Embed Size (px)
Citation preview

107
Structure-Activity Studies of Bradykinin andRelated Peptides
B2-Receptor Antagonists
Nour-Eddine Rhaleb, Sabine T616maque, Noureddine Rouissi, St6phane Dion,
Daniela Jukic, Guy Drapeau, and Domenico Regoli
Thirty-seven compounds were tested as antagonists of kinin B2- and B,-reeeptors to identify thechemical changes required to obtain antagonism, improve antagonist affinity, and eliminateresidual agonistic activities. Apparent affinity of antagonists was evaluated in terms of pA2 onthe rabbit jugular vein, the dog carotid and renal arteries, the hamster urinary bladder, theguinea pig ileum, the rat vas deferens, the guinea pig trachea, and the rabbit aorta, usingbradykinin and desArg'-bradykinin as B?- and B,-receptor activators. Replacement of Pro7 ofbradykinin with D-Phe leads to antagonism; substitution of Pro3 by Hyp and extension of thepeptide chain at the N-terminal with a D-Arg residue improves the affinity of antagonists;acetylation of N-terminal amine function reduces residual agonistic activity; these changes,combined with the replacement of Phe* by Leu as in Ac-D-Arg[Hyp3,D-Phe7,Leu8]-bradykinin,led to potent full B^-receptor antagonists. Affinity of antagonists differs markedly betweenhighly sensitive (rabbit jugular vein, dog carotid and renal artery), moderately sensitive(hamster urinary bladder, guinea pig ileum, and rat vas deferens), and insensitive preparations(the guinea pig trachea) in which antagonists act as potent stimulants. High concentrations ofantagonists block bradykinin completely in the rabbit jugular vein but not in the guinea pigileum, suggesting that kinins stimulate the moderately sensitive tissues by two mechanisms, ofwhich only one is blocked by antagonists. It thus appears that kinins act on various Bj-receptorsubtypes or by different action mechanisms. (Hypertension 1991;17:107-115)
Early classification of kinin receptors into B!and B2 subtypes was based on each ago-nist's order of potency and on the finding
that only B,-receptors could be selectively blockedby antagonists such as [Leu8]desArg9-bradykinin([Leu^desArg'-BK).20 Lack of B2-receptor antago-nists left the B^-receptor area still poorly defined.B2-receptor antagonists were first reported byVavrek and Stewart1; since then, a large number ofsimilar compounds have been prepared and testedin various laboratories.2"4
Extensive use of such compounds has been made invitro and in vivo to study B^-receptor pharmacologyand biological roles2-3-5 despite the fact that thesecompounds 1) are potent histamine (and possiblyprostaglandin) releasers,6-7 2) maintain partial agonis-tic activities,8 and 3) are not selective for B2-receptors
From the Medical School, University of Sherbrooke, Sher-brooke, Quebec, Canada.
Supported by the Medical Research Council of Canada. D.R. isa Career Investigator of the Medical Research Council of Canada.
Address for reprints: Domenico Regoli, MD, Department ofPharmacology, Medical School, University of Sherbrooke, Sher-brooke, Quebec J1H 5N4, Canada.
Received May 15, 1990; accepted in revised form July 24, 1990.
since they also act on B,-receptors.2 Attempts havebeen made910 to improve the affinity of the antago-nists and at the same time reduce the residual agonis-tic effects to provide adequate tools for a betterdefinition of the Brreceptor system, which appears tobe more complex than initially thought. Indeed, theexistence of multiple B2-receptor subtypes has beenpostulated by various authors.3-8-1 •
The present study focuses on antagonists. Thirty-seven compounds were tested to systematically ex-plore the usefulness of the various chemical changesthat are made to obtain antagonism, improve antag-onist affinity, reduce residual agonistic activity, andeliminate histamine release. An attempt has alsobeen made to reevaluate the bioassay organs cur-rently used in kinin pharmacology to provide sensi-tive and reliable tests for studying B2-receptors. Thecompounds have also been characterized with re-spect to their specificity for the kinins, their selectiv-ity for the B2-receptors, and their ability to act ascompetitive antagonists.
MethodsIsolated organs used in the present study are the
dog carotid artery (DCA), the dog renal artery
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from

108 Hypertension Vol 17, No 1, January 1991
(DRA), the rabbit jugular vein (RJV), the rabbitaorta (RA), the rat vas deferens (RVD), the hamsterurinary bladder (HUB), the guinea pig ileum (GPI),and the guinea pig trachea (GPT). The organs weretaken from animals that were killed by stunning andexsanguination, except the dogs, which were deeplyanesthetized to death by administration of pentobar-bital (50 mg/kg i.p.). Rings of DCA and DRA werecarefully dissected to preserve the endothelium12;strips of RA were prepared by the method ofFurchgott and Bhadrakom13; helical strips of RJVwere obtained with the procedure described byGaudreau et al.14 Both the prostatic and epididymalportions of RVD were used and handled according toTousignant et al15; strips of the dome of the hamsterurinary bladder were prepared with the methoddescribed by Vane16 for the rat stomach; longitudinalstrips of the GPI were obtained by the method ofRang17; strips of the GPT were prepared according toConstantine.18 The tissues were suspended in 10-mlorgan baths containing warm Krebs solution (37°C)oxygenated with a 95% Oz-5% CO2 mixture for DCA,DRA, RJV, RA, GPI, GPT, and RVD and Tyrode'ssolution at 32°C for the HUB. The arterial vessels, thevas deferens, and the trachea were stretched with aninitial tension of 2 g, whereas the isolated veins,urinary bladder, and ileum were given 0.5 g of tension.Changes of tension produced by the various agentswere measured with Grass isometric transducers (FT03C, Grass Instrument Co., Quincy, Mass.), calibratedto 1 g=30 mm. Contractions or relaxations weredisplayed on a Grass polygraph (model 7D). Beforetesting the drugs, the preparations were allowed toequilibrate for 60-120 minutes. During the equilibra-tion period, the tissues were repeatedly washed andtension was adjusted each 15 minutes.
Experimental Protocols
Repeated applications of double and single dosesof BK or of desArg'-BK were made in the absenceand presence of various kinin-related peptides toevaluate the apparent affinity of antagonists, in termsof pA2 (—log of the concentration of antagonist thatreduces the effect of a double dose of agonist to thatof a single dose).19
All kinin antagonists were initially applied totissues at concentrations of 10-100 ^.g/ml to mea-sure their potential agonistic activities in compari-son with BK or desArg9-BK. Residual effects ofantagonists will be expressed semiquantitatively,except for the compounds that showed aE (intrinsicactivity) of 0.4 or higher. The contractile effects ofkinins and their analogues were measured on theRJV and HUB treated with captopril (1.1 xlO"6 M)to prevent the degradation of peptides by kininaseII.20 The relaxant effects of kinins on DCA andDRA were evaluated in the presence of intactendothelium after contraction of the tissue to 1 g oftension with norepinephrine (2.0 xlO"8 M). Ethyl-enediaminetetraacetic acid (EDTA) (5.9 xlO"5 M)and indomethacin (2.8xlO"6 M) were added to the
solution to prevent the oxidation of the catechol-amine and block the synthesis of inhibitory prosta-glandins that reduce the contractile effect of nor-epinephrine or potentiate the relaxing effects of thekinins. Removal of endothelium was assessed by theabsence of a relaxant response of the vessels toacetylcholine (2.6xlO"6 M). The contractile effectsof kinins on the GPI were measured in the presenceof atropine (2.2xlO~6 M) to prevent the effect ofendogenous acetylcholine. The contractile effects ofkinins in the RA were measured after a 5-hourincubation period to obtain measurable and stableresponses21 to desArg'-BK.
Peptides and Other Agents
Primary structures of kinin-related peptides areshown in Table 1. All peptides shown in Table 1 aswell as substance P and angiotensin II (Ang II) wereprepared by solid-phase synthesis with the proceduredescribed by Drapeau and Regoli22 and thepseudopeptides with the method of Sasaki and Coy23
as modified by Drapeau et al.24 Peptides were puri-fied by high-performance liquid chromatography andtheir structure was assessed by fast atomic bombard-ment (FAB) mass spectrometry. Other drugs usedwere norepinephrine, indomethacin, atropine, andacetylcholine (Sigma Chemical Co., St. Louis, Mo.),EDTA (Fisher, Ottawa, Ontario, Canada), ascorbicacid (Baker, Dorval, Quebec, Canada), captopril (SQ14225, [r>3-mercapto-2-methylpropanoyl]-L-proline(S,S), supplied by Squibb Canada), and mergetpa(D,L-2-mercaptomethyl-3-guanidinoethylthiopro-panoic acid) purchased from Calbiochem-Behring,San Diego, Calif.
