Embed Size (px)
Citation preview

B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
ava i l ab l e a t www.sc i enced i rec t . com
www.e l sev i e r. com/ loca te /b ra in res
Research Report
Adenosine treatment delays postischemic hippocampal CA1loss after cardiac arrest and resuscitation in rats
Kui Xua, Michelle A. Puchowicza, W. David Lustc, Joseph C. LaMannaa,b,⁎aDepartment of Anatomy, Case Western Reserve University, School of Medicine, Cleveland, OH 44106, USAbDepartment of Neurology, Case Western Reserve University, School of Medicine, Cleveland, OH 44106, USAcDepartment of Neurosurgery, Case Western Reserve University, School of Medicine, Cleveland, OH 44106, USA
A R T I C L E I N F O
⁎ Corresponding author. Department of AnatoOH 44106-4938, USA. Fax: +1 216 368 1144.
E-mail address: [email protected] (J.C. LaMan
0006-8993/$ – see front matter © 2005 Elsevidoi:10.1016/j.brainres.2005.11.060
A B S T R A C T
Article history:Accepted 11 November 2005Available online 17 January 2006
Resuscitation from cardiac arrest results in reperfusion injury that leads to increasedpostresuscitation mortality and delayed neuronal death. One of the many consequences ofresuscitation from cardiac arrest is a derangement of energy metabolism and the loss ofadenylates, impairing the tissue's ability to regain proper energy balance. In this study, weinvestigated the effects of adenosine (ADO) on the recovery of the brain from 12 min ofischemia using a rat model of cardiac arrest and resuscitation. Compared to the untreatedgroup, treatment with adenosine (7.2 mg/kg) initiated immediately after resuscitationincreased the proportion of rats surviving to 4 days and significantly delayed hippocampalCA1neuronal loss. Brain blood flowwas increased significantly in the adenosine-treated rats1 h after cardiac arrest and resuscitation. Adenosine-treated rats exhibited less edema incortex, brainstem and hippocampus during the first 48 h of recovery. Adenosine treatmentsignificantly lowered brain temperature during recovery, and a part of the neuroprotectiveeffects of adenosine treatment could be ascribed to adenosine-induced hypothermia. Withthis dose, adenosine may have a delayed transient effect on the restoration of the adenylatepool (AXP = ATP + ADP + AMP) 24 h after cardiac arrest and resuscitation. Our findingssuggested that improved postischemic brain blood flow and ADO-induced hypothermia,rather than adenylate supplementation, may be the two major contributors to theneuroprotective effects of adenosine following cardiac arrest and resuscitation. Althoughadenosine did not prevent eventual CA1 neuronal loss in the long term, it did delay neuronalloss and promoted long-term survival. Thus, adenosine or specific agonists of adenosinereceptors should be evaluated as adjuncts to broaden the window of opportunity in thetreatment of the reperfusion injury following cardiac arrest and resuscitation.
© 2005 Elsevier B.V. All rights reserved.
Keywords:Transient global ischemiaDelayed neuronal deathBrain edemaBrain blood flowReperfusion injuryBrain hypothermiaNeuroprotection
1. Introduction
Cardiac arrest and resuscitation produce global ischemia andreperfusion damage to the brain, which lead to high mortalityand delayed neuronal death (Crumrine and LaManna, 1991;
my, Case Western Reserv
na).
er B.V. All rights reserved
Crumrine et al., 1991; Hoxworth et al., 1999; LaManna et al.,1995). Extracellular adenosine levels increase significantlyduring conditions of metabolic stress, such as ischemia,hypoxia and seizures. Adenosine has been proposed as anendogenous neuroprotective molecule (Phillis, 1989) and
e University, School of Medicine, 10900 Euclid Avenue, Cleveland,
.

Table 2 – Survival rates (4 days) after cardiac arrest andresuscitation
Experimentalgroup
Number of deaths Survival rate
b1day
1–2days
2–3days
3–4days
4 days/total
Untreated 9 4 2 0 55% (18/33)60 min ADO 3 3 0 0 57% (8/14)90 min ADO 1 0 1 0 86% (12/14)a
ADO-temp corrected 1 0 1 1 70% (7/10)2-chloroadenosine 1 2 2 0 64% (9/14)
a Indicates significantly different from the untreated group(Wilcoxon (Gehan) survival analysis, P b 0.05).
