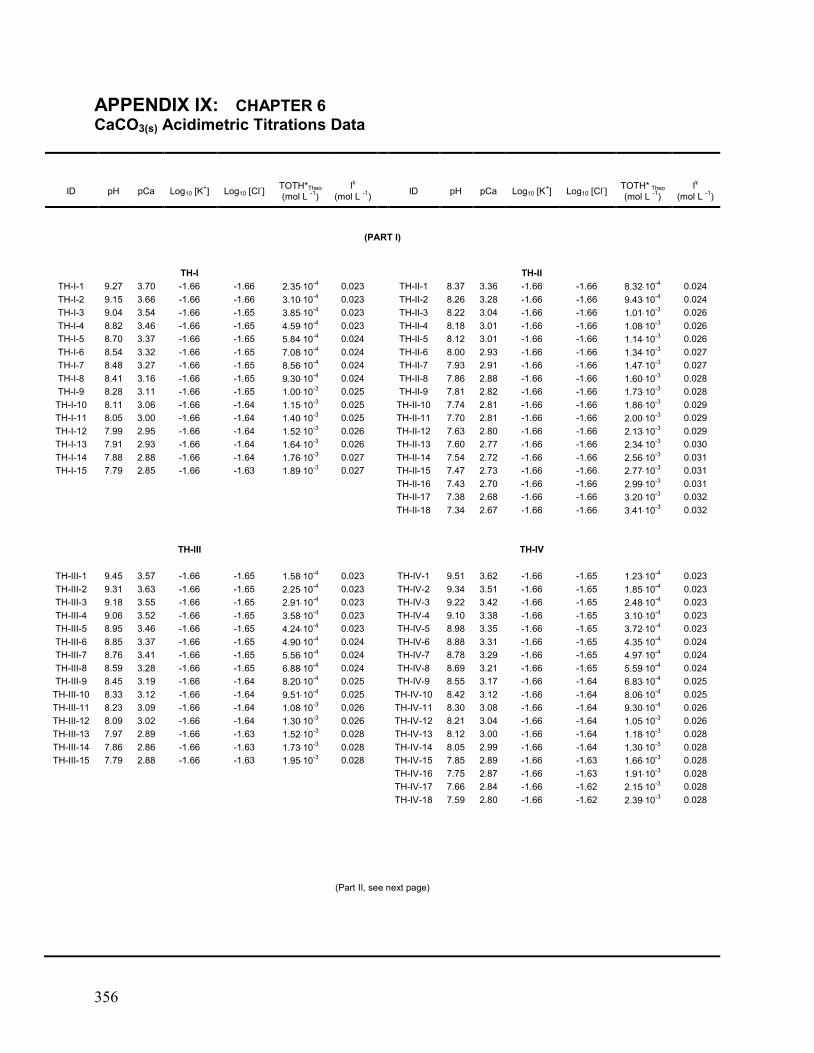
356
APPENDIX IX: CHAPTER 6
CaCO3(s) Acidimetric Titrations Data
ID pH pCa Log10 [K+] Log10 [Cl
-]
TOTH*Theo (mol L
-1)
I¥
(mol L -1
) ID pH pCa Log10 [K
+] Log10 [Cl
-]
TOTH* Theo (mol L
-1)
I¥
(mol L -1
)
(PART I)
TH-I TH-II
TH-I-1 9.27 3.70 -1.66 -1.66 2.3510-4
0.023 TH-II-1 8.37 3.36 -1.66 -1.66 8.3210-4
0.024
TH-I-2 9.15 3.66 -1.66 -1.66 3.1010-4
0.023 TH-II-2 8.26 3.28 -1.66 -1.66 9.4310-4
0.024
TH-I-3 9.04 3.54 -1.66 -1.65 3.8510-4
0.023 TH-II-3 8.22 3.04 -1.66 -1.66 1.0110-3
0.026
TH-I-4 8.82 3.46 -1.66 -1.65 4.5910-4
0.023 TH-II-4 8.18 3.01 -1.66 -1.66 1.0810-3
0.026
TH-I-5 8.70 3.37 -1.66 -1.65 5.8410-4
0.024 TH-II-5 8.12 3.01 -1.66 -1.66 1.1410-3
0.026
TH-I-6 8.54 3.32 -1.66 -1.65 7.0810-4
0.024 TH-II-6 8.00 2.93 -1.66 -1.66 1.3410-3
0.027
TH-I-7 8.48 3.27 -1.66 -1.65 8.5610-4
0.024 TH-II-7 7.93 2.91 -1.66 -1.66 1.4710-3
0.027
TH-I-8 8.41 3.16 -1.66 -1.65 9.3010-4
0.024 TH-II-8 7.86 2.88 -1.66 -1.66 1.6010-3
0.028
TH-I-9 8.28 3.11 -1.66 -1.65 1.0010-3
0.025 TH-II-9 7.81 2.82 -1.66 -1.66 1.7310-3
0.028
TH-I-10 8.11 3.06 -1.66 -1.64 1.1510-3
0.025 TH-II-10 7.74 2.81 -1.66 -1.66 1.8610-3
0.029
TH-I-11 8.05 3.00 -1.66 -1.64 1.4010-3
0.025 TH-II-11 7.70 2.81 -1.66 -1.66 2.0010-3
0.029
TH-I-12 7.99 2.95 -1.66 -1.64 1.5210-3
0.026 TH-II-12 7.63 2.80 -1.66 -1.66 2.1310-3
0.029
TH-I-13 7.91 2.93 -1.66 -1.64 1.6410-3
0.026 TH-II-13 7.60 2.77 -1.66 -1.66 2.3410-3
0.030
TH-I-14 7.88 2.88 -1.66 -1.64 1.7610-3
0.027 TH-II-14 7.54 2.72 -1.66 -1.66 2.5610-3
0.031
TH-I-15 7.79 2.85 -1.66 -1.63 1.8910-3
0.027 TH-II-15 7.47 2.73 -1.66 -1.66 2.7710-3
0.031
TH-II-16 7.43 2.70 -1.66 -1.66 2.9910-3
0.031
TH-II-17 7.38 2.68 -1.66 -1.66 3.2010-3
0.032
TH-II-18 7.34 2.67 -1.66 -1.66 3.4110-3
0.032
TH-III TH-IV
TH-III-1 9.45 3.57 -1.66 -1.65 1.5810-4
0.023 TH-IV-1 9.51 3.62 -1.66 -1.65 1.2310-4
0.023
TH-III-2 9.31 3.63 -1.66 -1.65 2.2510-4
0.023 TH-IV-2 9.34 3.51 -1.66 -1.65 1.8510-4
0.023
TH-III-3 9.18 3.55 -1.66 -1.65 2.9110-4
0.023 TH-IV-3 9.22 3.42 -1.66 -1.65 2.4810-4
0.023
TH-III-4 9.06 3.52 -1.66 -1.65 3.5810-4
0.023 TH-IV-4 9.10 3.38 -1.66 -1.65 3.1010-4
0.023
TH-III-5 8.95 3.46 -1.66 -1.65 4.2410-4
0.023 TH-IV-5 8.98 3.35 -1.66 -1.65 3.7210-4
0.023
TH-III-6 8.85 3.37 -1.66 -1.65 4.9010-4
0.024 TH-IV-6 8.88 3.31 -1.66 -1.65 4.3510-4
0.024
TH-III-7 8.76 3.41 -1.66 -1.65 5.5610-4
0.024 TH-IV-7 8.78 3.29 -1.66 -1.65 4.9710-4
0.024
TH-III-8 8.59 3.28 -1.66 -1.65 6.8810-4
0.024 TH-IV-8 8.69 3.21 -1.66 -1.65 5.5910-4
0.024
TH-III-9 8.45 3.19 -1.66 -1.64 8.2010-4
0.025 TH-IV-9 8.55 3.17 -1.66 -1.64 6.8310-4
0.025
TH-III-10 8.33 3.12 -1.66 -1.64 9.5110-4
0.025 TH-IV-10 8.42 3.12 -1.66 -1.64 8.0610-4
0.025
TH-III-11 8.23 3.09 -1.66 -1.64 1.0810-3
0.026 TH-IV-11 8.30 3.08 -1.66 -1.64 9.3010-4
0.026
TH-III-12 8.09 3.02 -1.66 -1.64 1.3010-3
0.026 TH-IV-12 8.21 3.04 -1.66 -1.64 1.0510-3
0.026
TH-III-13 7.97 2.89 -1.66 -1.63 1.5210-3
0.028 TH-IV-13 8.12 3.00 -1.66 -1.64 1.1810-3
0.028
TH-III-14 7.86 2.86 -1.66 -1.63 1.7310-3
0.028 TH-IV-14 8.05 2.99 -1.66 -1.64 1.3010-3
0.028
TH-III-15 7.79 2.88 -1.66 -1.63 1.9510-3
0.028 TH-IV-15 7.85 2.89 -1.66 -1.63 1.6610-3
0.028
TH-IV-16 7.75 2.87 -1.66 -1.63 1.9110-3
0.028
TH-IV-17 7.66 2.84 -1.66 -1.62 2.1510-3
0.028
TH-IV-18 7.59 2.80 -1.66 -1.62 2.3910-3
0.028
(Part II, see next page)
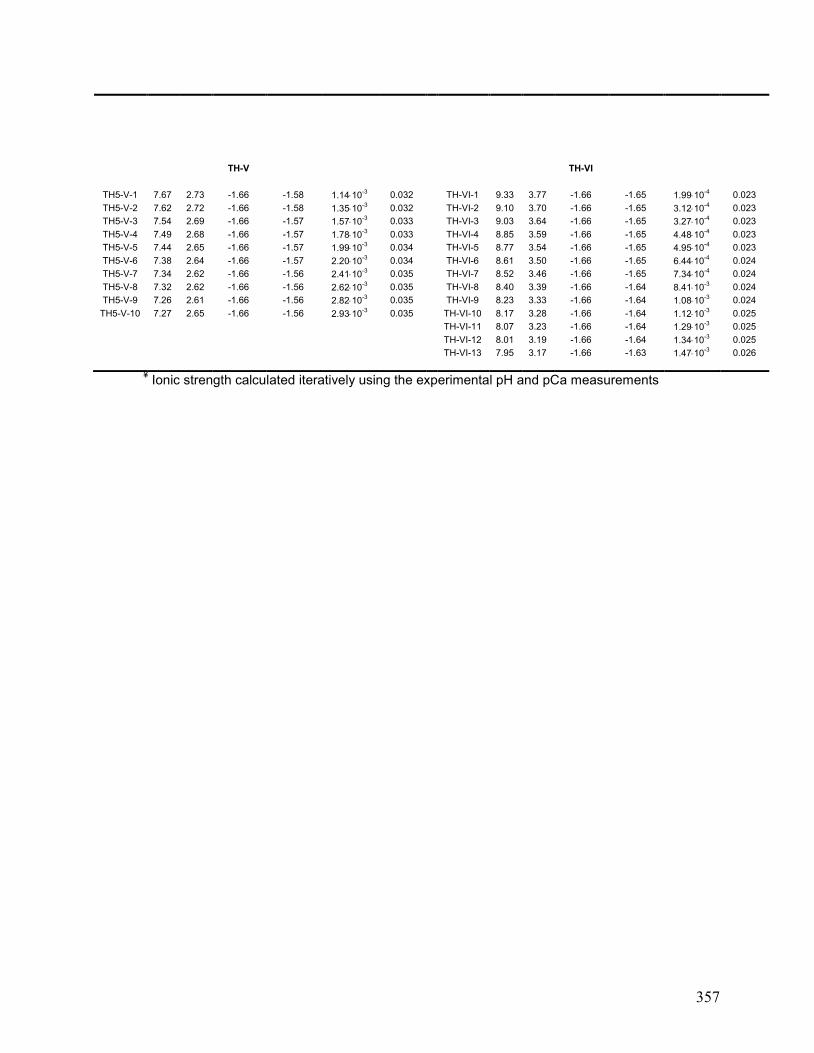
357
TH-V TH-VI
TH5-V-1 7.67 2.73 -1.66 -1.58 1.1410-3
0.032 TH-VI-1 9.33 3.77 -1.66 -1.65 1.9910-4
0.023
TH5-V-2 7.62 2.72 -1.66 -1.58 1.3510-3
0.032 TH-VI-2 9.10 3.70 -1.66 -1.65 3.1210-4
0.023
TH5-V-3 7.54 2.69 -1.66 -1.57 1.5710-3
0.033 TH-VI-3 9.03 3.64 -1.66 -1.65 3.2710-4
0.023
TH5-V-4 7.49 2.68 -1.66 -1.57 1.7810-3
0.033 TH-VI-4 8.85 3.59 -1.66 -1.65 4.4810-4
0.023
TH5-V-5 7.44 2.65 -1.66 -1.57 1.9910-3
0.034 TH-VI-5 8.77 3.54 -1.66 -1.65 4.9510-4
0.023
TH5-V-6 7.38 2.64 -1.66 -1.57 2.2010-3
0.034 TH-VI-6 8.61 3.50 -1.66 -1.65 6.4410-4
0.024
TH5-V-7 7.34 2.62 -1.66 -1.56 2.4110-3
0.035 TH-VI-7 8.52 3.46 -1.66 -1.65 7.3410-4
0.024
TH5-V-8 7.32 2.62 -1.66 -1.56 2.6210-3
0.035 TH-VI-8 8.40 3.39 -1.66 -1.64 8.4110-3
0.024
TH5-V-9 7.26 2.61 -1.66 -1.56 2.8210-3
0.035 TH-VI-9 8.23 3.33 -1.66 -1.64 1.0810-3
0.024
TH5-V-10 7.27 2.65 -1.66 -1.56 2.9310-3
0.035 TH-VI-10 8.17 3.28 -1.66 -1.64 1.1210-3
0.025
TH-VI-11 8.07 3.23 -1.66 -1.64 1.2910-3
0.025
TH-VI-12 8.01 3.19 -1.66 -1.64 1.3410-3
0.025
TH-VI-13 7.95 3.17 -1.66 -1.63 1.4710-3
0.026
¥ Ionic strength calculated iteratively using the experimental pH and pCa measurements
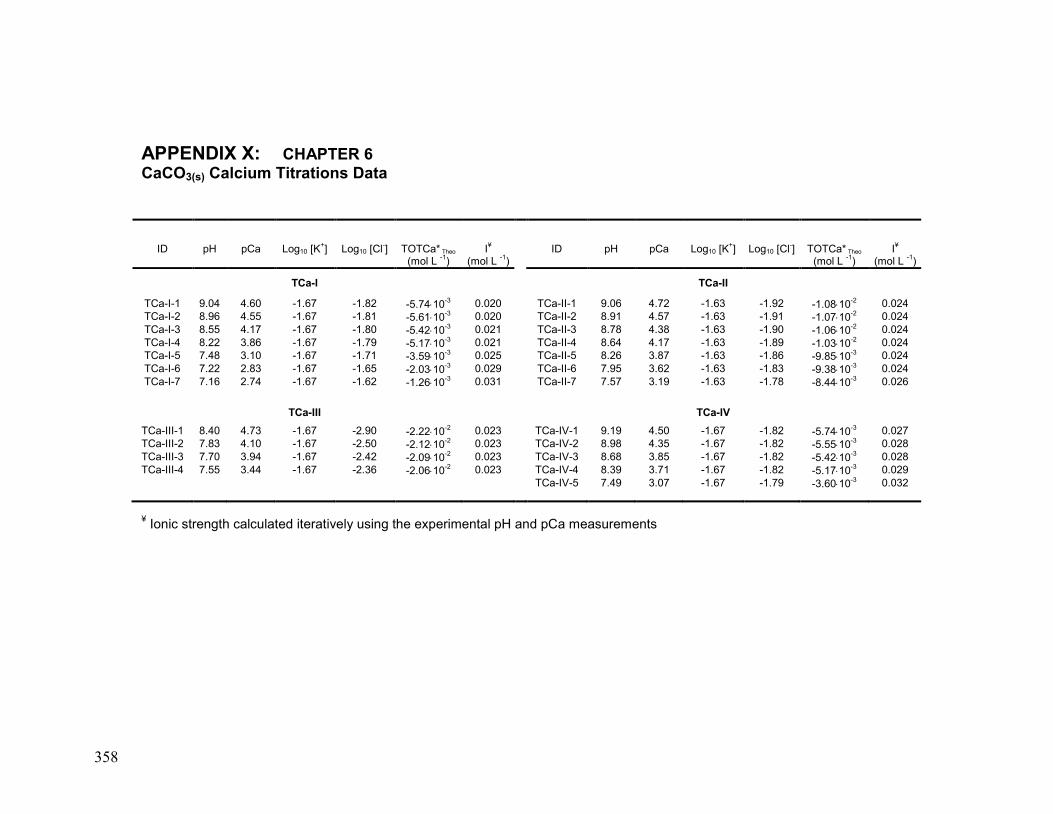
358
APPENDIX X: CHAPTER 6 CaCO3(s) Calcium Titrations Data
ID pH pCa Log10 [K+] Log10 [Cl
-] TOTCa* Theo
(mol L -1)
I¥
(mol L -1)
ID pH pCa Log10 [K+] Log10 [Cl
-] TOTCa* Theo
(mol L -1)
I¥
(mol L -1)
TCa-I TCa-II
TCa-I-1 9.04 4.60 -1.67 -1.82 -5.7410-3 0.020 TCa-II-1 9.06 4.72 -1.63 -1.92 -1.0810
-2 0.024
TCa-I-2 8.96 4.55 -1.67 -1.81 -5.6110-3 0.020 TCa-II-2 8.91 4.57 -1.63 -1.91 -1.0710
-2 0.024
TCa-I-3 8.55 4.17 -1.67 -1.80 -5.4210-3 0.021 TCa-II-3 8.78 4.38 -1.63 -1.90 -1.0610
-2 0.024
TCa-I-4 8.22 3.86 -1.67 -1.79 -5.1710-3 0.021 TCa-II-4 8.64 4.17 -1.63 -1.89 -1.0310
-2 0.024
TCa-I-5 7.48 3.10 -1.67 -1.71 -3.5910-3 0.025 TCa-II-5 8.26 3.87 -1.63 -1.86 -9.8510
-3 0.024
TCa-I-6 7.22 2.83 -1.67 -1.65 -2.0310-3 0.029 TCa-II-6 7.95 3.62 -1.63 -1.83 -9.3810
-3 0.024
TCa-I-7 7.16 2.74 -1.67 -1.62 -1.2610-3 0.031 TCa-II-7 7.57 3.19 -1.63 -1.78 -8.4410
-3 0.026
TCa-III TCa-IV
TCa-III-1 8.40 4.73 -1.67 -2.90 -2.2210-2 0.023 TCa-IV-1 9.19 4.50 -1.67 -1.82 -5.7410
-3 0.027
TCa-III-2 7.83 4.10 -1.67 -2.50 -2.1210-2 0.023 TCa-IV-2 8.98 4.35 -1.67 -1.82 -5.5510
-3 0.028
TCa-III-3 7.70 3.94 -1.67 -2.42 -2.0910-2 0.023 TCa-IV-3 8.68 3.85 -1.67 -1.82 -5.4210
-3 0.028
TCa-III-4 7.55 3.44 -1.67 -2.36 -2.0610-2 0.023 TCa-IV-4 8.39 3.71 -1.67 -1.82 -5.1710
-3 0.029
TCa-IV-5 7.49 3.07 -1.67 -1.79 -3.6010-3 0.032
¥ Ionic strength calculated iteratively using the experimental pH and pCa measurements

359
APPENDIX XI: CHAPTER 6
Methods and Calculations Preparation and standardization of titrant solutions
All solutions were prepared using analytical grade reagents and high purity deionized
(Milli-Q®
, ~ 18 Mohm cm) water. Hydrochloric acid solutions were prepared from
32% HCl and standardized using three gravimetrically prepared
tris(hydroxymethyl)methylamine (TRIS) solutions that were kept refrigerated. The
precision of this standardization was better than 0.1 %. The calcium titrant stock
solutions and calcium standards were prepared from CaCl2H2O crystals and kept
refrigerated before use. These solutions were standardized by volumetric titration with
EGTA. The EGTA titrant solution was standardized using Copenhagen IAPSO
standard seawater. The precision of this determination was better than 0.4 %.
Chemical analyses of CaCO3 suspensions
Alkalinity measurements were carried out using a Radiometer TTT85 titration system
with standardized HCl. The end-point of the titration was identified by the first-
derivative method. The precision of the analysis was better than ± 0.4% and the limit
of detection was of 0.6 mmol kg-1
. Total calcium concentrations were measured by
Flame Atomic Absorption Spectrophotometry (FAAS, AAnalyst 100TM
800 Perkin
Elmer) using external standards (i.e., diluted from a 1000 ppm Certified Standard).
The detection limit of this analysis was 3 μg L-1
with a reproducibility of ± 5%. pH
measurements were performed with a Schott N6980 pH combination electrode,
suitable for concentrated suspensions, and calibrated against four NIST-traceable pH
buffer solutions (4.01, 7.00, 10.00 and 11.00) at 25 0.5°C with a precision of ±
0.002 pH units. Its Nernstian behavior was always very similar (59.2 1.1 mV/log10
aH+) to the theoretical value at 25°C. Calcium ion activities were measured with a
combination Orion 97-20 ionplus® ion selective electrode calibrated with CaCl2
standards (5∙10-6
to 0.015 M) prepared in 0.02 M KCl solutions following the
manufacturer‟s recommendation to achieve a precision of ± 4% at 25 ± 1°C.
Carbonate ion activities were determined using a combination ELIT Ion 8091 ion
selective electrode initially calibrated by two methods: A) against NaHCO3 standards,

