Embed Size (px)
Citation preview

Optimering i organisk syntes; Betingelser- System-Syntesvägar
av
Lars Hansson
Akademisk avhandling
Som med tillstånd av rektorsämbetet vid Umeå universitet, för erhållande av filosofie doktorsexamen vid matematisk- naturvetenskaplig fakultet i Umeå, framlägges till offentlig granskning vid kemiska institutionen, hörsal B, Lu 0, torsdagen den 31 maj 1990, kl. 10.00.
Organisk kemi Umeå universitet Umeå 1990


Optimering i organisk syntes; Betingelser-System-Syntesvägar
Lars Hansson 1990
Avdelningen för organisk kemi Umeå universitet S-901 87 Umeå

Tryckt av Umeå Universitets tryckeri Umeå 1990
ISBN 91-7174-488-6

Title:
Author:
Abstract:
Keywords:
Optimization in Organic Synthesis; Conditions, Reaction systems, Synthetic Routes (Summary in Swedish, Papers in English)
Lars Hansson, Department of Organic Chemistry, University of Umeå, Umeå, S-901 87 Sweden
This thesis deals with different optimization problems encountered in organic synthesis. The use of response surface, sequential simplex and PLS techniques, for simultanious optimization of yield and suppression of side reactions is investigated. This is illustrated by an example of enamine synthesis, were a side reaction was a serious problem.
The problem of efficient screening to find suitable catalysts and solvents in new reactions is also investigated. Here, the use of principal properties as selection criterion, is demonstrated with a new process for the silylation of a,ß-unsaturated ketones. The extension of the new method to bis silylation of 1,2- and 1,3-diketones is demonstrated.
The total synthesis of (±)-geosmin is investigated by an approach aimed to reduce the number of necessary steps involved. The suggested strategy, is to find compatible solvents through several transformations in the sequence to accomplish one-pot multistep reactions. In this context an improved method for the preparation of 1,10-dimethyl-l(9)-octalone-2 was established. Comparison with previously reported total syntheses of (±)-geosmin was done.
Optimization, Response surface method, PLS method, Silylation, Enolether, Unsaturated ketones, Diketones, Geosmin synthesis, One- pot procedure
ISBN: 91-7174-488-6


Innehåll
1 Förteckning över de uppsatser som ingår iavhandlingen 9
2 Inledning 112.1 Experimentella betingelser 112.2 Reaktionssystem 112.3 Syntesvägar 122.4 Kort summering av 2.1-3 13
3 Optimering av experimentella betingelser närparasitreaktioner stör. 15
3.1 Optimering av enaminsyntes med responsytemodell 153.2 Optimering av enaminsyntes med simplex och
sammanvägda responser 163.3 Optimering av enaminsyntes med PLS 183.4 Jämförelse av metoderna under 3.1-3 203.5 Slutsatser 21
4 Sållning för att etablera nya reaktionssystem 234.1 Syntes av silyloxydiener från a,ß-omättade ketoner 234.2 Urval av lösningsmedel och katalysatorer 244.3 Sållningsexperiment 264.4 Optimala betingelser för systemet 284.5 Syntes av 1,2- och 1,3-bis-silyloxydiener från 1,2- och
1,3-diketoner 314.6 Slutsatser 33
5 Effektiviscring av syntesvägar via "one-pot" förfarande 355.1.1 Syntes av l,10-dimetyl-l(9)-oktalon-2 355.1.2 Regioselektivitet vid alkylering av 2-metylcyklo-
hexanon 375.1.3 Epoxidering av l,10-dimetyl-l(9)-oktalon-2 38
7

5.1.4 Diastereoselektivitet vid epoxidering av 1,10-dimetyl-l(9)-oktalon-2 38
5.2 Urval av möjliga förfaranden för "one-pot" syntes av (±)-geosmin från l,10-dimetyl-l(9)-oktalon-2 a-epoxid 39
5.3 Sållning av reagens för derivatisering av den intermediära epoxyalkoxiden 41
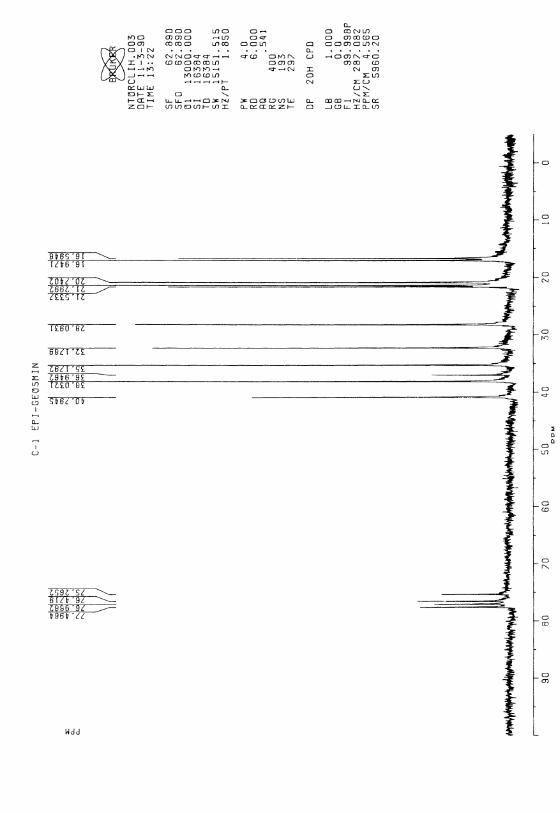
5.4 Jämförelse med tidigare totalsynteser av geosmin 425.5 Syntes av C-l epimeren av (±)-geosmin 435.6 Slutsatser 44
6 Slutord 45
7 Referenser 47
8 Appendix 53
Numreringen av kemiska föreningar börjar om med (1) under varje ny huvudrubrik och under 4.5.
8

I Förteckning över de uppsatser som ingår i avhandlingen
Denna avhandling är en sammanfattning av fem uppsatser, vilka hädanefter kommer att refereras till med romerska siffror.
I. Rolf Carlson, Lars Hansson and Torbjörn Lundstedt "Optimization in Organic Synthesis. Strategies When the Desired Reaction is Accompanied by Parasitic Side Reactions. An Example with Enamine Synthesis."Acta Chem. Scand. B 40 (1986) 444 )
II Lars Hansson and Rolf Carlson"Efficient Synthesis of Silyloxy Dienes from a,ß-Unsaturated Ketones."Acta Chem. Scand. 43 (1988) 188
III Lars Hansson and Rolf Carlson"Synthesis of Bis-Silyloxydienes from Diketones"Acta Chem. Scand. 43 (1989) 304
IV Lars Hansson, Rolf Carlson and Anna-Lena Sjöberg "Synthesis of (±)-Geosmin. Part 1; On the Synthesis and Epoxidation of l,10-dimethyl-l(9)-octalone-2"Acta Chem. Scand. XX (1990) XXX Insänt för publicering.
V Lars Hansson and Rolf Carlson"Synthesis of (±)-Geosmin. Part 2; A One-Pot Four-Step Conversion of l,10-dimethyl-l(9)-octalone-2 a-epoxide to (±)-Geosmin"Acta Chem. Scand. XX (1990) XXX Insänt för publicering.
9

10

2 Inledning
2.1 Experimentella betingelserSyntes av nya kemiska föreningar utgör en omfattande del av arbetet i organisk kemi. Anledningarna till att framställa nya substanser är många. De kan t.ex. vara intressanta ur farmakologisk synpunkt, som referensmaterial eller som startmaterial i tillverkningsprocesser. När en ny kemisk förening ska syntetiseras utnyttjas vanligen någon känd metod, men i många fall visar det sig emellertid, att tillämpningen av kända metoder på nya substrat inte ger acceptabla resultat. Bristerna i resultatet består oftast i låga kemiska utbyten och/eller i att svårseparerade biprodukter förorenar den önskade produkten. Metoder för optimering (faktorförsök, responsytemodeller, stegvis simplex-optimering) av de experimentella betingelserna under vilka en syntesreaktion genomförs är väletablerade tekniker [1]. Dessa metoder har tidigare huvudsakligen använts för optimering av en respons, kemiskt utbyte. Optimering med statistiska metoder av flera responser samtidigt har beskrivits för en del kromatografiska applikationer och FIA (flow injection analysis) [2]. Optimering av syntesreaktioner - med hjälp av sådana metoder - för att maximera utbytet av önskad produkt samtidigt med undertryckande av parasitreaktioner har emellertid inte tidigare beskrivits. En sådan tillämpning diskuteras och exemplifieras med optimeringen av en enaminsyntes som stördes av en parasitreaktion [I].
2.2 ReaktionssystemI de fall där en önskad transformation inte låter sig genomföras med tidigare beskrivna metoder, krävs naturligtvis nya metoder. Kostnadsaspekter och säkerhetsaspekter kan utgöra andra skäl att söka nya eller modifierade metoder, även om de befintliga metoderna ger goda resultat ur teknisk/kemisk synvinkel. En hypotes om en ny reaktion ligger oftast till grund för en ny metod. När en sådan hypotetisk reaktion skall provas i praktiken krävs effektiva urvalsmetoder för såväl lösningsmedel [3] som katalysatorer [4].
11