Concentrated solutions (1-5 mg/ml) of all agentswere prepared in distilled water and stored at -20°Cuntil use. Solutions of indomethacin were made inTrizma base (0.02 M)(Sigma). Ascorbic acid (5%)was added to the solutions of norepinephrine toprevent oxidation.
ResultsThe results obtained with three series of com-
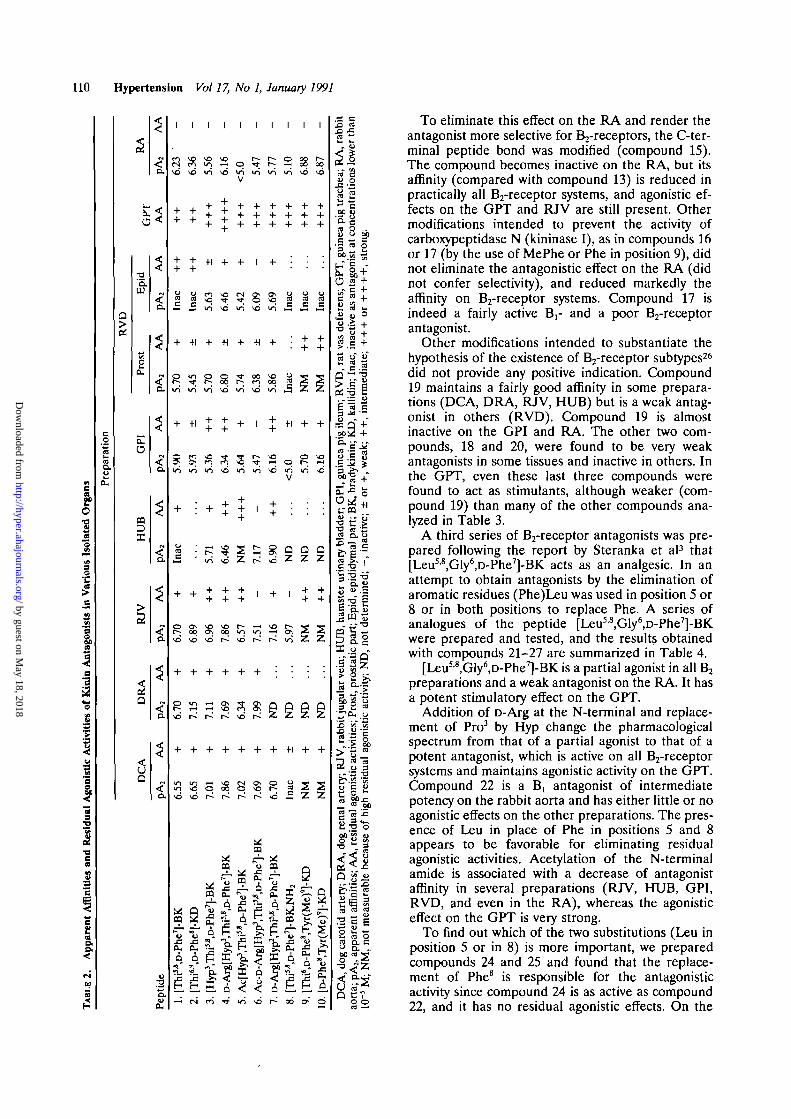
pounds designed to improve the affinity of antago-nists as well as to eliminate residual agonistic effectare presented in Tables 2, 3, and 4.
The first series (Table 2) contained analogues of[Thi58,D-Phe7]-BK, which was initially considered tobe a pure antagonist1 and later shown to act as partialagonist in some preparations (for instance theRVD).815 Indeed, this compound maintains someagonistic effect in all preparations except the RA andis an agonist of average potency on GPT. [Thi5|8,D-Phe'J-BK reduces or blocks the effect of BK in allpreparations except the HUB, the epididymal part ofthe RVD, and the GPT. It is fairly active as anantagonist on the B,-receptor (RA). Similar resultswere observed with the kallidin analogue [Thi6r9,D-Phe8)-kallidin, which was found to be slightly moreactive than compound 1.
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from

Rhaleb et al Bradykinin Receptor Antagonists 109
TABLE 1. Structures of Bradykinin or Kallidln Analogues
Bradykinin (BK): H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH
Kallldin (KD): H-Lys-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH
1. [Thi5JI,D-Phe7]-BK
2. [Thiw>D-Phe8]-ICD
3. [HypYThi^.D-Phe^-BK
4. D-Arg[Hyp3,Thi5-»,D-Phe7]-BK
5. Ac[Hyp3,ThiM,D-Phe7]-BK
6. Ac-D-Arg[Hyp3,ThiM,D-Phe7]-BK
7. D-Arg[Hyp2,Thi3'8>D-Phe7]-BK
8. [Thi'-'.D-Phe^-BK.NHz
9. [Thi6>D-Phe8,Tyr(Me)9]-KD
10.[D-Phe8,Tyr(Me)9]-KD
11. [D-Phe7]-BK
12. [Hyp'.D-Phe^-BK
13. D-Arg[Hyp3,D-Phe7]-BK
14. Ac-D-Arg[Hyp3,D-Phe7]-BK
15. D-Arg[Hyp3,D-Phe7,Phe8
^(CHrNH)Arg9]-BK
16. D-Arg[Hyp3,D-Phe7,MePhe']-BK
17. D-Arg[Hyp3,D-Phe7,Phe']-BK
18. [D-Phe17]-BK
19. D-Arg[Phc2,Hyp3,D-Phe7]-BK
20. [D-Phe^Hypi-BK
21. [Leu^Gly'.D-Phe^-BK
22. D-Arg[Hyp3>Leu",Gly6
)D-Phe7]-BK
23. Ac-D-Arg[Hyp3,Leu5-8,Gly6,D-Phe7]-BK
24. D-ArgtHyp3,Gly6,D-Phe7,Leu"]-BK
25. Ac-D-Arg[Hyp3,Gty')D-Phe7,Leu8]-BK
26. D-Arg[Hyp3,Leu5,Gly6,D-Phe7]-BK
27. D-Arg[Hyp3,Uu!'8,l>Phe7]-BK
28. D-Arg(Hyp3)D-Phe7,Leu8]-BK
29. Ac-D-Arg[Hyp3,D-Phe7,Leu8]-BK
30. D-ArgtHypM^u^.Gly'l-BK
31. [Leu^desArg'-BK
32. [Leu']desArglo-KD
33. Sar[Leu8]desArg9-BK
34. D-ArgfLeu^desArg'-BK
35. [Uu^desAig'-BICNHz
36. Sar[Leus]desArg'-BK.NH2
37. D-Arg[Hyp3,D-Phe7]desArg9-BK
The replacement of Pro3 by Hyp (compound 3)gives a compound that 1) maintains affinity on theB2-receptor systems,2526 2) becomes active also onthe HUB and RVD (epididymal part), 3) showsreduced affinity on the Brreceptor (the RA), and4) is more active than compounds 1 and 2 as astimulant of the GPT.
Extension of the chain at the amino end with aD-Arg residue (compound 4) is associated with animportant (generally around a log unit) increase ofaffinity in all B2-receptor preparations.2526 Com-pound 4 is a very potent stimulant of the GPT andshows important residual agonistic effects also on theRJV, HUB, and GPI.
jV-Acetylation of compound 3, as in compound 5, haspractically no effect, whereas the acetylation of com-pound 4, as in compound 6 (Ac-r>Arg[Hyp3,Thi5'8,D-Phe7]-BK) is associated with a slight decrease ofaffinity in some preparations (DCA, RJV, GPI, RVD)and with a modest increase of affinity in others (DRA,HUB). However, the most important effect of Af-acet-ylation is the elimination or the marked reduction ofthe residual agonistic effect especially on the RJV,HUB, GPI, and RVD. Compound 6 maintains goodactivity (as agonist) on the GPT and is less active thancompound 4 in the RA.
The replacement by Hyp of Pro2 instead of Pro3
(compound 7) gives no advantage. Compared withcompound 4, compound 7 is less active on the RJV,HUB, GPI, RVD, and RA. Compound 7 showssimilar residual agonistic activity as compound 4 inall preparations and is a potent stimulant of the GPT.
Other chemical modifications were tested to pro-tect the antagonist from degradation by C-terminalamidation (compound 8) or improve B2-receptor
affinity by the use of Tyr(Me) in position 9 of thekallidin antagonist (compounds 9 and 10). The re-sults were disappointing since the above changesmarkedly reduced antagonistic activity in all prepa-rations except the RA. Agonistic effect on the GPTwas maintained.