360
prepared in 0.02 M KCl solutions, covering a relatively wide range of CO32-
ion
concentrations (3∙10-6
to 0.012 M) and B) against pre-equilibrated calcite suspensions
prepared in 0.02 KCl at different initial Ca:HCO3-:CO3
2- ratios (achieved with
additions of CaCl2, KHCO3 and/or K2CO3) to cover a range of carbonate ion
concentrations similar to the one of method A (2∙10-6
to 0.008 M). In method A, CO32-
ion activities in the standards were estimated from thermodynamic equilibrium
calculations performed iteratively using the Newton-Raphson method implemented in
an in-house Matlab©
subroutine using alkalinity and pH measurements as input. In
contrast, in method B, CO32-
ion activities were estimated using the Ca2+
ion activities
measured with the ISE in the equilibrated calcite suspensions, just before the
calibration of the CO32-
ISE, and application of the solubility product relationship:
where a represent the activity of the specified ion and K°sp stands for the
thermodynamic solubility product of calcite at 25°C. Method B provided better
(Nernstian) and more reproducible calibration slopes and was, therefore, adopted for
the routine calibration of the CO32-
ISE. Thermodynamic constants used in all
calculations of this study are given in Table 1 in the main text. The optimum
operational pH, temperature and analyte concentration ranges were respected for the
three ISEs. Nevertheless, it must be noted that the CO32-
ISE only performed to
specifications in a few preliminary titrations (see below). We suspect that the presence
of CaCO3(s) particles in our experiments may significantly decrease the operational
life expectancy of this ISE. Consequently, this ISE was only used in preliminary
titrations to validate the experimental protocol described below, estimate the re-
equilibration time required after discrete titrant additions, evaluate titration system
drift, and monitor the calcite saturation state. In all other experiments, CO32-
ion
activities were derived from the Ca2+
ISE activities and application of Equation R1 (in
analogy to the CO32-
ISE calibration by method B).
2
02
3aCa
spKaCO
(R1)

361
Calcite specimen
All titrations were carried out with Baker “Instra-analyzed flux reagent” grade
calcium carbonate powder that was size-separated by settling through a 3 m x 0.1 m
Plexiglas®
tube filled with Millli-Q®
water. The middle third portion of the settled
CaCO3 was freeze-dried and used for all experiments reported in this paper. The
average grain size was estimated at 3-7 m based on Stoke‟s Law and corroborated by
numerous Scanning Electron Microscopy (SEM) images. X-ray diffraction and SEM
analyses of this material confirmed that the powder was composed of at least 99%
calcite. The specific surface area of the size-separated fraction was of 0.46 (± 0.02) m2
g-1
,
as determined by the multiple-point N2-BET method
with an Autosorbed-1
Physisorption Analyzer. This parameter was determined before and after one
acidimetric and one calcium titration to check for variations resulting from the
dissolution of the finest particles and/or possible calcite re-precipitation. The specific
surface area of the titrated solids was, within the uncertainty of our measurements,
identical to the starting material, and therefore, the original value was used in further
calculations. To minimize possible surface irregularities (such as step edges and
kinks), a fraction of the calcite powder was aged in Milli-Q®
water for about one year.
This is a common procedure used in sorption studies to “heal” carbonate mineral
surface defects (e.g., steps, dislocations and point defects) and minimize the
heterogeneity of surface site energies upon re-crystallisation. Furthermore, this pre-
treatment, through Ostwald ripening, allows the dissolution of smaller CaCO3
particles and precipitation onto larger particles, and hence, a narrowing of the particle
size distribution. Before use in surface titrations, the calcite powder was exhaustively
rinsed with Milli-Q®
water to remove adsorbed impurities, oven-dried at 70C, and
kept in a desiccator. For the sake of accuracy, the solubility product of our calcite
substrate was verified by equilibrating a series of calcite suspensions in Milli -Q®
water in centrifuge tubes under constant stirring for 10 days. After this time, pH,
alkalinity and the total calcium concentration (Ca, determined by AAS) were
measured and the Ca2+
and CO32-
activities calculated with MINEQL+ v.4.6 software.
The measured log10 Kºsp was –8.48 0.04, in excellent agreement with the value
selected by the National Institute of Standards and Technology.
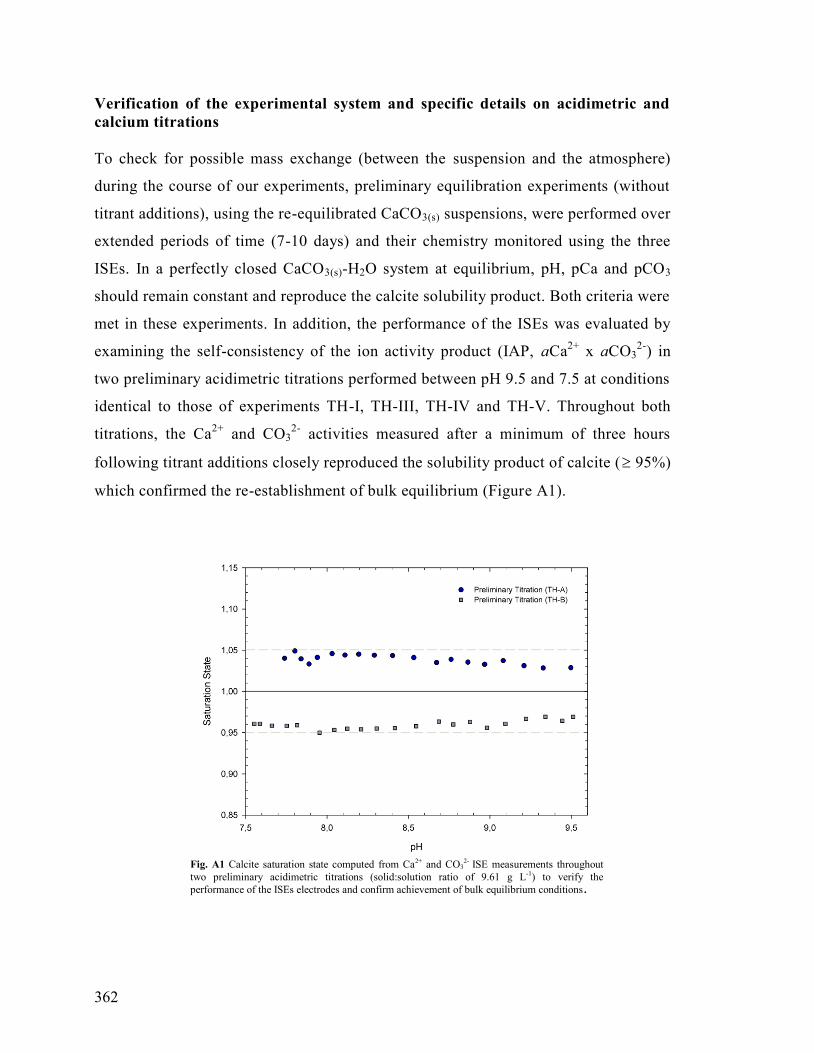
362
Verification of the experimental system and specific details on acidimetric and
calcium titrations
To check for possible mass exchange (between the suspension and the atmosphere)
during the course of our experiments, preliminary equilibration experiments (without
titrant additions), using the re-equilibrated CaCO3(s) suspensions, were performed over
extended periods of time (7-10 days) and their chemistry monitored using the three
ISEs. In a perfectly closed CaCO3(s)-H2O system at equilibrium, pH, pCa and pCO3
should remain constant and reproduce the calcite solubility product. Both criteria were
met in these experiments. In addition, the performance of the ISEs was evaluated by
examining the self-consistency of the ion activity product (IAP, aCa2+
x aCO32-
) in
two preliminary acidimetric titrations performed between pH 9.5 and 7.5 at conditions
identical to those of experiments TH-I, TH-III, TH-IV and TH-V. Throughout both
titrations, the Ca2+
and CO32-
activities measured after a minimum of three hours
following titrant additions closely reproduced the solubility product of calcite ( 95%)
which confirmed the re-establishment of bulk equilibrium (Figure A1).
Fig. A1 Calcite saturation state computed from Ca2+ and CO3
2- ISE measurements throughout
two preliminary acidimetric titrations (solid:solution ratio of 9.61 g L-1) to verify the
performance of the ISEs electrodes and confirm achievement of bulk equilibrium conditions.

363
Upon confirmation of the performance of the experimental system, acidimetric
titrations were conducted by stepwise addition of varying volumes (0.3 to 1.2 mL) of
standardized 0.1 M HCl solutions to pre-equilibrated calcite suspensions prepared at
different initial chemical conditions (Table 2 in main text). The ISE measurements
were recorded at least three hours after each titrant addition but longer time intervals
(up to 12 hours) were also investigated to confirm full restoration of bulk equilibr ium.
Complete titrations required from 4 to 5 days. Similarly, calcium titrations were
conducted by stepwise additions of varying volumes (0.05 to 2 mL) of a standardized
0.4 M CaCl2 solution to pre-equilibrated calcite suspensions prepared at different
initial chemical conditions (Table 2 in main text). The initial conditions for these
titration were carefully chosen to allow for detectable changes in Ca2+
activities upon
CaCl2 additions and to expand the range of total calcium concentration (Ca) covered
by each titration experiment. To this end, the initial composition of the suspensions
was varied by additions of HCl, KHCO3 and/or Na2CO3 solutions to set their
respective initial pH, Ca and CO2 before the first equilibration period (Table 2).
Preliminary calcium titrations revealed that a period of at least 18 hours is necessary
to obtain stable ISE readings after each discrete CaCl2 addition. Hence, data for these
experiments were acquired after at least 24 hours of equilibration following each
CaCl2 addition. Complete titrations required between 5 and 8 days.
Additional titrations were conducted using dilute calcite suspensions (0.2 g L-1
equivalent to 0.9 m2 L
-1) prepared in a 0.02 M KCl solution (Experiments TH-VI and
TCa-IV, respectively) under conditions identical to at least one acidimetric and one
calcium surface titration experiment. They served to: i) evaluate “mass effects”
influencing sorption behavior by properly accounting for dissolution (acidimetric
titrations) and precipitation (calcium titrations) while the surface available for
adsorption reactions is low (i.e., bulk reactions dominate over surface interactions),
and ii) detect possible “background effects” (e.g., adsorption) associated to the
titration system. Since the bulk kinetics of dissolution and precipitation are
proportional to the reactive surface area of the mineral, equilibration times for the
“blank” runs were longer than for equivalent titrations performed at higher
solid:solution ratios, and therefore, data were only recorded once stable ISEs readings

364
were obtained (after 7 hours for acidimetric and 30 hours for calcium titrations).
After each titration experiment, the ISEs were re-calibrated to verify their
performance and evaluate the electrode drift. In all cases, electrode drift was < 3%,
and thus, considered acceptable. To prevent carry-over contamination from preceding
experiments, the reaction vessel and its components were acid-washed (with 5% v/v
HCl solutions) and rinsed with Milli-Q®
water before each titration and the pH and
pCa of the Milli-Q®
water stored in the fully-assembled reaction vessel were
monitored for several hours to confirm the absence of contaminants (i.e., H+, Ca
2+)
possibly adhering to components of the reaction vessel. Low pH and relatively high
CO2 conditions allowing for moderate to high carbonic acid concentrations in the
experimental system were avoided to prevent the formation of CO2(g) nuclei inside the
reaction vessel and ensure that the CO2 an proton mass conservation conditions
required by our titration protocol were met (see main text). For instance, CO2 bubble
nucleation was observed in some titrations carried out to a pH of approximately 6.7. A
judicious selection of initial pH, Ca and CO2 conditions (Table 2 in main text)
guaranteed that sufficiently low levels of carbonic acid were maintained throughout
our titrations to prevent CO2(g) bubble formation while covering a pH range from 7.1
to 9.7. The maximum concentrations of H2CO3* registered at the end of our
experiments was ~ 410-4
M.

365
APPENDIX XII: CHAPTER 6
Referencing of Data to the ZNRSC
As explained in the main text, to properly compute net sorption densities from titration
experiments not initiated at the ZNRSC, the initial extent of proton occupancy of the
calcite sample (subsequently subjected to acidimetric or calcium titrations) must be
considered. By re-arranging equation 11 (with n=1) and using the initial pH and pCa
values measured after the second equilibration period in each experiment initiated away
from the ZNSRC, the respective initial occupancy ratios (Ratioocc
) can be calculated
according to:
which is equally expressed in mole fractions or molar concentrations of “exchangeable
lattice species” at the beginning of the experiment (identified with the superscript “0”).
The total molar concentration of cation exchangeable sites, (CaCO3)2(exc)TOT
, available
in each experiment is obtained from:
Ca(HCO3)2(exc)0 + (CaCO3)2(exc)
0= ECSD·A·S =
(CaCO3)2(exc)TOT
(II)
Thus, the initial molar proton occupancy, Ca(HCO3)2(exc)o, at the beginning of each
experiment is given by:
occ
occ
Ratio
Ratio
1
])CaCO[(])Ca(HCO[
TOT2(exc)30
2(exc)3
(III)
02(exc)3
02(exc)3
0)CaCO(
0)HCO(Ca
2
2
)CaCO(
)Ca(HCO
X
X)(
(exc)23
(exc)23
aCa
KaHRatio Excocc
(I)

366
Finally, the corrected adsorption data (moles L-1
), Hadscorr
, used in FITEQL
optimizations are obtained with:
Hadscorr
= (app
·A·S) + 2 Ca(HCO3)2(exc)0 (IV)
This correction was refined iteratively by averaging the Kex and ECSD obtained from data
sets of experiments TH-I, TH-III, TH-IV and TH-VI with those obtained with corrected
adsorption data from experiments TH-II and TH-V. Using the average Kex and ECSD
values, sorption data of the latter experiments were re-adjusted (Ratioocc
and
Ca(HCO3)2(exc)0 were re-calculated) and Kex and ECSD re-optimized as before. This
procedure was performed until the estimated H+ads
corr values and those calculated in the
preceding optimization converged to within ± 0.5 %. (Note that Hnet
is obtained by
dividing Hadscorr
by A·S). This correction applies to proton and calcium titration data
because, as explained in the main text, net proton uptake was observed in both types of
titration experiments.

367
APPENDIX XIII: CHAPTER 6
Equilibrium Speciation Calculations involving Ion Exchange
Speciation calculations, including reaction 3, were performed with an in-house Matlab©
subroutine integrating the Newton-Raphson iterative method where, in contrast to
MINEQL+ v4.6, the mass action law and mass balance matrices are decoupled to specify
suitable stoichiomeric coefficients (reaction 3) for the principal chemical components H+
and Ca2+
For illustrative purposes, the former, formulated in terms of the Tableau
method,31
is displayed in Table A-I. The Matlab subroutines can be obtained upon
request to the lead author.
The relevant mass balance equations specified in the code are as follows:
HHet = TOTH + 2 [Ca(HCO3)2(exc)] (V)
CaHet= TOTCa + [(CaCO3)2(exc)] (VI)
where HHet and CaHet are the calculated mass balances for proton and calcium
involving the aqueous and the solid phase, TOTH and TOTCa are the quantities defined
in Table 3 and the species in brackets are molar concentrations of the specified solid
phase species. Equations V and VI are subjected to the following constraints provided all
exchangeable cation sites are unreacted and available for proton uptake:
HHet = TOTH = CA - CB (VII)
= Total known excess or deficit of protons in the system
and,
CaHet = Ca + [(CaCO3)2(exc)]TOT
(VIII)
Reaction 3 (see main text) is added to the mass action law matrix with stoichiometric
coefficients compatible with Equations VII and VIII. As in all previous calculations with
MINEQL+, the stoichiometric coefficients of all other chemical species (aqueous phase)

368
remain identical in both the mass action law and mass balance matrices. This procedure
allows to properly compute equilibrium speciation of a carbonate system involving
reaction 3 as formulated in terms of the principal chemical component31
(CaCO3)2(exc). If
protons already occupy a fraction of the lattice exchangeable sites in the calcite powder,
the HHet and the CaHet constraints must be modified to consider the amount of
[Ca(HCO3)2(exc)] already present in the calcite specimen. For example, in the case of an
originally pure, hydrogen-free, calcite specimen subsequently treated with a “pre-
treatment” acid solution (acid leaching) and later subjected to different solution
conditions, [Ca(HCO3)2(exc)]0 and [(CaCO3)2(exc)]
0 are first estimated from speciation
calculations using the calibrated cKex value, and the known chemical composition of the
“pre-treatment” solution. This value is then used to modify the HTheo and the CaTheo
constraints imposed to the equilibrium speciation problem (via the mass balance matrix)
as follows:
HTheo = TOTHNew + 2 [Ca(HCO3)2(exc)]0 (IX)
CaTheo = CaNew + [(CaCO3)2(exc)]0 (X)
where TOTHNew and CaNew are known quantities and pertain to the experimental solution
to which the calcite powder is subjected after “pre-treatment” (before calcite immersion)
and [(CaCO3)2(exc)]0 = [(CaCO3)2(exc)]
TOT - [Ca(HCO3)2(exc)]. Note that twice the value
of [Ca(HCO3)2(exc)] must be added to TOTHNew
to properly account for the excess in
protons present in the system, whereas [Ca(HCO3)2(exc)] must be subtracted from
[(CaCO3)2(exc)]TOT
to account for the Ca2+
equivalents (removed during “pre-treatment”)
that are no longer present in the new CaCO3(s)-H2O system. The stoichiometric
coefficients of principal components H+ and Ca
2+ defining the formation of exchangeable
species in the mass balance matrix are those defined by equations V and VI (i.e., 2 and 1
respectively).