Utvcklingen av ett nytt reaktionssystem för silylering av a,ß-omättade ketoner åskådliggör, hur ett sådant urvalsförfarande kan utföras[II] med hjälp av principalegenskaper [5]. Det funna reaktionssystemets användning för bissilylering av 1,2- och 1,3-diketoner diskuteras också [III].
2.3 SyntesvägarOptimering av syntesvägar är till stor del en fråga om att hitta korta reaktionssekvenser mellan tillgängliga startmaterial och målstrukturerna. Detta är ett område som har undersökts och utvecklats av framför allt Corey [6] och Hendrickson [7], Datoriserade sökrutiner, som utför retrosyntetiska dissektioner av målstrukturernas kolskelett, används i bägge fallen [8]. Detta kombineras med kompilering av kända kemiska reaktioner ur en databas, med de synton som retrosyntesen givit upphov till. Hendricksons program söker dessutom funktionalitetsoptimala vägar, d.v.s. vägar där varje enskilt steg i sekvensen, där kolskelettet konstrueras också ska ge upphov till de funktionaliteter som behövs för nästa konstruktionssteg eller är del av den slutliga strukturen. En annan datorbaserad strategi för undersökning av syntesvägar, som inte baseras på kunskap om kända kemiska reaktioner, har utvecklats av Ugi [9]. Metoden bygger på en topologisk beskrivning av startmaterial och målstruktur. Gemensamt för dessa datorstrategier är dock att de saknar förmåga att identifiera, om två eller flera på varandra följande reaktioner kan utföras, utan att de intermediära produkterna isoleras ("one-pot" reaktion). Möjligheten att utföra sådana reaktioner är till stor del beroende på, om man kan finna lösningsmedel, som är kompatibla med de reagens, som används i de olika delstegen i sekvensen. Principen illustreras med en totalsyntes av en naturprodukt, där några olika "one-pot" förfaranden undersöktes [IV,V],
12

2.4 Kort summering 2.1-3I avhandlingen diskuteras optimering på tre olika "nivåer". Den första "nivån" rör metoder att optimera de experimentella betingelserna för en specifik kemisk reaktion. I de flesta kemiska reaktioner kan samverkanseffekter mellan olika styrvariabler förväntas [10]. Detta innebär att systemen ofta är för komplexa för att effektivt kunna undersökas genom experiment, där en variabel i taget varieras. Komplexiteten är dock inte större, än att olika statistisk/matematiska modeller av systemen kan ge mycket värdefull information.
Den andra "nivån" rör hjälpmedel för strukturerat urval av testsytem, när ett nytt eller modifierat reaktionssystem ska utvecklas. Detta är ett problem av högre komplexitet, där de använda hjälpmedlen är av kvalitativ, snarare än av kvantitativ natur.
Den tredje "nivån" behandlar ett ännu komplexare problem, optimering av reaktionssekvenser, där den beskrivna principen utgörs av identifiering av kompabilitet mellan lösningsmedel och reagens över hela syntetiska sekvenser. Detta är en kvalitativ metod som är väl lämpad för kompilering av information med datorunderstöd och skulle kunna vara ett komplement till de datorbaserade strategier som nämnts ovan.
13

14
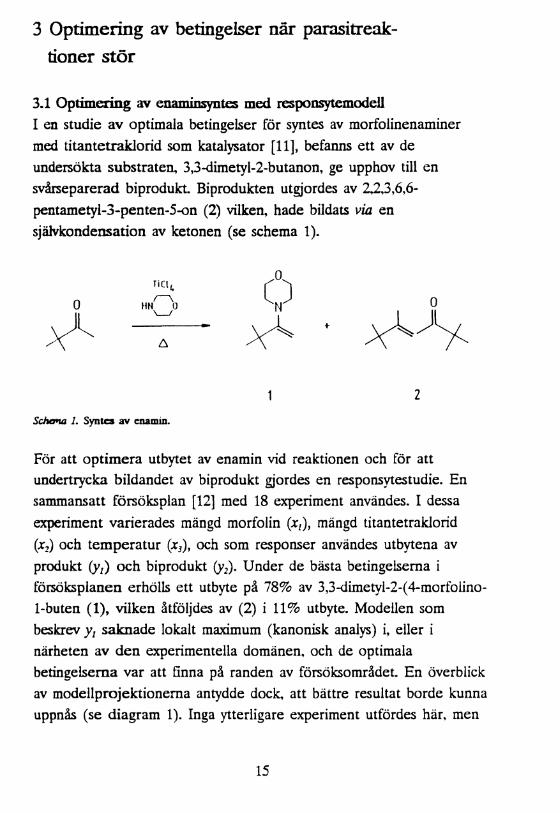
3 Optimering av betingelser när parasitreaktioner stör
3.1 Optimering av enaminsyntes med responsytemodell I en studie av optimala betingelser för syntes av morfolinenaminer med titantetraklorid som katalysator [11], befanns ett av de undersökta Substraten, 3,3-dimetyl-2-butanon, ge upphov till en svårseparerad biprodukt Biprodukten utgjordes av 2,2,3,6,6- pentametyl-3-penten-5-on (2) vilken, hade bildats via en självkondensation av ketonen (se schema 1).
A
1 2Schema 1. Syn tea av enamin.
För att optimera utbytet av enamin vid reaktionen och för att undertrycka bildandet av biprodukt gjordes en responsytestudie. En sammansatt försöksplan [12] med 18 experiment användes. I dessa experiment varierades mängd morfolin (xt), mängd titantetraklorid (x,) och temperatur (x5), och som responser användes utbytena av produkt (y;) och biprodukt (y2). Under de bästa betingelserna i försöksplanen erhölls ett utbyte på 78% av 3,3-dimetyl-2-(4-morfolino-1-buten (1), vilken åtföljdes av (2) i 11% utbyte. Modellen som beskrev yl saknade lokalt maximum (kanonisk analys) i, eller i närheten av den experimentella domänen, och de optimala betingelserna var att finna på randen av försöksområdet. En överblick av modellprojektionema antydde dock, att bättre resultat borde kunna uppnås (se diagram 1). Inga ytterligare experiment utfördes här, men
15
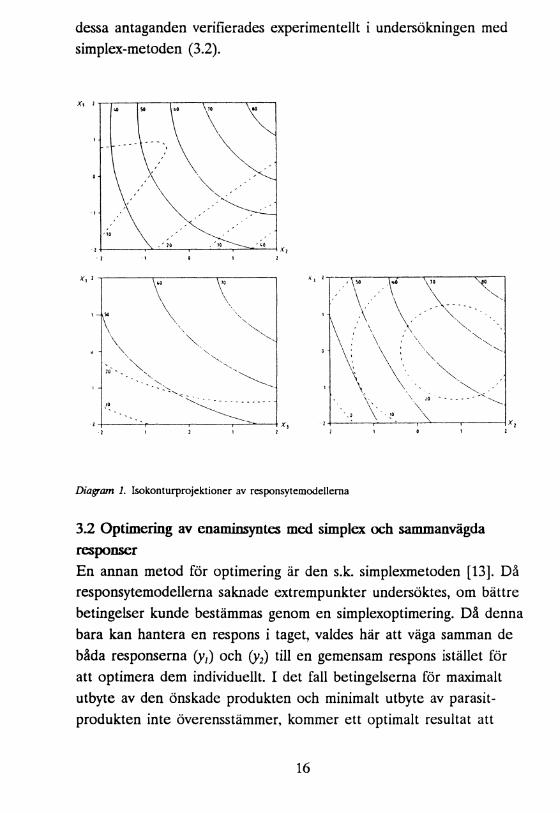
dessa antaganden verifierades experimentellt i undersökningen med simplex-metoden (3.2).
Diagram 1. Isokonturprojektioner av responsytemodellema
3.2 Optimering av enaminsyntes med simplex och sammanvägda responserEn annan metod för optimering är den s.k. simplexmetoden [13]. Då responsytemodellema saknade extrempunkter undersöktes, om bättre betingelser kunde bestämmas genom en simplexoptimering. Då denna bara kan hantera en respons i taget, valdes här att väga samman de båda responsema (y7) och (y2) till en gemensam respons istället för att optimera dem individuellt. I det fall betingelserna för maximalt utbyte av den önskade produkten och minimalt utbyte av parasitprodukten inte överensstämmer, kommer ett optimalt resultat att
16
utgöra en kompromiss mellan delresultaten. För att erhålla en sammanvägd respons, där betydelsen av de individuella responserna kan tillskrivas olika vikter, valdes Harringtons preferensfunktion’ D [14] för transformationen (se ekv.l och ekv.2).
D—(d!dy..d.n)lln (ekv.l)
d—expt-exptco+cyj (ekv.2)
D, den sammanlagda preferensen’ definieras som det geometriska medelvärdet av de individuella preferenserna dt och kan anta värden mellan 0 och 1. Med c0 och c, paramétreras de individuella funktionerna utifrån en valfri defintion av vad som anses vara ett bra. respektive dåligt resultat för de ifrågavarande responserna. Diagram 2 visar grafer av de individuella funktionerna med de parametrar som använts i enaminexemplet.
Diagram 1 dL och di som funktion av yL respektive yL
* Preferensfunktion är en fri översättning av desirability function.
17