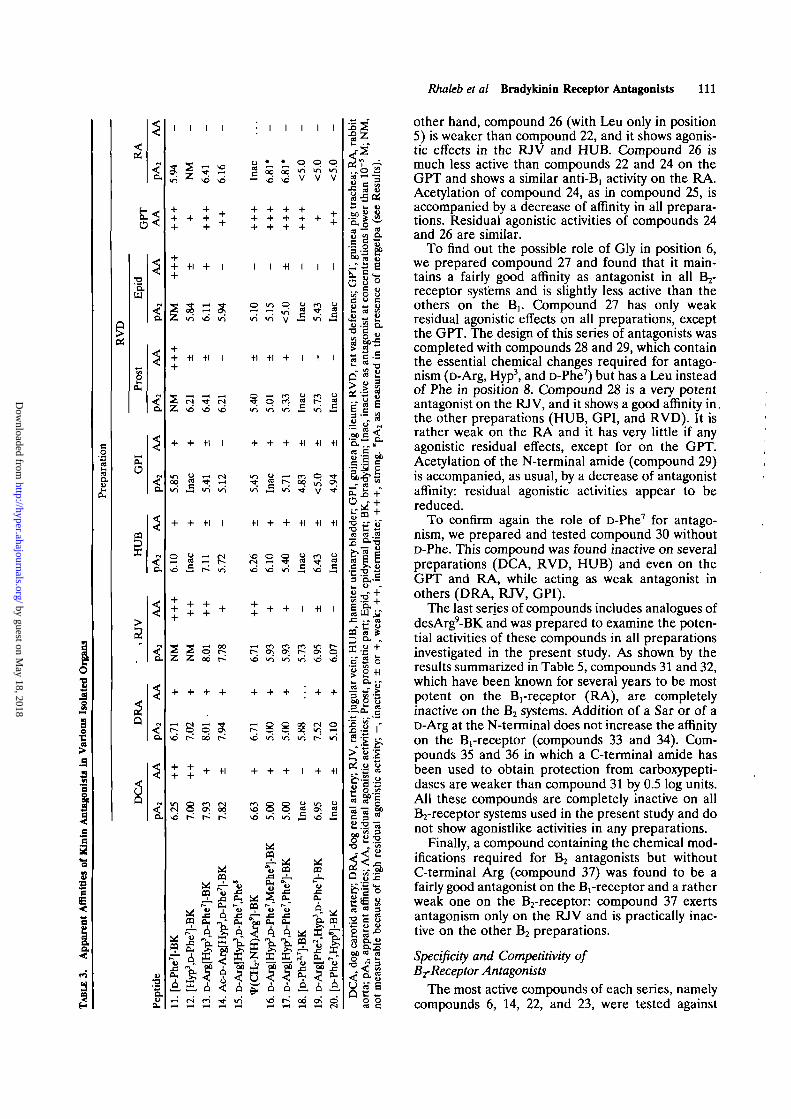
A second series of compounds, analogues of[D-Phe7]-BK, which contain the chemical change thatconfers antagonistic properties to BK,1 were pre-pared and tested. The results presented in Table 3show that [r>Phe7]-BK is a weak antagonist in severalB2-receptor systems, for instance the DCA, DRA,HUB, and GPI, but it is fairly active on the B,-receptor (RA). However, [D-Phe7]-BK has strongresidual agonistic activities in all preparations, par-ticularly the RJV and RVD. It is also a potentstimulant of the GPT. To improve antagonistic activ-ities and eliminate agonistic activities, the samemodifications used in the other series (Table 2) weremade in [D-Phe7]-BK. The replacement of Pro3 byHyp (compound 12) has a favorable influence since itincreases affinity, at least in some preparations(DCA, DRA, RVD); it also diminishes activity in theRJV and GPT and practically eliminates the B,-receptor antagonism. When a D-Arg is added at theN-terminal (compound 13), the compound acts as anantagonist in all preparations except the GPT; it is apotent antagonist showing pA2 values of around 8.0in several tissues, and it shows less agonistic effectthan compound 11. When compound 13 is acetylated,as in compound 14, affinity as antagonist is main-tained, residual agonistic effects are markedly de-creased or eliminated, and agonistic activity on theGPT is reduced. However, compound 14 remainsactive on the Brreceptor system.
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from
110 Hypertension Vol 17, No 1, January 1991
cm1Iis
Var
ioi
aa*a
•3
a•g
s
|I9.$
(dua
l
I
and
1
J
Aff
ini
2
App
are
3
ion
CO
CTJ
Q.
a
0OS
T3
Epi
rost
cu
Cu
o
ca
>
Q
5Q
<
a
<
a
%
r*
Q.
i
i
a
o.
Dtid
e
Cu
1
avd
++
+
Inac
+
o
In
+
8Ln
+
Inac
6.70
+
o
vd
+
invd
mu
x :CuA
1
i
++
+
Inac
+1
i n
i n
+1
S
6.89
+
v2vd
uj=
CU
1
1
i ni n
+1
5.63
+
o
+
vo
Ln
+
5.71
++
6.96
+
+
o
m
uXIcu
1
1
VO
vd
+
++
+
vo
•H
svd
+
VO
+
6.46
++
7.86
v£)
+
vg
CO
u
1i
i
o
V
+
+
lO
+
+
3
+
++
6.57
+
vd
+
rau
CU
is"
I
Acf
H
i n
1
i n
+
1
gvo
•H
00
vd
1
• jun
1
7.17
1
7.51
+
VO
CQ
CU
!
Ac-
D-
vd
1
+
s
+
vgi n
+
vo
VO
+
6.90
7.16
g
+
o
vd
UiCQ
^ ^
X!Cu
A.
I
i
oi n
Inac
u
•5
+1
o
<5.
g
1
5.97
Q
z
+1
3B1
K
CO
DX!CU
1CO
1
§gvd
Inac
+
+
oI O
g
++
z
g
+
z
2
Me)
9 ]
6;
"iCL.
aio<
1
00vd
Inac
+
z
+
VO
vo
g
++
z
g
+
z
0)
[D-P
h10
.
bit
X!
RA
, ra
low
er i
oX!
rac
u
'5.s.5So
rens
; G
U
i,
rat
Q
02
63
ile
o
tral
§c
8ra«
as a
nta
o>
inac
tna
c,
N -
•5
all
i i
Q"opi<JO. •_-
u5
ifCuO
•ora
Errar!
«r u
rii
x:
UB
,
I
lar
vein
;
a.a
abbi
ra
rena
l
I5
m
part
;
*cd
I
, epi
dii
pid,
UJ
par
t;
u
, pro
stat
i'ro
st
cu
ties
;:ti
vi
M
M'HS>
dual
(re
si
! *Q
arte
ryJo
g ca
rot
5
ties;
affi
nil
cK
s.D.ra
1a;
pA
ora
oil
2
++t - l
o
+
iate
;
"21
+S3
+"
ictiv
e
eg
r
line
d;
Gu
•a
o
QZ
acti
:icm
ist
f
dual
resn
x.
o
beca
ubl
eas
ura
u
MM
, no
t i
o
To eliminate this effect on the RA and render theantagonist more selective for Bj-receptors, the C-ter-minal peptide bond was modified (compound 15).The compound becomes inactive on the RA, but itsaffinity (compared with compound 13) is reduced inpractically all B2-receptor systems, and agonistic ef-fects on the GPT and RJV are still present. Othermodifications intended to prevent the activity ofcarboxypeptidase N (kininase I), as in compounds 16or 17 (by the use of MePhe or Phe in position 9), didnot eliminate the antagonistic effect on the RA (didnot confer selectivity), and reduced markedly theaffinity on B2-receptor systems. Compound 17 isindeed a fairly active B,- and a poor B2-receptorantagonist.
Other modifications intended to substantiate thehypothesis of the existence of B2-receptor subtypes26
did not provide any positive indication. Compound19 maintains a fairly good affinity in some prepara-tions (DCA, DRA, RJV, HUB) but is a weak antag-onist in others (RVD). Compound 19 is almostinactive on the GPI and RA. The other two com-pounds, 18 and 20, were found to be very weakantagonists in some tissues and inactive in others. Inthe GPT, even these last three compounds werefound to act as stimulants, although weaker (com-pound 19) than many of the other compounds ana-lyzed in Table 3.
A third series of B2-receptor antagonists was pre-pared following the report by Steranka et al3 that[Leu5i8,Gly6,D-Phe7]-BK acts as an analgesic. In anattempt to obtain antagonists by the elimination ofaromatic residues (Phe)Leu was used in position 5 or8 or in both positions to replace Phe. A series ofanalogues of the peptide [Leu58,Gly6,D-Phe7]-BKwere prepared and tested, and the results obtainedwith compounds 21-27 are summarized in Table 4.