369
APPENDIX XIV: CHAPTER 6
Tableau-based Formulation for the CaCO3(s)-KCl-H2O System (Mass Action Law Matrix)
Principal Components
Aqueous Phase Species H
+ Ca
2+ K
+ Cl
- CaCO3(s) (CaCO3)2(exc) Log K° (25 C)
H+ 1 0
Ca2+
1 0
K+ 1 0
Cl- 1 0
CO32-
-1 1 Log K°sp
OH- -1 Log K°w
HCO3- 1 -1 1 Log K°sp + Log K°HCO3
H2CO3* 2 -1 1 Log K°sp + Log K°HCO3+ Log K°H2CO3*
CaOH+ -1 1 Log K°CaOH
CaCO3(aq) 1 Log K°sp + Log K°CaCO3°
CaHCO3+ 1 1 Log K°sp + Log K°CaHCO3 + Log K°HCO3
CaCl+ 1 1 Log K°CaCl
KCl 1 1 Log K°KCl
Solid Phase Species
(CaCO3)2 (exc) 1 0
Ca(HCO3)2(exc) 2 -1 1 Log Kex
SUM
(mol / L) CA - CB Ca K Cl CO2 ECSD·S·A

APPENDIX XV:
Matlab SUBROUTINES
(CHAPTERS 2, 3, 4, and 6)

371
1. GENETIC ALGORITHM-BASED OPTIMIZATION OF INTRINSIC
FORMATION CONSTANTS (CONSTANT CAPACITANCE MODEL)
Chapters: 2, 3 and 4 %%%%%%%%%%%%%%%%%%%%%% CALL EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%%%% EQUIL %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% INPUT FILE : EQUIL Surface Complexation Model %% %% Calibration of SCM reactions for Goethite in 0.7 M NaCl solutions. %% Data taken from Gao and Mucci, 2001, GCA vol 65, 2361-2378 %% Constant Capacitance Model %% %% %% Adrián Villegas-Jiménez %% %% Earth and Planetary Sciences, McGill University %% Montreal, CANADA %% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %%%%%%%%%%%%%%%%%% DEFINITION OF CHEMICAL EQUILIBRIUM %%%%%%%%%%%%%%%%% % Names of aqueous components (Always enter H component first) format short aqcomp=[sym('H'),sym('Na'),sym('Cl')]; surcomp=[sym('S1')]; component=horzcat(aqcomp,surcomp), aqnvar=length(aqcomp)'; surnvar=length(surcomp)'; nvar=length(component)'; aqcomp_charge=[1;1;-1]; surcomp_charge=[0] % Reference surface comp_charge=vertcat(aqcomp_charge,surcomp_charge), comp_charge2=aqcomp_charge'; % Names of Species aqspecies=[sym('H'); sym('Na'); sym('Cl'); sym('OH'); sym('NaOH')]; naqspecies=length(aqspecies), surspecies=[sym('S1'); sym('S2'); sym('S3')]; nsurspecies=length(surspecies), nsurspecies_C1=3, species=vertcat(aqspecies,surspecies), nspec2=length(species), Number_Species=nspec2; % Stoichiometry AQSST1=[1,0,0; 0,1,0; 0,0,1; -1,0,0; -1,1,0]; AQSSS=length(AQSST1) SST3=AQSST1'; SURSST=[0,0,0; 1,0,0; -1,0,0];

372
SURSSTH=[0; 1; -1]; SURSST2=[1; 1; 1]; numsurface=length(SURSST2) ST4=[1; 1; 1]; % Correction to stoichiometric coefficient in surface reaction SCoef=[1; 1; 1]; AQSST=horzcat(AQSST1,zeros(naqspecies,surnvar)); SST=AQSST1; SURSST3=vertcat(zeros(naqspecies,1),SURSST2); size_SST=size(SST); % Mass balance of surface species derived from each reaction SPECSSP=SURSST2 % Thermodynamic or Apparent Formation Constants log_K=[0; 0; 0; -13.68; -14.25]; log_Ksup=[0]; log_Kadj=zeros((length(SURSST2)-length(log_Ksup)),1); paramnum=length(log_Kadj); log_KC=log_K; log_KC=vertcat(log_K,log_Ksup) % Charges of Species AQSPCHARGE=[ 1; 1; -1; -1; 0]; LLLE=length(AQSPCHARGE) % Define overall charge present in the cluster of surface species SURSPCHARGE=[ 0; 1; -1]; MAT_CHARGE2=AQSPCHARGE; SDM=2.96e-6 %Specify surface sites densities SA=27.7; %Specify specific surface area MVR=7.93 %Specify mass/volume ratio Convert=(SA*MVR); S=[1] % Surface sites concentrations in terms of molar fraction ns=1; % Define number of reactive sites sorb=1; % Define number of adsorbates % Electrostatic Factor % Constant Capacitance Model (CCM) EFO=[ 0; 1; -1]';

373
EFO1=EFO*(-38.9256) EFO2=horzcat(zeros(1,naqspecies),EFO1)'; SURSST5=vertcat(zeros(naqspecies,surnvar)); %%%%%%%%%%%%%%%%%%%%% ENTER EXPERIMENTAL DATA %%%%%%%%%%%%%%%%%%%%% pH=['enter vertical vector of pH values'] IS=['enter vertical vector of Ionic Strength values'] SCHARGE_P=['enter vertical vector of surface charge values: proton adsorption densitites (mol/ m2 units)'] Convert=['enter vertical vector containing conversion factors (m2/L units) corrected by dilution'] Scores3=['enter vertical vector with free aqueous component concentrations (log10 of molar units)']; %%%%%%%%%%%%% End of experimental Data %%%%%%%%%%%%%%%%%%%%%%%%%%%%% Species=Scores3; [a,b]=size(Scores3); DATSIZE=a % Reassignations nads=1; nspec=naqspecies; pH=pH_1; SCHARGE=SCHARGE_P.*Convert; IonicS=IS; fitcon_num=aqnvar; fitpar_num=paramnum; nvar=aqnvar; pH2=pH_1; I=IS; % Transformation of pH to molar proton concentrations using the Davies Equation
for i=1:DATSIZE act_coef2=10.^(-(0.5115*(1.^2))*((sqrt(IS(i))/(1+(sqrt(IS(i)))))-(0.3*IS(i)))); pH=-log10(10.^(-pH)./act_coef2);
end fixvar=-pH_1; pH=fixvar; % CONSTANT CAPACITANCE MODEL SigmaElec=SCHARGE_P.*9.649e4
for i=1:DATSIZE POTENTIAL(i,1)=(SigmaElec(i)/(IS(i)^0.5));
end % Specify adjustable parameters Param=[sym('K1'),sym('K2'),sym('Rel_Abundance'),sym('Site_density'),sym('Capacitance')]; CompSurf=[sym('S1')]; nparam=length(Param); ncs=length(CompSurf); Adj=horzcat(Param,CompSurf); nadj=length(Adj); Coulomb=zeros(DATSIZE,1);
for i=1:DATSIZE Coulomb(i)=-38.9256*POTENTIAL(i);
end Species=Species'; AQSPCHARGE=AQSPCHARGE'; Species=abs(10.^(Species)); % Calculate activity coefficients of aqueous species Gama=zeros(aqnvar,DATSIZE); Activ=zeros(aqnvar,DATSIZE);
for j=1:DATSIZE for i=1:aqnvar Gama(i,j)=10.^(-(0.5115*((AQSPCHARGE(i)).^2))*((sqrt(IS(i)))/(1+(sqrt(IS(i)))))-(0.3*(IS(i)))); Activ(i,j)=Species (i,j)*Gama(i); end
end
MassH=SCHARGE; save EQUIL_DATA; save fitdol %%%%%%%%%%%%%%%%%%% CLOSE AND SAVE EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%% %%%%%%%%%%%%%%%%%% CALL FITGEN MAIN SUBROUTINE %%%%%%%%%%%%%%%%%%%%

374
function [stats,pop,elitechrome,Constants] =FITGEN %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % FITGEN is the main subroutine to perform stochastic optimizations of Surface Complexation Model parameters % based on a %% genetic algorithm % It defines the GA parameters, solution space, number and length of chromosomes and calls all required % subroutines % % 10/04/02 Earth and Planetary Sciences, McGill University % Adrián Villegas-Jiménez % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ t=cputime; for k=1:1 % Set number of GA optimization of a single data set (up to 3) contador=k; load EQUIL_DATA load fitdol; save Electrostatics fitval=[]; resid2=1e100; Ecart2=1e56; OPTIM=1; save check resid2 OPTIM save check2 Ecart2 save output2 fitval varchrome=[]; minval=[]; maxval=[]; fprintf('PRESS ANY KEY TO CONTINUE \n'); pause clc home % fprintf('xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx \n'); fprintf('\n'); fprintf(' \t \t \tFITGEN A computer routine for the calculation of intrinsic surface parameters from experimental data \n'); fprintf('\n'); fprintf('xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx \n'); % fprintf(' Default chromosome length calculated from analytical concentrations \n'); fprintf('\n'); nvar=input('Specify the number of parameters to be optimized in the objective function >'); cc=input('Specify chromosome length and assign lower and upper limits for each parameter? (1 for YES, 2 for NO) >'); cc=2
if cc==1 for j=1:nadj Parameter=Adj(j) fprintf('\n'); VC=input('Enter now the size of the chromosome for the given parameter >'); fprintf('\n'); varchrome=horzcat(varchrome,VC); minv=input('Enter now the minimum value for the value of the given parameter >'); minval=horzcat(minval,minv); maxv=input('Enter now the maximum value for the value of the given parameter >'); maxval=horzcat(maxval,maxv); end else %1st GA Run minval=[-25000,-25000,...... Number of adjustable parameter] % Define minimum boundary value maxval=[25000,25000,........ Number of adjustable parameter] % Define maximum boundary value sizevec=[15,15,10,....] % Define number of bits required by each section of the chromosome (adjustable parameter)
if contador==2
%2nd GA Run minval=[-25000,-25000,...... Number of adjustable parameter] % Define minimum boundary value maxval=[25000,25000,........ Number of adjustable parameter] % Define maximum boundary value sizevec=[15,15,10,....] % Define number of bits required by each section of the chromosome (adjustable parameter) else end
if contador==3 %3rd GA Run

375
minval=[-25000,-25000,...... Number of adjustable parameter] % Define minimum boundary value maxval=[25000,25000,........ Number of adjustable parameter] % Define maximum boundary value sizevec=[15,15,10,....] % Define number of bits required by each section of the chromosome (adjustable parameter)
else end
end fprintf('\n'); fprintf('\n'); fprintf(' \t General Parameters of the Genetic Algorithm \n') fprintf(' \t PRESS ANY KEY TO CONTINUE\n'); pause fprintf(' \t Parameters must be entered as follows:\n') fprintf('\t [population size, number of generations\n') fprintf(' \t mutation rate, crossover rate,\n') fprintf(' \t type of crossover (1=single point 2=two points, 3=three points)\n') fprintf('\n'); fprintf('\n'); Conf=input('Enter the GA parameters now e.g. [10,1000,0.01,0.25,1] ) >'); prnlevel=1; ffunc=1; gray=1; popsize=Conf(1); popsize1=popsize; numgen=Conf(2); pm=Conf(3); px=Conf(4); xtype=Conf(5); ndim=sum(varchrome); ndim2=sum(varchromeb); % set GA configuration clc home elite = 2; gray = 1; numxover = 0; nummut = 0; nstat = 7; test = rem(popsize,2); if elite == 1 if (test == 0) popsize = popsize + 1; end else if (test ~= 0) popsize = popsize + 1; end end % Initialize population randomly pop = [(rand(popsize,ndim)<0.5)]; % % Optimize for a given number of generations tnumgen =numgen; SS=1; cgn=0; child=0; childcount=0; Fuse=1; for cgn = 1:tnumgen % % Compute fitness function for each member of the population % cgn=cgn+1; %%%%%%%%%%%%%%%%%%%% CALL FITLOG SUBROUTINE %%%%%%%%%%%%%%%%%%

376
[SS,SB,scores,Residual,vars1,Constants,resid,poss] = FITLOG(pop,numgen,ffunc,varchrome,varchromeb,maxval,maxvalb,minval,minvalb,nvar,cgn,childcount,child,xtype,Fuse,contador); function [SS,SB,scores,Residual,vars1,Constants,resid,poss] = FITLOG(pop,numgen,ffunc,varchrome,varchromeb,maxval,maxvalb,minval,minvalb,nvar,cgn,childcount,child,xtype,Fuse,contador) %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % FITLOG computes fitness scores and performs all calculations required by the objective function % It decodes binary-strings and splits chromosome into predetermined sections for each variable % % Subroutine designed for the optimization of multiple SCM parameters from surface protonation and adsorption data % Data fits for one or two adsorbates can be fitted within the Constant Capacitance Model % % The chromosome has to be splitted into nvar sections (one section for each master specie or principal component) % the corresponding lengths can be either be calculated in the input file or can be specified directly by the user % Solution is coded in nominal values and tested in logarithmic units % % 10/04/02 Earth and Planetary Sciences, McGill University % Adrián Villegas-Jiménez % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ global Data Plothandle load fitdol load check load check2 load Electrostatics [popsize,ndim] = size(pop); var=[]; gray=1;
if xtype ==5 if cgn >1 gray=2 vars=child'; end
end % Optimizing intrinsic parameters
if gray == 1 count=1; fin=0; start=1; maxval=maxval;
for j=1:nparam varchrome2=(varchrome(j)); minval2=(minval(j)); maxval2=(maxval(j)); pow_two = 2.^(0:varchrome2); maxintval = ((2^varchrome2)-1); range =maxval2-minval2; start = start+fin; fin= fin + varchrome2;
for i = 1:popsize tvars(1:varchrome2) = pop(i,start:fin); % % now decode binary number to real number (scale maxval to minval) %
real = 0;
for k = 1:varchrome2 real = real + pow_two(k)*tvars(varchrome2-k+1); end % % Takes integer value and converts to a real number (genotype to phenotype transformation)

377
% vars1(i,j) = (range*(real/maxintval)) + minval2; end start=1; end else end vars1=10.^(vars1./1000); format long E np=nparam; alpha=vars1(:,nparam)'; guess=zeros(aqnvar,DATSIZE);
for j=1:DATSIZE for i=1:aqnvar guess(i,j)=Activ(i,j); end
end quihubo=guess; act=ones(surnvar,DATSIZE); guess=(vertcat(guess,act))'; guess=log10(guess); unito=(ones(1,DATSIZE))'; guess=horzcat(guess,unito); Konstants1=zeros(popsize,np);
for i=1:popsize for j=1:np Konstants1(i,j)=vars1(i,j); end
end % Express constants in terms of an operational reference state as suggested by Dr D. Sverjensky % Konstants=Konstants1*(1/stdstate); Ksurcomp=ones(popsize,surnvar); Konstants=log10(horzcat(Ksurcomp,Konstants1))‟; % Estimating free concentrations of surface components icc=0; surconc=zeros(DATSIZE,surnvar); static=zeros(nsurspecies,1); EFO2=EFO';
for j=1:popsize jcc=np; icc=icc+1;
for i=1:DATSIZE
Electro=Coulomb(i,1); alpha=vars1(icc,jcc); UF=vars1(icc,jcc-1); UF=vars1(icc,jcc-1)*Convert(i,1); FRACT(1,1)=abs(vars1(icc,jcc-2)); FRACT(1,2)=1-FRACT(1,1); FF2=Konstants(1:nsurspecies,j);
for k=1:nsurspecies static(k,1)=log10(exp(Electro*alpha*EFO2(k,1))); end FF=FF2+static; SURSST3=horzcat(SURSST,FF); adivina=guess(i,(1:nvar+(ns+1)))'; adivina2=(10.^((SURSST3*adivina)./SCoef))'; surface=adivina2*SURSST4; unos=ones(1,surnvar); surface2=surface; FRACT(1,1:ns);

378
surconc(i,1:surnvar,icc)=(UF*FRACT(1,1:ns))./surface2; end
end
consur=surconc; icc=0; quihubo=quihubo'; M=zeros(DATSIZE,nvar+2,popsize); anexo=ones(DATSIZE,1);
for k=1:popsize MB1=log10(horzcat(quihubo,surconc(1:DATSIZE,1:surnvar,k))); MB1=horzcat(MB1,anexo); M(1:DATSIZE,1:(nvar+(ns+1)),k)=MB1(1:DATSIZE,1:(nvar+(ns+1)),1);
End
MB1=M; MB2=zeros(DATSIZE,popsize); MB7=zeros(DATSIZE,surnvar,popsize); MB6=zeros(surnvar,popsize); CB3=zeros(DATSIZE,popsize); CB4=zeros(DATSIZE,popsize); SB_residual=zeros(surnvar,popsize); adivina5=zeros(DATSIZE,popsize,nsurspecies);
if surnvar>1 SURFACE=zeros(DATSIZE,surnvar,popsize);
else SURFACE=zeros(DATSIZE,popsize);
end %loop for calculating surface species concentrations vale=[];
for j=1:popsize jcc=np; icc=icc+1;
for i=1:DATSIZE Electro=Coulomb(i,1); alpha=vars1(icc,jcc); FF2=Konstants(1:nsurspecies,j);
for k=1:nsurspecies static(k,1)=log10(exp(Electro*alpha*EFO2(k,1))); end
FF=FF2+static; SURSST3=horzcat(SURSST,FF); adivina=MB1(i,(1:nvar+ns+1),icc)'; adivina2=((SURSST3*adivina)./SCoef)'; % estimate concentration of surface species adivina4=10.^(adivina2); adivina5(i,icc,1:nsurspecies)=adivina4; surface2=(adivina4*SURSST4); MB7(i,1:surnvar,icc)=surface2; % Mass balance for the xth chromosome CB=adivina4*SURSPCHARGE; MH=adivina4*SURSSTH; MBH(i,icc)=MH; Up_Charge(i,icc)=CB/Convert(i,1); % Charge balance for the xth chromosome CB4(i,icc)=MBH(i,icc)/Convert(i,1);
if sorb > 1 GUESS_ADS(i,icc)=(adivina4*SURADS); else end end
end
for j = 1:popsize

379
for i=1:DATSIZE
if sorb == 1 % %Compute proton component residuals % scores_proton(i,j)=(MBH(i,j)-MassH(i))^2; scores(i,j)=(scores_proton(i,j)); else %
%Compute individual residuals % scores_proton(i,j)=(MBH(i,j)-MassH(i))^2; scores_ads(i,j)=(GUESS_ADS(i,j)-TOTADS(i))^2; % % Normalize residuals %
perc1=1/((exp(scores_proton(i,j)))+abs(log10(scores_proton(i,j)))); perc2=1/((exp(scores_ads(i,j)))+abs(log10(scores_ads(i,j)))); scores1(i,j)=perc1+perc2; % cumulative residual scores2(i,j)=perc1+perc2; % cumulative residual end
end
end
Sum_Scores1=(sum(scores1)); Sum_Scores2=(sum(scores2)); [min1,poss1]=min(Sum_Scores1); [min2,poss2]=min(Sum_Scores2); resid=min1; Residual=resid; poss=poss2; poss1=poss2; sitios=adivina5(1:DATSIZE,poss1,1:nsurspecies);
for i=1:DATSIZE site(1:DATSIZE,1:surnvar)=MB7(i,1:surnvar,poss1)./Convert(i,1);
end scores1=MBH(1:DATSIZE,poss1); scores=Sum_Scores1; KK=vars1'; Update=Up_Charge(:,poss1).*9.649e4; % Select "best" surface charge (C/m2) superficie=adivina5(1:DATSIZE,poss1,1:nsurspecies);
for i=1:DATSIZE super(i,1:nsurspecies)=superficie(i,1,1:nsurspecies);
end super_balance=sum(super');
if surnvar>1 Site_balance=(SURFACE(1:DATSIZE,1:surnvar,poss1));
else Site_balance=(SURFACE(1:DATSIZE,poss1))./Convert;
end
for i=1:DATSIZE super(i,1:nsurspecies)=superficie(i,1,1:nsurspecies);
end super_balance=sum(super'); Constants=KK(:,poss1); KONSTANTES=log10(Constants); Coulomb2=Coulomb; % Update surface charge from surface speciation %
if cgn >=0.5*numgen

380
for i=1:DATSIZE
POTENTIAL(i,1)=(Update(i,1)/(IonicS(i,1)^0.5)); end Coulomb=zeros(DATSIZE,1);
for i=1:DATSIZE Coulomb(i)=-38.9256*POTENTIAL(i); end save Electrostatics Coulomb
end
if resid <= resid2 results=CB4(:,poss1); OPTIM=results; generation=cgn resid2=resid; Ks=log10(Constants) SUMA_SB=sum(Site_balance)/DATSIZE; Site_balance=sum(site); if contador==1
save output Ks resid2 scores1 results site generation OPTIM Update Coulomb POTENTIAL Site_balance poss cgn scores1 Site_balance adivina2 MB6 SUMA_SB sitios
else end if contador==2
save output3 Ks resid2 scores1 results site OPTIM Update Coulomb Ads POTENTIAL Site_balance poss cgn scores1 Site_balance adivina2 MB6 SUMA_SB sitios
else end
if contador==3 save output4 Ks resid2 scores1 results site OPTIM Update Coulomb Ads POTENTIAL Site_balance poss cgn scores1 Site_balance adivina2 MB6 SUMA_SB sitios
else end
save check resid2 SCHARGE_P=SCHARGE_P(1:DATSIZE) pH2=pH_1(1:DATSIZE);
drawnow plot(pH2,SCHARGE_P,pH2,SCHARGE_P,'o',pH2,results,pH2,results,'*') xlabel ('pH') ylabel ('Surface Charge Density (moles/m2)') title ('Surface Complexation Model for Goethite')
else end %%%%%%%%%%%%%%%%%%%%% END FITLOG SUBROUTINE %%%%%%%%%%%%%%%%%%%% SS=SS'; SS=scores; [minva,possmb]=min(SS); [topfit,topi] = min(SS); ms = mean(SS); sd = std(SS); fprintf('%i \t %8.2f \t %8.2f \t %8.10f \n',cgn,ms,sd,topfit); elitechrome = pop(poss,:); fittest=elitechrome; %%%%%%%%%%%%%%%% %%%%% CALL EVAL_GA SUBROUTINE %%%%%%%%%%%%%%%%%%%% mate = EVAL_GA(pop,scores,elite); function mateset = EVAL_GA(pop,scores,elite)

