Embed Size (px)
Citation preview

WMC-79, a potent agent against colon cancers, inducesapoptosis through a p53-dependent pathway
Teresa Kosakowska-Cholody,1
W. Marek Cholody,1 Anne Monks,2
Barbara A. Woynarowska,3
and Christopher J. Michejda1
1Molecular Aspects of Drug Design, Structural BiophysicsLaboratory, Center for Cancer Research; 2Screening TechnologiesBranch, Laboratory of Functional Genomics, Science ApplicationsInternational Corporation, National Cancer Institute at Frederick,Frederick, Maryland; and 3Department of Radiation Oncology,University of Texas Health Science Center, San Antonio, Texas
AbstractWMC-79 is a synthetic agent with potent activityagainst colon and hematopoietic tumors. In vitro, theagent is most potent against colon cancer cells thatcarry the wild-type p53 tumor suppressor gene (HCT-116 and RKO cells: GI50 <1 nmol/L, LC50 f40 nmol/L).Growth arrest of HCT-116 and RKO cells occurs at theG1 and G2-M check points at sublethal concentrations(10 nmol/L) but the entire cell population was killed at100 nmol/L. WMC-79 is localized to the nucleus whereit binds to DNA. We hypothesized that WMC-79 bindingto DNA is recognized as an unrepairable damage in thetumor cells, which results in p53 activation. Thistriggers transcriptional up-regulation of p53-dependentgenes involved in replication, cell cycle progression,growth arrest, and apoptosis as evidenced by DNAmicroarrays. The change in the transcriptional profile ofHCT-116 cells is followed by a change in the levels ofcell cycle regulatory proteins and apoptosis. Therecruitment of the p53-dependent apoptosis pathwaywas suggested by the up-regulation of p53, p21, Bax,DR-4, DR-5, and p53 phosphorylated on Ser15; down-regulation of Bcl-2; and activation of caspase-8, -9, -7,and -3 in cells treated with 100 nmol/L WMC-79.Apoptosis was also evident from the flow cytometricstudies of drug-treated HCT-116 cells as well as fromthe appearance of nuclear fragmentation. However,whereas this pathway is important in wild-type p53colon tumors, other pathways are also in operationbecause colon cancer cell lines in which the p53 gene ismutated are also affected by higher concentrationsof WMC-79. [Mol Cancer Ther 2005;4(10):1617–27]
IntroductionThe bisimidazoacridones are bifunctional antitumoragents with strong selectivity against colon cancers (1, 2).Recent studies of the effect of bisimidazoacridones onsensitive colon tumors cells revealed that these com-pounds act as cytostatic agents that completely arrest cellgrowth at G1 and G2-M check points but do not triggercell death even at high concentrations (10 Amol/L; ref. 3).The chemical structure of bisimidazoacridones is symmet-rical in that it consists of two imidazoacridone moietiesheld together by linkers of various lengths and rigidities.We recently reported on the synthesis of unsymmetricalvariants of the original bisimidazoacridones (4). WMC-79(Fig. 1), a compound consisting of an imidazoacridonemoiety linked to a 3-nitronaphthalimide moiety via1,4-bispropenopiperazine linker, was found to be a potentbut selective cytotoxic agent in a variety of tumor celllines (4). However, it was more toxic against tumor celllines that carry the wild-type p53 tumor suppressor gene.The p53 protein is a tightly regulated transcription factor
that is elevated in response to DNA damage and has acritical function in maintaining the integrity of the genome.p53-driven cell cycle arrest prevents cells with altered DNAfrom proliferating and p53-controlled apoptosis selectivelyeliminates severely damaged cells (5–8). Whether the cellenters growth arrest or undergoes apoptosis depends onthe final integration of incoming signals with antagonisticeffects on cell growth. Many factors affect the cellularresponse to activated p53. These include cell type,oncogenic status of the cell, survival stimuli, intensity ofstress signals, level of p53 expression, and interaction ofp53 with specific proteins (9).We hypothesized that WMC-79 binding to DNA is
recognized as a damage that is not readily repaired in thetumor cells and which results in the activation of p53.The aim of this study was to investigate the molecular
mechanism by which WMC-79 induces growth arrest andapoptosis in the sensitive HCT-116 and RKO colon cancercell lines and to determine the role of p53 in thismechanism.
Materials andMethodsChemicalsAll mammalian cell culture reagents and trypan blue
were purchased from Life Technologies, Invitrogen Cor-poration (Grand Island, NY). Other reagents were fromSigma-Aldrich (St. Louis, MO).
Cell CultureThe human colon cancer cell lines HCT-116 p53+/+, HCT-
116 p53�/�, HCT-116 p21+/+, and HCT-116 p21�/� werea generous gift from Dr. Bert Vogelstein (Johns HopkinsUniversity, Baltimore, MD) and were maintained in
Received 5/24/05; revised 7/11/05; accepted 7/25/05.
Requests for reprints: Christopher J. Michejda, Molecular Aspects of DrugDesign, Structural Biophysics Laboratory, Center for Cancer Research,National Cancer Institute at Frederick, Frederick, MD 21702.Phone: 301-846-1216; Fax: 301-846-6231. E-mail: [email protected]
Copyright C 2005 American Association for Cancer Research.
doi:10.1158/1535-7163.MCT-05-0170
1617
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from

McCoy’s 5A media. Human colon carcinoma HCT-116(p53+/+), RKO (p53+/+), HCT-15 (p53+/�), COLO-205(p53�/�), and HT-29 (p53�/�) cells were purchased fromthe American Type Culture Collection (Rockville, MD).HCT-116, RKO, and HT-29 cells were grown in DMEM;COLO-205 and HCT-15 cells were grown in RPMI 1640. Allmedia were supplemented with 10% heat-inactivated fetalbovine serum, 2 mmol/L L-glutamine, 100 units/mLpenicillin, and 100 Ag/mL streptomycin. The cells weregrown at 37jC in a humidified atmosphere consisting of5% CO2 and 95% air.
Drugs and Drug Preparation ProcedureStock solution of WMC-79 synthesized in our laboratory
(4) was freshly prepared by dissolving the free base formof the compound in 2 equivalents of methanesulfonic acid(as 10 mmol/L water solution) and then diluted with waterto a final concentration of 500 Amol/L. This solution wasused to prepare 2 Amol/L working solution and its 10-foldserial dilutions in appropriate complete tissue culturemedia.
CellViability3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium
Bromide Assay. Cellular growth in the presence orabsence of experimental agents was determined usingthe 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT)–based CellTiter96 Non-Radioactive CellProliferation Assay (Promega, Madison, WI) according tothe instruction of the manufacturer with small modifica-tions as previously described (4, 10). Trypan blue exclusionassay was used to determine the number of live/dead cellsin HCT-116 cultures exposed to WMC-79.Fluorescence-Activated Cell Sorting. Tumor cells in
exponential phase of growth were seeded at a density of0.5 � 106 to 1 � 106 cells in 25 or 75 cm2 T flasks, allowed toattach for 24 hours, and then exposed to 10 or 100 nmol/LWMC-79. At appropriate intervals, drug-treated andcontrol cells (attached and floating) were collected andwashed twice in ice-cold PBS containing 1% fetal bovineserum. The cells were fixed in 70% ethanol and stored at�20jC until all time points had been collected. Fixed cellswere rinsed twice in ice-cold PBS containing 10% fetalbovine serum, treated with RNase A (1 unit/106 cells) for30 minutes at 37jC, and stained overnight with propidiumiodide (50 Ag/mL) at 4jC. Cell cycle analysis was doneon a Beckman Coulter Epics XL-MCL flow cytometer(Fullerton, CA) with 10,000 events per collected sample.
Cellular Drug Localization by Confocal MicroscopyCells in logarithmic growth phase were harvested by
trypsinization and 50,000 to 100,000 cells were seeded in
35-mm glass-bottomed microwell dishes (MatTek Corpo-ration, Ashland, MA). The following day, cells werewashed and fresh medium containing 100 nmol/L WMC-79 was added. Cells were examined at different time pointsunder a Zeiss 410 laser scanning confocal microscope.Areas were imaged using appropriate laser lines for WMC-79 excitation (488 nm).