[Leu5'8,Gly6,D-Phe7]-BK is a partial agonist in all Bjpreparations and a weak antagonist on the RA. It hasa potent stimulatory effect on the GPT.
Addition of D-Arg at the N-terminal and replace-ment of Pro3 by Hyp change the pharmacologicalspectrum from that of a partial agonist to that of apotent antagonist, which is active on all B2-receptorsystems and maintains agonistic activity on the GPT.Compound 22 is a B, antagonist of intermediatepotency on the rabbit aorta and has either little or noagonistic effects on the other preparations. The pres-ence of Leu in place of Phe in positions 5 and 8appears to be favorable for eliminating residualagonistic activities. Acetylation of the N-terminalamide is associated with a decrease of antagonistaffinity in several preparations (RJV, HUB, GPI,RVD, and even in the RA), whereas the agonisticeffect on the GPT is very strong.
To find out which of the two substitutions (Leu inposition 5 or in 8) is more important, we preparedcompounds 24 and 25 and found that the replace-ment of Phe8 is responsible for the antagonisticactivity since compound 24 is as active as compound22, and it has no residual agonistic effects. On the
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from
Rhaleb et al Bradykinin Receptor Antagonists 111
•5.
u
eo•3
1.3
I
3
o
I
1 1 1 1 I I I I I
++
++ +1 + I+
H &<
i n ' ^ o in
+ + +1 I
in »5 in in
© aI s P
r-^ppCT;« S «i h
o * *
i i
•-; ". m a T en in y »-3 in M
+ +1 +1 I +1 +1 + I I I+
« M Tf (S ^ Q ^ i S 1 " * :^r i ^^5 ^^^ ^^^ ^^^ ^^^ ^^^ ^H3 ^^^
+ + + +1 +1 -H
in y i-̂ c<i o•* g I-; 00 ^in fcS "n -̂ t V
+ + +1 I +1 + + -H -H +1
I -H I
t**" ON ON r*^ ON
p ^ p p
+ -H + + + I + +1
r̂ r- r* vo in, 5 « 01 J
m' ,3 \6 —
^ -8CQ OH
^ 4
11II
IX4
oa^7
2 0-
CQpr-
5
03
II pp g> * g? ^•^ < A % A
> 1
rit
J3 • - ffl
ga*..3
5£5sI .§ r
• ^ ed
Q iS3
• B S S S
o D.3W) ™ cdO J fa
other hand, compound 26 (with Leu only in position5) is weaker than compound 22, and it shows agonis-tic effects in the RJV and HUB. Compound 26 ismuch less active than compounds 22 and 24 on theGPT and shows a similar anti-Bi activity on the RA.Acetylation of compound 24, as in compound 25, isaccompanied by a decrease of affinity in all prepara-tions. Residual agonistic activities of compounds 24and 26 are similar.
To find out the possible role of Gly in position 6,we prepared compound 27 and found that it main-tains a fairly good affinity as antagonist in all B -̂receptor systems and is slightly less active than theothers on the B]. Compound 27 has only weakresidual agonistic effects on all preparations, exceptthe GPT. The design of this series of antagonists wascompleted with compounds 28 and 29, which containthe essential chemical changes required for antago-nism (D-Arg, Hyp3, and D-Phe7) but has a Leu insteadof Phe in position 8. Compound 28 is a very potentantagonist on the RJV, and it shows a good affinity in,the other preparations (HUB, GPI, and RVD). It israther weak on the RA and it has very little if anyagonistic residual effects, except for on the GPT.Acetylation of the N-terminal amide (compound 29)is accompanied, as usual, by a decrease of antagonistaffinity: residual agonistic activities appear to bereduced.
To confirm again the role of D-Phe7 for antago-nism, we prepared and tested compound 30 withoutD-Phe. This compound was found inactive on severalpreparations (DCA, RVD, HUB) and even on theGPT and RA, while acting as weak antagonist inothers (DRA, RJV, GPI).
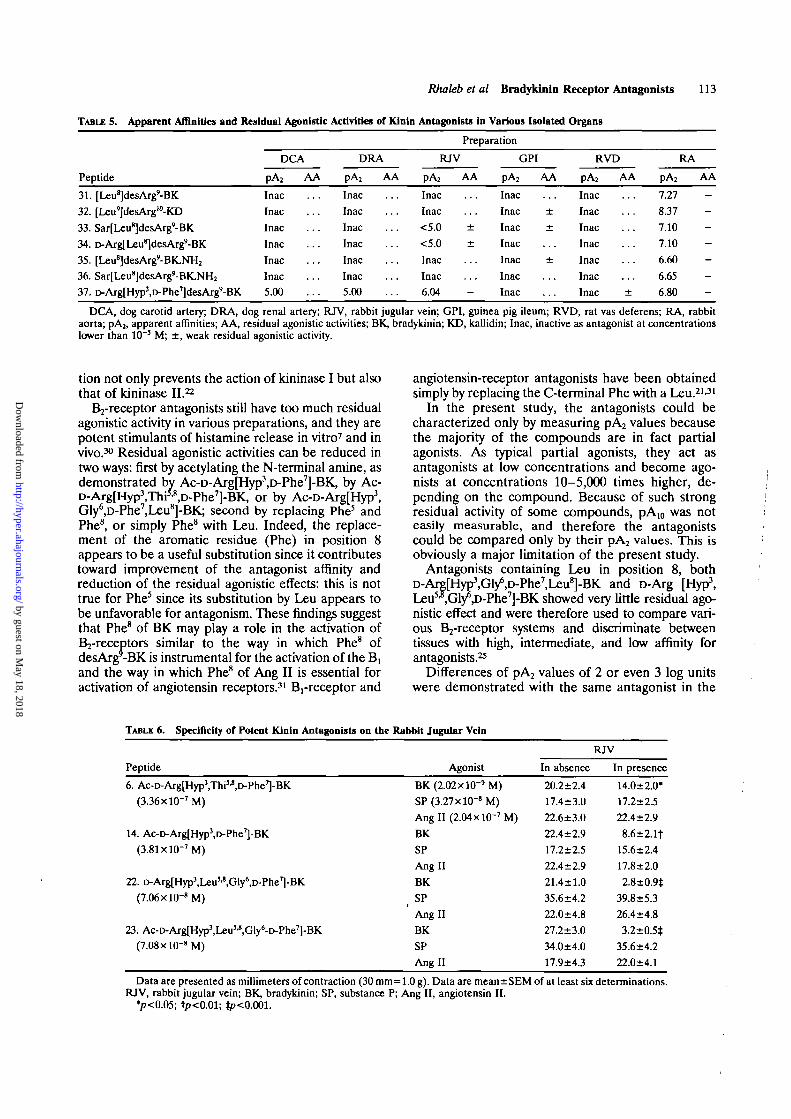
The last series of compounds includes analogues ofdesArg'-BK and was prepared to examine the poten-tial activities of these compounds in all preparationsinvestigated in the present study. As shown by theresults summarized in Table 5, compounds 31 and 32,which have been known for several years to be mostpotent on the Brreceptor (RA), are completelyinactive on the B2 systems. Addition of a Sar or of aD-Arg at the N-terminal does not increase the affinityon the Brreceptor (compounds 33 and 34). Com-pounds 35 and 36 in which a C-terminal amide hasbeen used to obtain protection from carboxypepti-dases are weaker than compound 31 by 0.5 log units.All these compounds are completely inactive on allB2-receptor systems used in the present study and donot show agonistlike activities in any preparations.
Finally, a compound containing the chemical mod-ifications required for B2 antagonists but withoutC-terminal Arg (compound 37) was found to be afairly good antagonist on the Brreceptor and a ratherweak one on the Bj-receptor: compound 37 exertsantagonism only on the RJV and is practically inac-tive on the other B2 preparations.
Specificity and Competitivity ofBrReceptor Antagonists
The most active compounds of each series, namelycompounds 6, 14, 22, and 23, were tested against
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from

112 Hypertension Vol 17, No 1, January 1991
1
•H I
2 S;
+i i i i
s
I I I
vd
+1 I I
a.vd
•H +1
06 (N -H
+1 +1
I +1 +1 -H
vd Z vo VO vo ,5
in m r~ o> <->H iq oq vq «vd vo vd vd £
+1 +1 -H I I
vo r~- r*̂ do *~Hi/^ \ ^ \ ^ i/^ i/^
-H +1 +1 I
VO vo VO f- vo J3
-H -H +1 +1 + -H -H I I
C ^ o o o o o o o d - U o o o o o o v d
•H +) -H • +1 +1
^ 4 SOH ^ vo
+1 +1 +1 -H -H -H -H I
3vo t^ vo vo
OQ *
m £ oa ^ oa
i Q 3 " • »CU « " ^ 4> ^
i3- o "« °r A.r^ *̂ cu « x .
m *_L . ca
OQ
o
o
A 4J A < AN i*i t̂ in «:H N N n M
I x l lA A ^ A
«!?
s!I| | ; •
O § Sg 8 +w "S "̂ "o *- +
« § G
j s , r
ill
•S3 IS 8 5
.̂ to (J
i l l
Si oS « a
^3
at
other stimulants (substance P and Ang II) of the RJVto determine their specificity for the kinins. As shownby the results summarized in Table 6, all compoundsare active against BK and do not affect the responsesof the isolated vein to substance P or Ang II.