Simuleringar baserade på responsfunktionerna från responsyte- optimeringen med D som en sammansatt responsvariabel gav en konvergerade simplex (21-32 exp.). Replikat med sex olika start- simplexar gav alla samma konvergenspunkt (£>=0,492-0,501). Den modifierade simplexmetoden enligt Neider och Mead [13b] användes vid dessa simuleringar.
Som ytterligare villkor för simplexoptimeringen tillskrevs alla föreslagna experiment med orimliga koordinater £>=0, d.v.s för x, <0,5 ekv., x2< 3.0 ekv. (mindre än stökiometriska mängder) och x3> 120 °C (högre än lösningsmedlets kokpunkt). Simuleringar med y, > 105% tillskrevs också £>=0. I koordinaterna för konvergenspunkten predikterades y; = 104% och y2=10,3% utifrån responsfunktionerna. Experimentellt erhölls utbyten på 86,5% respektive 7,8%, vilket är en förbättring jämfört med resutatet under 3.1. Den ytterligare information som erhölls genom simplex-optimeringen, ansågs dock otillräcklig. Därför vidtogs optimeringar med andra metoder (3.3).
3.3 Optimering av enam insyn tes med PLS PLS [15] är en metod som möjliggör bestämning av kvantitativa samband mellan två eller flera block av data i datamatriser, där en blockindelning gjorts. Genom att först beräkna en principalkom- ponentliknande modell för varje block kan sedan de kvantitativa sambanden mellan modellkomponenenterna i de olika blocken bestämmas. Som dataset i PLS-optimeringen utnyttjades här xh x2 och Xj men även xf och xpcj från experimenten i försöksplanen (3.1) i en matris (X) för att beskriva variationen i experimentella betingelser. I (X) inkluderades även motsvarande data från experimenten under (3.2). I en matris (Y) beskrevs variationen i de båda responsvariablerna yt och y2. PLS korrelation av (X) och (Y) resulterade i två signifikanta (korsvalidering) [15] komponenter i båda matriserna. I (Y) beskrevs 90,4% av den totala variansen i matrisen. Den erållna modellen användes sedan för prediktion av xh x2 och x3, näry; = 100% och y2=0%. Modellen predikterade en reaktionstem- peratur ^=132 °C, vilket är högre än kokpunkten för lösningsmedlet.
18

Förutsägelse av (Y) från (X) med x,=120 °C, men i övrigt som i den första prediktionen utfördes. Här erhölls en prediktion om y,=95,8% respektive y2=0,5%. Två experiment utförda med de i beräkningarna angivna koordinaterna gav y, =92,6%, y2=3,0%, respektive yt =94,3%, y2=2,0%. Diagram 3 nedan åskådliggör schematiskt principen för PLS-korrelationen.
r*-x» X
Diagram 3. Schematisk beskrivning av PLS-korrelationen.
19

3.4 Jämförelse av metoderna under 3.1-3 I tabell 1 jämförs de olika optimeringsmetoderna från 3.1-3.
Tabell 1. Jämförelse av optimeringsmetoder
Metod Predikterat utbyte(%) Funnet utbyte(%)
(1) (2) (1) (2)
3.1. - - 77,8 11,0
3.2. 104,0 10,3 86,5 7,8
3.3. 95,6 0,5 92,6 3,0
3.3 95,6 0,5 94,3 2,0
Prediktion av optimala betingelser för enaminsyntesen fungerade utmärkt med PLS-metoden. De experimentella resultaten stämmer väl med prediktionerna. Även om ingen explicit beräkning av optimala betingelser kunde göras utifrån responsytemodellen, gav projektionerna av responsfunktionerna information om, i vilka riktningar som bättre resultat kunde uppnås. Några experiment gjordes inte på dessa grunder, men experimenten under 3.2 och 3.3 verifierade riktigheten i gjorda antaganden om de undersökta variablernas inflytande. Därför blir jämförelsen i tabellen missvisande, när det gäller responsytemetodik. I PLS kan både (Y) predikteras från (X) och (X) från (Y). Prediktioner av (X) från (Y) kan inte åstadkommas med responsytemodeller, då (X) vanligen innehåller lier parametrar än (Y) och regression bara kan utföras med en respons i taget. PLS klarar också av att hantera dataset med fler variabler än experiment [15]. Vid simplex-simuleringarna kunde simplexcn med
20

hjälp av vissa vilkor tvingas att konvergera i den experimentella domänen. Metoden gav dock föga ytterligare information som inte redan erhållits från projektionerna av responsfunktionerna.
3.5 SlutsatserUtgångspunkten i alla optimeringsförfaranden av experimentella betingelser bör vara en statistisk försöksplan. Försök utförda i enlighet med sådana planer ger information som är lika användbar för beräkning av responsytemodeller som för behandling med PLS. Kombinationen statistisk försöksplan-PLS [16] har här visat sig vara ett mycket kraftfullt sätt att optimera en kemisk syntesreaktion.
21


4 Sållning för att etablera reaktionssystem
4.1 Syntes av trimetylsilyloxydiener från a,B-omättade ke toner Ett stort antal metoder för syntes av silylenoletrar från mättade ketoner har beskrivits i den kemiska litteraturen [17]. Metoderna för syntes av de motsvarande silyloxydienerna (silyldienoletrar) från a,ß-omättade ketoner är däremot få. House-metoden [18] (trimetyl- klorosilan, trietylamin i dimetyformamid) har använts för syntes av2-trimetylsilyloxy-l,3-butadien (lb) i ett något modifierat förfarande [19], men utbytet är måttligt. Katalytiska metoder, där zinkklorid eller natriumjodid används, har också beskrivits [20].En generell metod för silylering av både mättade och omättade ketoner är deprotonering av ketonen med litium diisopropylamid i ett aprotiskt lösningsmedel [17], följd av O-silylering genom behandling med trimetylklorosilan. Kostnadsskäl gör dock denna metod olämplig för arbete i större skala. De katalytiska metoderna är i vissa fall effektiva men har en del begränsningar [20, II].
För att undersöka möjligheten att andra katalysatorer och/eller lösningsmedel skulle kunna ge ett bättre reultat än de hittills beskrivna, gjordes ett sållningsexperiment där lösningsmedel och katalysatorer varierades (Schema 2). Urvalsförfarandet för dessa beskrivs under 4.2.
0 OTMSiösninqsm./kah
TMSCl/TEA
Schema 2 Syntes av silyloxydiener
23