Western Blot AnalysisImmunoblot analysis of cell protein lysates was done
according to the protocol of the manufacturer (Santa CruzBiotechnology, Inc., Santa Cruz, CA). Briefly, cells werelysed on ice for 30 to 60 minutes in radioimmunopreci-pitation assay buffer (1� PBS, 1% igepal, 0.5% sodiumdeoxycholate, 0.1% SDS) with freshly added inhibitors(10 Ag/mL phenylmethylsulfonyl fluoride, 50 Ag/mLaprotinin, and 1 mmol/L sodium orthovanadate). Celllysate was passed through a 21-gauge needle followed bycentrifugation at 10,000 � g for 10 minutes at 4jC. Proteinconcentration was determined using Bio-Rad protein assay(Bio-Rad Laboratories, Hercules, CA). Samples weremixed with 2� Laemmli buffer, denaturated at 100jC for3 minutes, and proteins were separated by electrophoresis(NuPAGE 4-12% Bis-Tris Gel, Invitrogen, Life Technologies,Carlsbad, CA). Separated proteins were transferred topolyvinylidene difluoride membrane (Millipore, Bedford,MA) and subjected to immunoblottingwith various primaryantibodies. Positive antibody reactions were visualized witha horseradish peroxidase–conujugated secondary antibodyand an enhanced chemiluminescence detection system(Amersham Pharmacia Biotech, Little Chalfont, UnitedKingdom) according to the protocol of the manufacturer.The membrane was then deprobed and reprobed with ananti-actin antibody to confirm that all samples containedsimilar amounts of proteins. TBS-0.05% Tween 20 was usedas a wash buffer; 5% nonfat dry milk (Bio-Rad Laboratories)was dissolved in TBS-0.05% Tween 20 and was used as ablocking solution. The following antibodies were used inthis study: mouse anti-Bax (Ab-3), mouse anti-Bcl-2 (Ab-1),mouse anti–cyclin D1 (Ab-3), mouse anti-E2F1 (Ab-1),mouse anti-Mdm2 (Ab-2), mouse anti-p21 (Ab-1), mouseanti-p53 (Ab-6), mouse anti-pRb (Ab-5), rabbit anti-caspase-7 (AB-1), rabbit anti-DR4 (AB-1), rabbit anti-DR5 (AB-2),rabbit anti-phospho-p53(Ser15) (Ab-3; Oncogene ResearchProducts, Boston, MA), goat anti-actin (C-11), goat anti-caspase-8 (C-20), rabbit anti-caspase-9 (PharMingen, SanDiego, CA), mouse anti–cyclin A, mouse anti–cyclin B1,rabbit anti–cyclin E (Biosource International, Camarillo,CA), and rabbit anti-phospho-Cdc2(Tyr15) (Cell SignalingTechnology, Beverly, MA). All secondary antibodies (horse-radish peroxidase conjugates) and Cruz Marker molecularweight standards were from Santa Cruz Biotechnology.
Gene Expression ProfilingHuman HCT-116 colon adenocarcinoma cells at 60%
confluency were exposed to 100 nmol/L WMC-79 for3, 12, 24, and 72 hours. Total RNAs from tested anduntreated cells were isolated using RNeasy Mini Kit(Qiagen, Valencia, CA) according to the instructions of themanufacturer. RNA was checked for purity and stability
Figure 1. Chemical structure of WMC-79.
WMC-79 Induces Apoptosis in Colon Cancer Cells1618
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from

by gel electrophoresis. Drug-induced gene expressionchanges were evaluated by competitive hybridization ofequal amounts of control versus drug-treated cDNAusing 20K oligonucleotide microarrays (Hs-OperonV2-vB1.2p17-092903) purchased from the Advanced Tech-nology Center, Center for Cancer Research, NationalCancer Institute (Gaithersburg, MD) and data wereanalyzed through the Computer Information TechnologyCenter mAdb website. Samples from two separateexperiments were evaluated; experiment 1 involved 24and 72 hours of exposure to the drug whereas experi-ment 2 involved 3, 12, and 24 hours of treatment. Eachsample was run on a single microarray (duplicate 24-hourtreatment). Gene expression changes common to all treat-ment conditions were selected based on those genes show-ing >3-fold change in expression in any two of the fivearrays.
ResultsActivity ofWMC-79 against Human ColonTumor Cell