381
%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % Select most fit members of population for mating subset % using tournament selection and eliteist strategy (if turned on) % scores % % function mateset = EVAL_GA(pop,scores,elite) % EVAL_GA performs selection of chromsomes (by stochastic tournament) % Version 1.1 5/5/2005 Adrián Villegas-Jiménez % Earth and Planetary Sciences, McGill University % Modified from the original “MUTATE “ Matlab subroutine Version 1.0 by Ron Shaffer 1/23/96 % TSELECT tournament mating selection % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % set constants % [popsize,ndim] = size(pop) % % With elistist strategy on you only need to choose a mating set with popsize - 1 % members in it b/c the last spot is save for the elite chromosome % %if elite == 1 % popsize = popsize - 2; %end % % % compute vector of random integers % randlist = [round(rand((popsize*2),1)*popsize+0.5)]; % % Begin tournament selection % count = 0; for i = 1:popsize count = count + 2; cmo = count - 1; % % 2 randomly chosen chromosomes from population will compete for inclusion % in mating subset according to the fitness values obtained from the mass % balance equation (minimization) % if scores(randlist(count)) < scores(randlist(cmo)) mateset(i,1:ndim) = pop(randlist(count),1:ndim); else mateset(i,1:ndim) = pop(randlist(cmo),1:ndim); end end %%%%%%%%%%%%%%%%%%% END EVAL_GA SUBROUTINE %%%%%%%%%%%%%%%%%%% % perform crossover if required % %%%%%%%%%%%%%%%%%%% CALL XOVER_GA SUBROUTINE %%%%%%%%%%%%%%%%%%% [new,xcount] = XOVER_GA(old,px,xtype,Residual,scores1,vars1,nvar) function [new,xcount] = XOVER_GA (old,px,xtype,Residual,scores1,vars1,nvar)

382
%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % XOVER_GA: Performs crossover operations of Genetic Algorithm-Based Fitting of Surface Complexation Model (SCM) % Parameters % Version 1.1 5/5/2005 Adrián Villegas-Jiménez % Earth and Planetary Sciences, McGill University % % Modified from the original “XOVER” Matlab subroutine by Ron Shaffer % Version 1.0 1/23/96 Ron Shaffer % Version 1.1 2/27/96 Ron Shaffer % added options for two-point and uniform crossover % Version 1.2 6/24/96 Ron Shaffer % fixed bug in 2-point crossover discovered by % Mr. Radovan Cemes ([email protected]). Crossover % points are now sorted before swapping is performed. % % new -- new population of chromosomes % xcount-- # of times crossover was performed % old -- input population of chromosomes % px -- crossover probability % xtype -- type of crossover % % Several types of xover can be performed according to an inequality constraint % determined by the residual value as suggested by Gen et al, 1996. % % Residual--Difference between estimated values and total concentrations % of components excepting H % -- 1) 1-point crossover % -- 2) 2-point crossover % -- 3) Uniform crossover % -- 4) Randomised and/or crossover as suggested by Bill Keller from University of Sussex % -- 5) Direction based crossover as suggested by Michalewicz et al, 1994 % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % set constants % [popsize,ndim] = size(old); halfpop = floor(popsize/2); xcount = 0; % % loop through chromosomes determining whether xover should be performed % and if so performing single-point crossover. % if xtype == 1 randlist = rand((halfpop),1); for i = 1:halfpop x = (i*2) - 1; xpo = x + 1; new(x,1:ndim) = old(x,1:ndim); new(xpo,1:ndim) = old(xpo,1:ndim); if (randlist(i) < px) xcount = xcount + 1; xpoint = round((rand * ndim)+0.5); new(xpo,1:xpoint)=old(x,1:xpoint); new(x,1:xpoint) = old(xpo,1:xpoint); end end end % % two-point crossover % if xtype == 2 randlist = rand((halfpop),1); for i = 1:halfpop x = (i*2)-1; xpo = x+1;
383
new(x,1:ndim) = old(x,1:ndim); new(xpo,1:ndim) = old(xpo,1:ndim); if (randlist(i) < px) xcount = xcount + 1; [xpoint] = sort(round((rand(1,2) * ndim)+0.5)); new(xpo,xpoint(1):xpoint(2)) = old(x,xpoint(1):xpoint(2)); new(x,xpoint(1):xpoint(2)) = old(xpo,xpoint(1):xpoint(2)); end end end % % uniform crossover % if xtype == 3 for i = 1:halfpop x = (i*2)-1; xpo = x+1; new(x,1:ndim) = old(x,1:ndim); new(xpo,1:ndim) = old(xpo,1:ndim); for j = 1:ndim test = rand; if test < px xcount = xcount + 1; new(xpo,j) = old(x,j); new(x,j) = old(xpo,j); end end end end % Randomised and/or crossover if xtype == 4 for i = 1:halfpop x = (i*2)-1; xpo = x+1; new(x,1:ndim) = old(x,1:ndim); new(xpo,1:ndim) = old(xpo,1:ndim); for j = 1:ndim test = rand; if test <= px xcount = xcount + 1; if old(x,j)==1 new(x,j) = 1; new(xpo,j) = 1; else new(x,j) = 0; new(xpo,j) = 1; end else if old(xpo,j)==1 new(x,j) = 0; new(xpo,j) = 1; else new(x,j) = 0; new(xpo,j) = 0; end end end
xcount = xcount + 1;
if old(x,j)==1 if old(xpo,j)==1; new(x,j) = 1; new(xpo,j) = 1; else new(x,j) = 1; new(xpo,j) = 0; end

384
else if old(xpo,j)==1 new(x,j) = 1; new(xpo,j) = 0; else new(x,j) = 0; new(xpo,j) = 0; end end end else end % Direction based xover. It uses the values of the objective function in determining the direction of genetic search. It distinguishes between the binary and the numeric representations scores1=scores1; vars1=vars1;
if xtype == 5 new = zeros(nvar,popsize);
for i = 1:nvar for j = 1:popsize test = rand; if j==popsize if scores1(i,popsize) <= scores1(i,1) xcount = xcount + 1; new(i,j)=abs((test*(vars1(i,j)-vars1(i,1)))+ vars1(i,j)); else xcount = xcount + 1; new(i,j)=abs((test*(vars1(i,1)-vars1(i,j)))+ vars1(i,1)); end else
if scores1(i,j) <= scores1(i,(j+1)) xcount = xcount + 1; new(i,j)=abs((test*(vars1(i,j)-vars1(i,(j+1))))+ vars1(i,j)); else xcount = xcount + 1; new(i,j)=abs((test*(vars1(i,(j+1))-vars1(i,j)))+ vars1(i,(j+1))); end end end end
else end %%%%%%%%%%%%%%%%%%% END XOVER_GA SUBROUTINE %%%%%%%%%%%%%%%%%% numxover = numxover + txnum; child=child; % perform mutation % %%%%%%%%%%%%% %%%%%%% CALL MUT_GA SUBROUTINE %%%%%%%%%%%%%%%%%%% [new,nmut] = MUT_GA(pop,pm,Residual)

385
function [new,nmut] = MUT_GA(pop,pm,Residual) %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % MUT_GA: performs mutation on population of chromsomes % Version 1.1 5/5/2005 Adrián Villegas-Jiménez % Earth and Planetary Sciences, McGill University % % Modified from the original MUTATE Matlab subroutine by Ron Shaffer % version: 1.0 % date: 1/23/96 % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % set constants % %if Error2 <=0.5 % pm = 0.01 %end [popsize,ndim] = size(pop); nmut = 0; % % loop through population testing whether to mutate % for i = 1:popsize for j = 1:ndim test = rand; if test < pm pop(i,j) = abs(pop(i,j)-1); nmut = nmut + 1; end end end % % return new population % new = pop; % % Children + elite chromosome (if elitism turned on) form the new generation % %%%%%%%%%%%%%%%%%%% END MUT_GA SUBROUTINE %%%%%%%%%%%%%%%%%%% nummut = nummut + tnmut; pop = child; if elite == 2 pop=vertcat(pop,fittest); else end [t,s]=size(pop); Population_Size=t end end %%%%%%%%%%%%%%%%%%% END FITGEN MAIN SUBROUTINE %%%%%%%%%%%%%%%%%%

386
2. GENETIC ALGORITHM-BASED OPTIMIZATION OF INTRINSIC
FORMATION CONSTANTS (TRIPLE LAYER MODEL)
Chapter 2 %%%%%%%%%%%%%%%%%%%%%% CALL EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%%%% EQUIL %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% Input file : EQUIL Surface Complexation Model %% %% Calibration of SCM reactions for Goethite in 0.01 M NaCl solutions. %% Data taken from Villalobos and Leckie, 2001, GCA, vol 235, 15-32 %% Triple Layer Model %% %% Adrián Villegas-Jiménez %% Earth and Planetary Sciences, McGill University %% Montreal, CANADA %% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %%%%%%%%%%%%%%% DEFINITION OF CHEMICAL EQUILIBRIUM %%%%%%%%%%%%%%%%%%% %Names of aqueous components (Always enter H component first) format short aqcomp=[sym('H'),sym('Na'),sym('Cl')]; surcomp=[sym('S1')]; aqcomp_charge=[1;1;-1]; surcomp_charge=[0] aqspecies=[sym('H'); sym('Na'); sym('Cl'); sym('OH')]; surspecies=[sym('S1'); sym('S2'); sym('S3'); sym('S4'); sym('S5')]; component=horzcat(aqcomp,surcomp); aqnvar=length(aqcomp)'; surnvar=length(surcomp)'; ncs=surnvar; nvar=length(component)'; naqspecies=length(aqspecies); comp_charge=vertcat(aqcomp_charge,surcomp_charge); comp_charge2=aqcomp_charge'; nsurspecies=length(surspecies); species=vertcat(aqspecies,surspecies); nspec2=length(species); Number_Species=nspec2; % % Stoichiometry % AQSST1=[1,0,0; 0,1,0; 0,0,1; -1,0,0]; AQSSS=length(AQSST1); SST3=AQSST1'; SURSST=[0,0,0,1; -1,0,0,1; 1,0,0,1; -1,1,0,1; 1,0,1,1];

387
logSup=[0; 0; 0; 0]; SURSST2=[1; 1; 1; 1; 1]; numsurface=length(SURSST2) SURADSST=[0,0,0; -1,0,0; 1,0,0; -1,0,0; 1,0,0]; ST4=[1; 1; 1; 1; 1]; SCoef=ST4; %Matrix to define stoichiometry of adsorbed species SURSST7 = [0,0,0,1; -1,0,0,1; 1,0,0,1; -1,1,0,1; 1,0,1,1]; AQSST=horzcat(AQSST1,zeros(naqspecies,surnvar)); SST=AQSST1; SURSST3=vertcat(zeros(naqspecies,ncs),SURSST2); size_SST=size(SST); % Mass balance of surface species derived from each reaction SPECSSP=SURSST2 layers=3; nads=1 % %Thermodynamic Formation Constants % log_K=[0; 0; 0; -14]; log_Ksup=[0]; log_Kadj=zeros((length(SURSST2)-length(log_Ksup)),1); paramnum=length(log_Kadj); log_KC=log_K; log_KC=vertcat(log_K,log_Ksup) % %Charges of Species % AQSPCHARGE=[1; 1; -1; -1]; LLLE=length(AQSPCHARGE) %Define overall charge associated with each electrostatic plane by the adsorbed species N_Planes=3

388
% Matrix is defined as follows rows represent surfacespecies and columns are surface reactive sites % SURCHARGE contains charges directly associated with the surface whereas STERNCHARGE contains those present at % the Stern Layer SURCHARGE= [0; -1; 1; -1; 1]; STERNCHARGE=[0; 0; 0; 1; -1]; DUMMY= [0,0,0; -1,0,0; 1,0,0; -1,1,0; 1,-1,0]; dummy=2; SURSPCHARGE=horzcat(SURCHARGE,STERNCHARGE); Stoichiometry=horzcat(SURADSST,SURSPCHARGE); % Merge Charge Vectors MAT_CHARGE2=AQSPCHARGE; % %Surface sites densities (mol/m2) SDM=3.819E-06 %2.3 sites/nm2 SDM2=1.6603E-05 %10 sites/nm2 SA=70; % Specify specific surface area (m2/g) MVR=12.6 % Specify mass/volume ratio (g/L) DIEL=78.5; % Dielectric constant of solvent (water) S=[1] % Surface sites concentrations in terms of molar fraction %Capacitance=2; % Electrostatic Factor EFO=SURSPCHARGE*(-38.9256); SURSST5=vertcat(zeros(naqspecies,surnvar)); %%%%%%%%%%%% ENTER EXPERIMENTAL DATA %%%%%%%%%%%%%%%%%%%%%%%%%%%%% pH=['enter vertical vector of pH values'] IS=['enter vertical vector of Ionic Strength values'] SCHARGE_P=['enter vertical vector of surface charge values: proton adsorption densitites (mol/ m2 units)'] Convert=['enter vertical vector containing conversion factors (m2/L units) corrected by dilution'] Scores3=['enter vertical vector with free aqueous component concentrations (log10 of molar units)']; %%%%%%%%%%%%% END OF EXPERIMENTAL DATA %%%%%%%%%%%%%%%%%%%%%%%%%%%%% fixed=pH; pH_Fix=1; S1=length(pH); I=0.01; varnum=1; a=length(pH); UF=[SDM*SA*MVR]; DATSIZE=a;
for i=1:DATSIZE MADS_C1(i)=SCHARGE_P(i)*(SA*MVR); end TOT_ADS=horzcat(MADS_C1',(zeros(23,2))); Charge_initial=SADS_C1;

389
for i=1:DATSIZE Convert(i)=(SA*MVR); end
for i=1:DATSIZE if SADS_C1(i,1)<=0 psi(i)=0; else psi(i)=1; end end SADS2=SADS_C1; nads=1;pH_1=pH; nspec=naqspecies Z=1; %Specify valence of symmetric electrolyte (NaCl) % Transformation of pH to molar proton concentrations
for i=1:DATSIZE act_coef2=10.^(-(0.5115*(1.^2))*((sqrt(IS(i))/(1+(sqrt(IS(i)))))-(0.3*IS(i)))); %Davis Equation pH(i,1)=log10((10.^(-pH(i,1)))./act_coef2)
end format long % INPUT Background symmetric electrolyte: NaCl BGND=0.01; Species_Conc=zeros(aqnvar-1,DATSIZE); for j=1:DATSIZE Species_Conc(1:aqnvar-1,j)=BGND; end Species_Conc=Species_Conc'; Species_Conc=horzcat(pH,log10(Species_Conc)); pH2=pH; %Calculate activity coefficients of aqueous species for i=1:DATSIZE IEq(i,1)=I; end Activ=zeros(aqnvar,DATSIZE); Gama2=zeros(aqnvar,DATSIZE); for j=1:DATSIZE for i=1:aqnvar Gama2(i,1)=(-(0.5115*((aqcomp_charge(i)).^2))*((sqrt(IS(j))/(1+(sqrt(IS(j)))))-(0.3*IS(j)))); Activ(i,j)=10.^(Species_Conc(j,i)+Gama2(i,1)); end end EM=3; Activ=abs(Activ); Species_Conc; save fitdol save EQUIL_DATA %%%%%%%%%%%%%%%%%%% CLOSE AND SAVE EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%% %%%%%%%%%%%%%%%%%%%% CALL FITGEN MAIN SUBROUTINE %%%%%%%%%%%%%%%%%%

390
function [stats,pop,elitechrome,Constants] =FITGEN %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % FITGEN is the main subroutine to perform stochastic optimizations, via a genetic algorithm, of Surface % Complexation Model parameters coupled with multi-layer electrostatic models % It defines the GA parameters, solution space, number and length of chromosomes and calls all required % subroutines % % 05/05/04 Earth and Planetary Sciences, McGill University % Adrián Villegas-Jiménez % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ clear save optimized t=cputime; load EQUIL_DATA % Specify file containing the definition of the geochemical equilibrium and aqueous speciation load results% Specify file containing aqueous speciation results ncs=1; %Specify number of surface sites dummy=1; fitval=[]; resid2=1e100; Ecart2=1e56; save check resid2; save check2 Ecart2; save output2 fitval; save update Ecart2 varchrome=[]; minval=[]; maxval=[]; fprintf('PRESS ANY KEY TO CONTINUE \n'); clc home fprintf('xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx \n'); fprintf('\n'); fprintf(' \t \t \tFITGEN A computer pseudocode for the calculation of intrinsic surface parameters from experimental data \n'); fprintf('\n'); fprintf('xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx \n'); % fprintf(' Default chromosome length calculated from analytical concentrations \n'); fprintf('\n'); fprintf(' \t General Parameters of the Genetic Algorithm \n') fprintf(' \t PRESS ANY KEY TO CONTINUE\n'); pause fprintf(' \t Parameters must be entered as follows:\n') fprintf('\t [population size, number of generations\n') fprintf(' \t mutation rate, crossover rate,\n') fprintf(' \t type of crossover (1=single point 2=two points, 3=three points)\n') fprintf('\n'); fprintf('\n'); Conf=[100,100,0.02,0.25,1]; prnlevel=1; ffunc=1; gray=1; popsize=Conf(1); popsize1=popsize; numgen=Conf(2); pm1=Conf(3); pm2=pm1; pm3=pm1; px1=Conf(4); px2=pm1; px3=px1; xtype=Conf(5); % % set GA configuration % clc home elite = 2; gray = 1; numxover = 0; nummut = 0; nstat = 7; test = rem(popsize,2);
if elite == 2 if (test == 0)

391
popsize = popsize + 1; end
else if (test ~= 0) popsize = popsize + 1; end
end % % % Initialize population randomly % fprintf('\n');
switch EM % Specify number of electrostatic planes
case 1 %Constant Capacitance Model N_Planes=1; fprintf('\n'); SCA=input('Experimental surface charge data available? (1=YES 2=NO >'); fprintf('\n');
if SCA ==1 MAT_Vari=1;
% %Define interval of intensive variables (log K's) in Matrix A minval=[-25000,-25000,...... Number of adjustable parameters] % Minimum
%boundary value maxval=[25000,25000,........ Number of adjustable parameter] % Define maximum
%boundary value sizevec=[15,15,10,....] % Define number of bits required by each section of the
%chromosome (adjustable parameter) nparam=length(sizevec); np=nparam; varchrome=sizevec; ndim=sum(varchrome); pop = [(rand(popsize,ndim)<0.5)]; else MAT_Vari=1; % % Define interval of intensive variables (log K's) in Matrix A minval=[-25000,-25000,...... Number of adjustable parameters] % Define minimum
%boundary value maxval=[25000,25000,........ Number of adjustable parameters] % Define
%maximum boundary value sizevec=[15,15,10,....] % Define number of bits required by each section of the chromosome (adjustable parameter)
nparam=length(sizevec); np=nparam; varchrome=sizevec; ndim=sum(varchrome); pm=1/ndim; %Define initial values for electrostatic components exp(zFY/RT) in Matrix C logef=[-1];
for i=1:DATSIZE MAT_C(i,1)=logef(1); end
pop = [(rand(popsize,ndim)<0.5)]; end
case 2 % Basic Stern Model N_Planes=2; MAT_Vari=2; %Define interval of intensive variables (log K's) in Matrix B minval=[-2000,-2000,-2000,-2000,-2000,-2000]; maxval=[2000,2000,2000,2000,2000,2000]; sizevec=[11,11,11,11,11,11]; nparam=length(sizevec); np=nparam; varchrome1=sizevec; ndim=sum(varchrome1); %Define interval of intensive variables (Capacitances) in Matrix B minval2=[20,20];
maxval2=[4000,4000];

392
sizevec2=[12,12]; nparam2=length(sizevec2); np2=nparam2; varchrome2=sizevec2; ndim2=sum(varchrome2); pm=1/ndim; pm2=1/ndim2; np2=nparam2; N_Cap=nparam2;
%Define initial values for electrostatic components exp(-zFY/RT) in Matrix A logef=[-1,-0.5,-0.2];
for i=1:DATSIZE
MAT_C(i,1:3)=logef(1:3); end
pop = [(rand(popsize,ndim)<0.5)]; pop2 = [(rand(popsize,ndim2)<0.5)];
case 3 % Triple Layer Model N_Planes=3; MAT_Vari=2; % Define interval of intensive variables (log K's) in Matrix B minval=[-25000,-25000,...... Number of adjustable parameter] % Define minimum boundary
%value maxval=[25000,25000,........ Number of adjustable parameter] % Define maximum boundary
%value sizevec=[15,15,10,....] % Define number of bits required by each section of the chromosome
%(adjustable parameter)
np=length(maxval); sizevec=[20,20,15,15,1,1];
paramnum=length(minval); nparam=length(sizevec); np=nparam; varchrome1=sizevec; lenchrome=sum(varchrome1); % Define interval of intensive variables (Capacitances) in Matrix C minval2=[-10,-800]; maxval2=[200,-500]; sizevec2=[10,10]; varchrome2=sizevec2; lenchrome2=sum(varchrome2);
% Define interval of extensive variables (Capacitances) in Matrix A minval3=[-4000,-2000,-1000]; maxval3=[4000,2000,1000]; sizevec3=[12,11,10]; varchrome3=sizevec3; lengthchrome3=sum(varchrome3); ndim3=sum(varchrome3); nparam2=length(sizevec2); np2=nparam2; N_Cap=nparam2; pm=1/lenchrome; pm2=1/lenchrome2; ndim1=sum(varchrome1); ndim2=sum(varchrome2); ndim=ndim1+ndim2; for k=1:DATSIZE pop4(1:popsize,1:ndim3,k)= [(rand(popsize,ndim3)<0.5)]; end
pop1=[(rand(popsize,ndim1)<0.5)]; pop2=[(rand(popsize,ndim2)<0.5)];
otherwise error('UNDEFINED ELECTROSTATIC MODEL (Re-initialize FITGEN)'); end % % Optimize for a given number of generations % tnumgen =numgen; SS=1; cgn=0; child=0; childcount=0; Fuse=1; varza10=0; dummy=2; varchrome=varchrome2';