Compound 27 was tested extensively to determinethe type of antagonism (competitive or noncompeti-tive) that it exerts on the RJV and on the GPI. Thisseries of experiments was undertaken in an attemptto find an explanation for the differences of antago-nist affinities that were observed with several com-pounds between the RJV and the other preparations,particularly the HUB and GPI.
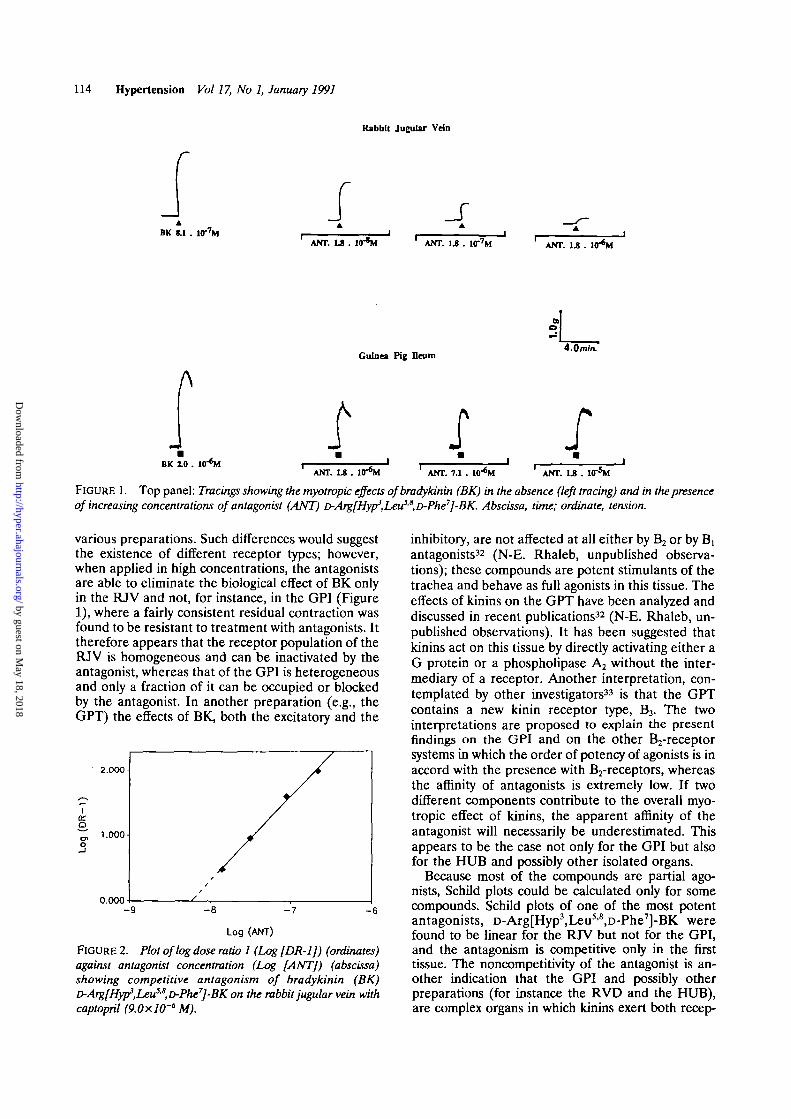
Increasing concentrations of antagonist were testedin the RJV and the GPI to occupy the receptorpopulation and eliminate (by the high concentrations)the myotropic effect of BK. This was possible only inthe RJV. An important residual effect was found to bepresent in the GPI even in the presence of highestconcentrations of antagonists, as shown in Figure 1.When elaborated according to Schild27 and plottedagainst agonist dose ratios, the antagonist concentra-tions that were used in the RJV fit a perfect right linewith a slope near unity (Figure 2); however, the dataobtained on the GPI did not fit into a Schild plot.
DiscussionThe results summarized in this article indicate that
potent and specific antagonists for the kinin B2-receptor can be obtained by replacing Pro7 withD-Phe in the nonapeptide bradykinin as suggested byVavrek and Stewart.1 This substitution is essential tochange the pharmacological spectrum from agonistto antagonist; however, [D-Phe^-BK is a weak antag-onist showing residual agonistic effects in variousbiological assays. Initially, the affinity of this com-pound was increased by replacing Phe and Phe8 withthe unnatural amino acid thienylalanine (Thi).1 Re-cent findings3-25'26 as well as the present resultsindicate that Phe5 and Phe8 need not be replacedwith Thi. Similar pA2 values were measured in thepresent study with r>Arg[Hyp3,Thi5'8,D-Phe7]-BK andwith D-Arg[Hyp3,D-Phe^-BK, suggesting that Thi5J!
does not play any role for antagonism, at least invitro. Two other chemical changes, one in position 3(the substitution of Pro3 by Hyp) and especially theaddition of a D-Arg at the N-terminal, appear to bevery useful to improve antagonist affinity, as shownbefore by Stewart and Vavrek.26
B2-receptor antagonists are not selective becausethey can be transformed by kininase I into C-terminaldesArg metabolites and thus become fairly potentanti-Bi.2 Although B,-receptors may not be present inmany organs in healthy animals and humans, they maybe generated in pathological conditions28 and espe-cially during treatment with kininase II inhibitors.29
Interference by B,-receptors can be eliminated bytreating the animal or the tissues with mergetpa, acarboxypeptidase N inhibitor34 or by the use of apseudopeptide bond, for instance ^-CH2-NH be-tween the C-terminal Phe and Arg. Such a modifica-
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from
Rhaleb et al Bradytdnin Receptor Antagonists 113
TABLE 5. Apparent Affinities and Residual Agonistic Activities of Kinln Antagonists in Various Isolated Organs
Preparation
Peptide
DCA
pA2 AA
InacInacInacInacInacInac5.00
DRA
pA2 AA
InacInacInacInacInacInac5.00
RJV
pA2 AA
InacInac<5.0 ±<5.0 ±InacInac6.04
GPI
pA2 AA
InacInac ±Inac ±InacInac ±InacInac
RVD
pA2 AA
InacInac
InacInacInac
InacInac ±
RA
pA2 AA
7.278.377.107.106.606.656.80
31. [Leu^desArg'-BK32. [Leu']desArg'°-KD33. Sar[Leu8]desArg'-BK34. D-ArgtLeu^desArg'-BK35. [Leu8]desArg'-BK.NH2
36. Sar[Leu8]desArg'-BK.NH2
37. D-ArgtHyp'.D-Phe'JdesArg'-BK
DCA, dog carotid artery; DRA, dog renal artery; RJV, rabbit jugular vein; GPI, guinea pig ileura; RVD, rat vas deferens; RA, rabbitaorta; pA2, apparent affinities; AA, residual agonistic activities; BK, bradykinin; KD, kallidin; Inac, inactive as antagonist at concentrationslower than 10"3 M; ±, weak residual agonistic activity.
tion not only prevents the action of kininase I but alsothat of kininase II.22
B2-receptor antagonists still have too much residualagonistic activity in various preparations, and they arepotent stimulants of histamine release in vitro7 and invivo.30 Residual agonistic activities can be reduced intwo ways: first by acetylating the N-terminal amine, asdemonstrated by Ac-D-Arg[Hyp3,D-Phe7]-BK, by Ac-D-Arg[Hyp3,ThP-8,D-Phe7]-BK, or by Ac-D-Arg[Hyp3,Gly6,D-Phe7,Leu8]-BK; second by replacing Phe5 andPhe8, or simply Phe8 with Leu. Indeed, the replace-ment of the aromatic residue (Phe) in position 8appears to be a useful substitution since it contributestoward improvement of the antagonist affinity andreduction of the residual agonistic effects: this is nottrue for Phe5 since its substitution by Leu appears tobe unfavorable for antagonism. These findings suggestthat Phe8 of BK may play a role in the activation ofB2-receptors similar to the way in which Phe8 ofdesArgr-BK is instrumental for the activation of the B,and the way in which Phe8 of Ang II is essential foractivation of angiotensin receptors.31 Brreceptor and
angiotensin-receptor antagonists have been obtainedsimply by replacing the C-terminal Phe with a Leu.21-31
In the present study, the antagonists could becharacterized only by measuring pA2 values becausethe majority of the compounds are in fact partialagonists. As typical partial agonists, they act asantagonists at low concentrations and become ago-nists at concentrations 10-5,000 times higher, de-pending on the compound. Because of such strongresidual activity of some compounds, pA10 was noteasily measurable, and therefore the antagonistscould be compared only by their pA2 values. This isobviously a major limitation of the present study.