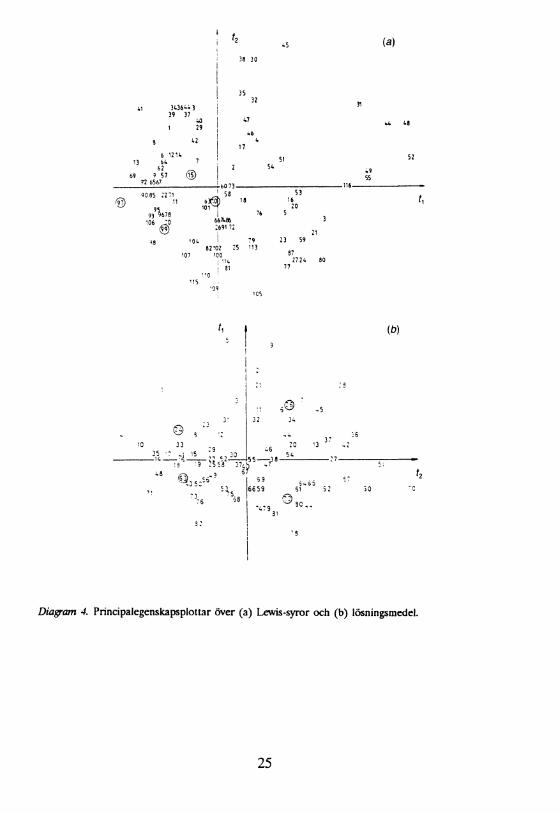
4.2 Urval av lösningsmedel och katalysatorerDet som ytterst avgör reaktiviteten hos kemiska system är de fysikalisk/kemiska egenskaperna hos de enskilda molekylerna på mikroskopisk nivå. Mått på dessa egenskaper (laddningsfördelningar, konformationer, steriska hinder) är inte tillgängliga genom direkta mätningar, vare sig på lösningsmedel eller på katalysatorer. Däremot kan ett stort antal makroskopiska egenskaper, som kan härledas till de mikroskopiska egenskaperna, kvantifieras med enkla metoder (kokpunkt, smältpunkt, dielektricitetskonstant). I arbeten av Carlson et al. har man genom att sammanställa ett stort antal fysikalisk/ kemiska parametrar för lösningsmedel [3] och katalysatorer [4] och sedan utföra principalkomponentanalys (PCA) [15, 21] på dessa data erhållit två dimensionella kartor (två PC i båda fallen), som beskriver den systematiska variationen i egenskaper hos dessa typer av kemikalier. Dessa kartor över principalegenskaper [22] är ett effektivt hjälpmedel vid urval, då god täckning av den möjliga variationen i fysikalisk/kemiska egenskaper önskas. Urvalen av lösningsmedel och Lewis-syror för sållningsförsök vid siloxylering visas med cirklar i diagram 4.
24
(a)
34.36^; 3 39 37 W
29
;2
69
6 121;13 6; 7
62 -v9 $7 (15)
?2 6567
32
Ll46
17
©9085 2271
93^678 106 JO ©
-*■60 73 — S3
,‘]9 ,s66X86 2691 72
; 955
82102 25 113107 100
(b)3
2 3
10n---Til -4.8
© ,‘J29
15 - C7 30
3;
13 © sV ÖT^p55 J*59
6659
© 3C , ,
1 8
Diagram 4. Principalegenskapsplottar över (a) Lewis-syror och (b) lösningsmedel.
25

En jämn spridning i urvalet av lösningsmedel syns i plotten, medan urvalet av katalysatorer gjordes i nedre vänstra kvadranten av plotten. Orsaken till att en liten spridning av katalysatorer valdes är vetskapen om att kraftigt oxofila Lewissyror till höger (dtantetraklorid, tenntetraklorid) klyver kisel-syre bindningen i silylenoletrar [23]. Syrorna i detta område är dessutom kraftigt polariserande och kunde förväntas ge problem med polymerisering av Substraten, vilket senare verifierades i sållningsexperimentet.
4.3 SållningsexperimentDet urspungliga målet för studien var att finna en effektiv metod för syntes av (lb) genom silylering av metylvinylketon (la). Därför valdes denna keton som testsubstrat vid sållningen. Som katalysatorer användes också natriumjodid och litiumbromid, då natriumjodid hade visat sig effektivt i liknande reaktioner [20b, 24] och litiumbromid för att åstadkomma variation av både metall och halogenid. Några data som möjliggör samma typ av urval som i 4.2 ovan finns inte tillgängliga för alkalimetallernas halogenider. Resultatet av sållningen visas i tabell 2.
26

Tabell 2. Utbyte(%) av (lb) med olika kombinationer av katalysatorer och lösningsmedel
Lösningsmedel' MeCN THF Bensen
Katalysator^
Cui 6,3 4,3 5,4
ZnCl2 8,0 5,7 5,9(12,5)“
Znl2 15,3 10,4 10,8
LiBr 0 61,6 6,1
Nal 5,4(5,3)" 8,2 0
a Reaktionen utförd under samma betingelser som anges i ref. 20a vid syntes av
1 -metoxy-3-trimetylsilyloxy-l ,3,-butadien. b Reaktionen utförd under samma betingelser som anges i ref. 24 vid syntes av
silylenoletrar från mättade ketoner.c Dimetylsulfoxid ingick ursprungligen i lösningsmedelssetet men uteslöts då
sulfoxiden deoxygenerades under betingelserna. d FeClj ingick ursprungligen i setet av katalysatorer men uteslöts då inget
utbyte av (lb) observerades i något av lösningsmedlen, p.g.a. polymerisering av substratet.
Sållningsexperimentet visar tydligt, att kombinationen litiumbromid/ tetrahydrofuran är ett utvecklingsbart system. För att bestämma optimala betingelser för reagenset litiumbromid/trimetylklorosilan/tri- etylamin i tetrahydrofuran undersöktes betingelserna med responsytemetodik, vilket beskrivs under 4.4.
27