393
varchromeb=varchrome1'; minvalb=minval; maxvalb=maxval; pop=pop1; [popsize,ndim] = size(pop); var=[]; gray=1; % if xtype ==5 if cgn >1 gray=2 vars=child'; end end for cgn = 1:tnumgen if cgn~=1 & MAT_Vari~=1 vars=vars1; else end
MAT_B=zeros(popsize,(N_Planes-1)); gener=cgn; %% BUILT-IN SUBROUTINE %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% Binary encoding of adjustable parameters (Matrix A and Matrix B) %% %% Compute fitness function for each member of the population %% %% Computes fitness scores for Genetic Algorithm %% Splits chromosome into predetermined sections for each variable %% %% 05/05/04 Earth and Planetary Sciences, McGill University %% Adrián Villegas-Jiménez %% %% The chromosome has to be splitted into nvar sections (one section for each master specie or principal component) %% the corresponding lengths can be either be calculated in the input file or can be specified directly by the user %% Solution is coded in nominal values but tested in logarithmic units %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %%% Optimizing master variables concentrations %%%
for i=1:MAT_Vari if i==1 varchrome4=varchrome1; nparam4=np; minval4=minval; maxval4=maxval; vars=0; tvars=0; pop3=pop1; else varchrome4=varchrome2; nparam4=np2; minval4=minval2; maxval4=maxval2; vars1=vars; vars=0; tvars=0; pop3=pop2; end
if gray == 1 count=1; fin=0; start=1;
for j=1:nparam4
varchrome_op=(varchrome4(j)); minval_op=(minval4(j)); maxval_op=(maxval4(j)); pow_two = 2.^(0:varchrome_op);
maxintval_op = ((2^varchrome_op)-1);

394
range =maxval_op-minval_op; start = start+fin;
fin= fin + varchrome_op;
for i = 1:popsize tvars(1:varchrome_op) = pop3(i,start:fin); % % now decode binary number to real number (scale maxval to minval) % real = 0;
for k = 1:varchrome_op real = real + pow_two(k)*tvars(varchrome_op-k+1); end % % Takes integer value and converts to a real number (genotype to
% phenotype transformation) % vars(i,j) = (range*(real/maxintval_op)) + minval_op; end start=1; end else end
end format long E vars1=(vars1./1000); vars2=10.^(vars./1000); MAT_A=vars1; %Matrix encoding log K values MAT_B=vars2; %Matrix encoding capacitance values % vars2=zeros(popsize,dummy,DATSIZE); for h=1:DATSIZE
if gray == 1 count=1;
fin=0; start=1;
for j=1:dummy+1 varchrome33=(varchrome3(j)); minval33=(minval3(j)); maxval33=(maxval3(j)); pow_two = 2.^(0:varchrome33); maxintval = ((2^varchrome33)-1); range =maxval33-minval33; start = start+fin; fin= fin + varchrome33;
for i = 1:popsize tvars(1:varchrome33) = pop4(i,start:fin,h); %
% now decode binary number to real number (scale maxval to minval) % real = 0;
for k = 1:varchrome33 real = real + pow_two(k)*tvars(varchrome33-k+1); end % % Takes integer value and converts to a real number (genotype to phenotype
%transformation) % vars33(i,j,h) = ((range*(real/maxintval)) + minval33); end start=1; end
else end end

395
vars33=(vars33./1000); MAT_C=vars33; format long E vars=zeros(popsize,nparam,DATSIZE); static=zeros(popsize,DATSIZE,nsurspecies); [%%%%%%%%%%%%%%%%%%%% CALL FITLOG SUBROUTINE %%%%%%%%%%%%%%%%%%% scores,scoress,minss1,poss1,poss2] = FITLOG(MAT_A,MAT_B,MAT_C,EM,N_Cap,popsize,numgen,nparam,np,np2,gener,N_Planes,ncs,cgn,tnumgen); function [scores,scoress,minss1,poss1,poss2] = FITLOG(MAT_A,MAT_B,MAT_C,EM,N_Cap,popsize,numgen,nparam,np,np2,gener,N_Planes,ncs,cgn,tnumgen); %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % FITLOG computes fitness scores and performs all calculations required by the objective function % % Subroutine designed for the optimization of multiple SCM parameters from surface protonation and adsorption data % Data fits for one or two adsorbates is fitted within the scope of multi-layer electrostatic models % % The chromosome has to be splitted into nvar sections (one section for each master specie or principal component) % the corresponding lengths can be either be calculated in the input file or can be specified directly by the user % Solution is coded in nominal values but tested in logarithmic units % % 1/12/07 Earth and Planetary Sciences, McGill University % Adrián Villegas-Jiménez % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ load EQUIL_DATA load check load optimized load update % -------------------------------------------------------------- Section 1 --------------------------------------------------------------------------------- % Compute Charges from Electrostatics % ----------------------------------------- --------------------------------------------------------------------------------------------------------------------- for i=1:popsize for h=1:DATSIZE for j=1:N_Planes % Number of electrostatic layers MAT_C2(h,j,i)=-log(10^(MAT_C(i,j,h)))*0.02569; end end end MAT_D=MAT_C2; % COMPUTE MULTI-LAYER CHARGES ACCORDING TO ELECTROSTATICS switch EM % Specify type of electrostatic model
case 1 % Constant Capacitance Model for i=1:popsize Charge_Elec_O(1:DATSIZE,j)=MAT_C2(1:DATSIZE,j)*MAT_B(i,1); end case 2 % Basic Stern Model for i=1:popsize Charge_Elec_O(1:DATSIZE,i)=(MAT_C2(1:DATSIZE,1)-MAT_C2(1:DATSIZE,2))*MAT_B(i,1); Charge_Elec_D(1:DATSIZE,i)=MAT_B(i,2)*(MAT_C2(1:DATSIZE,3)-MAT_C2(1:DATSIZE,2)) ; Charge_Elec_B(1:DATSIZE,i)=-Charge_Elec_O(1:DATSIZE,1)-Charge_Elec_D(1:DATSIZE,1);
end case 3 % Triple Layer Model (Charge calculated in molar units) for i=1:popsize for j=1:DATSIZE Charge_Elec_O(j,i)=(MAT_C2(j,1)-MAT_C2(j,2))*MAT_B(i,1)*(Convert(j,1)/9.649e4);
% Gouy-Chapman Equation at 25 Celsius Charge_Elec_D(j,i)=(-0.1174*(IEq(j,1))^0.5)*(sinh(Z*19.46*MAT_C2(j,3))) *(Convert(j,1)/9.649e4);

396
Charge_Elec_B(j,i)=(((MAT_C2(j,2)-MAT_C2(j,3))*MAT_B(i,2))-Charge_Elec_O(j,i)) *(Convert(j,1)/9.649e4);
end end otherwise end load update Capaci=MAT_B(1,1:N_Planes-1); % minimize with respect to residuals of the B-Layer if EM~= 1
for i=1:DATSIZE %Diffuse Potential in Volts MAT_D(i,3)=(asinh ((Charge_Elec_D(i,1)*(9.649e4/Convert(j,1))) /(-0.1174*(IEq(i,1)^0.5))))/19.4635; MAT_D(i,2)=(((Charge_Elec_O(i,1)*(9.649e4/Convert(i)))+(Charge_Elec_B(i,1)*(9.649e4/Convert(i))))/Capaci(1,2))+MAT_D(i,3);
MAT_D(i,1)=((Charge_Elec_O(i,1)*(9.649e4/Convert(i)))/Capaci(1,1))+MAT_D(i,2); %Surface Potential in Volts
end else end % Compute charge associated with each electrostatic plane guess=zeros(aqnvar,DATSIZE); for j=1:DATSIZE
for i=1:aqnvar guess(i,j)=Activ(i,j); end end quihubo=guess; act=ones(surnvar,DATSIZE); guess=(vertcat(guess,act))'; guess=log10(guess); unito=(ones(1,DATSIZE))'; for i=1:N_Planes-1
guess=horzcat(guess,unito); end Konstants1=zeros(popsize,np); for i=1:popsize
for j=1:np Konstants1(i,j)=10^(MAT_A(i,j)); end end %Express constants in terms of the 1 Molar reference state as suggested by Dr D. Sverjensky %Konstants=Konstants1*(1/stdstate); Ksurcomp=ones(popsize,surnvar); Konstants2=log10(Konstants1)'; for j=1:popsize
log_K1(1:nsurspecies,j)=vertcat(log_Ksup,Konstants2(1:paramnum,j)); end %Estimating free concentrations of surface components icc=0; surconc=zeros(DATSIZE,surnvar); % --------------------------------------------------------------------------------------------------------------------------------------------------------------- % ---------------------------------- Loop for calculating the free surface components concentrations -------------------------------------- % --------------------------------------------------------------------------------------------------------------------------------------------------------------- statica=zeros(nsurspecies,N_Planes-1); for j=1:popsize
jcc=np; icc=icc+1;
for i=1:DATSIZE
UF=10^(MAT_A(icc,jcc)); UF=10^(MAT_A(icc,jcc))*Convert(i,1);
FRACT(1,1)=abs(10^(MAT_A(icc,jcc-1)));

397
FRACT(1,2)=1-FRACT(1,1); FF2=log_K1(1:nsurspecies,j);
for nsite=1:ncs switch nsite
case 1 x=1; y=1;
case 2 x=nsurspecies+1;
y=2; case 3 x=(2*nsurspecies)+1; y=3; case 4 x=(3*nsurspecies)+1; y=4; otherwise end
kk=0;
for k=x:((nsurspecies+x)-1) kk=kk+1;
for nplane=1:N_Planes-1 statica(kk,nplane,icc)=log10(exp(MAT_D(i,nplane,icc)*EFO(k,nplane))); end end
statica2(1:nsurspecies,i)=sum(statica(1:nsurspecies,1:N_Planes-1,j)')'; end
FF=FF2+statica2(1:nsurspecies,i); SURSST3=horzcat(SURSST,FF); adivina=guess(i,(1:nvar+ncs))'; adivina2=10.^((SURSST3*adivina))'; surface=adivina2*SURSST2; surface2=surface; surconc(i,1:surnvar,icc)=(UF*FRACT(1,1:ncs))./surface2;
end end consur=surconc; quihubo=quihubo'; M=zeros(DATSIZE,nvar+2,popsize); anexo=ones(DATSIZE,1); for k=1:popsize MB1=horzcat((log10(quihubo)),log10(surconc(1:DATSIZE,1:surnvar,k))); MB1=horzcat(MB1,anexo); M(1:DATSIZE,1:nvar+1,k)=MB1(1:DATSIZE,1:nvar+1,1); end SB_residual=zeros(surnvar,popsize); if surnvar>1
SURFACE=zeros(DATSIZE,surnvar,popsize); else
SURFACE=zeros(DATSIZE,popsize); end MB1=M; % --------------------------------------------------------------------------------------------------------------------------------------------------------------- % ---------------------------------- Loop for calculating the surface species concentrations -------------------------------------------------- % --------------------------------------------------------------------------------------------------------------------------------------------------------------- icc=0; static=zeros(nsurspecies,N_Planes-1);

398
for j=1:popsize jcc=np;
icc=icc+1;
for i=1:DATSIZE FF2=log_K1(1:nsurspecies,j);
for nsite=1:ncs switch nsite
case 1 x=1; y=1; case 2 x=nsurspecies+1; y=2; case 3 x=(2*nsurspecies)+1; y=3; case 4 x=(3*nsurspecies)+1; y=4; otherwise end
kk=0;
for k=x:((nsurspecies+x)-1) kk=kk+1;
for nplane=1:N_Planes-1
static(kk,nplane)=log10(exp(MAT_D(i,nplane,icc)*EFO(k,nplane))); end end static2(1:nsurspecies,i)=sum(static(1:nsurspecies,1:N_Planes-1)')'; end FF=FF2+static2(1:nsurspecies,i); SURSST3=horzcat(SURSST,FF);
adivina=MB1(i,(1:nvar+1),icc)'; % log adivina2=(SURSST3*adivina)'; %estimate concentration of surface species adivina4=10.^(adivina2);% Nominal concentration of surface species Stoi=Stoichiometry'; adivina5=adivina4'; ADSB(i,1:aqnvar+dummy)=(Stoi*adivina5)'; sitios(i,1:surnvar,icc)=adivina4*ST4; CB(i,1:N_Planes-1)=(adivina4*SURSPCHARGE); SUR_SPEC(i,1:nsurspecies,icc)=adivina2(1:nsurspecies); % Compute charges CHARGE_MAT(i,1:N_Planes-1,j)=(adivina4*SURSPCHARGE); CB4_0(i,j)=abs(CB(i,1)-(Charge_Elec_O(i,j))); % Charge density balance for the xth chromosome
if EM ~=1 CHARGE_DIFFUSE(i,j)=((-0.1174*(IEq(i,1)).^0.5)*(sinh(Z*19.46*MAT_C2(1,3)))
*(Convert(i)/9.649e4)); CB4_B(i,j)=abs(CB(i,2)-(Charge_Elec_B(i,j))); % Charge density balance for the
%xth chromosome CB4_D(i,j)=abs(CHARGE_DIFFUSE(i,j)-Charge_Elec_D(i,j)); else end
if EM ~=1 CHARGE_MAT(i,N_Planes-2,j)=CB(i,1); CHARGE_MAT(i,N_Planes-1,j)=CB(i,2); CHARGE_MAT(i,N_Planes,j)=(-CB(i,1)-CB(i,2)); % Compute charge in diffuse
%layer else end modelled(i,1:nads,j)=ADSB(i,1:nads)/Convert(i,1); for h=1:nads TOT1=horzcat(TOT_ADS(i,1:aqnvar),Charge_Elec_O(i,j)); TOT2=horzcat(TOT1,Charge_Elec_B(i,j));
MB7(i,1:aqnvar,j)=(ADSB(i,1:aqnvar)-TOT_ADS(i,1:aqnvar)).^2; % Mass Balance

399
TOTT1=horzcat(Charge_Elec_O(i,j),Charge_Elec_B(i,j)); % Electrostatics MB8(i,1:dummy,j)=(ADSB(i,aqnvar+1:aqnvar+dummy)-TOTT1).^2; % end end
end % --------------------------------------------------------------------------------------------------------------------------------------------------------------- % ---------------------------------- Microloop to compute scores and select best chromosome ------------------------------------------ % ---------------------------------------------------------------------------------------------------------------------------------------------------------------
SKOR1=MB7; %Mass Balance Adsorbed Species SKOR2=MB8;
for i=1:popsize
for j=1:DATSIZE RESIDUAL(j,i)=sum(SKOR1(j,1:aqnvar,i)); RESIDUAL2(j,i)=sum(SKOR2(j,1:N_Planes-1,i)); end
end
if layers==0 scores1=RESIDUAL; scores2=RESIDUAL2;
else
for j=1:popsize for i=1:DATSIZE perc1=1/((exp(RESIDUAL(i,j)))+abs(log10(RESIDUAL(i,j)))); perc2=1/((exp(RESIDUAL2(i,j)))+abs(log10(RESIDUAL2(i,j)))); scores1(i,j)=perc1+perc2; scores2(i,j)=perc2; end
end scores1=sum(scores1); scores2=sum(scores2); scores3=scores1+scores2; end Sum_Scores=scores; [minss1,poss1]=min(scores1); %Find best chromosome for the intensive variables scores=scores1; resid=minss1; [minss2,poss2]=min(scores2); % Find best chromosome for the extensive variables scoress=scores2; residual=minss2; RES_1=CB4_0(1:DATSIZE,poss1); RES_2=CB4_B(1:DATSIZE,poss1); RES_3=CB4_D(1:DATSIZE,poss1); MB11=MB1(1:DATSIZE,(1:nvar+1),poss1); RES2=horzcat(RES_1,RES_2)'; RES2=abs(horzcat(RES2',RES_3)'); Sp_Conc5=10.^(SUR_SPEC(1:DATSIZE,1:nsurspecies,poss1)); %molar concentration mod=modelled(1:DATSIZE,1:nads,poss1); % molar density Capaci=MAT_B(poss1,1:N_Planes-1); % minimize with respect to residuals of the B-Layer for m=1:popsize
Charge_0calc(1:DATSIZE,m)=(CHARGE_MAT(1:DATSIZE,1,poss1)).*(9.649e4./Convert); %Coulombs/m2 Charge_Bcalc(1:DATSIZE,m)=(CHARGE_MAT(1:DATSIZE,2,poss1)).*(9.649e4./Convert); %Coulombs/m2 Charge_Dcalc(1:DATSIZE,m)=(CHARGE_MAT(1:DATSIZE,3,poss1)).*(9.649e4./Convert); %Coulombs/m2 end % --------------------------------------------------------------------------------------------------------------------------------------------------------------- % ------------------------------ Compute potentials from electrostatics and computed charges ---------------------------------------- % --------------------------------------------------------------------------------------------------------------------------------------------------------------- if cgn > numgen
for i=1:DATSIZE Charge_Elec_O(i,1:popsize)=Charge_0calc(i,1:popsize); %Coulombs/m2
Charge_Elec_B(i,1:popsize)=Charge_Bcalc(i,1:popsize); %Coulombs/m2

400
Charge_Elec_D(i,1:popsize)=Charge_Dcalc(i,1:popsize); %Coulombs/m2 end
MAT_D=MAT_C2; save update MAT_D
for j=1:popsize
for i=1:DATSIZE MAT_D(i,3,j)=(asinh(Charge_Dcalc(i,j)/(-0.1174*(IEq(i,1)^0.5))))/19.4635;%Diffuse
%Potential in Volts MAT_D(i,2,j)=(-Charge_Dcalc(i,j)/Capaci(1,2))+MAT_D(i,3,j);%Stern Potential in
%Volts MAT_D(i,1,j)=(Charge_0calc(i,j)/Capaci(1,1))+MAT_D(i,2,j); %Surface Potential in
%Volts POT_D(i,j)=(asinh(Charge_Dcalc(i,poss1)/(-0.1174*(IEq(i,1)^0.5))))/19.4635;
%Diffuse Potential in Volts
POT_B(i,j)=(-Charge_Dcalc(i,poss1)/Capaci(1,2))+POT_D(i,1); %Stern Potential in Volts
POT_0(i,j)=(Charge_0calc(i,poss1)/Capaci(1,1))+POT_B(i,1); %Surface Potential in Volts
end end
MAT_C1=horzcat(POT_0(1:DATSIZE,poss1),POT_B(1:DATSIZE,poss1)); results2=horzcat(MAT_C1,POT_D(1:DATSIZE,poss1)) MATEO=log10(exp(-38.9256*results2)) MAT_C=zeros(popsize,3,DATSIZE);
for i=1:popsize
for k=1:DATSIZE MAT_C(i,1:3,k)=MATEO(k,1:3); end
end
save update MAT_C else
results2(DATSIZE,1:3)=MAT_C(poss1,1:3,DATSIZE) end for i=1:DATSIZE
site(1:DATSIZE,1:surnvar)=sitios(i,1:surnvar,poss1)./Convert(i,1); end mod=modelled(1:DATSIZE,1,poss1); K=log_K1(1:nsurspecies,poss1); if resid <= resid2 iternum=cgn; volts=MAT_C2(1:DATSIZE,1:N_Planes,poss1) capaci=MAT_B(poss1,1:2) resid2=resid; save output K resid2 site mod volts capaci iternum save check resid2 drawnow plot(pH_1,SADS_C1,pH_1,SADS_C1,'o',pH_1,mod(1:DATSIZE,1),pH_1,mod(1:DATSIZE,1),'*') xlabel ('pH') ylabel ('Surface Charge Density (moles/m2)') title ('Surface Complexation Model for Goethite') else end if cgn == numgen results=MAT_C2; %Matrix containing initial potential values results=results2 results2=log10(exp(-38.9256*potential)); MAT_C22=(exp(-38.9256*results));%Calculate electrostatic factors MAT_C2=MAT_C22; MATT=MAT_C22'; optim_old=MATT(1:N_Planes,1:DATSIZE); PSII=results; %nominal values of electrostatic potential MAT_C=log10(MAT_C2); %Log of electrostatic factor