Lines Depends on the Status of p53The inhibition of cell proliferation and/or cell cytotox-
icity induced by WMC-79 was measured by MTT assay,trypan blue exclusion, and cell cycle analysis. Initialexperiments in colon cancer cell lines indicated that thosecarrying the wild-type p53 gene (HCT-116 and RKO) werethe most sensitive, especially at the TGI and LC50 levels(Table 1). In follow-up experiments with isogenic HCT-116cell lines engineered to express or be null for either p53 orp21 (HCT-116 p53+/+, HCT-116 p53�/�, HCT-116 p21+/+,and HCT-116 p21�/�), HCT p53�/� cells were f10-foldresistant to WMC-79 as compared with HCT-116 p53+/+
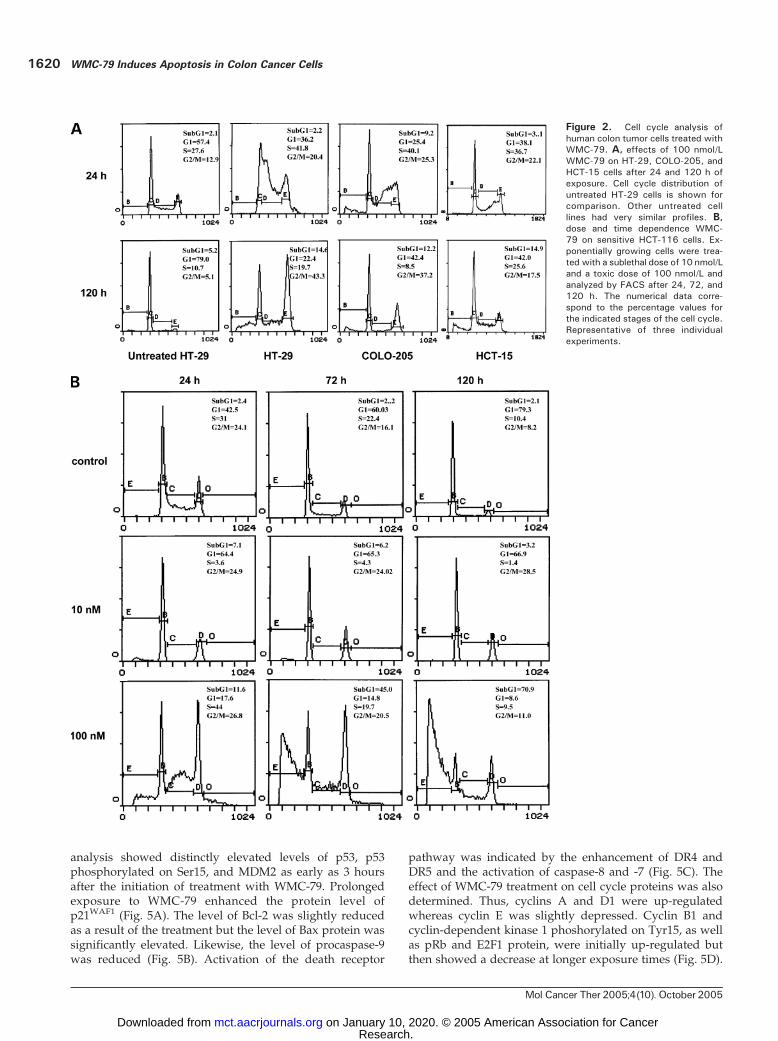
whereas HCT-116 p21�/� cells were most sensitive to thistreatment (Table 1). As evidenced by fluorescence-activatedcell sorting (FACS) analysis, 24-hour treatment of HT-29,COLO-205, HCT-15, and HCT-116 colon cancer cells with100 nmol/L WMC-79 caused p53-independent transient
accumulation of cells in S phase (Fig. 2) but prolongedexposure to the drug led to G2-M growth arrest in cellswith mutated p53 gene (Fig. 2A) and apoptosis in cells withwild-type p53 (Fig. 2B).The p53 wild-type cell lines, HCT-116 and RKO, were
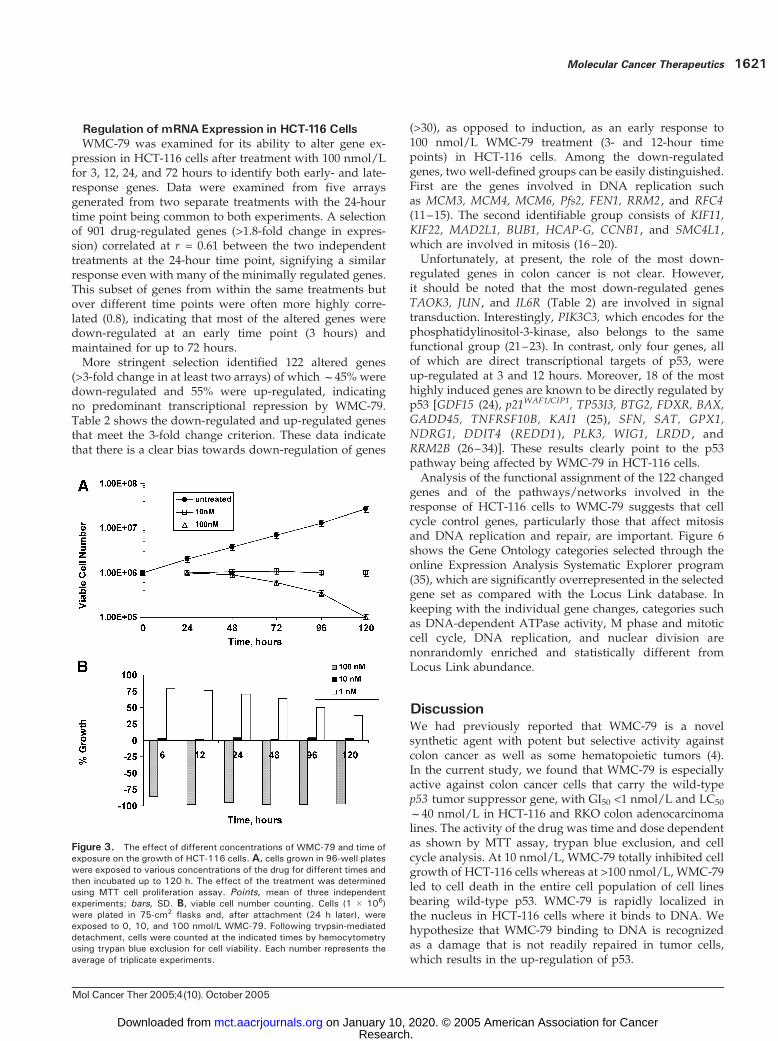
further used for a more extensive evaluation of thecytotoxic properties of WMC-79. A detailed study of theeffect of WMC-79 treatment on dose and duration oftreatment was carried out. WMC-79 at 10 nmol/L totallyinhibited cell growth without evidence of cell death evenafter 120 hours of exposure (Figs. 3A and B and 2B).However, the cell growth arrest seemed to be irreversibleas 6-hour exposure followed by 120-hour incubation indrug-free medium resulted in the same level of growthinhibition as 120-hour continuous drug exposure (Fig. 3A).Because the MTT viability assay cannot distinguishbetween total growth arrest and equilibrium betweengrowth and death, we did additional experiments in whichHCT-116 cultures exposed to 10 or 100 nmol/L WMC-79were analyzed by counting of trypan blue–stained cells atvarious time points (Fig. 3B). At 10 nmol/L WMC-79, cellnumber was constant for the duration of the experiment(the number of dead cells was negligible). FACS analysis ofHCT-116 cells exposed to 10 nmol/L WMC-79 (Fig. 2B)showed an apparent cell growth arrest at G1 and G2-Mphases with complete depletion of S phase. This growtharrest persisted for the rest of the experiment with nodistinct evidence of cell death at the 120-hour time point.In contrast, 100 nmol/L WMC-79 was clearly cytotoxic
to these cells (Figs. 3A and B and 2B). As evidenced byFACS analysis, we observed initial accumulation of HCT-116 cells in S phase during the first 24 hours (from 31% forcontrol to 44% for treated cells), a marked decreased in G1
phase (from 42.5% to 17.6%), and then massive cell death,which was evidenced by the appearance of sub-G1 cellpopulation. This fraction increased steadily with time ofexposure to the drug (Fig. 2B). Furthermore, a 6-hourexposure to 100 nmol/L WMC-79 followed by drugwashout was sufficient to induce exactly the same cytotoxicresult as that with continuous treatment with the drug(Fig. 3A). The cytotoxic effect of WMC-79 at 100 nmol/Lwas also confirmed by cell counting (Fig. 3B).
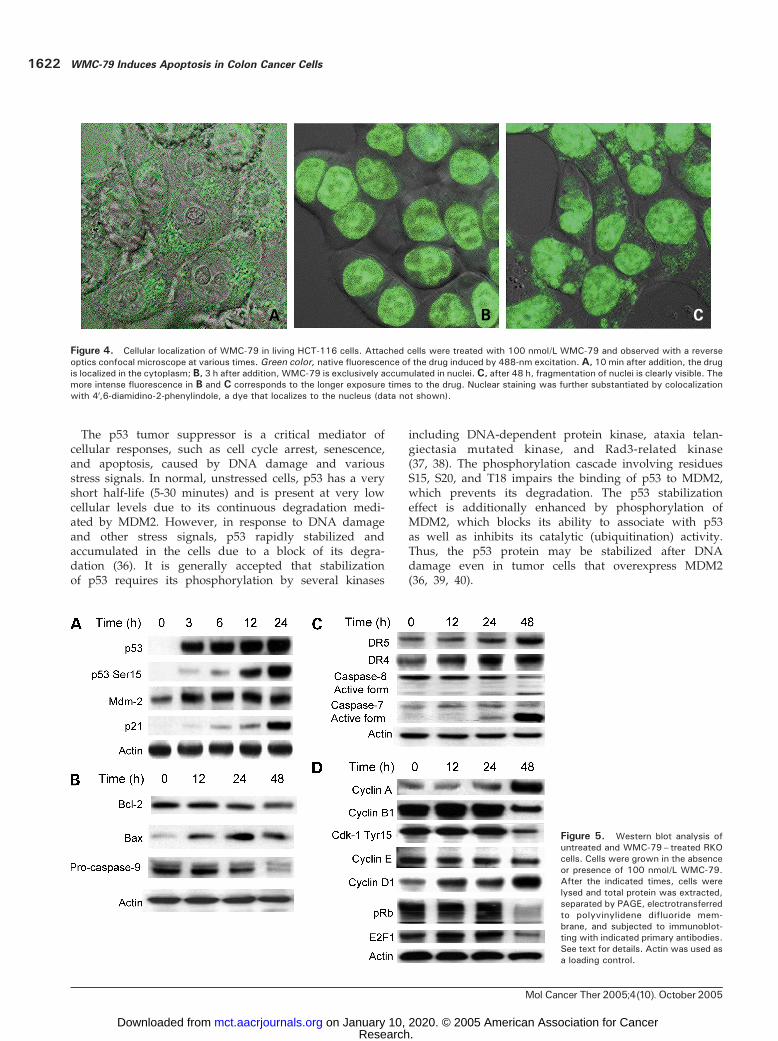
Cellular Drug LocalizationTo study WMC-79 localization in target cells, we took
advantage of the intrinsic fluorescence of the drug thatallowed direct visualization by confocal microscopy.Figure 4 presents the distribution of WMC-79 in HCT-116cells. The drug easily crosses the cellular membrane and,within hours, accumulates in the nucleus where it staysuntil cell death, which for this cell line (HCT-116) is visibleafter 48 hours (fragmentation of nucleus).