4.4 Optimala betingelser för systemetFör att bestämma en optimal temperatur och en optimal mängd litiumbromid utfördes nio experiment i en försöksplan enligt Doehlert [25]. Den beräknade modellen uppvisade ett maximum i den undersökta domänen. En reaktionstemperatur på 40 °C och användandet av 2 ekvivalenter litiumbromid gav ett utbyte på 80 % i extrempunkten (se "trådnätsrepresentationen" av responsytemodellen i diagram 5). Mängderna av trimetylklorosilan och trietylamin som användes var 1,2 respektive 1,3 ekvivalenter.
Ekvlv. UBr
Temperatur *C
Diagram 5. Responsyta (Doehlert designen).
Då fullständig omsättning av startmaterialet inte erhölls under betingelserna ovan, undersöktes inflytandet av LiBr/TMSCl/TEA reagensets stökiometri vid 40 °C i en sammansatt försöksplan med 18 experiment. Den beräknade modellen saknade extrempunkt men predikterade kvantitativt utbyte inom den experimentella domänen. Vid ett stökiometriskt förhållande av LiBr/TMSCl/TEA/keton=2/1,5/1,5/1 erhölls (lb) i 98 % utbyte (gaskromatografiskt bestämt).
28
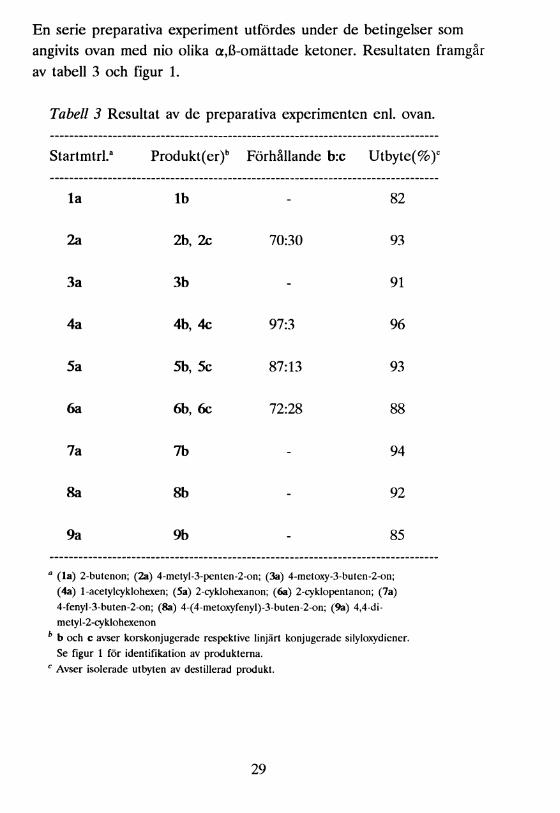
En serie preparativa experiment utfördes under de betingelser som angivits ovan med nio olika a,ß-omättade ketoner. Resultaten framgår av tabell 3 och figur 1.
Tabell 3 Resultat av de preparativa experimenten enl. ovan.
Startmtrl.3 Produkt(er)b Förhållande b:c Utbyte(%)°
la lb - 82
2a 2b, 2c 70:30 93
3a 3b - 91
4a 4b, 4c 97:3 96
5a 5b, 5c 87:13 93
6a 6b, 6c 72:28 88
7a 7b - 94
8a 8b - 92
9a 9b - 85
a (la) 2-butenon; (2a) 4-metyl-3-penten-2-on; (3a) 4-metoxy-3-buten-2-on; (4a) 1-acetylcyklohexen; (5a) 2-cyklohexanon; (6a) 2-cyklopentanon; (7a) 4-fenyl-3-buten-2-on; (8a) 4-(4-metoxyfenyl)-3-buten-2-on; (9a) 4,4-di- metyl-2-cyklohexenon
b b och c avser korskonjugerade respektive linjärt konjugerade silyloxydiener.Se figur 1 för identifikation av produkterna.
c Avser isolerade utbyten av destillerad produkt.
29
OTMS
OTMS
^b
OTMS
Uc
OTMS OTMS
5b
OTMS
6b
OTMS
6c
OTMS
OTMS OTMS
Jör^8b 9b
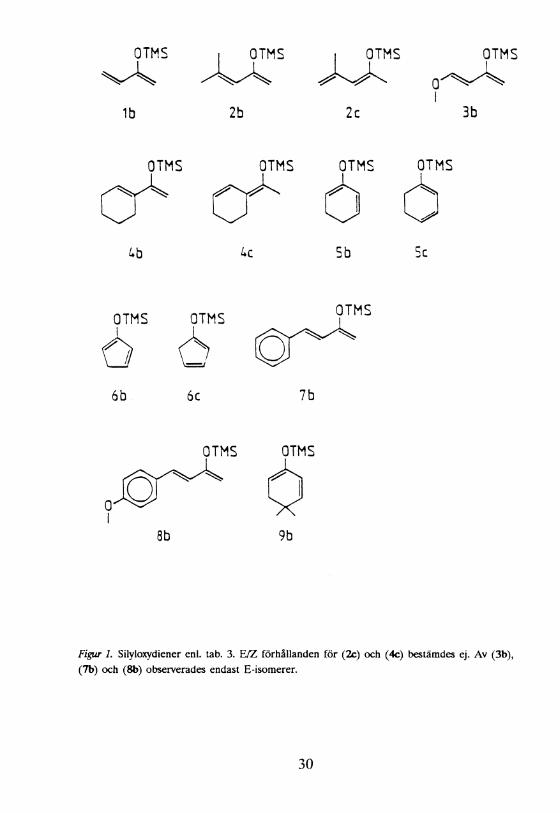
Figur 1. Silyloxydiener enl. tab. 3. E/Z förhållanden för (2c) och (4c) bestämdes ej. Av (3b), (7b) och (8b) observerades endast E-isomerer.
30
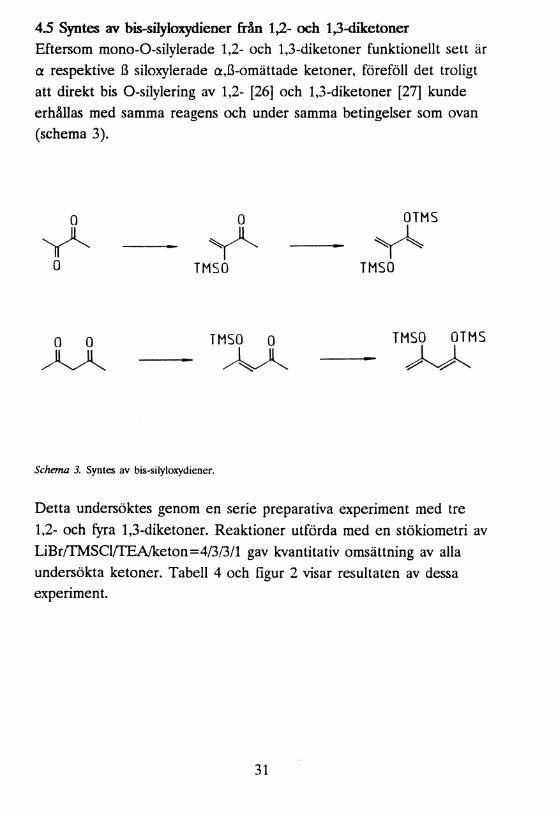
4.5 Syntes av bis-silyloxydiener från 1,2- och 1,3-diketoner Eftersom mono-O-silylerade 1,2- och 1,3-diketoner funktionellt sett är a respektive ß siloxylerade a,ß-omättade ketoner, föreföll det troligt att direkt bis O-silylering av 1,2- [26] och 1,3-diketoner [27] kunde erhållas med samma reagens och under samma betingelser som ovan (schema 3).
0 0 OTMS
TMSO TMSO
0 0AA
TMSO 0 TMSO OTMS
Schema 3. Syntes av bis-silyloxydiener.
Detta undersöktes genom en serie preparativa experiment med tre 1,2- och fyra 1,3-diketoner. Reaktioner utförda med en stökiometri av LiBr/TMSCl/TEA/keton=4/3/3/l gav kvantitativ omsättning av alla undersökta ketoner. Tabell 4 och figur 2 visar resultaten av dessa experiment.
31

Tabell 4 Resultat av de preparativa experimenten enl. ovan.
Startmtrl.3 Produkt(er)b Utbyte(%)c
la lb 84
2a 2b 93
3a 3b 93
4a 4b 93
5a 5b, 5c 93
6a 6b 94
7a 7b 62
a (la) 2,3-butandion; (2a) 2,3-pentandion; (3a) 3-metyl-l,2-cyklopentandion; (4a) 2,4-pentandion; (5a) 2-acetylcyklohexanon; (6a) l-fenyl-l,3-butandion; (7a) 1,3- cyklohexandion.
b Se figur 2 för identifikation av produkterna. c Avser isolerade utbyten av destillerad produkt.
32

OTMS Q TM SQTMSI
!
OTMS
1b
i. b
6b
QTMS
7b
Figur Z Bis-silyloxydiener enl. tab. 4. E/Z förhållanden:(4b) 50/50; (6b) 95/5. Stereoisomerin för (2b) och (5b) bestämdes ej.
4.6 SlutsatserPrincipalegenskapspiottarna är ett värdefullt hjälpmedel för att göra strukturerade urval av testkandidater vid sållningsförfaranden. Dessa ger en möjlighet att göra urvalen på sådant sätt, att en reell och god spridning i fysikalisk/kemiska egenskaper hos de utvalda testkandidatema erhålls. Principalegenskapsbeskrivningar av andra typer av kemiska komponenter än lösningsmedel och Lewis syror har rapporterats [22]. Dessa rör aminer, ketoner, aldehyder och zeoliter. Det vore dock önskvärt med liknande karaktäriseringar av flera typer av katalysatorer (t.ex. hydrogeneringskatalysatorer, alkalimetall- halogenider) i många sållningssammanhang. Att det är nödvändigt att variera flera egenskaper hos systemet samtidigt, framstår tydligt i exemplet ovan.
33


5 Effektivisering av syntesvägar via "one-pot"- reaktioner
5.1.1 Syntes av l,10-dimetyl-l(9)-oktalon-2I undersökningen av "one-pot" förfaranden (5.3) behövdes 1,10- dimetyl-l(9)-oktalon-2 a-epoxid (6) som substrat och därmed även dess prekursor, l,10-dimetyl-l(9)-oktalon-2 (1). Då dessa föreningar utgör mellanprodukter i en totalsyntes av geosmin, var det betydelsefullt att en effektiv metod valdes för synteserna. Syntesen av (1) har beskrivits via Robinson-annulering av 2-metyl-2-(3-oxopentyl)- cyklohexanon. 3-Oxoalkyl Substituenten har här införts genom alkylering av 2-metylcyklohexanon (2) i 2-position med olika3-pentanoner med lämnande grupper i ß-position [29], Alkylering med etylvinylketon (3) och andra syntetiska ekvivalenter [30] till de ß-substituerade 3-pentanonerna har också använts. Vid dessa förfaranden har både alkyleringssteget och ringslutningen utförts under såväl sura som basiska betingelser [31].
De metoder som beskrivits i litteraturen och som föreföll mest attraktiva för syntes i större skala var den syrakatalyserade reaktionen mellan (2) och l-kloro-3-pentanon (4) [29b] och alkylering av iminen från (2) och a-metylbensylamin med (3) utan tillsats av sur eller basisk katalysator [30b, 32]. Metoder där basiska katalysatorer utnyttjas för alkylering verkade vara mindre lämpade för användning i större skala, men en metod där litiumenolat används undersöktes i elt modifierat förfarande [IV, 30c]. Alkylering med (3) under sura betingelser verkade också intressant, då en analog reaktion mellan (2) och metylvinylketon har beskrivits [33]. I reaktionerna under sura betingelser undergår den intermediära alkyleringsprodukten ringslutning och dehydratisering in situ, medan reaktionen med imin först kräver hydrolys av den alkylerade iminen följd av ringslutning och dehydratisering i ytterligare ett steg (se schema 4). En jämförelse av dessa metoder gjordes, och syntesen av (1) via alkylering av (2) med (3) under sura betingelser befanns vara effektivast. Betingelserna
35
för denna reaktion - som inte var beskriven tidigare - bestämdes experimentellt med hjälp av en statistisk försöksplan [34] (se Appendix). Det observerades även att samtliga undersökta metoder gav upphov till den strukturisomera föreningen l,8-dimetyl-l(9)- oktalon-2 (5) som biprodukt [IV]. (se 5.1.2).
ci o>1*,C6C6> A
-M7U oJyV
<51
fVfl
Tnr,r.t.
n*,n2oo o
NaUft,fteUM,A- n2o m ♦ (si
Schema 4. Syntes av l,10-dimetyl-l(9)-oktalon-2.
I referenserna 29b och 33 användes överskott av alkyleringsmedlet för att omsätta (2) så fullständigt som möjligt. I den ovan nämda försöksplanen användes (3) också i överskott (1,25 ekvivalenter). Orsaken till att ingen variation av överskottet (3) gjordes i försöksplanen var att beräkning av utbytet baserat, på (2) verkade tveksam mot bakgrund av att (3) är ca: 20 gånger dyrare än (2) beräknat på molär basis. Detsamma gäller för (4) som i ref. 29c används i överskott. Därför gjordes istället en undersökning av reaktionen med varierande överskott av (2), där utbytet baserades på den dyrare (3). Att använda överskott av (2) borde dessutom leda till lägre grad av polyalkylering [35] av (2).
36