401
KK_1=log_K1(1:nsurspecies,poss1); save optimized MAT_C KK_1 Sp_Conc6=Sp_Conc5'; quihubo2=quihubo; MAT_C3=zeros(DATSIZE,N_Planes); Charge_0=zeros(DATSIZE,1); Charge_B=zeros(DATSIZE,1); Charge_D=zeros(DATSIZE,1); save microdata Charge_Elec_O; Charge_Elec_B; Charge_Elec_D; Charge_0calc; Charge_Bcalc; Charge_Dcalc; % Switch to Newton-Raphson microiterations %%%%%%%%%%%%%%%%%%%%% END FITLOG SUBROUTINE %%%%%%%%%%%%%%%%%%%% % fprintf('%i \t %8.2f \t %8.2f \t %8.10f \n',cgn,minss1)
if elite == 2 elitechrome1 = pop1(poss1,:); elitechrome2 = pop2(poss1,:); for i=1:DATSIZE elitechrome3(1,1:ndim3,i)=pop4(poss2,1:ndim3,i); end else end % select most fit members of population for mating subset % using tournament selection and eliteist strategy (if turned on) %%%%%%%%%%%%%%%% %%%%% CALL EVAL_GA SUBROUTINE %%%%%%%%%%%%%%%%%%%% [mateset1, mateset2,mateset3] = EVAL_GA(pop1,pop2,pop4,scores,scoress,elite); function [mateset1,mateset2,mateset3] = EVAL_GA(pop1,pop2,pop4,scores,scoress,elite); %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % Select most fit members of population for mating subset % using tournament selection and eliteist strategy (if turned on) % scores % % function mateset = EVAL_GA(pop,scores,elite) % EVAL_GA performs selection of chromsomes (by stochastic tournament) % Version 1.1 5/5/2005 Adrián Villegas-Jiménez % Earth and Planetary Sciences, McGill University % Modified from the original “MUTATE “ Matlab subroutine Version 1.0 by Ron Shaffer 1/23/96 % TSELECT tournament mating selection % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % set constants % [popsize,ndim1] = size(pop1); [popsize,ndim2] = size(pop2); [popsize,ndim3,DATSIZE] = size(pop4); % % With elistist strategy on you only need to choose a mating set with popsize - 1 % members in it b/c the last spot is save for the elite chromosome % if elite == 2 popsize1 = popsize - 1; popsize2 = popsize - 1;

402
popsize3 = popsize - 1; end % % compute vector of random integers % randlist1 = [round(rand((popsize1*2),1)*popsize2+0.5)]; randlist2 = [round(rand((popsize2*2),1)*popsize2+0.5)]; randlist3 = [round(rand((popsize3*2),1)*popsize3+0.5)]; %. % Begin tournament selection % count = 0; for i = 1:popsize1 count = count + 2; cmo = count - 1; % % 2 randomly chosen chromosomes from population % will compete for inclusion in mating subset % if scores(randlist1(count)) > scores(randlist1(cmo)) mateset1(i,1:ndim1) = pop1(randlist1(count),1:ndim1); else mateset1(i,1:ndim1) = pop1(randlist1(cmo),1:ndim1); end if scores(randlist2(count)) > scores(randlist2(cmo)) mateset2(i,1:ndim2) = pop2(randlist2(count),1:ndim2); else mateset2(i,1:ndim2) = pop2(randlist2(cmo),1:ndim2); end for k=1:DATSIZE if scoress(randlist3(count)) < scoress(randlist3(cmo)) mateset3(i,1:ndim3,k) = pop4(randlist3(count),1:ndim3,k); else mateset3(i,1:ndim3,k) = pop4(randlist3(cmo),1:ndim3,k); end end end %%%%%%%%%%%%%%%%%%% END EVAL_GA SUBROUTINE %%%%%%%%%%%%%%%%%%% % perform crossover if required % %%%%%%%%%%%%%%%%%%%%%%% CALL XOVER_GA SUBROUTINE %%%%%%%%%%%%%%% [child1, child2, child3, xcount] = XOVER_GA(mateset1,mateset2,mateset3,px1,px2,px3,xtype);

403
function [child1, child2, child3, xcount] = XOVER_GA(mateset1,mateset2,mateset3,px1,px2,px3,xtype); %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % XOVER_GA: Performs crossover operations of Genetic Algorithm-Based Fitting of Surface Complexation Model (SCM) % Parameters % Version 1.1 5/5/2005 Adrián Villegas-Jiménez % Earth and Planetary Sciences, McGill University % % Modified from the original “XOVER” Matlab subroutine by Ron Shaffer % Version 1.0 1/23/96 Ron Shaffer % Version 1.1 2/27/96 Ron Shaffer % added options for two-point and uniform crossover % Version 1.2 6/24/96 Ron Shaffer % fixed bug in 2-point crossover discovered by % Mr. Radovan Cemes ([email protected]). Crossover % points are now sorted before swapping is performed. % % new -- new population of chromosomes % xcount-- # of times crossover was performed % old -- input population of chromosomes % px -- crossover probability % xtype -- type of crossover % % Several types of xover can be performed according to an inequality constraint % determined by the residual value as suggested by Gen et al, 1996. % % Residual--Difference between estimated values and total concentrations % of components excepting H % -- 1) 1-point crossover % -- 2) 2-point crossover % -- 3) Uniform crossover % -- 4) Randomised and/or crossover as suggested by Bill Keller from University of Sussex % -- 5) Direction based crossover as suggested by Michalewicz et al, 1994 % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % set constants % old1=mateset1; old2=mateset2; old3=mateset3; [popsize1,ndim1] = size(old1); [popsize2,ndim2] = size(old2); [popsize3,ndim3,DATSIZE] = size(old3); halfpop1 = popsize1/2; halfpop2 = popsize2/2; halfpop3 = popsize3/2; xcount = 0; px=px1; if xtype == 1 randlist = rand((halfpop1),1); for i = 1:halfpop1 x = (i*2) - 1; xpo = x + 1; new1(x,1:ndim1) = old1(x,1:ndim1); new1(xpo,1:ndim1) = old1(xpo,1:ndim1); if (randlist(i) < px1) xcount = xcount + 1; xpoint = round((rand * ndim1)+0.5); new1(xpo,1:xpoint)=old1(x,1:xpoint); new1(x,1:xpoint) = old1(xpo,1:xpoint); end end end child1=new1; if xtype == 1 randlist = rand((halfpop2),1);
404
for i = 1:halfpop2 x = (i*2) - 1; xpo = x + 1; new2(x,1:ndim2) = old2(x,1:ndim2); new2(xpo,1:ndim2) = old2(xpo,1:ndim2); if (randlist(i) < px) xcount = xcount + 1; xpoint = round((rand * ndim2)+0.5); new2(xpo,1:xpoint)=old2(x,1:xpoint); new2(x,1:xpoint) = old2(xpo,1:xpoint); end end end child2=new2; if xtype == 1 randlist = rand((halfpop3),1); for k=1:DATSIZE for i = 1:halfpop3 x= (i*2) - 1; xpo = x + 1; new3(x,1:ndim3,k) = old3(x,1:ndim3,k); new3(xpo,1:ndim3,k) = old3(xpo,1:ndim3,k); if (randlist(i) < px3) xcount = xcount + 1; xpoint = round((rand * ndim3)+0.5); new3(xpo,1:xpoint,k)=old3(x,1:xpoint,k); new3(x,1:xpoint,k) = old3(xpo,1:xpoint,k); end end end end child3=new3; % % two-point crossover % if xtype == 2 randlist = rand((halfpop),1); for i = 1:halfpop x = (i*2)-1; xpo = x+1; new(x,1:ndim) = old(x,1:ndim); new(xpo,1:ndim) = old(xpo,1:ndim); if (randlist(i) < px) xcount = xcount + 1; [xpoint] = sort(round((rand(1,2) * ndim)+0.5)); new(xpo,xpoint(1):xpoint(2)) = old(x,xpoint(1):xpoint(2)); new(x,xpoint(1):xpoint(2)) = old(xpo,xpoint(1):xpoint(2)); end end end % % uniform crossover % if xtype == 3 for i = 1:halfpop x = (i*2)-1; xpo = x+1; new(x,1:ndim) = old(x,1:ndim); new(xpo,1:ndim) = old(xpo,1:ndim); for j = 1:ndim test = rand; if test < px xcount = xcount + 1; new(xpo,j) = old(x,j); new(x,j) = old(xpo,j);

405
end end end end % Randomised and/or crossover if xtype == 4 for i = 1:halfpop x = (i*2)-1; xpo = x+1; new(x,1:ndim) = old(x,1:ndim); new(xpo,1:ndim) = old(xpo,1:ndim); for j = 1:ndim test = rand; if test <= px xcount = xcount + 1; if old(x,j)==1 if old(xpo,j)==1 new(x,j) = 1; new(xpo,j) = 1; else new(x,j) = 0; new(xpo,j) = 1; end else if old(xpo,j)==1 new(x,j) = 0; new(xpo,j) = 1; else new(x,j) = 0; new(xpo,j) = 0; end end else xcount = xcount + 1; if old(x,j)==1 if old(xpo,j)==1; new(x,j) = 1; new(xpo,j) = 1; else new(x,j) = 1; new(xpo,j) = 0; end else if old(xpo,j)==1 new(x,j) = 1; new(xpo,j) = 0; else new(x,j) = 0; new(xpo,j) = 0; end end end end end end %%%%%%%%%%%%%%%%%%% END XOVER_GA SUBROUTINE %%%%%%%%%%%%%%%%%% % % perform mutation % %%%%%%%%%%%%% %%%%%%% CALL MUT_GA SUBROUTINE %%%%%%%%%%%%%%%%%%% [newmut1, newmut2, newmut3, tnmut] = MUT_GA(child1, child2, child3, pm1, pm2, pm3); nummut = nummut + tnmut;

406
function [newmut1, newmut2, newmut3, nmut] = MUT_GA(pop1, pop2, pop4, pm1, pm2, pm3) %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % % MUT_GA: performs mutation on population of chromsomes % Version 1.1 5/5/2005 Adrián Villegas-Jiménez % Earth and Planetary Sciences, McGill University % % Modified from the original MUTATE Matlab subroutine by Ron Shaffer % version: 1.0 % date: 1/23/96 % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ [popsize1,ndim1,DATSIZE] = size(pop1); [popsize2,ndim2,DATSIZE] = size(pop2); [popsize3,ndim3,DATSIZE] = size(pop4); nmut = 0; % % loop through population testing whether to mutate % for i = 1:popsize1 for j = 1:ndim1 test = rand; if test < pm1 pop1(i,j) = abs(pop1(i,j)-1); nmut = nmut + 1; end end end for i = 1:popsize2 for j = 1:ndim2 test = rand; if test < pm2 pop2(i,j) = abs(pop2(i,j)-1); nmut = nmut + 1; end end end for k=1:DATSIZE for i = 1:popsize3 for j = 1:ndim3 test = rand; if test < pm3 pop4(i,j,k) = abs(pop4(i,j,k)-1); nmut = nmut + 1; end end end end % return new population % newmut1=pop1; newmut2=pop2; newmut3 = pop4; %%%%%%%%%%%%%%%%%%% END MUT_GA SUBROUTINE %%%%%%%%%%%%%%%%%%% % Children + elite chromosome (if elitism turned on) form the new generation % pop1 = newmut1; pop2 = newmut2; pop4 = newmut3;

407
if elite == 2 popi=popsize; pop1(popi,:) = elitechrome1; pop2(popi,:) = elitechrome2; for k=1:DATSIZE pop4(popi,1:ndim3,k)=elitechrome3(1,1:ndim3,k); end else end end %%%%%%%%%%%%%%%%%% END FITGEN MAIN SUBROUTINE %%%%%%%%%%%%%%%%%%%

408
3. INPUT FILE FOR GENETIC ALGORITHM-BASED OPTIMIZATION OF
INTRINSIC FORMATION CONSTANTS BASED UPON THE CONSTANT
CAPACITANCE MODEL AND THE SINGLE-SITE SCHEME
Chapters 3 and 4 %%%%%%%%%%%%%%%%%%%%%% CALL EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%%%% EQUIL %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% INPUT FILE: EQUIL Surface Complexation Model %% %% Calibration of SCM reactions for NiCO3(s) (Gaspeite) %% Reactions written in terms of a single generic surface site %% Constant Capacitance Model %% %% Adrián Villegas-Jiménez %% Earth and Planetary Sciences, McGill University %% Montreal, CANADA %% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %%%%%%%%%%%%%%%%%% DEFINITION OF CHEMICAL EQUILIBRIUM %%%%%%%%%%%%%%%%% %Names of aqueous components (Always enter H component first) format short aqcomp=[sym('H'),sym('Na'),sym('Cl'),sym('Ni')]; surcomp=[sym('S1')]; component=horzcat(aqcomp,surcomp) aqnvar=length(aqcomp)'; surnvar=length(surcomp)'; nvar=length(component)'; aqcomp_charge=[1;1;-1;2]; surcomp_charge=[0] % Reference surface charge comp_charge=vertcat(aqcomp_charge,surcomp_charge) %Names of Species aqspecies=[sym('H'); sym('Na'); sym('Cl'); sym('Ni')]; naqspecies=length(aqspecies) surspecies=[sym('S1'); sym('S2'); sym('S3'); sym('S4'); sym('S5'); sym('S6')]; nsurspecies=length(surspecies); species=vertcat(aqspecies,surspecies); nspec=length(species); Number_Species=nspec; %Stoichiometry AQSST1=[1,0,0,0; 0,1,0,0; 0,0,1,0; 0,0,0,1]; SURSST=[0,0,0,0,1; 1,0,0,0,1; -1,0,0,0,1; -2,0,0,0,1; -1,1,0,0,1; -1,0,0,1,1];

409
SURSST2=[1; 1; 1; 1; 1; 1]; SURSST4=SURSST2; SURSSTH=[0; 1; -1; -2; -1; -1]; SCoef=[ 1; 1; 1; 1; 1; 1]; AQSST=horzcat(AQSST1,zeros(naqspecies,surnvar)); SST=vertcat(AQSST,SURSST) size_SST=size(SST) %Mass balance of surface species derived from each reaction SPECSSP=SURSST2; % %Thermodynamic Formation Constants % log_K=[0; 0; 0; 0]; log_Ksup=[0]; ncs=1 log_Kadj=zeros((length(SURSST2)-length(log_Ksup)),1); paramnum=length(log_Kadj); log_KC=log_K; % % Charges of Aqueous Species % AQSPCHARGE=[1; 1; -1; 2]; % % Define charges of surface species % SURSPCHARGE=[0; 1; -1; -2; 0; 1]; % Electrostatic Factor % Constant Capacitance Model (CCM) EFO=[ 0; 1; -1; -2; 0; 1]'; MAT_CHARGE2=vertcat(AQSPCHARGE,SURSPCHARGE);

410
SDM=9.99e-6 %Surface sites densities SA=0.23165; %Specify specific surface area % Conversion factor to express in terms of reference state as postulated by Dr Sverjensky,2002 %rsa=3.74; %rsd=5.83e18; %fsa=10; %fsd=1e18; %stdstate=(fsa*fsd)/(rsd*rsa); SD=[1];%Molar Fraction %%%%% End of definition of chemical equilibrium %%%%%%%%%%%%%%%%%%%%%% %%%%%%%%%%%% ENTER EXPERIMENTAL DATA %%%%%%%%%%%%%%%%%%%%%%%%%%%%% pH=['enter vertical vector of pH values'] IS=['enter vertical vector of Ionic Strength values'] SCHARGE_P=['enter vertical vector of surface charge values: proton adsorption densitites (mol/ m2 units)'] Convert=['enter vertical vector containing conversion factors (m2/L units) corrected by dilution'] Scores3=['enter vertical vector with free aqueous component concentrations (log10 of molar units)']; %%%%%%%%%%%%% End of experimental Data %%%%%%%%%%%%%%%%%%%%%%%%%%%%% format long Species=Scores3; [a,b]=size(Scores3); DATSIZE=a % Reassignations nads=1; nspec=naqspecies; pH=pH_1; SCHARGE=SCHARGE_P.*Convert; IonicS=IS; fitcon_num=aqnvar; fitpar_num=paramnum; nvar=aqnvar; pH2=pH_1; I=IS; %Transformation of pH to molar proton concentrations using the Davies Equation for i=1:DATSIZE
act_coef2=10.^(-(0.5115*(1.^2))*((sqrt(IS(i))/(1+(sqrt(IS(i)))))-(0.3*IS(i)))); pH=-log10(10.^(-pH)./act_coef2); end fixvar=-pH_1; pH=fixvar; % CONSTANT CAPACITANCE MODEL SigmaElec=SCHARGE_P.*9.649e4 for i=1:DATSIZE
POTENTIAL(i,1)=(SigmaElec(i)/(IS(i)^0.5)); end % Specify adjustable parameters Param=[sym('K1'),sym('K2'),sym('K3'),sym('K4'),sym('K5'),sym('Rel_Abundance'),sym('Site_density'),sym('Capacitance')]; CompSurf=[sym('S1')]; nparam=length(Param); ncs=length(CompSurf); Adj=horzcat(Param,CompSurf); nadj=length(Adj); Coulomb=zeros(DATSIZE,1); for i=1:DATSIZE
Coulomb(i)=-38.9256*POTENTIAL(i); end Species=Species'; AQSPCHARGE=AQSPCHARGE'; Species=abs(10.^(Species));

411
%Calculate activity coefficients of aqueous species Gama=zeros(aqnvar,DATSIZE); Activ=zeros(aqnvar,DATSIZE); for j=1:DATSIZE
for i=1:aqnvar Gama(i,j)=10.^(-(0.5115*((AQSPCHARGE(i)).^2))*((sqrt(IS(i)))/(1+(sqrt(IS(i)))))-(0.3*(IS(i)))); Activ(i,j)=Species (i,j)*Gama(i); end end MassH=SCHARGE; ns=1; save EQUIL_DATA save fitdol %%%%%%%%%%%%%%%%%%% CLOSE AND SAVE EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%%

412
4. CALCULATION OF AQUEOUS SPECIATION FROM ALKALINITY AND
pH MEASUREMENTS (NEWTON-RAPHSON METHOD)
Chapters 4 and 6 %%%%%%%%%%%%%%%%%%%%%% CALL EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%%%% EQUIL %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% INPUT FILE: EQUIL Aqueous Equilibrium Problem %% %% Determination of aqueous speciation from alkalinity and pH measurements %% Carbonate Standards for the calibration of the carbonate ion selective electrode %% %% 05/05/2004 Adrián Villegas-Jiménez %% Earth and Planetary Sciences, McGill University %% Montreal, CANADA %% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %%%%%%%%%%%%%%%%%% DEFINITION OF CHEMICAL EQUILIBRIUM %%%%%%%%%%%%%%%%% %Names of aqueous components (Always enter H component first) format short aqcomp=[sym('Alk'),sym('K'),sym('Cl'),sym('H2CO3')]; component=aqcomp; aqnvar=length(aqcomp)'; nvar=length(component)'; aqcomp_charge=[1;1;-1;0]; comp_charge=aqcomp_charge; comp_charge2=comp_charge'; aqcomp_charge=comp_charge; %Names of Species aqspecies=[sym('H'); sym('K'); sym('Cl'); sym('H2CO3'); sym('OH'); sym('HCO3'); sym('CO3')]; naqspecies=length(aqspecies); nCO3=7; AQSST1=[1,0,0,0; 0,1,0,0; 0,0,1,0; 0,0,0,1; -1,0,0,0; -1,0,0,1; -2,0,0,1]; species=aqspecies; nspec=length(species); Number_Species=nspec; num_alk=zeros(nspec,1) num_alk2=zeros(nspec,1) for i=1:nspec if AQSST1(i,1)< 0 num_alk(i,1)=AQSST1(i,1); else num_alk2(i,1)=AQSST1(i,1); end end %Stoichiometry Aqueous=AQSST1; AQSST=AQSST1; SST=AQSST; size_SST=size(SST) AQSST1=SST; %