Western Blot AnalysisWe examined the effect of WMC-79 on expression of
various cell cycle regulators and apoptosis-regulatingproteins in RKO and HCT-116 cells. The time-dependenteffects of exposure to 100 nmol/L WMC-79 on the proteinlevels in RKO cells are shown in Fig. 5A to D, and verysimilar results were seen for HCT-116 cells. Western blot
Table 1. Activity of WMC-79 against selected human coloncancer cell lines
Cell line p53 status GI50*(nmol/L)
TGIc
(nmol/L)LC50
b
(nmol/L)
HCT-116 p53+/+ <1 10 F 1 40 F 10HCT-116 p53�/� 4 F 0.4 90 F 11 400 F 50HCT-116 p53+/+ p21+/+ <1 10 F 1 100 F 15HCT-116 p53+/+ p21�/� <1 10 F 1 35 F 5RKO p53+/+ <1 10 F 1 35 F 5HCT-15 p53+/� 3.3 F 0.5 100 F 15 430 F 60COLO-205 p53�/� 2.2 F 0.3 120 F 20 450 F 50HT-29 p53�/� 1.7 F 0.2 100 F 20 500 F 70
NOTE: The GI50, TGI, and LC50 values are the average of at least threeindependent determinations.*GI50, concentration of the drug resulting in inhibition of cell growth to50% of controls.cTGI, concentration of the drug resulting in total growth inhibition.bLC50, concentration of the drug required to reduce the initial cell numberby 50%.
Molecular Cancer Therapeutics 1619
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from
analysis showed distinctly elevated levels of p53, p53phosphorylated on Ser15, and MDM2 as early as 3 hoursafter the initiation of treatment with WMC-79. Prolongedexposure to WMC-79 enhanced the protein level ofp21WAF1 (Fig. 5A). The level of Bcl-2 was slightly reducedas a result of the treatment but the level of Bax protein wassignificantly elevated. Likewise, the level of procaspase-9was reduced (Fig. 5B). Activation of the death receptor
pathway was indicated by the enhancement of DR4 andDR5 and the activation of caspase-8 and -7 (Fig. 5C). Theeffect of WMC-79 treatment on cell cycle proteins was alsodetermined. Thus, cyclins A and D1 were up-regulatedwhereas cyclin E was slightly depressed. Cyclin B1 andcyclin-dependent kinase 1 phoshorylated on Tyr15, as wellas pRb and E2F1 protein, were initially up-regulated butthen showed a decrease at longer exposure times (Fig. 5D).
Figure 2. Cell cycle analysis ofhuman colon tumor cells treated withWMC-79. A, effects of 100 nmol/LWMC-79 on HT-29, COLO-205, andHCT-15 cells after 24 and 120 h ofexposure. Cell cycle distribution ofuntreated HT-29 cells is shown forcomparison. Other untreated celllines had very similar profiles. B,dose and time dependence WMC-79 on sensitive HCT-116 cells. Ex-ponentially growing cells were trea-ted with a sublethal dose of 10 nmol/Land a toxic dose of 100 nmol/L andanalyzed by FACS after 24, 72, and120 h. The numerical data corre-spond to the percentage values forthe indicated stages of the cell cycle.Representative of three individualexperiments.
WMC-79 Induces Apoptosis in Colon Cancer Cells1620
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from
Regulation ofmRNAExpression in HCT-116 CellsWMC-79 was examined for its ability to alter gene ex-
pression in HCT-116 cells after treatment with 100 nmol/Lfor 3, 12, 24, and 72 hours to identify both early- and late-response genes. Data were examined from five arraysgenerated from two separate treatments with the 24-hourtime point being common to both experiments. A selectionof 901 drug-regulated genes (>1.8-fold change in expres-sion) correlated at r = 0.61 between the two independenttreatments at the 24-hour time point, signifying a similarresponse even with many of the minimally regulated genes.This subset of genes from within the same treatments butover different time points were often more highly corre-lated (0.8), indicating that most of the altered genes weredown-regulated at an early time point (3 hours) andmaintained for up to 72 hours.More stringent selection identified 122 altered genes
(>3-fold change in at least two arrays) of which f45% weredown-regulated and 55% were up-regulated, indicatingno predominant transcriptional repression by WMC-79.Table 2 shows the down-regulated and up-regulated genesthat meet the 3-fold change criterion. These data indicatethat there is a clear bias towards down-regulation of genes
(>30), as opposed to induction, as an early response to100 nmol/L WMC-79 treatment (3- and 12-hour timepoints) in HCT-116 cells. Among the down-regulatedgenes, two well-defined groups can be easily distinguished.First are the genes involved in DNA replication suchas MCM3, MCM4, MCM6, Pfs2, FEN1, RRM2 , and RFC4(11–15). The second identifiable group consists of KIF11,KIF22, MAD2L1, BUB1, HCAP-G, CCNB1 , and SMC4L1 ,which are involved in mitosis (16–20).Unfortunately, at present, the role of the most down-
regulated genes in colon cancer is not clear. However,it should be noted that the most down-regulated genesTAOK3, JUN , and IL6R (Table 2) are involved in signaltransduction. Interestingly, PIK3C3, which encodes for thephosphatidylinositol-3-kinase, also belongs to the samefunctional group (21–23). In contrast, only four genes, allof which are direct transcriptional targets of p53, wereup-regulated at 3 and 12 hours. Moreover, 18 of the mosthighly induced genes are known to be directly regulated byp53 [GDF15 (24), p21WAF1/CIP1, TP53I3, BTG2, FDXR, BAX,GADD45, TNFRSF10B, KAI1 (25), SFN, SAT, GPX1,NDRG1, DDIT4 (REDD1 ), PLK3, WIG1, LRDD , andRRM2B (26–34)]. These results clearly point to the p53pathway being affected by WMC-79 in HCT-116 cells.Analysis of the functional assignment of the 122 changed
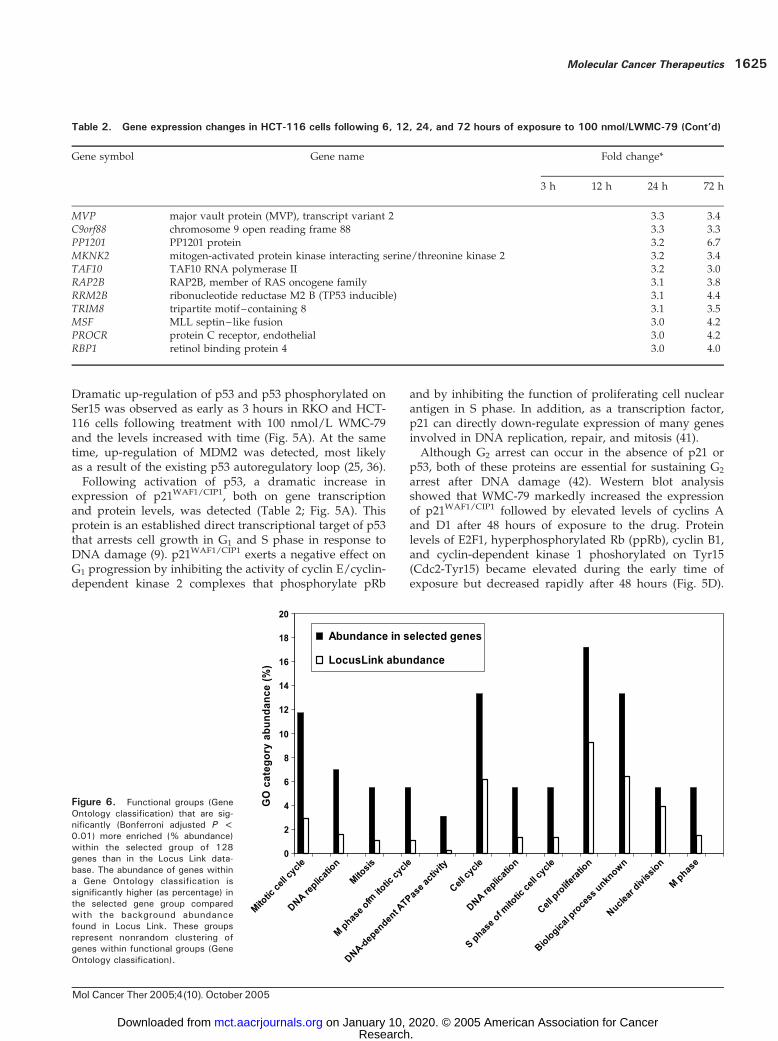
genes and of the pathways/networks involved in theresponse of HCT-116 cells to WMC-79 suggests that cellcycle control genes, particularly those that affect mitosisand DNA replication and repair, are important. Figure 6shows the Gene Ontology categories selected through theonline Expression Analysis Systematic Explorer program(35), which are significantly overrepresented in the selectedgene set as compared with the Locus Link database. Inkeeping with the individual gene changes, categories suchas DNA-dependent ATPase activity, M phase and mitoticcell cycle, DNA replication, and nuclear division arenonrandomly enriched and statistically different fromLocus Link abundance.