Antagonists containing Leu in position 8, bothD-Arg(Hyp3,Gly6,D-Phe7,Leu8]-BK and r>Arg [Hyp3,Leu5i8,Gly*,D-Phe7]-BK showed very little residual ago-nistic effect and were therefore used to compare vari-ous Bj-receptor systems and discriminate betweentissues with high, intermediate, and low affinity forantagonists.25
Differences of pA2 values of 2 or even 3 log unitswere demonstrated with the same antagonist in the
TABLE 6. Specificity of Potent Klnin Antagonists on the Rabbit Jugular Vein
Peptide
6. Ac-D-Arg[Hyp3,Thi5J!,D-Phe7]-BK(3.36xlO"7M)
14. Ac-D-Arg[Hyp3,D-Phe7]-BK(3.81xl0"7M)
22. D-ArglHyp'.Leu^Gly'.D-Phe'J-BK(7.06x10-'M)
23. Ac-D-ArglHypM^u^Gly'-D-Phe'l-BK(7.08x10-* M)
Agonist
BK (2.02x10-'M)SP (3.27x10-'M)AngII(2.04xl0-7M)BK
SP
Ang IIBK
SP
Ang nBK
SP
Ang II
In absence
20.2±2.417.4±3.022.6±3.022.4±2.917.2±2.522.4±2.921.4+1.035.6±4.222.0±4.827.2±3.034.0±4.017.9±4.3
RJV
In presence
14.0±2.0*17.2+.2.522,4+2.98.6±2.1f
15.6±2.417.8±2.02.8±0.9*
39.8±5.326.4+4.83.2±0.5t
35.6±4.222.0+4.1
Data are presented as millimeters of contraction (30 mm=RJV, rabbit jugular vein; BK, bradykinin; SP, substance P;
•p<0.05; tp<0.01; lp<0.001.
1.0 g). Data are mean±SEM of at least six determinations.Ang II, angiotensin II.
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from
114 Hypertension Vol 17, No 1, January 1991
Rabbit Jugular Vdn
r
BK S.1 . 1<T7M
f _JANT. L8 . l<r*M ' ANT. 1.8 . 1(T7M ANT. 1.8 . 1<T*M
Guinea Pig Deurn4.0m/n.
A
BK 2.0 . Kr*MANT. L8 . 1<T*M ANT. 7.1 . W*M ANT. L8 .
FIGURE 1. Top panel: Tracings showing the myotropic effects ofbradykinin (BK) in the absence (left tracing) and in the presenceof increasing concentrations of antagonist (ANT) D-Arg[Hyp3,LeuSjS,D-Phe7]-BK. Abscissa, time; ordinate, tension.
various preparations. Such differences would suggestthe existence of different receptor types; however,when applied in high concentrations, the antagonistsare able to eliminate the biological effect of BK onlyin the RJV and not, for instance, in the GPI (Figure1), where a fairly consistent residual contraction wasfound to be resistant to treatment with antagonists. Ittherefore appears that the receptor population of theRJV is homogeneous and can be inactivated by theantagonist, whereas that of the GPI is heterogeneousand only a fraction of it can be occupied or blockedby the antagonist. In another preparation (e.g., theGPT) the effects of BK, both the excitatory and the
2.000
Io
1.000-
0.000
Log (ANT)
FIGURE 2. Plot of log dose ratio 1 (Log [DR-1J) (ordinates)against antagonist concentration (Log [ANT]) (abscissa)showing competitive antagonism of bradykinin (BK)D-Arg[Hyp3,Leu5S,D-Phe7]-BK on the rabbit jugular vein withcaptopril (9.0xlO'6 M).
inhibitory, are not affected at all either by B2 or by B]antagonists32 (N-E. Rhaleb, unpublished observa-tions); these compounds are potent stimulants of thetrachea and behave as full agonists in this tissue. Theeffects of kinins on the GPT have been analyzed anddiscussed in recent publications32 (N-E. Rhaleb, un-published observations). It has been suggested thatkinins act on this tissue by directly activating either aG protein or a phospholipase A2 without the inter-mediary of a receptor. Another interpretation, con-templated by other investigators33 is that the GPTcontains a new kinin receptor type, B3. The twointerpretations are proposed to explain the presentfindings on the GPI and on the other B2-receptorsystems in which the order of potency of agonists is inaccord with the presence with B2-receptors, whereasthe affinity of antagonists is extremely low. If twodifferent components contribute to the overall myo-tropic effect of kinins, the apparent affinity of theantagonist will necessarily be underestimated. Thisappears to be the case not only for the GPI but alsofor the HUB and possibly other isolated organs.
Because most of the compounds are partial ago-nists, Schild plots could be calculated only for somecompounds. Schild plots of one of the most potentantagonists, D-Arg[Hyp3,Leu5'8,D-Phe7]-BK werefound to be linear for the RJV but not for the GPI,and the antagonism is competitive only in the firsttissue. The noncompetitrvity of the antagonist is an-other indication that the GPI and possibly otherpreparations (for instance the RVD and the HUB),are complex organs in which kinins exert both recep-
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from

Rhaleb et at Bradykinln Receptor Antagonists 115
tor-mediated (blocked by antagonists) and non-recep-tor-mediated effects, which are not influenced by theantagonists presently available.
Acknowledgments
We acknowledge the excellent secretarial work ofCecile Theberge and the technical assistance ofMartin Boussougou, Richard Laprise, and MarieBattistini. Catherine Jouneaux and Francois Nantelcontributed to experimental work and Francois Nan-tel prepared all the tables.