413
%Thermodynamic Formation Constants log_K=[0; 0; 0; 0; -14; -6.35; -16.68]; log_KC=log_K; %Charges of Species AQSPCHARGE=[ 1; 1; -1; 0; -1; -1; -2]; MAT_CHARGE2=AQSPCHARGE; save EQUILIBRIA %%%%%%%%%%%%%%%% END DEFINITION OFCHEMICAL EQUILIBRIUM %%%%%%%%%%%%%%%% %%%%%%%%%%%%%%%%%%%%% ENTER EXPERIMENTAL DATA %%%%%%%%%%%%%%%%%%% %Serial Data % Define initial ionic strength pH_1=['enter vertical vector of pH values'] IS=['enter vertical vector of Ionic Strength values'] %Total Analytical Concentrations of Components (Enter as follows: AC=TOTH, TOTK, TOTCl, TOTCO3) AC=['enter matrix with total principal components concentrations (nominal units), component H takes the negative of the experimental alkalinity']; TOT=AC; DATSIZE=length(AC); pH=-pH_1; fixnumber=1; varnum=fixnumber; pH_Fix=1;ISS=IS; pHeq=pH; act_coef2=zeros(DATSIZE,1); fixcon=zeros(DATSIZE,1); z=1; FIXA=1; %Transformation of pH to molar proton concentrations using the Davies Equation for i=1:DATSIZE
act_coef2(i,1)=10.^(-(0.5115*(1.^2))*((sqrt(IS(i))/(1+(sqrt(IS(i)))))-(0.3*IS(i)))) fixcon(i,1)=-log10(10.^(-pH(i))./act_coef2(i,1)) end fixcon=fixcon'; H=fixcon; fixed=fixcon; save EQUIL_DATA %%%%%%%%%%%%%%%%%%% CLOSE AND SAVE EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%%

414
%%%%%%%%%%%%%%%%%%%% CALL ALKA MAIN SUBROUTINE %%%%%%%%%%%%%%%%%%% ALKA %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % ALKA: Solution of aqueous speciation problems using the Newton-Raphson method % Carbonate system determined by alkalinity and pH measurements % % Version 1.0 05/05/2004 Adrián Villegas-Jiménez % Earth and Planetary Sciences, McGill University % Montreal, Canada % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ load EQUIL_DATA comp=aqcomp; aqcomp2=aqcomp'; ident=zeros(aqnvar,1); for i=1:aqnvar ident(i,1)=i; end comp=comp'; Master_Species=horzcat(ident,comp); TOT=zeros(aqnvar,1); ident=zeros(aqnvar,1); TP=input('Do you want to enter data? (1 for YES, 2 for NO) >'); switch TP
case 1 compvar=input('Do you want to perform a titration using one of the master species? (1 for YES, 2 for NO) >');
if compvar==1
Type=2; minvar=input('Enter the minimum value for titrating master specie (-log units) \n>'); maxvar=input('Enter the maximum value for titrating master specie (-log units) \n >');
if maxvar < minvar fprintf('ERROR! : Specified minimum concentration is higher than maximum value') break else end
intervar=input('Enter the concentration intervals for titration (-log units)\n >'); DATSIZE=(maxvar-minvar)/intervar; fixcomp=minvar; fixed=zeros(DATSIZE,1);
for i=1:DATSIZE fixcomp=fixcomp+intervar; fixed(i,1)=fixcomp; end
if intervar > maxvar fprintf('ERROR! : Titration interval higher than maximum value assigned to titrating specie') break else end
Master_Species fixnumber=input('What master species do you want to choose as the titrant (choose a number)? \n'); fixname=Master_Species(fixnumber,2); varnum=fixnumber; z=comp_charge(fixnumber,1); for i=1:nvar var=comp(i,1) if i~=fixnumber TOT(i,1)=input('Enter now the total analytical concentrations of master species >'); else TOT(fixnumber,1)=minvar; end end

415
fixed=vertcat(TOT(fixnumber,1),fixed); TOT=TOT';
pH=TOT(1,1); AC=zeros(DATSIZE+1,nvar);
for i=1:DATSIZE+1 for j=1:nvar if j==fixnumber fixed(i,1);
AC(i,fixnumber)=fixed(i,1); else
AC(i,j)=TOT(1,j); end
end end
pH=AC(:,1);
else
z=1; fixnumber alk=1; varnum=fixnumber; Type=1; DATSIZE=1; pH=input('Enter the equilibrium pH >\n') TOT(1,1)=pH fixed=pH;
for i=1:nvar fprintf('Aqueous Component \n') var=comp(i,1) TOT(i,1)=input('Enter now the total analytical concentrations for the given speciation problem >');
end
AC=TOT; TOT=TOT';
end
ISS=input('Enter the estimated value of ionic strength >')
for i=1:DATSIZE IS(i,1)=ISS; end case 2
Type=3; load EQUIL_DATA TOT=AC; fixnumber=varnum; otherwise end Equil=input('Is this an alkalinity problem? (if yes press 1, otherwise press any other key >'); Initial_Comp=zeros(1,nvar-1); Y=input('Are initial concentrations different than total analytical concentrations? (if yes press 1, otherwise press any other key >');
if Y==1
F=input('Enter now the factor by which you want to divide the total analytical concentrations >'); else F=1; end initials=zeros(DATSIZE,aqnvar); for m=1:DATSIZE for j=1:aqnvar
if j~=varnum initials(m,j)=abs(TOT(m,j))/F; else initials(m,varnum)=abs(TOT(m,j))/F; %0
end

416
if initials(m,j)==0 initials(m,j)=1e-4;
else end end
if FIXA==3 & Calcite==1
initials(m,varnum)=1; else end end Scores3=zeros(DATSIZE,nvar); Residuals=zeros(DATSIZE,nvar); Species=zeros(DATSIZE,nspec); IonicS=zeros(DATSIZE,1); fixcon=zeros(DATSIZE,1); fixa=input('Fixed activities of at least 1 component? (if yes press 1, otherwise press any other key >'); I_Corr=input('Include ionic strength corrections? (if yes press 1, otherwise press any other key >'); %Transformation of pH or master variable's acitvity to molar proton concentrations using the Davies Equation AI=input('Known activities but no total concentrations? (if yes press 1, otherwise press any other key >'); if AI==1
Act_Fix=1; else
Act_Fix=2; end if I_Corr==1
for i=1:DATSIZE z=aqcomp_charge(varnum,1); act_coef2(i,1)=10.^(-(0.5115*(z.^2))*((sqrt(IS(i)))/(1+(sqrt(IS(i))))-(0.3*IS(i))));
if TP==1 & Type==2 break else fixcon(i,varnum)=-log10(10.^(-fixed(i))./act_coef2(i,1)); end end else end Spc=zeros(DATSIZE,naqspecies); AC2=zeros(aqnvar,DATSIZE); for i=1:DATSIZE
for j=1:aqnvar if j~=varnum AC2(j,i)=initials(i,j); else AC2(varnum,i)=10^(-fixcon(i,1)); end end
Spc(i,1:naqspecies)=10.^((SST*log10(AC2(1:aqnvar,i)))+log_K)'; FI(i,1)=0.5*(Spc(i,1:naqspecies)*(MAT_CHARGE2.^2)); Ratio(i,1)=FI(i,1)/0.5; end for i=DATSIZE
[F]=min(TOT(i,1)); end OPTIONS(1) = 0; OPTIONS(2) = 1e-5; OPTIONS(3) = 1e-10; num_iter=0; if Act_Fix==1
optimum=raphson2(initials,fixcon,fixnumber,fixa,num_iter,FI,varnum,AC2,Equil,Ratio,I_Corr); else %%%%%%%%%%%%%%%%%%% CALL ALKALINITY SUBROUTINE %%%%%%%%%%%%%%%%%%% optimum=alkalinity(initials,fixcon,fixnumber,fixa,num_iter,FI,varnum,AC2,Equil,Ratio,I_Corr);

417
function [optima] =alkalinity(initials,fixcon,fixnumber,fixa,num_iter,FI,varnum,AC2,Equil,Ratio,I_Corr); %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ % Alkalinity computes jacobians based on the Newton-Raphson and Gauss-Elimination method % Input data includes carbonate alkalinity and pH measurements % % Version 1.0 05/05/2004 Adrián Villegas-Jiménez % Earth and Planetary Sciences, McGill University % Montreal, Canada % %^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ load EQUIL_DATA format long if num_iter==0
results=initials; else end num_iter=num_iter+1; Ratio=FI(1,1)/0.5; %---------------------------------------------------------------------------------------------------------------------------------------------------------------- %------------------------------------------- Call Newton-Raphson after a given number of generations ------------------------------------ %---------------------------------------------------------------------------------------------------------------------------------------------------------------- maxiter=100; % Vector with initial concentrations for M=1:DATSIZE
niter=0; h=0;
for i=1:aqnvar h=h+1; optim_old(i,1)=results(M,i); h=h-1; end
counter=0; xtol = 1000*eps; ftol = 1e6*eps; error = 2*xtol; ISS=IS;
if num_iter==1 & pH==1
IEq=zeros(a,1); Activ=zeros(nvar,a); Species=zeros(nspec,a); else end
xtol = 1000000*eps; xtol = eps; ftol = 1e6*eps; error = 2*xtol; ISS=ISS(M,1);
while niter<=maxiter
counter=0+1; niter=niter+1 % Transformation of molar concentrations to activities using the Davies Equation gamma=zeros(nspec,nvar);
for k=1:nspec for j=1:aqnvar z=comp_charge2(1,j); act_coef2=(-(0.5115*(z^2))*((sqrt(ISS))/(1+(sqrt(ISS)))-(0.3*ISS))); gamma(k,j)=(act_coef2*SST(k,j)); end end

418
if I_Corr==1 if pH_Fix==1 z=1; act_coef_H=10.^(-(0.5115*(z^2))*((sqrt(ISS)/(1+(sqrt(ISS))))-(0.3*ISS))); fixvar=((10^(pH(M,1)))./act_coef_H) else end
for i=1:nspec for j=1:nvar if gamma(i,j)==1 gamma(i,j)=0; else end end end
Korr1=(sum(gamma'))'; Korr2=zeros(nspec,1);
if I_Corr==1
for i=1:nspec if MAT_CHARGE2(i,1)~=0 Korr2(i,1)=(-(0.5115*(MAT_CHARGE2(i,1)^2))
%*((sqrt(ISS)/(1+(sqrt(ISS))))-(0.3*ISS))); else end end else end
log_K1=(log_K+Korr1)-Korr2; %corrected log K for ionic strength else fixvar=10^(pH(M,1)); log_K1=log_K; end optim_old2=optim_old; % Mass Balance Equations (Minimum Energy Condition) if pH_Fix==1
optim_old(varnum,1)=fixvar; else end
optim_old3=log10(optim_old) Sp_Conc2=(SST*optim_old3)
for i=1:nspec Sp_Conc5(i,1)=10.^(Sp_Conc2(i,1)+log_K1(i)); end
%Estimate ionic strength Sp_Conc4=(Sp_Conc5'); ISS3=0.5*(Sp_Conc4*(MAT_CHARGE2.^2)); % update ionic strength ISS2=ISS; Ionic_Strength=ISS2(1,1);
%if ISS2 <= 1.1(Ratio) & I_Corr==1 if niter < 0.5*maxiter ISS=ISS3; Ratio=ISS2/0.5; else end
MB=(Sp_Conc4*SST); Mass=MB;
%Definition of Charge Balance (Electroneutrality Condition) Electro_Neut2=(Sp_Conc4*MAT_CHARGE2);
%---------------------------------------------------------------------------------------------------------------------------------------------------------------- %----------------------------------------------------- Calculate Jacobian Matrix ----------------------------------------------------------- %----------------------------------------------------------------------------------------------------------------------------------------------------------------
419
switch FIXA case 1
for pivote=2:aqnvar for i=1:nspec for j=2:aqnvar if j==pivote k=pivote; else k=j; end Jacob(i,j-1)=(Sp_Conc5(i,1)*SST(i,pivote)*SST(i,k))/(Sp_Conc5(k,1)); end end Deriva(pivote-1,1:nvar-1)=sum(Jacob); end case 2 for pivote=1:nvar for i=1:nspec for j=1:nvar if j==pivote k=pivote; Jacob(i,j)=(Sp_Conc5(i,1)*SST(i,pivote)
*SST(i,k))/(Sp_Conc5(k,1)); else k=j;
Jacob(i,j)=(Sp_Conc5(i,1)*SST(i,pivote) *SST(i,k))/(Sp_Conc5(k,1));
end end end Deriva(pivote,1:nvar)=sum(Jacob); end case 3 for pivote=1:aqnvar-1 for i=1:nspec for j=1:aqnvar-1 if j==pivote k=pivote; else k=j; end Jacob(i,j)=(Sp_Conc5(i,1)*SST(i,pivote)*SST(i,k))/(Sp_Conc5(k,1)); end end Deriva(pivote,1:aqnvar-1)=sum(Jacob); end otherwise end Der=Deriva; optim_old=10.^(optim_old3); Calculo=Mass(1:aqnvar); Fixed=AC(M,1:aqnvar);
if FIXA~=3 Fixed(1,varnum)=Calculo(1,varnum); else end switch FIXA
case 1 residuos=(Mass(2:aqnvar) - AC(M,2:aqnvar)); case 2 residuos=(Mass - AC(M,1:aqnvar)); case 3 a=Mass(1:aqnvar-1) b=AC(M,1:aqnvar-1) residuos=(Mass(1:aqnvar-1) - AC(M,1:aqnvar-1)); otherwise end % solves the system of linear equations (Gauss-Elimination Method)

420
residuos=residuos'; factor=Der \ residuos; % computes total inorganic concentration if Equil==1 H=sum((Sp_Conc5(1:nspec)).*(-num_alk2)) TOTCO2=sum((Sp_Conc5(1:nspec)).*(num_alk)) H2CO3=(AC(M,1)+H)/(TOTCO2/Sp_Conc5(nvar)); optima=optim_old; optima(aqnvar)=H2CO3; optima=log10(optima); Sp_Conc6=(SST*optima);
for i=1:nspec Sp_Conc7(i,1)=10.^(Sp_Conc6(i,1)+log_K1(i)); end CO3=Sp_Conc7(nCO3,1); TOTCO2=sum(Sp_Conc7.*(SST(1:nspec,aqnvar))); AC(M,aqnvar)=TOTCO2; else end switch FIXA
case 1 optim_new=(optim_old(2:aqnvar) - factor); optima=optim_new;
for i=1:aqnvar-1 if optim_new(i,1)==0 optima(i,1)=(optim_old2(i,1)); else end if optim_new(i,1) < 0 optima(i,1)=(optim_old2(i,1)/10); else end if optim_new(i,1) > 0 optima(i,1)=(optim_new(i,1)); else end end
case 2 optim_new=(optim_old - factor);
for i=1:aqnvar if optim_new(i,1)==0 optima(i,1)=optim_new(i,1); else end if optim_new(i,1) < 0 optima(i,1)=optim_old(i,1)/10; else end if optim_new(i,1) > 0 optima(i,1)=optim_new(i,1); else end end case 3 optim_new=(optim_old(1:aqnvar-1) - factor);
for i=1:aqnvar-1 if optim_new(i,1)==0 optima(i,1)=(optim_new(i,1)); else end if optim_new(i,1) < 0 optima(i,1)=(optim_old(i,1)/10); else end if optim_new(i,1) > 0 optima(i,1)=(optim_new(i,1)); else end end
otherwise end
optim_old2=optima; optim_new=zeros(aqnvar,1);

421
switch FIXA
case 1 optim_old2 optim_new(2:aqnvar,1)=optim_old2; optim_new(varnum,1)=(fixvar); case 2 optim_new(1:aqnvar,1)=optim_old2; case 3 optim_new(1:aqnvar-1,1)=optim_old2 optim_new(aqnvar,1)=1;
otherwise end
error= norm(optim_new - optim_old) optim_old=optim_new; optim_old2=optim_old(1:aqnvar); optim_old=optim_old2; IEq(M,1)=ISS; Calc(M,1:aqnvar)=Calculo; Fix(M,1:aqnvar)=Fixed; ERR(M,1)=error; Kons(1:nspec,M)=log_K1; Optimum(1:aqnvar,M)=optim_old; Num(M,1)=niter; ICal(M,1)=ISS3;
if FIXA==3
Species=(SST*log10(optim_old)); else
Species=(SST*log10(optim_old)); end
for i=1:nspec
Spec(i,1)=10.^(Species(i,1)+log_K1(i)); end
zet=zeros(nspec,1); gamma=zeros(nspec,nvar);
for k=1:nspec
zet(k,1)=AQSPCHARGE(k,1)'; act_coef2(k,M)=10^(-(0.5115*(zet(k,1)^2))*((sqrt(IEq(M,1)))/(1+(sqrt(IEq(M,1))))- (0.3*IEq(M,1)))); end
Activ(1:nspec,M)=act_coef2(1:nspec,M).*Spec(1:nspec,1);
if Equil==1 CO2(M,1)=TOTCO2;
Car(M,1)=CO3; else
Car=0; CO2=0;
end
save results IEq Calc Fix ERR CO2 Kons Car Optimum Num Activ ICal end
end %%%%%%%%%%%%%%%%%%% END ALKALINITY SUBROUTINE %%%%%%%%%%%%%%%%%%%% end %%%%%%%%%%%%%%%%%%%% END ALKA MAIN SUBROUTINE %%%%%%%%%%%%%%%%%%%%

422
5. CALCULATION OF pH FROM E0, JH AND JOH VALUES (NEWTON-
RAPHSON METHOD)
Chapter 4 pH %%%%%%%%%%%%%%%%%%%% CALL pH MAIN SUBROUTINE %%%%%%%%%%%%%%%%%%%% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% %% pH function: Computes the pH values by the Newton-Raphson method using the Nerstian equation %% with accurately-calibrated electrode parameters (standard electromotive force, %% junction potential coefficients) as input %% %% 05/05/2005 Adrián Villegas-Jiménez %% Earth and Planetary Sciences, McGill University %% Montreal, CANADA %% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ function [results] =pH % %% Enter Experimental Data and Activity Coefficients E=['enter vertical vector of EMF values']; acoef=['enter vertical vector of activity coefficients for the H+ ion']; DATSIZE2=length(E); %%% Enter Parameters for the Combination pH glass electrode EM1='enter alkaline-end EMF' joh='enter alkaline-end EMF' EM2='enter acid-end EMF' jh='enter acid-end EMF' % Define maximum number of iterations MAXiter='enter maximum number of iterations'; range='enter number pH values to be determined with the alkaline-end electrode parameters'; % Evaluate function and compute derivatives for i=1:DATSIZE
activ=acoef(i); H=1e-13; % Guess free proton concentration (initial value) old=H; res=10; if i<range EM=EM1; else EM=EM2; end for k=1:MAXiter Y=-E(i,1)+(EM+(59.2*log10(H))+(jh*H)+(joh*(1e-14/(H*(activ^2))))); Y2=59.2*(1/(H*2.30258509299405))+jh+joh; Z(i,1)=(EM+(59.2*log10(H))+(jh*H)+(joh*(1e-14/(H*(activ^2))))); factor=(Y / Y2); new=(old - factor); old=new; H=new; results(i)=new; error(i,1)=Y; end end pH=-log10(results'.*acoef); save results pH error Z %%%%%%%%%%%%%%%%%%%%% END pH MAIN SUBROUTINE %%%%%%%%%%%%%%%%%%%

423
6. CHEMICAL EQUILIBRIUM SPECIATION INVOLVING ION-
EXCHANGE REACTIONS (NEWTON-RAPHSON METHOD)
Chapter 6 %%%%%%%%%%%%%%%%%%%%%% CALL EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%%%% EQUIL %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% INPUT FILE: EQUIL Aqueous Equilibrium Problem %% %% Definition of chemical equilibrium of the CaCO3(s)-KCl-H2O system %% %% %% 10/07/2007 Adrián Villegas-Jiménez %% Earth and Planetary Sciences, McGill University %% Montreal, CANADA %% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %%%%%%%%%%%%%%%%%% DEFINITION OF CHEMICAL EQUILIBRIUM %%%%%%%%%%%%%%%%% %Names of components format short aqcomp=[sym('H'),sym('Ca'),sym('K'),sym('Cl'),sym('CaCO3surf'),sym('CaCO3s')]; component=aqcomp; aqnvar=length(aqcomp)'; nvar=length(component)'; aqcomp_charge=[1;2;1;-1;0;0]; comp_charge=aqcomp_charge; comp_charge2=comp_charge'; aqcomp_charge=comp_charge; %Names of Species aqspecies=[sym('H'); sym('Ca'); sym('K'); sym('Cl'); sym('CO3'); sym('OH'); sym('H2CO3'); sym('HCO3'); sym('CaCO3'); sym('CaHCO3'); sym('CaOH'); sym('CaCl'); sym('KCl'); sym('CaCO3surf'); sym('H2CO3surf')]; naqspecies=length(aqspecies); AQSST1=[1,0,0,0,0,0; 0,1,0,0,0,0; 0,0,1,0,0,0; 0,0,0,1,0,0; 0,-1,0,0,0,1; -1,0,0,0,0,0; 2,-1,0,0,0,1; 1,-1,0,0,0,1; 0,0,0,0,0,1; 1,0,0,0,0,1; -1,1,0,0,0,0; 0,1,0,1,0,0; 0,0,1,1,0,0; 0,0,0,0,1,0; 2,-1,0,0,1,0];