209B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
suggested as a potential postischemic treatment approach(Marangos, 1990) because of its multiple interactions with thepathophysiological mechanisms activated during ischemiaand reperfusion (Rudolphi et al., 1992; Schubert et al., 1997;Sweeney, 1997; von Lubitz et al., 1995). Administration of anadenosine analog, 2-chloroadenosine was protective againstischemic cell loss in the rat hippocampus (Evans et al., 1987).The adenosine uptake blocker, propentofylline (HWA 285), haspreviously been shown to protect hippocampal CA1 pyramidalcells from ischemia-induced delayed hippocampal neuronaldeath (Andine et al., 1990). These observations suggest thatenhanced extracellular concentrations of adenosine protectthe brain from the ischemic and reperfusion insult. However,there has been no clinical use of adenosine in the treatment ofcerebral ischemia caused by cardiac arrest. One of the reasonsmight be fear of some of the disturbing side effects ofadenosine, e.g. bradycardia and hypotension (William, 1989).The other reason is probably because it has not been tested ina clinically relevant model of cardiac arrest and resuscitation.Also, no mechanistic studies have provided a rationale for itsputative salutary effect. In the present study, a rat model ofcardiac arrest and resuscitation has been used to evaluate theefficacy of adenosine, and the adenosine analogue and A1
receptor agonist, 2-chloroadenosine, as a postischemic treat-ment after transient global ischemia.
2. Results
2.1. The physiological variables
The physiological behavior of animals in this study wascoincident with that reported previously for this model of
Table 1 – Physiological variables of the long-term recovery rat
Variable Untreated (n = 33) ADO-treated (n = 27) AD
Body weight (g) 337 ± 40 328 ± 42MABP (mm Hg)Pre 110 ± 6 108 ± 71 h 84 ± 14a 84 ± 14a
Arterial pH (unit)Pre 7.41 ± 0.03 7.41 ± 0.031 h 7.35 ± 0.05a 7.38 ± 0.05a
PaO2 (mm Hg)Pre 93 ± 13 95 ± 101 h 98 ± 14 100 ± 13
PaCO2 (mm Hg)Pre 40 ± 3 41 ± 31 h 37 ± 5a 40 ± 5
Hematocrit (%)Pre 48 ± 3 48 ± 21 h 48 ± 2 48 ± 1
Glucoseplasma (mM)Pre 7.9 ± 1.2 7.8 ± 1.51 h 8.6 ± 2.0 7.8 ± 1.3
Lactateplasma (mM)Pre 2.2 ± 0.8 1.9 ± 0.51 h 2.8 ± 0.9a 2.2 ± 0.6
a Indicated the post-resuscitation value was significantly different (t test
cardiac arrest and resuscitation (Crumrine et al., 1991; Hox-worth et al., 1999). As seen in Table 1, there are no significantdifferences in any of the physiological variables among any ofthe experimental groups. Blood gases were taken 1 h afterresuscitation for adjustment of ventilation to achieve PaO2 andPaCO2 values in the normal range. Typically, the rats regainedtheir spontaneous respiration at 3 to 6 h after resuscitation.The 1 h PaO2 was higher (not significantly different) in all fourgroups of animals because higher oxygen concentration (about30%)was given at the beginning of the recovery period. ArterialpH was not completely recovered to baseline at 1 h recovery inmost rats. The pattern of blood pressure was consistent withthose earlier studies. The rats underwent hypertension(minutes) right after the recovery which was followed byhypotension (1–2 h). At 1 h recovery, the mean blood pressurereached about 70% of prearrest values. There was no severehypotension in the ADO-treated rats. The postresuscitationblood pressure of rats usually returned to the prearrest value
s
O temperature control (n = 10) 2-Chloroadenosine (n = 14)
322 ± 55 322 ± 60
110 ± 6 109 ± 683 ± 15a 77 ± 13a
7.42 ± 0.03 7.43 ± 0.037.39 ± 0.06 7.36 ± 0.06a
94 ± 11 94 ± 2099 ± 21 99 ± 13
43 ± 2 42 ± 540 ± 4 39 ± 4
48 ± 3 47 ± 348 ± 4 46 ± 3
8.1 ± 2.1 8.1 ± 1.58.9 ± 1.7 8.8 ± 2.0
1.8 ± 0.4 1.9 ± 0.52.6 ± 0.8a 2.7 ± 0.9a
, P b 0.05) from the pre-arrest value in the same experimental group.