References1. Vavrek RJ, Stewart JM: Competitive antagonists of bradyki-
nin. Peptides 1985;6:161-1642. Regoli D, Drapeau G, Rovero P, Dion S, Rhaleb N-E, BaraW
J, D'Orl6ans-Juste P, Ward P: Conversion of kinins and theirantagonists into B, receptor activators and blockers in isolatedvessels. Eur JPharmacol 1986;127:219-224
3. Steranka LR, Manning DC, Dehaas CJ, Ferkany JW, BaraskySA, Connor JR, Vavrek RJ, Stewart JM, Snyder SH: Brady-kinin as a pain mediator Receptors are localized to sensoryneurons, and antagonists have analgesia actions. Proc NatlAcad Sci USA 1988;85:3245-3249
4. Stewart JM, Vavrek RJ: Bradykinin competitive antagonistsfor classical kinin systems. Adv Exp Med Biol 1986;198A:537-542
5. Griesbacher T, Lembeck F: Effect of bradykinin antagonistson bradykinin-induced plasma extravasation, venoconstriction,prostaglandin E2 release, nociceptor stimulation and contrac-tion of the iris sphincter muscle in the rabbit. Br J Pharmacol1987;92:333-340
6. Devillier P, Renoux M, Giroud JP, Regoli D: Peptides andhistamine release from rat peritoneal mast cells. Eur J Phar-macol 1985;117:89-96
7. Devillier P, Renoux M, Drapeau G, Regoli D: Histaminerelease from rat peritoneal mast cells by kinin antagonists. EurJ Pharmacol 1988;149:137-140
8. Rifo J, Pourrat M, Vavrek RJ, Stewart JM, Huidobro-Toro J?:Bradykinin-receptor antagonists used to characterize the het-erogeneity of bradykinin-induced responses in the rat vasdeferens. Eur J Pharmacol 1987;142:305-312
9. Regoli D, Rhaleb N-E, Drapeau G, Dion S: Kinin receptorsubtypes. / Cardiovasc Pharmacol 1990a:15(suppl 6):S30-S38
10. Regoli D, Rhaleb N-E, Dion S, Drapeau G: New selectivebradykinin receptor antagonists and bradykinin Bj receptorcharacterization. TIPS 1990;ll:156-161
11. Plevin R, Owen PJ: Multiple B? kinin receptors in mammaliantissues. TIPS 1988;9:387-389
12. D'Orleans-Juste P, Dion S, Mizrahi J, Regoli D: Effects ofpeptides and nonpeptides on isolated arterial smooth muscles:Role of endothelium. Eur J Pharmacol 1985;114:9-21
13. Furchgott RF, Bhadrakom S: Reaction of strips of rabbit aortato epinephrine isopropyl-arterenol, sodium nitrate and otherdrugs. J Pharmacol Exp Thar 1953;108:124-143
14. Gaudreau P, Barabe J, St-Pierre S, Regoli D: Pharmacologicalstudies of kinins on venous smooth muscles. Can J Pharmacol1981;59:371-379
15. Tousignant C, Dion S, Drapeau G, Regoli D: Characterizationof pre and postjunctional receptors for neurokinins and kininsin the rat vas deferens. Ncuropeptides 1987;9:333-343
16. Vane JR: A sensitive method for the assay of 5-hydroxy-tryptamine. BrJ Pharmacol 1957;12:344-349
17. Rang HP: Stimulant actions of volatile anaesthesics on smooth'muscle. BrJ Pharmacol 1964;22:356-365
18. Constantine JW: The spirally cut tracheal strip preparation. /Pharm Pharmacol 1965;17:384-385
19. Schild HO: pA^ a new scale for the measurement of drugantagonism. Br J Pharmacol 1947;2:189-206
20. Regoli D, BaraW J: Pharmacology of bradykinin and relatedkinins. Pharmacol Rev 1980;32:l-46
21. Regoli D, BaraW J, Park WKJ Receptors for bradykinin inrabbit aortae. Can J Physiol Pharmacol 1977^5:855-867
22. Drapeau G, Regoli D: Synthesis of bradykinin analogs. Immu-nochemical Techniques: Part M. Chemotaxis and Inflammation.Methods Enzymol 1988;163:263-272
23. Sasaki Y, Coy DH: Solid-phase synthesis of peptides contain-ing the CH2-NH peptide bond isostere. Peptides 1987;8:119-121
24. Drapeau G, Rhaleb N-E, Dion S, Jukic D, Regoli D:[Phe"1i'(CHrNH)Arg'] Bradykinin, a B2 receptor selectiveagonist which is not broken down by either kininase I orkininase II. Eur J Pharmacol 1988;155:193-195
25. Regoli D, Rhaleb N-E, Drapeau G, Dion S, Tousignant C,D'Orlians-Juste P: Basic pharmacology of kinins: Pharmaco-logic receptors and other mechanisms, in Kinins V. New York,Plenum Press, 1989, pp 399-407
26. Stewart JM, Vavrek RJ: Design of bradykinin antagonists, inMarshall GR (ed): Proceedings of the 10th American PeptideSymposium. Escom Press, 1988, pp 433-437
27. Schild HO: pA, and competitive drug antagonism. Br J Phar-macol 1949;4:277-280
28. Regoli D, Marceau F, Lavigne J: Induction of Bi receptors forkinins in the rabbit by a bacterial lipopotysaccharide. Eur JPharmacol 1981;71:105-115
29. Nwator IAA, Whalley ET: Angiotensin converting enzymeinhibitors and expression of desArg'-BK (kinin Bi) receptorsin vivo. Eur J Pharmacol 1989;160:125-132
30. Lawrence ID, Warner JA, Cohan VL, Lichtenstein LM,Kagey-Sobotka A, Vavrek RJ, Stewart JM, Proud D: Induc-tion of histamine release from human skin mast cells bybradykinin analogs. Biochem Pharmacol 1989;38:227-233
31. Regoli D, Rioux F, Park WK: Pharmacology of angiotensin.Pharmacol Rev 1974;26:69-123
32. Rhaleb N-E, Dion S, D'Orl6ans-Juste P, Drapeau G, RegoliD, Browne RG: Bradykinin antagonism: Differentiationbetween peptide antagonists and antiinflammatory agents. EurJ Pharmacol 1988;151:275-279
33. Farmer SG, Burch RM, Meeker SA, Wilkins DE: Evidence fora pulmonary B3 BK receptor. Mol Pharmacol 1989;36:l-8
34. Plummer TH, Ryan TJ: A potent mercapto biproduct analoginhibitor of carboxypeptidase N. Biochem Biophys Res Com-mun 1981;98:448-454
KEY WORDS • Brkinin receptors • vascular smooth muscle •bradykinin • peptides
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from

HypertensionVol 15, No 6, Part 2, June 1990
267 Pages, Soft Cover
Proceedings of the Council forHigh Blood Pressure Research,
1989Hypertension
Cleveland, OhioSeptember 26-29,1989
Carlos M. Ferrario, MDGuest Editor
Order No 73-2075
$10 Domestic$15 International
Major Topics
Structural Adaptation
Hypertension in Blacks
The Kidney in Hypertension
CIBA Award for Hypertension Research
Sixth Annual Marion HypertensionResearch Clinical Fellowship Award: 1989
American HeartAssociation
Scientific PublishingCustomer Service
7320 Greenville AvenueDallas, TX 75231-4599
Tel (214) 706-1310Fax (214)691-2704
®
(doxazosin mesylateScored Tablets 1 mg, 2 mg, 4 mg, 8 mg
(J^^ RoerigA division of Pfizer Pharmaceuticals
©1991, Pfizer Inc. RDC069A91
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from

# ; • WN SAFE
/rlilHn-7Am UOl) sustained release(PIITIQZGITI rlUJ capsules
For hypertension
Hypertension control,not complaints
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from

90mgSR 120 mg SR
sustainedreleasecapsules
CARDIZEM SR(diltiazem HCI)For hypertension I *• • Unsurpassed efficacy• * Low side-effect profile• • Convenient bid dosage
BRIEF SUMMARYCAROIZEr SR(diltiazem hydrochloride)Sustained Release CapsulesCONTRAINDICATIONS
CARDIZEM is contramdicated in (1) patients with sick sinus syndrome exceptm the presence of a functioning ventricular pacemaker, (2) patients with second-or third-degree AV block except in the presence of a functioning ventricularpacemaker, (3) patients with hypotension (less than 90 mm Hg systolic),(4) patients who have demonstrated hypersensitivity to the drug, and (5) pa-tients with acute myocardial infarction and pulmonary congestion documentedby x-ray on admission
WARNINGS1 - Cardiac Conduction. CARDIZEM prolongs AV node refractory periods without
significantly prolonging sinus node recovery time, except in patients with sicksinus syndrome This effect may rarely result in abnormally slow heart rates(particularly in patients with sick sinus syndrome) or second- or third-degreeAV block (nine of 2,111 patients or 0.43%) Concomitant use ot diltiazem withbeta-blockers or digitalis may result m additive effects on cardiac conduc-tion A patient with Pnnzmetal's angina developed periods of asystole (2 to5 seconds) after a single dose of 60 mg of diltiazem
2. Congestive Heart Failure. Although diltia/em has 3 negative inotropic effectin isolated animal tissue preparations, hemodynamic studies in humans withnormal ventricular function have not shown a reduction in cardiac index norconsistent negative effects on contractility (dp/dt). An acute study of oraldiltiajem in patients with impaired ventricular function (ejection fraction24% •? 6%) showed improvement in indices of ventricular function withoutsignificant decrease in contractile function (dp'dt) Experience with the use otCARDIZEM (diltiazem hydrochlonde) in combination with beta-blockers inpatients with impaired ventricular function is limited Caution should beexercised when using this combination.
3. Hypotension. Decreases in blood pressure associated with CARDIZEM therapymay occasionally result m symptomatic hypotension
4. Acute Hepatic Injury. Mild elevations of transammases with and withoutconcomitant elevation in alkaline phosphatase and bilirubm have beenobserved in clinical studies Such elevations were usually transient andfrequently resolved even with continued diltiazem treatment In rare in-stances, significant elevations m enzymes such as alkaline phosphatase,LDH. SGO1. SGPT, and other phenomena consistent with acute hepatic injuryhave been noted These reactions tended to occur early after therapy initiation(I to 8 weeks) and have been reversible upon discontinuation of drug therapyThe relationship to CARDIZEM is uncertain in some cases, but probable insome. (See PRECAUTIONS)
PRECAUTIONSGeneral CARDIZEM (diltiazem hydrochlonde) is extensively metabolized by
the liver and excreted by the kidneys and in bile As with any drug given overprolonged periods, laboratory parameters should be monitored at regular inter-vals The drug should be used with caution in patients with impaired renal orhepatic function. In subacute and chronic dog and rat studies designed toproduce toxicity. high doses of diltiazem were associated with hepatic damagein special subacute hepatic studies, oral doses of 125 mg'kg and higher in ratswere associated with histologtcal changes in the liver which were reversible whenthe drug was discontinued In dogs, doses of 20 mg. kg were also associated withhepatic changes; however, these changes were reversible with continued dosing.