DiscussionWe had previously reported that WMC-79 is a novelsynthetic agent with potent but selective activity againstcolon cancer as well as some hematopoietic tumors (4).In the current study, we found that WMC-79 is especiallyactive against colon cancer cells that carry the wild-typep53 tumor suppressor gene, with GI50 <1 nmol/L and LC50
f40 nmol/L in HCT-116 and RKO colon adenocarcinomalines. The activity of the drug was time and dose dependentas shown by MTT assay, trypan blue exclusion, and cellcycle analysis. At 10 nmol/L, WMC-79 totally inhibited cellgrowth of HCT-116 cells whereas at >100 nmol/L, WMC-79led to cell death in the entire cell population of cell linesbearing wild-type p53. WMC-79 is rapidly localized inthe nucleus in HCT-116 cells where it binds to DNA. Wehypothesize that WMC-79 binding to DNA is recognizedas a damage that is not readily repaired in tumor cells,which results in the up-regulation of p53.
Figure 3. The effect of different concentrations of WMC-79 and time ofexposure on the growth of HCT-116 cells. A, cells grown in 96-well plateswere exposed to various concentrations of the drug for different times andthen incubated up to 120 h. The effect of the treatment was determinedusing MTT cell proliferation assay. Points, mean of three independentexperiments; bars, SD. B, viable cell number counting. Cells (1 � 106)were plated in 75-cm2 flasks and, after attachment (24 h later), wereexposed to 0, 10, and 100 nmol/L WMC-79. Following trypsin-mediateddetachment, cells were counted at the indicated times by hemocytometryusing trypan blue exclusion for cell viability. Each number represents theaverage of triplicate experiments.
Molecular Cancer Therapeutics 1621
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from
The p53 tumor suppressor is a critical mediator ofcellular responses, such as cell cycle arrest, senescence,and apoptosis, caused by DNA damage and variousstress signals. In normal, unstressed cells, p53 has a veryshort half-life (5-30 minutes) and is present at very lowcellular levels due to its continuous degradation medi-ated by MDM2. However, in response to DNA damageand other stress signals, p53 rapidly stabilized andaccumulated in the cells due to a block of its degra-dation (36). It is generally accepted that stabilizationof p53 requires its phosphorylation by several kinases
including DNA-dependent protein kinase, ataxia telan-giectasia mutated kinase, and Rad3-related kinase(37, 38). The phosphorylation cascade involving residuesS15, S20, and T18 impairs the binding of p53 to MDM2,which prevents its degradation. The p53 stabilizationeffect is additionally enhanced by phosphorylation ofMDM2, which blocks its ability to associate with p53as well as inhibits its catalytic (ubiquitination) activity.Thus, the p53 protein may be stabilized after DNAdamage even in tumor cells that overexpress MDM2(36, 39, 40).
Figure 4. Cellular localization of WMC-79 in living HCT-116 cells. Attached cells were treated with 100 nmol/L WMC-79 and observed with a reverseoptics confocal microscope at various times. Green color, native fluorescence of the drug induced by 488-nm excitation. A, 10 min after addition, the drugis localized in the cytoplasm; B, 3 h after addition, WMC-79 is exclusively accumulated in nuclei. C, after 48 h, fragmentation of nuclei is clearly visible. Themore intense fluorescence in B and C corresponds to the longer exposure times to the drug. Nuclear staining was further substantiated by colocalizationwith 4V,6-diamidino-2-phenylindole, a dye that localizes to the nucleus (data not shown).
Figure 5. Western blot analysis ofuntreated and WMC-79– treated RKOcells. Cells were grown in the absenceor presence of 100 nmol/L WMC-79.After the indicated times, cells werelysed and total protein was extracted,separated by PAGE, electrotransferredto polyvinylidene difluoride mem-brane, and subjected to immunoblot-ting with indicated primary antibodies.See text for details. Actin was used asa loading control.
WMC-79 Induces Apoptosis in Colon Cancer Cells1622
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from
Table 2. Gene expression changes in HCT-116 cells following 6, 12, 24, and 72 hours of exposure to 100 nmol/LWMC-79
Gene symbol Gene name Fold change*
3 h 12 h 24 h 72 h
A. Down-regulated genes
TAOK3 TAO kinase 3 18.2 16.7 26.6JUN v-jun sarcoma virus 17 oncogene homologue 11.6 8.5 11.8IL6R interleukin 6 receptor 10.5 11.8LOC90668 hypothetical protein BC008134 10.1 10.6 9.8INSIG2 insulin-induced gene 2 9.4 13.3 8.11PHF6 PHD finger protein 6 9.2 16.5 12.2AGTR2 angiotensin II receptor, type 2 6.2 10.4 7.8ZCCHC4 zinc finger, CCHC domain containing 4 5.8 4.4 4.8CCNJ cyclin J 5.2 5.5 4.7FBXW7 F-box and WD-40 domain protein 7 5.1 6.6 3.0FLJ13273 hypothetical protein FLJ13273 4.9 4.4 4.3CG018 hypothetical gene CG018 4.8 6.3 3.0GPHN gephyrin 4.6 29.1 8.2PEX5L peroxisomal biogenesis factor 5– like 4.5 4.3 4.3MCSP mitochondrial capsule selenoprotein 4.1 3.8 4.3LDB3 LIM domain binding 3 4.1 4.7 7.4CYP7A1 cytochrome P450, family 7, subfamily A, polypeptide 1 4.0 4.0SYNPO2 synaptopodin 2 3.7 3.9 3.0TIE1 tyrosine kinase with immunoglobulin-like and
epidermal growth factor– like domains 13.6 4.1
TCF15 transcription factor 15 (basic helix-loop-helix) 3.6 3.5JUB jub, ajuba homologue (Xenopuslaevis) 3.5 3.7 3.4PIK3C3 phosphoinositide-3-kinase, class 3 3.3 3.7PTPRZI protein tyrosine phosphatase, receptor-type, Z polypeptide 1 3.3 4.9 3.2GPHA2 glycoprotein hormone a 2 3.2 3.2 3.3STAR steroidogenic acute regulator 3.3 6.2 4.0ATP5S ATP synthase, H-transporting, mitochondrial F0 complex 3.2 6.3TFAP4 transcription factor AP-4 (activating enhancer binding protein 4) 4.7 3.5RAB43 RAB43, member RAS oncogene family 4.2 3.0PARP9 poly(ADP-ribose) polymerase family, member 9 4.1 5.7SEC31L2 SEC31-like 2 (S. cerevisiae) 3.8 4.3CCR2 chemokine (C-C motif) receptor 2 3.6 4.0MITF microphthalmia-associated transcription factor 3.3 4.1SLC19A1 solute carrier family 19 (folate transporter), member 1 3.3 3.4SMC4L1 SMC4 structural maintenance of chromosomes 4–like 1 (yeast) 3.3 3.0RRM2 ribonucleotide reductase M2 polypeptide 7.9 3.7PPP2R3A protein phosphatase 2 (formerly 2A), regulatory subunit B00, a 7.8 4.4Pfs2 DNA replication complex GINS protein PSF2 6.9IFNAR1 IFN (a, h, and N) receptor 1 5.3 3.9TUBB tubulin, h polypeptide 4.9 3.0PLA2G2E phospholipase A2, group IIE 4.5ZNF587 zinc finger protein 587 4.0 3.6KIF22 kinesin family member 22 3.9 4.6K-ALPHA-1 tubulin, a, ubiquitous 3.9 3.9BUB1 BUB1 budding uninhabited by benzimidazoles 1 homologue (yeast) 3.9MCM4 MCM4 minichromosome maintenance deficient 4 (S. cerevisiae) 3.9CCNB1 cyclin B1 3.8 4.4FEN1 flap structure–specific endonuclease 1 3.7H2AFFP H2A histone family, member F, pseudogene 3.6 3.0HIST1H2APS5 histone 1, H2a, pseudogene 5 3.6 3.0HCAP-G chromosome condensation protein G 3.6MCM3 MCM3 minichromosome maintenance deficient 3 (S. cerevisiae) 3.3MAD2L1 MAD2 mitotic arrest deficient– like 1 (yeast) 3.3
(Continued on the following page)
*No entry indicates gene expression change <3.