Fig. 1 – Hippocampal neuronal counts after cardiac arrestand resuscitation. * Indicates significant difference (P b 0.05)compared to the non-arrested controls; ** indicatessignificant difference (P b 0.05) compared to the untreated4-day recovery group.
210 B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
within 2–3 h of recovery. The hematocrit and levels ofpostresuscitation plasma glucose were similar to the prearrestvalues; however, the postresuscitation values of plasmalactate were significantly lower in the untreated group, theADO-temp group and the 2CADO group.
2.2. Overall survival rates after cardiac arrest andresuscitation
As seen in Table 2, 4-day overall survival rates weredetermined in resuscitated rats of the untreated group, ADO-treated and ADO-treated with temperature control groups.Neurologic deficits were observed in all these groups (Crumr-ine and LaManna, 1991; LaManna et al., 1995). The ADO-treated rats had better recovery from cardiac arrest andresuscitation, especially seen in the higher dose ADO-treatedgroup. They had fewer seizures, could walk and drink earlier
Fig. 2 – Temperature of temporalis muscle. ADO-treated group (lower temperature compared to the untreated group (n = 11) dura similar temperature compared to the untreated group at any ti
than the other groups. At the end of 4 days, the rats treatedwith ADO 7.2 mg/kg had a significantly better (85.7%, 12/14,P b 0.05, Wilcoxon (Gehan) survival analysis) survival ratecompared to the untreated rats (54.5%, 18/33) and ADO 4.8/kggroup (57.1%, 8/14). The survival rates were 70% (7/10) and64.3% (9/14) in ADO-Temp group and 2CADO group, respec-tively. Of those that did not survive for 4 days, most diedduring the first 2 days after resuscitation, suffering mostlyfrom respiratory failure or cardiovascular collapse.
2.3. Delayed hippocampal neuronal death
As seen in Fig. 1, the hippocampal CA1 neuron counts weredetermined in the non-arrested, the untreated and ADO-treated groups. At 4 days of recovery, the hippocampal CA1counts were 86 ± 4 (n = 6, mean ± SD) in the ADO 7.2 mg/kggroup, which was significantly more than the counts (21 ± 10,n = 10) in the untreated group and similar to that of the non-arrested controls (104 ± 11, n = 7). The CA1 cell count in ADO4.8 mg/kg group was 57 ± 23 (n = 8). ADO-treated,temperature-controlled rats were found to have 66 ± 22(n = 7) neurons, which was about two-thirds of that in thesame ADO-treated group without temperature correction.The neuron count in the 2CADO-treated group was 50 ± 34(n = 9). All the groups with treatment had significantly higherhippocampal CA1 counts than the untreated group. The cellcounts of CA2–CA3 pyramidal cell regions were not affectedby any conditions.
For the untreated and ADO 7.2mg/kg groups, rats were alsoallowed to recover for 30 days after resuscitation. Similar CA1neurons were found in the untreated group (3 ± 3, n = 3) andADO-treated group (9 ± 3, n = 3) at 30 days of recovery. The CA1counts in both groups were significantly lower as compared tothe non-arrested controls. There were also significantly lessCA1 neurons remaining in the 30-day recovery rats ascompared to the 4-day recovery rats in each group.
n = 9) and 2CADO-treated group (n = 8) had significantlying recovery (30 min and later). ADO-Temp group (n = 9) hadme point during recovery.

Table 3 – Extraction fraction of adenosine (n = 7)
Brain region Extractionfraction (%)
Blood flow(ml/min/g)
Frontal cortex 31 ± 8 1.44 ± 0.38Parietal cortex 32 ± 10 1.51 ± 0.33Hippocampus 35 ± 9 0.95 ± 0.29Brainstem 31 ± 9 1.37 ± 0.45Cerebellum 42 ± 7 0.90 ± 0.32
211B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
2.4. Temperature of temporalis muscle
The temperature of temporalis muscle was measured in theuntreated (n = 11), ADO-treated (7.2 mg/kg, n = 9), ADO-treated with temperature control (n = 9) and 2CADO-treated(n = 8) groups before arrest and the first 6 h afterresuscitation (see Fig. 2). As seen in Fig. 2, the meanprearrest temperature was similar in each group (37.5–37.7 °C). At the end of the 12 min cardiac arrest, thetemperature dropped about 2.5 °C in all groups comparedto their prearrest values, which were consistent withprevious studies (LaManna et al., 1995). After 30 min ofrecovery, the temperature in the ADO-treated group and2CADO group was significantly lower than that of theuntreated group (36.4 ± 0.3, mean ± SD). The difference wasmaintained even after the infusion was stopped. The 2CADOgroup had a significantly lower temperature compared to theADO-treated group during the 90 min of infusion, but thetemperature recovered faster than the ADO-treated afterinfusion stopped. At 6 h recovery, temperature in theuntreated group was 37.1 ± 0.4, which was nearly restoredto prearrest value, and was significantly higher than that ofADO-treated group (35.4 ± 0.4) and 2CADO group (36.5 ± 0.6).The temperature in the ADO-temperature control group wasmaintained similarly to the values of the untreated group forthe entire time of the measurement.