Resultaten av experimenten visas i tabell 5.
Tabell 5 Utbyte(%) av oktalon baserat på etylvinylketon när överskottet av 2-metylcyklohexanon (2) varieras
Exp. nr. Ekvivalenter (2) tid/tim. Utbyte(%)
1 1,0 12 57
2 2,0 10 75
3 2,5 8 86
4 3,0 8 82
5a 2,5 24 67
a Experiment med (4) under samma betingelser som i Exp. 4, med undantag av
reaktionstid.
I preparativa experiment kunde (1) isoleras i utbyten på 81-83%. Om utbytet i ett konventionellt förfarande [29b, 33] baseras på den dyrare komponenten, är utbytet endast 35-40%. Exp. 5 visar att (4) utnyttjas effektivare än i ref. 29b, när reaktionen utförs med överskott av (2).
5.1.2 Regioselektivitet vid alkylering 2-metylcyklohexanon Den produkt som erhölls i experimenten ovan var i samtliga fall en blandning av (1) och (5) i proportionerna 96:4, vilket väl överensstämmer med den termodynamiska jämviktsblandningen av de regioisomera enolerna från (2) [36]. Detta resultat var förvånade, då undersökningar av regioselektiviteten vid alkylering av (2) med metyl- akrylat under basiska betingelser har visat, att om man använder överskott av (2), erhålls minskad regioselektivitet [35]. Det kunde
37

också visas att vid alkylering av (2) med en ekvivalent metylakrylat under termodynamiska betingelser, erhölls högre selektivitet i alkyleringen än den utifrån enolatjämvikten förväntade. Vid reaktion med ett överskott av (2) (tre ekvivalenter) erhölls emellertid en produktsammansättning som svarade mot jämviktsläget för enolaten. Slutsatserna av dessa observationer var att 2,6-isomeren undergår ytterligare alkyleringar snabbare än 2,2-isomeren. Under betingelser med överskott av (2) minskas polyalkyleringen, och produktens sammansättning närmar sig jämviktsläget för enolaten. Experiment med andra alkyleringsmedel än metylakrylat gav samma resultat. Resultatet var oberoende av valt lösningsmedel (t-butylalkohol och bensen).
Det förefaller därför troligt att reaktiviteten i polyalkyleings- reaktionerna hos enolerna av 2,2- och 2,6-isomererna, sker med jämförbara hastigheter vid alkylering med (3) under sura betingelser.
5.1.3 Epoxidering av l,10-dimetyl-l(9)-oktalon-2 Epoxideringen av (1) har tidigare rapporterats med väteperoxid i metanol [37] och nyligen även med m-kloroperoxybensoesyra i diklormetan [38]. I det senare fallet finns inte fullständiga uppgifter om det experimentella förfarandet rapporterade, varför en serie experiment gjordes för att undersöka betydelsen av mängden oxidant reaktionen [IV]. Reaktionen med väteperoxid utfördes i ett något modifierat förfarande, jämfört med tidigare metoder [37, IV]. I båda fallen erhölls isolerade utbyten på ca: 90% som en blandning ava- och ß-epoxider, men i olika proportioner (se 5.1.4). Produkterna innehöll dessutom ca: 4% av epoxid från (5), se (5.1.1).
5.1.4 Diastereosclektivitet vid epoxidering av 1,10-dimetyl- l(9)-oktalon-2
Reaktionen med väteperoxid gav ett förhållande av a/ß på 60/40, vilket är i överensstämmelse med ref.x. Reaktionen med m-kloroper- oxobensoesyra gav ett a/ß förhållande på 92/8, vilket skiljer sig från resultaten i ref. 38, där ett förhållande på 96/4 har rapporterats.
38

Produktsammansättningen framgår av schema 5. Stereokemin hos epoxiden från (5) undersöktes ej.
Schema 4. Epoxidering av l,10-dimetyl-l(9)-oktaion-2.
5.2 Urval av möjliga förfaranden för "one-pot" syntes av (±)-geosmin från 1,10-dimetyl-1 (9)-oktalon-2 a-epoxid
a,ß-Epoxyketoner undergår ett antal reaktioner som ansågs vara intressanta som möjliga första länkar i ett "one-pot" förfarande för syntes av (±)-geosmin (7) direkt från (6). De reaktioner som avses beskrivs nedan tillsammans med det kompletta förfarandet.
a) Wharton-reaktionen [39], där behandling av epoxyketoner med hydrazinhydrat i alkohol resulterar i allyliska alkoholer. Reduktion av den olefiniska omättnaden skulle sedan kunna åstadkommas genom tillsats av väteperoxid till reaktionsblandningen, om stort överskott hydrazinhydrat använts i det föregående steget (hydrogenering via diimid [40] genererad in situ).
b) Reduktion av epoxyfunktionaliteten genom behandling med difenyldiselenid och natriumborhydrid i etanol [41], följd av behandling med etanditiol och en sur katalysator. Den önskade produkten skulle sedan kunna erhållas genom att
39

reaktionsblandningen behandlas med Raney-nickel.c) Selektiv reduktion av ketofunktionaliteten med litiumaluminium-
hydrid i tetrahydrofuran, följd av behandling av de bildade epoxyalkoxidema med /7-toluensulfonylklorid [30b, 38]. De epoxy- tosylat som bildas vore sedan möjliga att reducera direkt till (±)-geosmin genom ytterligare behandling med litiumaluminiumhydrid.
Gemensamt för a-c är att sekvenserna utförs i ett lösningsmedel utan att någon intermediär isoleras. Schema 6 visar a-c med formler.
Schema 6 Undersökta "one-pot" förfaranden.
Vid de experiment som utfördes för att i praktiken prova de föreslagna förfarandena, gjordes följande observationer:
a) Wharton-reaktionen gav högt utbyte av den förväntade 1,10- dimetyl-9-hydroxy-l-oktalinen (8) (>85%), men ingen reduktion av dubbelbindningen kunde observeras efter det att väteperoxid satts till reaktionsblandningen. Reduktionen av isolerad (8) undersöktes vidare
40