Molecular Cancer Therapeutics 1623
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from
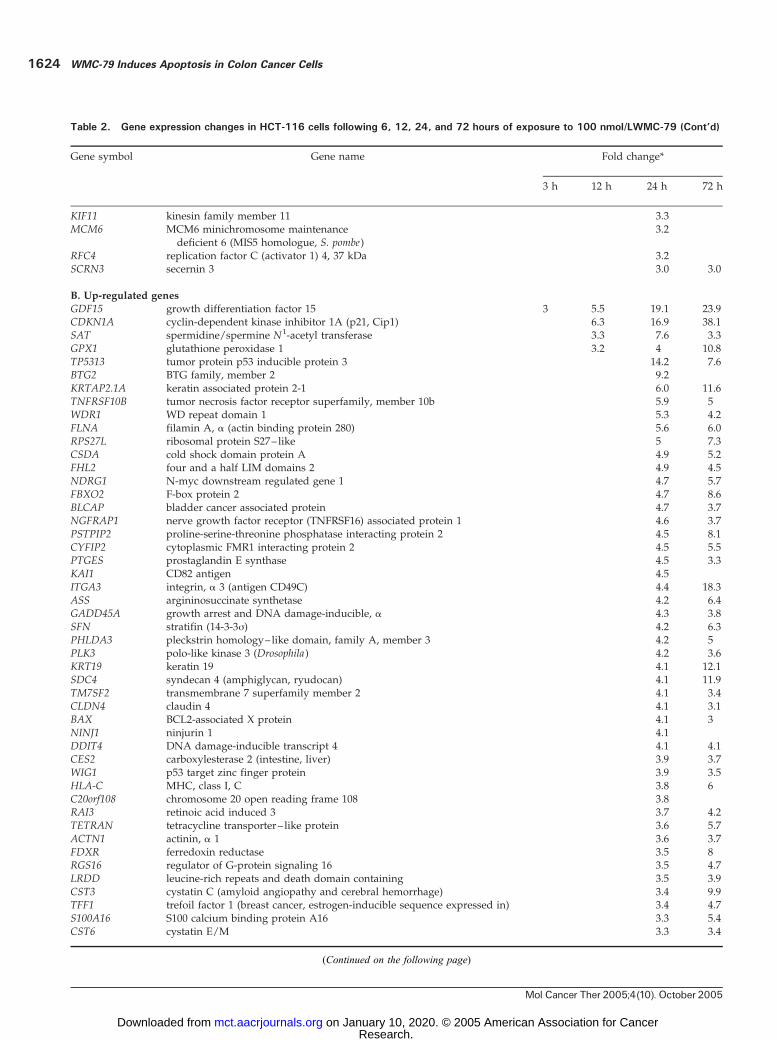
Table 2. Gene expression changes in HCT-116 cells following 6, 12, 24, and 72 hours of exposure to 100 nmol/LWMC-79 (Cont’d)
Gene symbol Gene name Fold change*
3 h 12 h 24 h 72 h
KIF11 kinesin family member 11 3.3MCM6 MCM6 minichromosome maintenance
deficient 6 (MIS5 homologue, S. pombe)3.2
RFC4 replication factor C (activator 1) 4, 37 kDa 3.2SCRN3 secernin 3 3.0 3.0
B. Up-regulated genes
GDF15 growth differentiation factor 15 3 5.5 19.1 23.9CDKN1A cyclin-dependent kinase inhibitor 1A (p21, Cip1) 6.3 16.9 38.1SAT spermidine/spermine N1-acetyl transferase 3.3 7.6 3.3GPX1 glutathione peroxidase 1 3.2 4 10.8TP5313 tumor protein p53 inducible protein 3 14.2 7.6BTG2 BTG family, member 2 9.2KRTAP2.1A keratin associated protein 2-1 6.0 11.6TNFRSF10B tumor necrosis factor receptor superfamily, member 10b 5.9 5WDR1 WD repeat domain 1 5.3 4.2FLNA filamin A, a (actin binding protein 280) 5.6 6.0RPS27L ribosomal protein S27– like 5 7.3CSDA cold shock domain protein A 4.9 5.2FHL2 four and a half LIM domains 2 4.9 4.5NDRG1 N-myc downstream regulated gene 1 4.7 5.7FBXO2 F-box protein 2 4.7 8.6BLCAP bladder cancer associated protein 4.7 3.7NGFRAP1 nerve growth factor receptor (TNFRSF16) associated protein 1 4.6 3.7PSTPIP2 proline-serine-threonine phosphatase interacting protein 2 4.5 8.1CYFIP2 cytoplasmic FMR1 interacting protein 2 4.5 5.5PTGES prostaglandin E synthase 4.5 3.3KAI1 CD82 antigen 4.5ITGA3 integrin, a 3 (antigen CD49C) 4.4 18.3ASS argininosuccinate synthetase 4.2 6.4GADD45A growth arrest and DNA damage-inducible, a 4.3 3.8SFN stratifin (14-3-3j) 4.2 6.3PHLDA3 pleckstrin homology– like domain, family A, member 3 4.2 5PLK3 polo-like kinase 3 (Drosophila) 4.2 3.6KRT19 keratin 19 4.1 12.1SDC4 syndecan 4 (amphiglycan, ryudocan) 4.1 11.9TM7SF2 transmembrane 7 superfamily member 2 4.1 3.4CLDN4 claudin 4 4.1 3.1BAX BCL2-associated X protein 4.1 3NINJ1 ninjurin 1 4.1DDIT4 DNA damage-inducible transcript 4 4.1 4.1CES2 carboxylesterase 2 (intestine, liver) 3.9 3.7WIG1 p53 target zinc finger protein 3.9 3.5HLA-C MHC, class I, C 3.8 6C20orf108 chromosome 20 open reading frame 108 3.8RAI3 retinoic acid induced 3 3.7 4.2TETRAN tetracycline transporter– like protein 3.6 5.7ACTN1 actinin, a 1 3.6 3.7FDXR ferredoxin reductase 3.5 8RGS16 regulator of G-protein signaling 16 3.5 4.7LRDD leucine-rich repeats and death domain containing 3.5 3.9CST3 cystatin C (amyloid angiopathy and cerebral hemorrhage) 3.4 9.9TFF1 trefoil factor 1 (breast cancer, estrogen-inducible sequence expressed in) 3.4 4.7S100A16 S100 calcium binding protein A16 3.3 5.4CST6 cystatin E/M 3.3 3.4
(Continued on the following page)
WMC-79 Induces Apoptosis in Colon Cancer Cells1624
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from
Dramatic up-regulation of p53 and p53 phosphorylated onSer15 was observed as early as 3 hours in RKO and HCT-116 cells following treatment with 100 nmol/L WMC-79and the levels increased with time (Fig. 5A). At the sametime, up-regulation of MDM2 was detected, most likelyas a result of the existing p53 autoregulatory loop (25, 36).Following activation of p53, a dramatic increase in
expression of p21WAF1/CIP1, both on gene transcriptionand protein levels, was detected (Table 2; Fig. 5A). Thisprotein is an established direct transcriptional target of p53that arrests cell growth in G1 and S phase in response toDNA damage (9). p21WAF1/CIP1 exerts a negative effect onG1 progression by inhibiting the activity of cyclin E/cyclin-dependent kinase 2 complexes that phosphorylate pRb
and by inhibiting the function of proliferating cell nuclearantigen in S phase. In addition, as a transcription factor,p21 can directly down-regulate expression of many genesinvolved in DNA replication, repair, and mitosis (41).Although G2 arrest can occur in the absence of p21 or
p53, both of these proteins are essential for sustaining G2
arrest after DNA damage (42). Western blot analysisshowed that WMC-79 markedly increased the expressionof p21WAF1/CIP1 followed by elevated levels of cyclins Aand D1 after 48 hours of exposure to the drug. Proteinlevels of E2F1, hyperphosphorylated Rb (ppRb), cyclin B1,and cyclin-dependent kinase 1 phoshorylated on Tyr15(Cdc2-Tyr15) became elevated during the early time ofexposure but decreased rapidly after 48 hours (Fig. 5D).