Dermatological events (see ADVERSE REACTIONS section) may be transientand may disappear despite continued use ot CARDIZEM However, skin eruptionsprogressing to erythema multiiorme and. or extoliative dermatitis have also beeninfrequently reported Should a dermatologic reaction persist, the drug should bediscontinued
Drug Interaction. Due to the potential for additive effects, caution and carefultitration are warranted in patients receiving CARDIZEM concomitantly with anyagents known to affect cardiac contractility and or conduction (See WARNINGS.)Pharmacologic studies indicate that there may be additive effects in prolongingAV conduction when using beta-blockers or digitalis concomitantly withCARDIZEM (SeeWARNINGS)
As with all drugs, care should be exercised when treating patients withmultiple medications. CARDIZEM undergoes biotransformation by cytochromeP-450 mined function oxidase Coadministration of CARDIZEM with other agentswhich follow the same route of biotranstormation may result m the competitiveinhibition of metabolism. Dosages of similarly metabolized drugs, particularlyIhose of low therapeutic ratio of in patients with renal and/or hepatic impairment,
may require adjustment when starting or stopping concomitantly administeredCARDIZEM to maintain optimum therapeutic blood levels.
Beta-blockers: Controlled and uncontrolled domestic studies suggest thatconcomitant use of CARDIZEM and beta-blockers or digitalis is usually welltolerated, but available data are not sufficient to predict the effects of concomi-tant treatment in patients with left ventricular dysfunction or cardiac conductionabnormalities
Administration of CARDIZEM (diltiazem hydrochlonde) com,omitant!y withpropranolol in five normal volunteers resulted in increased propranolol levels inall subjects and bioavailabihty of propranolol was increased approximately 50%If combination therapy is initiated or withdrawn m conjunction with propranolol,an adjustment in the propranolol dose may be warranted. (See WARNINGS )
Cimettdme. A study in six healthy volunteers has shown a significant increasein peak diltiazem plasma levels (58%) and area-under-t he-curve (53%) after aI-week course of cimetidme at 1,200 mg per day and diltiazem 60 mg per dayRamtidme produced smaller, nonsignificant increases The effect may be me-diated by cimetidme's known inhibition of hepatic cytochrome P-450, the enzymesystem probably responsible for the first-pass metabolism of diltiazem. Patientscurrently receiving diltiazem therapy should be carefully monitored for a changein pharmacological effect when initiating and discontinuing therapy with cimeti-dine. An adjustment in the diltiazem dose may be warranted.
Digitalis: Administration ot CARDIZEM with digoxin in 24 healthy male sub-jects increased plasma digoxin concentrations approximately 20%. Anotherinvestigator found no increase in digoxin levels in 12 patients with coronaryartery disease Since there have been conflicting results regarding the effect ofdigoxin levels, it is recommended that digoxin levels be monitored when initiat-ing, adjusting, and discontinuing CARDIZEM therapy to avoid possible over- orunder-digitahzation. (See WARNINGS.)
Anesthetics: The depression of cardiac contractility, conductivity, and auto-maticity as well as the vascular dilation associated with anesthetics may bepotentiated by calcium channel blockers When used concomitantly. anestheticsand calcium blockers should be titrated carefully
Carcinogenesis, Mutagenesis, Impairment of Fertility. A 24-month study inrats and a 21-month study in mice showed no evidence of carcinogemcity. Therewas also no mutagemc response in in vitro bacterial tests No intrinsic effect onfertility was observed in rats
Pregnancy. Category C. Reproduction studies have been conducted in mice,rats, and rabbits Administration of doses ranging from five to ten times greater(on a mg/kg basis) than the daily recommended therapeutic dose has resulted inembryo and fetal lethality These doses, in some studies, have been reported tocause skeletal abnormalities. In the perinatal/postnatal studies, there was somereduction in early individual pup weights and survival rates There was anincreased incidence of stillbirths at doses of 20 times the human dose or greater
There are no well-controlled studies in pregnant women, therefore, useCARDIZEM in pregnant women only if the potential benefit justifies the potentialrisk to the fetus.
Nursing Mothers. Diltiazem is excreted in human milk. One report suggeststhat concentrations in breast milk may approximate serum levels. If use ofCARDIZEM is deemed essential, an alternative method of infant feeding shouldbe instituted.
Pediatric Use. Safety and effectiveness in children have not been established
ADVERSE REACTIONSSerious adverse reactions have been rare in studies earned out to date, but it
should be recognized that patients with impaired ventricular function and cardiacconduction abnormalities have usually been excluded from these studies.
The adverse events described below represent events observed in clinical studiesot hypertensive patients receiving either CARDIZEM Tablets or CARDIZEM SRCapsules as well as experiences observed in studies of angina and dunng market-ing. The most common events in hypertension studies are shown m a table withrates in placebo patients shown for companson Less common events are listed bybody system, these include any adverse reactions seen in angina studies that werenot observed in hypertension studies In all hypertensive patients studied (over900), the most common adverse events were edema (9%). headache (8%),dizziness (6%), asthenia (5%), sinus bradycardia (3%). flushing (3%). and 1° AVblock (3%). Onty edema and perhaps bradycardia and dizziness were dose related.The most common events observed in clinical studies (over 2.100 patients) ofangina patients and hypertensive patients receiving CARDIZEM Tablets orCARDIZEM SR Capsules were de. greater than 1%) edema (54%). headache(4.5%). dizziness (3.4%), asthenia (2 8%), first-degree AV block (1.8%). flushing(17%), nausea (1 6%). bradycardia (1.5%). and rash (1.5%).
DOUBLE BUND PLACEBO CONTROLLEDHYPERTENSION TRIALS
Adverseheadache
AV block first degree
dimness
edema
bradycardia
ECG abnormality
asthenia
constipation
dyspepsia
nausea
palpitations
polyima
somnolence
alk phos increase
hypotension
insomnia
rash
AV block second degree
DiltiazemN=315
#pts(%)
38(12%)
24 (7.6%)
22(7%)
19 (6%)
19 (6%)
13(4.1%)
10(3.2%)
5(1.6%)
4(1.3%)
4 (1.3%)
4 (1.3%)
4(1.3%)
4(1.3%)
3(1%)
3(1%)
3(1%)
3(1%)
2 (0 6%)
Placebo11=211
#pts(%)
17(8%)
4(1.9%)
6 (2 8%)
2 (0.9%)
3(1.4%)
3(1.4%)
1 (0.5%)
2 (0.9%)
1 (0.5%)
2 (0 9%)
2 (0.9%)
2 (0.9%)-
1 (0.5%)
1 (0 5%)
1 (0.5%)
1 (0.5%)
In addition, the following events were reported infrequently (less than 1%) orhave been observed in angina trials. In many cases, the relation to drug isuncertainCardiovascular: Angina, arrhythmia, bundle branch block, tachycardia, ven
tncular extra systoles, congestive heart failure, syncopeNervous System: Amnesia, depression, gait abnormality, hallucinations, ner-
vousness, paresthesia. personality change, tinnitus, tremor,abnormal dreams
Gastrointestinal: Anorexia, diarrhea, dysgeusia. mild elevations of SGOT, SGPT.and LDH (see hepatic warnings) vomiting weight increasethirst,
uermatological. Petechiae. pruritus, photosensitivity, urticariaOther: Amblyopia, CPK increase, dyspnea, epistaxis, eye irritation,
hyperglycemia. sexual difficulties, nasal congestion, noctuna,osteoarticular pain, impotence, dry mouth.
The following postmarking events have been reported infrequently in pa-tients receiving CARDIZEM alopecia, gingival hyperplasia, erythema multiforme,and leukopema Definitive cause and effect relationship between these eventsand CARDIZEM therapy cannot yet be established.
Issued 1/89
flMARION MIRKFLL DOW INC.
P R 0 I) U f. T S D I V I S I O N
K A \ i A h i . I I 1 . M O 0 4 1 1 4
CSRAC210 0191 CO
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from

N E Rhaleb, S Télémaque, N Rouissi, S Dion, D Jukic, G Drapeau and D RegoliStructure-activity studies of bradykinin and related peptides. B2-receptor antagonists.
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 1991 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/01.HYP.17.1.107
1991;17:107-115Hypertension.
http://hyper.ahajournals.org/content/17/1/107World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer process is available in the
Request Permissions in the middle column of the Web page under Services. Further information about thisOffice. Once the online version of the published article for which permission is being requested is located, click
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertension Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on May 18, 2018
http://hyper.ahajournals.org/D
ownloaded from





