424
SST2=[ 1,0,0,0,0,0; 0,1,0,0,0,0; 0,0,1,0,0,0; 0,0,0,1,0,0; 0,-1,0,0,0,1; -1,0,0,0,0,0; 2,-1,0,0,0,1; 1,-1,0,0,0,1; 0,0,0,0,0,1; 1,0,0,0,0,1; -1,1,0,0,0,0; 0,1,0,1,0,0; 0,0,1,1,0,0; 0,1,0,0,1,0; 2,0,0,0,1,0]; [taille]=length(SST2); Ca_Diss=zeros(taille); CO2_MBSTT=zeros(taille); species=aqspecies; nspec=length(species); Number_Species=nspec; num_alk=zeros(nspec,1) num_alk2=zeros(nspec,1) for i=1:nspec if AQSST1(i,1)< 0 num_alk(i,1)=AQSST1(i,1); else num_alk2(i,1)=AQSST1(i,1); end end %Stoichiometry Aqueous=AQSST1; AQSST=AQSST1; SST=AQSST; size_SST=size(SST) AQSST1=SST; % Thermodynamic or Apparent Formation Constants % log_K=[0; 0; 0; 0; -8.48; -14; 8.2; 1.85; -5.28; 3.11; -12.85; 0.2; -0.5; 0; „enter log10(Kexc)‟]; % Solid phases Kps=-8.48; Fix_solid=1; Solid_Act=1; log_KC=log_K;

425
%Charges of Species AQSPCHARGE=[ 1; 2; 1; -1; -2; -1; 0; -1; 0; 1; 1; 1; 0; 0; 0]; MAT_CHARGE2=AQSPCHARGE; save EQUILIBRIA % Total Analytical Concentrations of Components (Enter as follows: AC=TOTH, TOTK, TOTCl, TOTCO3) and specify a % value of “1” for the last component in all cases. % aqcomp=[sym('H'),sym('Ca'),sym('K'),sym('Cl'),sym('CaCO3surf'),sym('CaCO3s')] AC=[„enter experimental data either as a vertical vector or as a matrix for serial data„]; TOT=AC; DATSIZE=length(AC); varnum=5; FIXA=2; %Serial Data % Define initial ionic strength IS=[„enter ionic strength either as a vertical vector or as a matrix for serial data„]; ISS=IS; %Define fixed variable values (if applicable) pH_1=[„enter pH data as a vertical vector or as a matrix for serial data„]; Ca_Fix=pH_1; Calcium=1; CO3_Fix=Ca_Fix; nfix=2; fixact=horzcat(pH_1,Ca_Fix); pH_1=log10(pH_1); Ca_Fix=log10(Ca_Fix); CO3_Fix=log10(CO3_Fix); fixvar=5; pH=-pH_1; fixnumber=1; varnum2=2; varnum3=5; pH_Fix=2; pHeq=pH; act_coef2=zeros(DATSIZE,1); fixcon=zeros(DATSIZE,1); z=1; %Transformation of pH to molar proton concentrations using the Davies Equation for i=1:DATSIZE act_coef2(i,1)=10.^(-(0.5115*(1.^2))*((sqrt(IS(i))/(1+(sqrt(IS(i)))))-(0.3*IS(i)))); fixcon(i,1)=-log10(10.^(-pH(i))./act_coef2(i,1)); end fixcon=fixcon'; H=fixcon; fixed=fixcon; varnum=5; fixvariables=1 %fixnow fixvariables=2 %TOT Calcite=1; save EQUIL_DATA %%%%%%%%%%%%%%%%%%% CLOSE AND SAVE EQUIL SUBROUTINE %%%%%%%%%%%%%%%%%%

426
%%%%%%%%%%%%%%%%%%%% CALL ION_SPEC SUBROUTINE %%%%%%%%%%%%%%%%%%%% ION_SPEC %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% INPUT FILE: ION_SPEC Aqueous Equilibrium Problem %% %% File "ION_SPEC" uses the function "TOT" to estimate the equilibrium %% concentrations of dissolved and surface species %% %% 10/07/2007 Adrián Villegas-Jiménez %% Earth and Planetary Sciences, McGill University %% Montreal, CANADA %% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ load EQUIL_DATA comp=aqcomp; aqcomp2=aqcomp'; ident=zeros(aqnvar,1);
for i=1:aqnvar ident(i,1)=i;
end comp=comp'; Master_Species=horzcat(ident,comp); TOT=zeros(aqnvar,1); ident=zeros(aqnvar,1); TP=input('Do you want to enter data? (1 for YES, 2 for NO) >'); switch TP
case 1 compvar=input('Do you want to perform a titration using one of the master species? (1 for YES, 2 for NO) >');
if compvar==1 Type=2;
minvar=input('Enter the minimum value for titrating master specie (-log units) \n>'); maxvar=input('Enter the maximum value for titrating master specie (-log units) \n >');
if maxvar < minvar fprintf('ERROR! : Specified minimum concentration is higher than maximum value')
break else end
intervar=input('Enter the concentration intervals for titration (-log units)\n >');
DATSIZE=(maxvar-minvar)/intervar; fixcomp=minvar; fixed=zeros(DATSIZE,1);
for i=1:DATSIZE fixcomp=fixcomp+intervar; fixed(i,1)=fixcomp; end
if intervar > maxvar fprintf('ERROR! : Titration interval higher than maximum value assigned to titrating
%specie')
break else end fixnumber=input('What master species do you want to choose as the titrant (choose a
%number)? \n'); fixname=Master_Species(fixnumber,2); varnum=fixnumber; z=comp_charge(fixnumber,1);
for i=1:nvar var=comp(i,1)
if i~=fixnumber

427
TOT(i,1)=input('Enter now the total analytical concentrations of master species >');
else TOT(fixnumber,1)=minvar;
end end
fixed=vertcat(TOT(fixnumber,1),fixed); TOT=TOT'; pH=TOT(1,1); AC=zeros(DATSIZE+1,nvar);
for i=1:DATSIZE+1
for j=1:nvar if j==fixnumber fixed(i,1); AC(i,fixnumber)=fixed(i,1); else AC(i,j)=TOT(1,j); end end
end pH=AC(:,1);
else
z=1; fixnumber alk=1; varnum=fixnumber; Type=1; DATSIZE=1; pH=input('Enter the equilibrium pH >\n') TOT(1,1)=pH fixed=pH;
for i=1:nvar fprintf('Aqueous Component \n') var=comp(i,1) TOT(i,1)=input('Enter now the total analytical concentrations for the given speciation
problem >');
end
AC=TOT; TOT=TOT';
end
ISS=input('Enter the estimated value of ionic strength >')
for i=1:DATSIZE IS(i,1)=ISS; end
case 2 Type=3; load EQUIL_DATA TOT=AC; fixnumber=varnum; otherwise end Equil=input('Is this an alkalinity problem? (if yes press 1, otherwise press any other key >'); if Eq==1 Equil=1; else Equil=2; end Initial_Comp=zeros(1,nvar-1); Y=input('Are initial concentrations different than total analytical concentrations? (if yes press 1, otherwise press any other key >'); if Y==1 F=input('Enter now the factor by which you want to divide the total analytical concentrations >'); else F=1; end

428
initials=zeros(1,nvar-1); if Equil~=1
initials=zeros(DATSIZE,nvar-1); for m=1:DATSIZE for j=1:nvar-1 if j==1
k=2; else end
initials(m,j)=TOT(m,k)/F; k=k+1; end end else end initials=zeros(DATSIZE,aqnvar); for m=1:DATSIZE for j=1:aqnvar if j~=varnum initials(m,j)=abs(TOT(m,j))/F; else initials(m,varnum)=abs(TOT(m,j))/F; %0 end if initials(m,j)==0 initials(m,j)=1e-4; else end end end Scores3=zeros(DATSIZE,nvar); Residuals=zeros(DATSIZE,nvar); Species=zeros(DATSIZE,nspec); IonicS=zeros(DATSIZE,1); fixcon=zeros(DATSIZE,1); fixa=input('Fixed activities of at least 1 component? (if yes press 1, otherwise press any other key >'); I_Corr=input('Include ionic strength corrections? (if yes press 1, otherwise press any other key >'); %Transformation of pH or master variable's acitvity to molar proton concentrations using the Davies Equation AI=input('Known activities press 1 known total concentrations press any other key >'); if AI==1 Act_Fix=1; else Act_Fix=2; end initials=zeros(DATSIZE,aqnvar); size(initials); for m=1:DATSIZE for j=1:aqnvar if j~=varnum initials(m,j)=abs(TOT(m,j))/F; else initials(m,varnum)=abs(TOT(m,j))/F; end
if initials(m,j)==0 initials(m,j)=1e-4; else end
end end Scores3=zeros(DATSIZE,nvar); Residuals=zeros(DATSIZE,nvar); Species=zeros(DATSIZE,nspec); IonicS=zeros(DATSIZE,1); fixcon=zeros(DATSIZE,1);

429
if I_Corr==1 for i=1:DATSIZE
z=aqcomp_charge(varnum,1); act_coef2(i,1)=10.^(-(0.5115*(z.^2))*((sqrt(IS(i)))/(1+(sqrt(IS(i))))-(0.3*IS(i))));
if TP==1 & Type==2 break else
fixcon(i,varnum)=-log10(10.^(-fixed(i))./act_coef2(i,1)); end end else end Spc=0; AC2=zeros(aqnvar,DATSIZE); for i=1:DATSIZE for j=1:aqnvar if j~=varnum AC2(j,i)=initials(i,j); else AC2(varnum,i)=10^(-fixcon(i,1)); end end Spc(1,1:naqspecies)=10.^((SST*log10(AC2(1:aqnvar,i)))+log_K)'; FI(i,1)=0.5*(Spc(1,1:naqspecies)*(MAT_CHARGE2.^2)); Ratio(i,1)=FI(i,1)/0.5; end for i=DATSIZE
[F]=min(TOT(i,1)); end OPTIONS(1) = 0; OPTIONS(2) = 1e-5; OPTIONS(3) = 1e-10; num_iter=0; %%%%%%%%%%%%%%%%%%%% CALL TOT SUBROUTINE %%%%%%%%%%%%%%%%%%%% if Act_Fix==1
optimum=raphson2(initials,fixcon,fixnumber,fixa,num_iter,FI,varnum,AC2,Equil,Ratio,I_Corr); else optimum=tot(initials,fixcon,fixnumber,fixa,num_iter,FI,varnum,AC2,Equil,Ratio,I_Corr); function [optima] =tot(initials,fixcon,fixnumber,fixa,num_iter,FI,varnum,AC2,Equil,Ratio,I_Corr); %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ %% %% TOT computes jacobians based on the Newton-Raphson and Gauss-Elimination method %% %% %% 10/07/2007 Adrián Villegas-Jiménez %% Earth and Planetary Sciences, McGill University %% Montreal, CANADA %% %%^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ load EQUIL_DATA format long
if num_iter==0 results=initials;
else end num_iter=num_iter+1; Ratio=FI(1,1)/0.5; for M=1:DATSIZE

430
maxiter=10; niter=0; h=0; for i=1:aqnvar
h=h+1; optim_old(i,1)=results(M,i); h=h-1;
end counter=0; xtol = 1000*eps; ftol = 1e6*eps; error = 2*xtol; ISS=IS; if num_iter==1 & pH==1
IEq=zeros(a,1); Activ=zeros(nvar,a); Species=zeros(nspec,a); else end xtol = 10*eps; ftol = 1e6*eps; error = 1e10; ISS=ISS(M,1); while error>1e-14
counter=0+1; niter=niter+1; % Transformation of molar concentrations to activities using the Davies Equation gamma=zeros(nspec,nvar);
for k=1:nspec for j=1:aqnvar z=comp_charge2(1,j); act_coef2=(-(0.5115*(z^2))*((sqrt(ISS))/(1+(sqrt(ISS)))-(0.3*ISS))); gamma(k,j)=(act_coef2*SST(k,j)); end end if I_Corr==1 if FIXA==3 act_coef_H=10.^(-(0.5115*(z^2))*((sqrt(ISS)/(1+(sqrt(ISS))))-(0.3*ISS))); fixvar=((10^(pH_1(M,1)))./act_coef_H); else end for i=1:nspec for j=1:nvar if gamma(i,j)==1 gamma(i,j)=0; else end end end Korr1=(sum(gamma'))'; Korr2=zeros(nspec,1); for i=1:nspec if MAT_CHARGE2(i,1)~=0 Korr2(i,1)=(-(0.5115*(MAT_CHARGE2(i,1)^2))*((sqrt(ISS)/(1+(sqrt(ISS))))-(0.3*ISS))); else end end
log_K1=(log_K+Korr1)-Korr2; %corrected log K for ionic strength

431
else fixvar=10^(pH(M,1));
log_K1=log_K; end optim_old2=optim_old;
% Mass Balance Equations (Minimum Energy Condition) optim_old3=log10(optim_old); Sp_Conc2=(SST*optim_old3); for i=1:nspec
Sp_Conc5(i,1)=10.^(Sp_Conc2(i,1)+log_K1(i)); end
% Estimate ionic strength Sp_Conc4=(Sp_Conc5');
if niter<0.9*maxiter
ISS2=ISS; ISS=ISS2;
else ISS2=0.5*(Sp_Conc4*(MAT_CHARGE2.^2)); % update ionic strength Ionic_Strength=ISS2; ISS=ISS2;
end MB=(Sp_Conc4*SST); Mass=MB; Spec=Sp_Conc4';
%Definition of Charge Balance (Electroneutrality Condition) Electro_Neut2=(Sp_Conc4*MAT_CHARGE2);
% --------------------------------------------------------------------------------------------------------------------------------------------------------------- % ----------------------------------------------------- Calculate Jacobian Matrix ---------------------------------------------------------- % --------------------------------------------------------------------------------------------------------------------------------------------------------------- switch FIXA
case 1 for pivote=2:aqnvar for i=1:nspec for j=2:aqnvar if j==pivote k=pivote; Jacob(i,j-1)=(Sp_Conc5(i,1)*SST(i,pivote)*SST(i,k))/(Sp_Conc5(k,1)); else k=j; end Jacob(i,j-1)=(Sp_Conc5(i,1)*SST(i,pivote)*SST(i,k))/(Sp_Conc5(k,1)); end end
Deriva(pivote-1,1:nvar-1)=sum(Jacob); end
case 2
for pivote=1:nvar-1 for i=1:nspec
for j=1:nvar-1
if j==pivote k=pivote; Jacob(i,j)=(Sp_Conc5(i,1)*SST(i,pivote)*SST(i,k))/(Sp_Conc5(k,1)); else

432
k=j; Jacob(i,j)=(Sp_Conc5(i,1)*SST(i,pivote)*SST(i,k))/(Sp_Conc5(k,1)); end end end Deriva(pivote,1:nvar-1)=sum(Jacob); end
case 3 for pivote=2:aqnvar-1 for i=1:nspec for j=2:aqnvar-1 if j==pivote k=pivote; else k=j; end Jacob(i,j-1)=(Sp_Conc5(i,1)*SST(i,pivote)*SST(i,k))/(Sp_Conc5(k,1)); end end
Deriva(pivote-1,1:aqnvar-2)=sum(Jacob); end otherwise end % solves the system of linear equations (Gauss-Elimination Method) Der=Deriva; optim_old=10.^(optim_old3); Calculo=Mass(1:aqnvar); Fixed=AC(M,1:aqnvar); if FIXA~=3 Fixed(1,varnum)=Calculo(1,varnum); else Fixed(varnum,1)=Calculo(varnum,1); end switch FIXA case 1 residuos=(Mass(2:aqnvar) - AC(M,2:aqnvar)); case 2 BB=(Mass(1:aqnvar-1)) AA=AC(M,1:aqnvar-1) residuos=(Mass(1:aqnvar-1) - AC(M,1:aqnvar-1)); case 3 residuos=(Mass(2:aqnvar-1) - AC(M,2:aqnvar-1)); otherwise end residuos=residuos'; factor=Der / residuos;
if Equil==2 H=sum((Sp_Conc5(1:nspec)).*(-num_alk2)); TOTCO2=sum((Sp_Conc5(1:nspec)).*(num_alk)); H2CO3=(AC(M,1)+H)/(TOTCO2/Sp_Conc5(nvar)); optima=optim_old; optima(aqnvar)=H2CO3; optima=log10(optima);

433
Sp_Conc6=(SST*optima);
for i=1:nspec Sp_Conc7(i,1)=10.^(Sp_Conc6(i,1)+log_K1(i)); end CO3=Sp_Conc7(nspec,1); TOTCO2=sum(Sp_Conc7.*(SST(1:nspec,aqnvar))); AC(M,aqnvar)=TOTCO2; else end switch FIXA case 1 optim_new=(optim_old(2:aqnvar) - factor);
for i=1:aqnvar-1
if optim_new(i,1)==0 optima(i,1)=log10(AC(i+1,num_iter)); else end
if optim_new(i,1) < 0 optima(i,1)=log10(optim_old2(i,1)/10); else end
if optim_new(i,1) > 0 optima(i,1)=log10(optim_new(i,1)); else end end case 2 optim_old4=optim_old; optim_new=(optim_old(1:aqnvar-1) - factor);
for i=1:aqnvar-1 if optim_new(i,1)==0 optim_old4(i,1)=optim_new(i,1);
else end if optim_new(i,1) < 0 optim_old4(i,1)=optim_old(i,1)/10; else end
if optim_new(i,1) > 0 optim_old4(i,1)=optim_new(i,1); else end end
case 3
optim_old4=optim_old; optim_new=(optim_old(2:aqnvar-1) - factor)
for i=2:aqnvar-1 if optim_new(i-1,1)==0 optim_old4(i,1)=(optim_new(i-1,1)); else end
if optim_new(i-1,1) < 0 optim_old4(i,1)=(optim_old(i-1,1)/10); else end
if optim_new(i-1,1) > 0
optim_old4(i,1)=(optim_new(i-1,1)); else end
end otherwise end optim_old2=optim_old4; optim_new=zeros(aqnvar,1); optim_new=optim_old2;

434
error= norm(optim_new - optim_old); optim_old=optim_new; TOTCO2=sum(Spec.*CO2_MBSTT); optim_old2=optim_old(1:aqnvar); optim_old=optim_old2; TOTCa=sum(Sp_Conc5.*CaDiss); TOTCO2=sum(Spec.*CaDiss); IEq(M,1)=ISS; TOTCal(M,1)=TOTCa; TOTCar(M,1)=TOTCO2; Calc(M,1:aqnvar)=Calculo; Fix(M,1:aqnvar)=Fixed; ERR(M,1)=error; Kons(1:nspec,M)=log_K1; Optimum(1:aqnvar,M)=optim_old; Num(M,1)=niter;
if FIXA==3 Species=(SST*log10(optim_old));
else Species=(SST*log10(optim_old));
end
for i=1:nspec Spec(i,1)=10.^(Species(i,1)+log_K1(i));
end
zet=zeros(nspec,1); gamma=zeros(nspec,nvar);
for k=1:nspec zet(k,1)=AQSPCHARGE(k,1)'; act_coef2(k,M)=10^(-(0.5115*(zet(k,1)^2))*((sqrt(IEq(M,1)))/(1+(sqrt(IEq(M,1))))-(0.3*IEq(M,1))));
end
Activ(1:nspec,M)=act_coef2(1:nspec,M).*Spec(1:nspec,1); Conc(1:nspec,M)=log10(Spec(1:nspec,1)); Especies=Spec(1:nspec,1)'; Car=0; CO2=0;
save results IEq Calc Fix ERR CO2 Kons Conc Car Optimum Num Activ TOTCal TOTCar
end end end %%%%%%%%%%%%%%%%%%%%%% END TOT SUBROUTINE %%%%%%%%%%%%%%%%%%%%%%
fprintf('\n'); fprintf(' \t \t \tPROBLEM CONVERGED! Type: "load results" to see output data \n'); fprintf('\n'); %%%%%%%%%%%%%%%%%%%% END ION_SPEC SUBROUTINE %%%%%%%%%%%%%%%%%%%%





