under andra betingelser (se 5.5). Det kunde dock visas att liknande strukturer med mindre steriska hinder i närheten av dubbelbindningen (2,3 bindningen i l,10-dimetyl-l(9),2-hexalin) reducerades under betingelserna ovan. Bubbling av luft genom reaktionsblandningen i närvaro av koppar(II)-sulfat, kunde här ersätta väteperoxid [40].
b) Difenyldiselenid/natriumborhydrid-reagenset var oförmöget att reducera epoxiden under de betingelser som rapporterats i ref. 41 Inte heller om reaktionen genomfördes under mer drastiska betingelser (återloppskokning i EtOH), kunde den förväntade ketolen, tran.y-l,10-dimetyl-tran.y-9-hydroxy-dekalon-2 observeras. Experiment med ketol, framställd med andra metoder [37], visade att syntesen och reduktionen av den motsvarande ditioketalen kunde genomföras i närvaro av reagenset.
c) Den selektiva reduktionen av ketonen med litiumaluminium- hydrid var utan problem, medan derivatiseringen av epoxyalkoxiderna var långsam och ofullständig [V]. Behandling av epoxytosylaten i reaktionsblandningen med litiumaluminiumhydrid gav dock målstrukturen (±)-geosmin, trans-l,10-dimetyl-trarts-9-dekalol, men i lågt utbyte. För att om möjligt åstadkomma en effektivare derivatisering av substratet undersöktes några andra derivat och derivatiseringsmetoder.
5.3 Sållning av reagens för derivatisering av den intermediära epoxyalkoxiden
Då även alkylsulfat och andra sulfonsyraestrar än tosylat kan reduceras till alkan med litiumaluminiumhydrid [42], undersöktes derivatisering av alkoxiderna med metansulfonylklorid [43], pyridin- svaveltrioxidkomplex och klorsulfonsyra. Vid tosylering och mesylering användes 4-dimetylaminopyridin (DMAP) [44] för att om möjligt öka reaktionshastigheten och omsättningen av substratet. För att erhålla en totalbild av derivatiseringsmetodernas användbarhet utvärderades dessa genom att derivaten reducerades till (7) med litiumaluminiumhydrid. Resultaten av detta framgår av tabell 6.
41

Tabell 6 Utbyte(%) av (7) i "one-pot" reaktionen enligt c, när olika derivat används.
Exp. Reagens Tid/tim.a Omsättn.(%)b Utbyte(%)
1 TsCl, DMAP 170 80-90 14
2 MsCl, DMAP 72 90-95 23
3 CISOjH 1 100 40
4 SOrPy 2 100 55
5C S03-Py 2 100 33
a Avser tiden för derivatiseringen av substratet.b I experiment 1 och 2 uppskattades omsättningen till derivat genom kvantifiering
av återstående epoxyalkohol med gaskromatografi. I 3-5 utfördes motsvarande analys med tunnskiktskromatografi.
c I detta experiment behandlades (6) initiait med överskott av litiumaluminium- hydrid för att erhålla dialkoxider istället för epoxialkoxider.
I tabell 4 ovan framgår att pyridin-svaveltrioxidkomplex gav de bästa resultatet, både med avseende på utbyte och reaktionstid för derivatiseringen. Vid preparativa experiment erhölls isolerade utbyten på 52% av (7) efter kromatografi. En jämförelse med andra totalsynteser av (7) redovisas under 5.4.
5.4 Jämförelse med tidigare totalsynteser av (±)-geosmin I tabellen nedan jämförs tidigare rapporterade totalsynteser av (7) med den i avhandlingen beskrivna. De angivna utbytena baseras på de kommersiellt tillgängliga startmaterial som krävs för de olika vägarna. Antalet steg i de olika fallen framgår också i tabellen.
42

Tabell 7 Jämförelse av tidigare rapporterade totalsynteser av geosmin med resultaten i detta arbete.
Metod Referenser Antal steg Utbyte(%)a Startmtrl.b
1 45a, 45b 12 10 A
2 29b, 37 5 10 B
3 29b, 38 5 16 B
4 32, 30b 6 27 C,DC
5 IV,V 3 35 D
a Beräkningarna av utbyten i metod 2-5 baseras på B och D istället för 2-metyl- cyklohexanon då dessa är ca: 20 ggr dyrare än cyklohexanonen.
b (A) 1,3-dimetylcyklohexanon; (B) l-kloro-3-pentanon; (C) iminen av 2-metylcyklo- hexanon och a-metylbensylamin; (D) etylvinylketon.
c Eftersom stökiometriska mängder av C och D användes kan utbytet baseras på endera materialet.
5.5 Syntes av C-l epimeren av (±)-geosmin Eftersom försöken att reducera (8) med diimid, genererad in situ, misslyckades, utfördes några experiment med isolerat material. Hydrogenering vid ca: 4 atm. i Parr-apparat med palladium på kol eller platina(IV)oxid (Adam’s katalysator) i alkohol med eller utan tillsats av ättiksyra, gav ingen reduktion av substratet. När reduktionen utfördes i närvaro av små mängder svavelsyra, erhölls en blandning isomera 1,10-dimetyl-dekaliner. Detta resultat var inte oväntat, då allyliska alkoholer lätt undergår elimination av vatten under starkt sura betingelser. Den dien som uppstår efter en sådan elimination är mindre steriskt hindrad än (8), och dubbelbindningarna reduceras snabbt. Hydrogenolys av allylalkoholen, följd av reduktion
43

av en olefin, är en mindre trolig väg i detta fall (se nedan).När (8) utsattes för högtryckshydrogenering (95 atm., 100 ° C, Pt02)
i etanol, reducerades dubbelbindningen utan att någon hydrogenolys av hydroxyfunktionaliteten observerades. Den erhållna produkten var inte den förväntade (7), utan dess C-l epimer, cis-l,10-dimetyl-fram- 9-dekalol (9) [30a]. Den troliga orsaken till det stereokemiska resultatet av hydrogeneringen är att katalysatorn koordineras av hydroxygruppen och styr additionen av väte till samma sida av molekylen. Detta antagande styrks av att samma produkt erhålls vid hydrogenering av den isomera allyliska alkoholen med exocyklisk dubbelbindning [30a]. En annan tänkbar orsak vore, att reduktionen föregås av isomerisering av dubbelbindningen till en mindre hindrad position. Vid en sådan isomerisering skulle troligen en frans-relation erhållas mellan hydroxygruppen och C-l metylgruppen. Utbytet av (9) efter kromatografering var 82%. Produkten var kristallin och hade en smältpunkt på 24-25 °C. Denna förening har tidigare rapporterats som olja [30a].
5.6 SlutsatserÄven om två av de tre undersökta "one-pot" reaktionerna var ineffektiva i det specifika fallet som undersöktes, visar experimenten ovan, att dessa har en potential för liknande transformationer av andra substrat. Det förfarande (c) som gav den önskade produkten utgör tillsammans med den ovan beskrivna metoden för syntes av (1), en mycket kort och effektiv totalsyntes av (7).
Ett stort antal "one-pot" reaktioner har rapporterats i den kemiska litteraturen, men någon medveten strategi för systematiskt utvecklande av sådana reaktioner existerar inte. Databaserad kompilering av kända reaktioner och kompatibla lösningsmedel över hela syntetiska sekvenser skulle kunna vara en sådan strategi.
44

Slutord
Jag vill här uttrycka min tacksamhet till min vän och handledare Docent Rolf Carlson för hans stöd och vägledning under mitt arbete och för hans kritiska granskning av denna avhandling.
Jag vill även tacka alla kolleger på avdelningen för den hjälp de har givit mig i mitt arbete och för många lärorika diskussioner.
Vidare tackar jag den tekniska personalen på avdelningen, vars insats har underlättat mitt arbete mycket.
Slutligen tackar jag min kära Saide, utan vars tålamod, uppmuntran och stöd detta arbete inte varit genomförbart.
45


Referenser
1. Box, G. E. P., Hunter, W. G. och Hunter, J. S. Statistics for Experimenters, Wiley, New York (1978)
2. (a) Berridge, J. C. och Morrissey, E. G. /. Chromatogr. 316 (1984) 69; (b) Alonso, J., Bartroli, J., Codio, J. ochdel Valle, M. Anal. Lett. 20 (1987) 1247
3. Carlson, R., Lundstedt, T. och Albano, C. Acta Chem. Scand. B39 (1987) 79
4. Carlson, R., Lundstedt, T., Nordahl, Å. och Prochazka, M.Acta Chem. Scand. B40 (1986) 522
5. Hellberg, S., Sjöström, M., Skagerberg, B. och Wold, S. J. Med. Chem. 30 (1987) 1126
6. (a) Corey, E. J. Pure Appi. Chem. 14 (1961) 19; (b) Corey, E. J. Chem. Soc. Rev. 17 (1988) 111
7. (a) Hendrickson, J. B. Topics Current Chem. 62 (1976) 49 Springer Verlag, Berlin-Heidelberg-New York; (b) Hendrickson, J.B. Acc. Chem. Res. 19 (1986) 274
8. (a) Corey, E. J. Quart. Rev. 25 (1971) 455; (b) Hendrickson, J. B., Grier, D. L. och Toczko, A. G. J. Am. Chem. Soc. 107 (1985) 5228
9. Ugi, I., Bauer, J., Brandt, J., Friedrich, J., Gasteiger, J., Jochum,C. och Schubert, W. Angew. Chem. Int. Ed. Engl. 18 (1979) 111; (b) Bauer, J. och Ugi, I. J. Chem. Research (S) (1982) 298;
47