Table 2. Gene expression changes in HCT-116 cells following 6, 12, 24, and 72 hours of exposure to 100 nmol/LWMC-79 (Cont’d)
Gene symbol Gene name Fold change*
3 h 12 h 24 h 72 h
MVP major vault protein (MVP), transcript variant 2 3.3 3.4C9orf88 chromosome 9 open reading frame 88 3.3 3.3PP1201 PP1201 protein 3.2 6.7MKNK2 mitogen-activated protein kinase interacting serine/threonine kinase 2 3.2 3.4TAF10 TAF10 RNA polymerase II 3.2 3.0RAP2B RAP2B, member of RAS oncogene family 3.1 3.8RRM2B ribonucleotide reductase M2 B (TP53 inducible) 3.1 4.4TRIM8 tripartite motif–containing 8 3.1 3.5MSF MLL septin– like fusion 3.0 4.2PROCR protein C receptor, endothelial 3.0 4.2RBP1 retinol binding protein 4 3.0 4.0
Figure 6. Functional groups (GeneOntology classification) that are sig-nificantly (Bonferroni adjusted P <0.01) more enriched (% abundance)within the selected group of 128genes than in the Locus Link data-base. The abundance of genes withina Gene Ontology classification issignificantly higher (as percentage) inthe selected gene group comparedwith the background abundancefound in Locus Link. These groupsrepresent nonrandom clustering ofgenes within functional groups (GeneOntology classification).
Molecular Cancer Therapeutics 1625
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from

Transcription of cyclin B1 can be directly repressed by p53(43) and, indeed, we observed significant down-regulationof CCNB1 gene (Table 2). The time-dependent effect ofWMC-79 on the expression of various cell cycle regulatorsis in complete accord with cell cycle analysis by FACS(Fig. 2B). These results are consistent with a mechanism inwhich WMC-79 exerts cytotoxicity by direct induction ofapoptosis from S phase or arrest in G2-M phase andsubsequent induction of apoptosis by depletion of cyclinB1, as was recently reported (44).Numerous proapoptotic genes that are transcriptionally
activated by p53 have been identified, suggesting thatp53 apoptotic response is multifaceted (45). The BAX gene,a proapoptotic member of the Bcl-2 family, is an importanttarget for p53. This protein together with an additional p53target gene product (Noxa, PUMA, and p53AIPI) localizesto the mitochondria and induces the loss of the mitochon-drial membrane potential and the release of cytochrome c.Cytochrome c interacts with Apaf-1, resulting in theactivation of caspase-9, which in turn activates the effectorcaspase-3 (45, 46). Release of mitochondrial cytochromec and activation of caspase-3 are blocked by antiapoptoticBcl-2, another member of Bcl-2 family. It is clear thatoverexpression of Bcl-2 can block p53-mediated apoptosis.Bax binds to Bcl-2 and antagonizes its ability to blockapoptosis so that a p53-dependent Bax synthesis could tipthe scales toward apoptosis (9, 47). In summary, Bcl-2protects cells from the induction of apoptosis whereas Baxpromotes cell killing. In many studies, Bcl-2 gene expres-sion was down-regulated and Bax expression was up-regulated in apoptosis (7, 47). Western blot analysis (Fig. 5B)showed a time-dependent elevation of Bax protein leveland down-regulation of Bcl-2 and procaspase-9 proteins.This suggests that an increasing ratio of Bax/Bcl-2 proteinsplays an important role in the induction of apoptosis byWMC-79 in HCT-116.p53 has also been implicated in the membrane death
receptor–induced pathway of apoptosis. It can up-regulateexpression of TRAIL death receptors (DR4 and DR5; refs.48, 49) and activate the CD95 (APO-1/Fas) receptor/ligandsystem (50). We found that incubation of RKO cells with100 nmol/L WMC-79 induced elevation of DR4 and DR5proteins and activation of caspase-8 (Fig. 5C). Together, ourdata suggest two mechanisms by which p53 may induceapoptosis: one involving activation of caspase-9 by up-regulation of Bax and down-regulation of Bcl-2 proteins,and the second through the activation of death receptorsand caspase-8. Activated caspase-8 and caspase-9 can thenactivate the effectors caspase-3 and caspase-7. We reportedearlier (4) that in response to 100 nmol/L WMC-79, theactivity of caspase-3 was dramatically increased after 48hours and reached an f7-fold increase over the basal levelin untreated cells.In conclusion, our data show that WMC-79, a novel,
very potent cytotoxic agent, is much more effectiveagainst colon cancer cells that carry wild-type p53(f50% of all colon cancers) and, at concentration >100nmol/L, kills cells by induction of apoptosis. The p53-
dependent cell response and apoptotic pathway is evidentby a change in expression of a large number of p53-regulated genes and overexpression of p53 itself, its Ser15-phoshorylated form, p21WAF1/CIP1, Bax, DR4, and DR5;down-regulation of Bcl-2; and finally activation ofcaspase-9, -8, -3 and -7. Apoptotic cell death was alsoconfirmed by flow cytometry, cell morphology of WMC-79–treated colon cancer cells (Fig. 4), nuclear fragmenta-tion, and formation of nucleosomal ladders in leukemiacell lines (4). Preliminary in vivo experiments on HCT-116colon cancer xenografted in nude mice revealed goodactivity when the compound was administered i.v. (4).WMC-79 is somewhat related to the potent antitumoragent MLN944 (51), which also binds to DNA andapparently affects transcription. However, there arecurrently insufficient data to make a direct comparisonbetween the two agents.It should also be noted that WMC-79 is also cytotoxic to
tumor cells in which the p53 gene is either mutated or notexpressed. This suggests that other pathways leading to cellarrest and death are involved. Further experiments areneeded to elucidate those mechanisms.
Acknowledgments
We thank Dr. Bert Vogelstein for the HCT-116 p53+/+, p53�/� andHCT-116 p21+/+, p21�/� cells; Louise R. Finch and Refika B. Turnier forflow cytometric analysis; and John W. Connelly for assistance with theexpression arrays.
References
1. Hernandez L, Cholody WM, Hudson EA, Resau JH, Pauly G, MichejdaCJ. Mechanism of action of bisimidazoacridones, new drugs with potent,selective activity against colon cancer. Cancer Res 1995;55:2338–45.
2. Cholody WM, Hernandez L, Hassner L, Scudiero DA, Djurickovic DB,Michejda CJ. Bisimidazoacridones and related compounds: new antineo-plastic agents with high selectivity against colon tumors. J Med Chem1995;38:3043–52.
3. Cholody WM, Kosakowska-Cholody T, Michejda CJ. Bisimidazoacri-dones induce a potent cytostatic effect in colon tumor cells that sensitizesthem to killing by UCN-01. Cancer Chemother Pharmacol 2001;47:241–9.
4. Cholody WM, Kosakowska-Cholody T, Hollingshead MG, HariprakashaHK, Michejda CJ. A new synthetic agent with potent but selectivecytotoxic activity against cancer. J Med Chem 2005;48:4474–81.
5. Solary E, Droin N, Bettaieb A, Corcos L, Dimanche-Boitrel MT, GarridoC. Positive and negative regulation of apoptotic pathways by cytotoxicagents in hematological malignancies. Leukemia 2000;14:1833–49.
6. Meek DW. The p53 response to DNA damage. DNA Repair (Amst)2004;3:1049–56.
7. Chan SL, Yu VC. Proteins of the Bcl-2 family in apoptosis signalling:from mechanistic insights to therapeutic opportunities. Clin Exp PharmacolPhysiol 2004;31:119–28.
8. Bates S, Vousden KH. Mechanimsms of p53-mediated apoptosis. CellMol Life Sci 1999;55:28–37.
9. Sionov VR, Haupt Y. The cellular response to p53: the decisionbetween life and death. Oncogene 1999;18:6145–57.
10. Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-fluxanticancer drug screen using a diverse panel of cultured human tumor celllines. J Natl Cancer Inst 1991;83:757–66.
11. Gonzalez MA, Tachibana KE, Laskey RA, Coleman N. Control of DNAreplication and its potential clinical exploitation. Nat Rev Cancer 2005;5:135–41.
12. Wang SW, Asakawa K, Win TZ, Toda T, Norbury CJ. Inactivation ofthe pre-mRNA cleavage and polyadenylation factor Pfs2 in fission yeastcauses lethal cell cycle defects. Mol Cell Biol 2005;25:2288–96.