(c) Gasteiger, J. och Jochum, C. Topics Current Chem. 74 (1978) 95 Springer Verlag, Berlin-Heidelberg-New York
10. För exempel se; (a) Nilsson, Å. Akademisk avhandling, Umeå Universitet, Umeå (1985); (b) Lundstedt, T. Akademisk avhandling, Umeå Universitet, Umeå (1986); (c) Nordahl, Å. Akademisk avhandling, Umeå Universitet, Umeå (1990)
11. Carlson, R., Nilsson, Å. och Strömqvist, M. Acta Chem. Scand.B37 (1983) 7
12. Myers, R. H. Response Surface Methodology, Allyn and Bacon, Inc. Boston (1971)
13. (a) Spendley, W., Hext, G. R. och Himsworth, F. R.Technometrics 4 (1962) 441 (b) Nedler, J. A. och Mead, R. Computer J. (1965) 308
14. Harrington Jr., E. C. Ind. Quai Control. 21 (1965) 494
15. Wold, S., Albano, C., Dunn III, W. J., Edlund, U., Esbensen, K., Geladi, P., Hellberg, S., Johansson, E., Lindberg, W., och Sjöström, M. I: Kowalski, B. (red.) Proc. NATO Adv. Study in Chemometrics, Cosenza, Italien, September 1983
16. För exempel se ref. 10b
17. Colvin, E. W. Silicon in Organic Synthesis. Butterworths, London Boston, Sydney, Wellington, Durban, Toronto 1981
18. House, H. O., Czuba, L. J. Gall, M. och Oelmstead, H. D. J. Org. Chem. 34 (1969) 2324
19. Jung, M. E. och McCombs, C. A. Org. Synth. 58 (1976) 163
48

20. (a) Danishefsky, S. och Kitahara, T. J. J. Am. Chem. Soc. 96 (1974) 7807 (b) Cazeau, P., Duboudin, F., Babot, O. och Dunogues, J. Tetrahedron Lett. 41 (1987) 2089
21. Joliffe, I. T. Principal Component Analysis, Springer Verlag,New York (1986)
22. Prochazka, M. Akademisk avhandling, Umeå Universitet, Umeå (1990)
23. Ref. 17 sid. 220-222
24. Rochin, C., Babot, O., Dunogues, J. och Duboudin, F.Synthesis (1986) 667
25. Doehlert, D. H. Appi. Statistics 19 (1970) 231
26. (a) Reetz, M. T., och Neumeier, G. Chem. Ber. 112 (1979) 2209 (b) Simchen, G. och Kober, W. Synthesis (1976) 259
27. (a) Babot, O., Cazeau, P. och Duboudin, F. J. Organomet. Chem. 326 (1987) C 57(b) Torkelson, S. och Ainsworth, C. Synthesis (1977) 431
28. Principalegenskapsplottarna finns återgivna i ref. 22
29. (a) Masaiti, Y., Hirakura, M. och Seki, F. J. Org. Chem. 23 (1958) 841 (b) Zoretic, P. A., Branchaud, B. och Maestrone, T. Tetrahedron Lett. (1975) 527
30. (a) Marshall, J. A. och Hochstetler, A. R. J. Org. Chem. 31 (1966) 1020 (b) Revial, G. Tetrahedron Lett. 30 (1989) 4121
49

(c) Stotter, P. L., och Hill, K. A. /. Am. Chem. Soc. 96 (1974) 6524
31. Se ref. 29 och 30
32. Pfau, M., Revial, G., Guingant, A. och d’Angelo, J. Fourth European Symposium on Organic Chemistry Aix-en-provence, Frankrike, 1985
33. Heathcock, C. H. och Ellis, J. E., McMurry, J. E. och Coppolino, A. Tetrahedron Lett. (1971) 4595
34. Sjöberg, A-L. Projektarbete, Avd. för organisk kemi, Umeå Universitet, Vt 1989, Opublicerade resultat
35. House, H. O., Roelofs, W. L. och Trost, B. M. J. Org. Chem. 31 (1966) 646
36. House, H. O. och Kramar, V. /. Org. Chem. 28 (1963) 3362
37. Ayer, W. A., Browne, M. L. och Fung, S. Can. J. Chem. 54 (1976) 3276
38. Gosselin, P., Joulain, D., Laurin, P. och Rouessac, F. Tetrahedron Lett. 30 (1989) 2775
39. Wharton, P. S. och Bohlen, D. H. /. Org. Chem. 26 (1961) 3615
40. Miller, C. E. /. Chem. Ed. 42 (1965) 254
41. Miyashita, M., Suzuki, T. och Yoshikoshi, A. Tetrahedron Lett.28 (1987) 4293
42. (a) Corey, E. J. och Achiwa J. Org. Chem. 34 (1969) 3667
50

(b) Haines, T. H. Lipids 5 (1970) 149
43. Eschenmoser, A. och Frey, A. Helv. Chim. Acta. 35 (1952) 1660
44. Scriven, E. F. W. Chern. Soc. Rev. 12 (1983) 129
45. (a) Marshall, J. A. och Schaeffer, D. J. /. Org. Chem. 30 (1965) 3642(b) Marshall, J. A. och Hochstetler, A. R. J. Org. Chem. 33 (1968) 2593
51


10. Appendix
53


Opublicerade resultat från faktorförsök.(5.1.1)
Variabel Nivåer- +
X, Temperature 70 100
x2 Tillsatstid/h. 0,5 6
x3 Katalysatormängd/mol% 1,4 2,8
x4 Tillsatsvolym (toluen)/ml 10 20
Xj x2 x3 *4 Utbyte/% Reakt. tid/h.
- - 35 48
+ 36 26
+ 4- 35 48
+ + - 44 21
+ + 41 26
+ - + - 50 16
+ + - 44 19
+ + + + 56 16
Reaktionerna utfördes med 10 mmol 2-metylcykIohexanon och 12,5 mmol etylvinyl- keton. Följande effekter bestämdes; ßl =5,1, ß2=2,l, ß3=3,9, ß4=-0,6. Experiment i gradientvektorns riktning (*x=110 °C, x2=6,4 tim., *2=3,2 mol%, *4=14 ml), gav ett utbyte på 62%.
55


N~> o O 07 C 1 —I . NI <=*• X I *. •—* —1 CNI_( —* —.oX UJ LU Dt-Z 1— Œ. i—iz a t—
O O O LO O07 CO O —i LOCO CO O LO CO
CN) Csl O iCO CO OCO 00 LO OfOfO-H
K) CO CO LO
O —<o^rO C LO
o- ca o —* r\O K) 07 <=r —• CNJ
0_o \Li_ u_ —1 • a ^ NUCO CO (O CO t— CO X
:* a a co co loD_ ÛC Œ ûi Z I—
□XCJ
XOCNJ
XCO
XO CO CNJ LO O 07 CO CO LO C O 07 O LO K7
■-H o 07 r\ u- oo 07 CO LO
CNI 07 X LO
X CJ CJ \\ X
CD OD —• NU X X —I CO X X X CO
30£8 'fri
SSU '03m 9 "ÏÏ3 7^=3089 '03 /S83£ '13
3008 '63391 fr ' 0£
8803 ' fr £3fr36 ' fr £0039 *S£5631 *3£
COGOXCO
££Ifr 'fr3_6S3l_*93IS 66T9?986fr '33
o
CLCLo
LO
OCD
Or\
Wdd

EP I-
GE0
SM I
NNO o o co
OC LU LU DH 21 I— CL —< ZQH
O O O LO OCO CO O —i LO CO CO O LO CO
(N Csl O ^ <3- —<CO CO O CO CO LO
O NO NO —•NO CO CO LO
Q_O SLlLl^mQSiNJ
CO LO O CO H- CO X
O —<O <3- O C LO
^ co o no r\ O CO CO <3- —I CM
:* a o co co lu cl. oc cl ûc z: H-
aeuCJ
XOCM
eua
euO CO OJ LO O CO 00 CO O C O CO a LO CM
-lOcorN'tc CO 00 co
CN CO X LO
21 CJ CJ \\ 21
Q0 CD —< INtJ CU Cd _l O Lu X CL CO
r<^6 *9i
Z66l’\l
V.
Oco
IS9Z ’£{em '%l£986 '9Zt?96fr ‘U
fOco
Wdd






