13. Sharma S, Otterlei M, Sommers JA, et al. WRN helicase and FEN-1
WMC-79 Induces Apoptosis in Colon Cancer Cells1626
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from

form a complex upon replication arrest and together process branchmi-grating DNA structures associated with the replication fork. Mol Biol Cell2004;15:734–50.
14. Zhou B, Yen Y. Characterization of the human ribonucleotidereductase M2 subunit gene; genomic structure and promoter analyses.Cytogenet Cell Genet 2001;95:52–9.
15. Majka J, Burgers PM. The PCNA-RFC families of DNA clamps andclamp loaders. Prog Nucleic Acid Res Mol Biol 2004;78:227–60.
16. Hirokawa N, Takemura R. Kinesin superfamily proteins and theirvarious functions and dynamics. Exp Cell Res 2004;301:50–9.
17. Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV,Benezra R. Complete loss of the tumor suppressor MAD2 causespremature cyclin B degradation and mitotic failure in human somatic cells.Proc Natl Acad Sci U S A 2004;101:4459–64.
18. Ouyang B, Lan Z, Meadows J, et al. Human Bub1: a putative spindlecheckpoint kinase closely linked to cell proliferation. Cell Growth Differ1998;9:877–85.
19. Geiman TM, Sankpal UT, Robertson AK, et al. Isolation andcharacterization of a novel DNA methyltransferase complex linkingDNMT3B with components of the mitotic chromosome condensationmachinery. Nucleic Acids Res 2004;32:2716–29.
20. Schmiesing JA, Ball AR, Jr., Gregson HC, Alderton JM, Zhou S,Yokomori K. Identification of two distinct human SMC protein complexesinvolved in mitotic chromosome dynamics. Proc Natl Acad Sci U S A1998;95:12906–11.
21. Chen Z, Raman M, Chen L, Lee SF, Gilman AG, Cobb MH. TAO(thousand-and-one amino acid) protein kinases mediate signaling fromcarbachol to p38 mitogen-activated protein kinase and ternary complexfactors. J Biol Chem 2003;278:22278–83.
22. Minden A, Lin A, Smeal T, et al. c-Jun N-terminal phosphorylationcorrelates with activation of the JNK subgroup but not the ERKsubgroup of mitogen-activated protein kinases. Mol Cell Biol 1994;14:6683–8.
23. Chen RH, Chang MC, Su YH, Tsai YT, Kuo ML. Interleukin-6 inhibitstransforming growth factor-h-induced apoptosis through the phosphati-dylinositol 3-kinase/Akt and signal transducers and activators of tran-scription 3 pathways. J Biol Chem 1999;274:23013–9.
24. Yang H, Filipovic Z, Brown D, Breit SN, Vassilev LT. Macrophageinhibitory cytokine-1: a novel biomarker for p53 pathway activation. MolCancer Ther 2003;2:1023–9.
25. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat RevCancer 2002;2:594–604.
26. Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 14-3-3j positivelyregulates p53 and suppresses tumor growth. Mol Cell Biol 2003Oct;23:7096–107.
27. Maxwell PJ, Longley DB, Latif T, et al. Identification of 5-fluorouracil-inducible target genes using cDNA microarray profiling. Cancer Res 2003;63:4602–6.
28. Gladyshev VN, Factor VM, Housseau F, Hatfield DL. Contrastingpatterns of regulation of the antioxidant selenoproteins, thioredoxinreductase, and glutathione peroxidase, in cancer cells. Biochem BiophysRes Commun 1998;251:488–93.
29. Stein S, Thomas EK, Herzog B, et al. NDRG1 is necessary for p53-dependent apoptosis. J Biol Chem 2004;279:48930–40.
30. Ellisen LW, Ramsayer KD, Johannessen CM, et al. REDD1, adevelopmentally regulated transcriptional target of p63 and p53,links p63 to regulation of reactive oxygen species. Mol Cell 2002;10:995–1005.
31. Xie S, Wu H, Wang Q, et al. Genotoxic stress-induced activation ofPlk3 is partly mediated by Chk2. Cell Cycle 2002;1:424–9.
32. Hellborg F, Wiman KG. The p53-induced Wig-1 zinc fingerprotein is highly conserved from fish to man. Int J Oncol 2004;24:1559–64.
33. Tinel A, Tschopp J. The PIDDosome, a protein complex implicated inactivation of caspase-2 in response to genotoxic stress. Science2004;304:843–6.
34. Yanamoto S, Kawasaki G, Yoshitomi I, Mizuno A. Expression ofp53R2, newly p53 target in oral normal epithelium, epithelial dysplasia andsquamous cell carcinoma. Cancer Lett 2003;190:233–43.
35. Hosack DA, Dennis G, Jr., Sherman BT, Lane HC, Lempicki RA.Identifying biological themes within lists of genes with EASE. Genome Biol2003;4:R70.
36. Moll UM, Petrenko O. The MDM2-p53 Interaction. Mol Cancer Res2003;1:1001–8.
37. Woo RA, McLure KG, Lees-Miller SP, Rancourt DE, Lee PMK. DNA-dependent protein kinase acts upstream of p53 in response to DNAdamage. Nature 1998;394:700–4.
38. Koundrioukoff S, Polo S, Almouzni G. Interplay between chromatinand cell cycle checkpoints in the context of ATR/ATM-dependentcheckpoints. DNA Repair (Amst) 2004;3:969–78.
39. Freedman DA. Regulation of the p53 protein by the MDM2oncoprotein—thirty-eighth G.H.A. Clowes Memorial Award Lecture.Cancer Res 1999;59:1–7.
40. Caspari T. Checkpoints: how to activate p53? Curr Biol 2000;10:315–7.
41. Chang BD, Watanabe K, Broude EV, et al. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: implications for carcinogenesis,senescence, and age-related diseases. Proc Natl Acad Sci U S A 2000;97:4291–6.
42. Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 tosustain G2 arrest after DNA damage. Science 1998;282:1497–501.
43. Innocente SA, Lee JM. p53 is a NF-Y- and p21-independent, Sp1-dependent repressor of cyclin B1 transcription. FEBS Lett 2005;579:1001–7.
44. Yuan J, Yan R, Kramer A, et al. Cyclin B1 depletion inhibitsproliferation and induces apoptosis in human tumor cells. Oncogene2004;23:5843–52.
45. Balint E, Vousden KH. Activation and activities of the p53 tumorsuppressor protein. Br J Cancer 2001;85:1813–23.
46. Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM.Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem 1999;274:5053–60.
47. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell1997;88:323–31.
48. Guan B, Yue P, Clayman GL, Sun SY. Evidence that the deathreceptor DR4 is a DNA damage-inducible, p53-regulated gene. J CellPhysiol 2001;188:98–105.
49. Takimoto R, El-Deiry WS. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site.Oncogene 2000;19:1735–43.
50. Muller M, Wilder S, Bannasch D, et al. p53 activates the CD95(APO-1/Fas) gene in response to DNA damage by anticancer drugs. J ExpMed 1998;188:2033–45.
51. Sappal DS, McClendon AK, Fleming JA, et al. Biological character-ization of MLN944: a potent DNA binding agent. Mol Cancer Ther 2004;3:47–58.
Molecular Cancer Therapeutics 1627
Mol Cancer Ther 2005;4(10). October 2005
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from

2005;4:1617-1627. Mol Cancer Ther Teresa Kosakowska-Cholody, W. Marek Cholody, Anne Monks, et al. apoptosis through a p53-dependent pathwayWMC-79, a potent agent against colon cancers, induces
Updated version
http://mct.aacrjournals.org/content/4/10/1617
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/4/10/1617.full#ref-list-1
This article cites 48 articles, 21 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/4/10/1617.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://mct.aacrjournals.org/content/4/10/1617To request permission to re-use all or part of this article, use this link
Research. on January 10, 2020. © 2005 American Association for Cancermct.aacrjournals.org Downloaded from
![DNA vaccination in rhesus macaques induces potent immune … · 2009-10-02 · MHC-linked spontaneous control of viremia [herein and (29)]. Following peak viremia, several vaccinated](https://img.pdfslide.net/doc/110x75/5f4b888b6ae97e40910990dc/dna-vaccination-in-rhesus-macaques-induces-potent-immune-2009-10-02-mhc-linked.jpg)




