2.5. Extraction fraction of adenosine
Table 3 shows the extraction fraction of adenosine and bloodflow in different brain regions in the non-arrested rats. Theblood flow values in all regions were consistent with ourprevious report (LaManna and Harik, 1986) and our non-arrested control values measured by IAP autoradiography inthe present study. We found that the single pass extractionof adenosine in cerebral cortex, hippocampus and brainstem
Table 4 – Blood flow (mean ± SD, ml/g/min) after cardiac arres
Brainregions
Non-arrested
R
(n = 7) 1 h (n = 7)
Frontal cortex 1.48 ± 0.08 0.54 ± 0.11a
Parietal cortex 1.55 ± 0.09 0.49 ± 0.15a
Hippocampus 0.90 ± 0.22 0.32 ± 0.10a
Brainstem 1.26 ± 0.26 0.76 ± 0.21a
Cerebellum 0.87 ± 0.23 0.42 ± 0.14a
a Indicates significantly different from the values of the non-arrested co
was about 30%, whereas cerebellum had the highestadenosine extraction (∼45%). The results confirm significantuptake of adenosine at the blood–brain barrier (BBB).
2.6. Regional brain blood flow after resuscitation
Regional (frontal and parietal cortex, hippocampus, brainstemand cerebellum) brain blood flow was measured in non-arrested controls, untreated rats (1, 6, 24 and 48 h afterresuscitation, Table 4) and adenosine-treated rats (80 μg/kg/min, 1 h after resuscitation, Fig. 3). As seen in Table 4, at 1h recovery, blood flows recovered less than 40% in cortex andhippocampus and about 60% and 50% in brainstem andcerebellum, respectively. By day 2 of recovery, blood flows inall regions returned completely to baseline. With the treat-ment of adenosine, blood flow at 1 h after resuscitation wasincreased significantly in all regions compared to the untreat-ed group (Fig. 3). Especially, the brainstem blood flow of theadenosine-treated group was nearly returned to baseline.
2.7. Brain edema after cardiac arrest and resuscitation
As seen in Table 5, baseline water content of cortex was 80%.The water content of cortex was markedly increased at 1, 6and 24 h after resuscitation; with the adenosine treatment,brain edema was significantly depressed. At 48 h of recovery,the water content of cortex in both groups was nearlyrecovered to non-arrested control values. Baseline watercontent of the hippocampus was similar to that in the cortex.Brain edema in the hippocampus reached maximum at about6 h in the untreated rats, significantly higher than baseline.Also, ADO treatment suppressed the increased water contentin this region, the decrease was not significant. Baseline watercontent in the brainstemwas 78%, significantly lower than thebaseline of cortex. After resuscitation, the water content ofbrainstem seemed to follow the same trend as cortex. It onlyreached a level of statistical difference at 24 h postresuscita-tion, where the water content was almost 10% higher than thenon-arrested controls. Rats treatedwith adenosine showed nosignificant edema in the brainstem through the first 2 days ofrecovery from cardiac arrest.
2.8. Metabolite assays
AXP levels (nmol/mg dry weight tissue, Table 6) in thecortex, hippocampus and brainstem were assayed in non-
t and resuscitation
ecovery after cardiac arrest and resuscitationUntreated
6 h (n = 3) 24 h (n = 3) 48 h (n = 3)
0.89 ± 0.19a 0.96 ± 0.34 1.49 ± 0.180.99 ± 0.28 1.09 ± 0.58 1.58 ± 0.050.73 ± 0.28 0.61 ± 0.17 0.99 ± 0.130.93 ± 0.07 0.92 ± 0.34 1.18 ± 0.150.73 ± 0.05 0.70 ± 0.34 0.91 ± 0.21
ntrols.
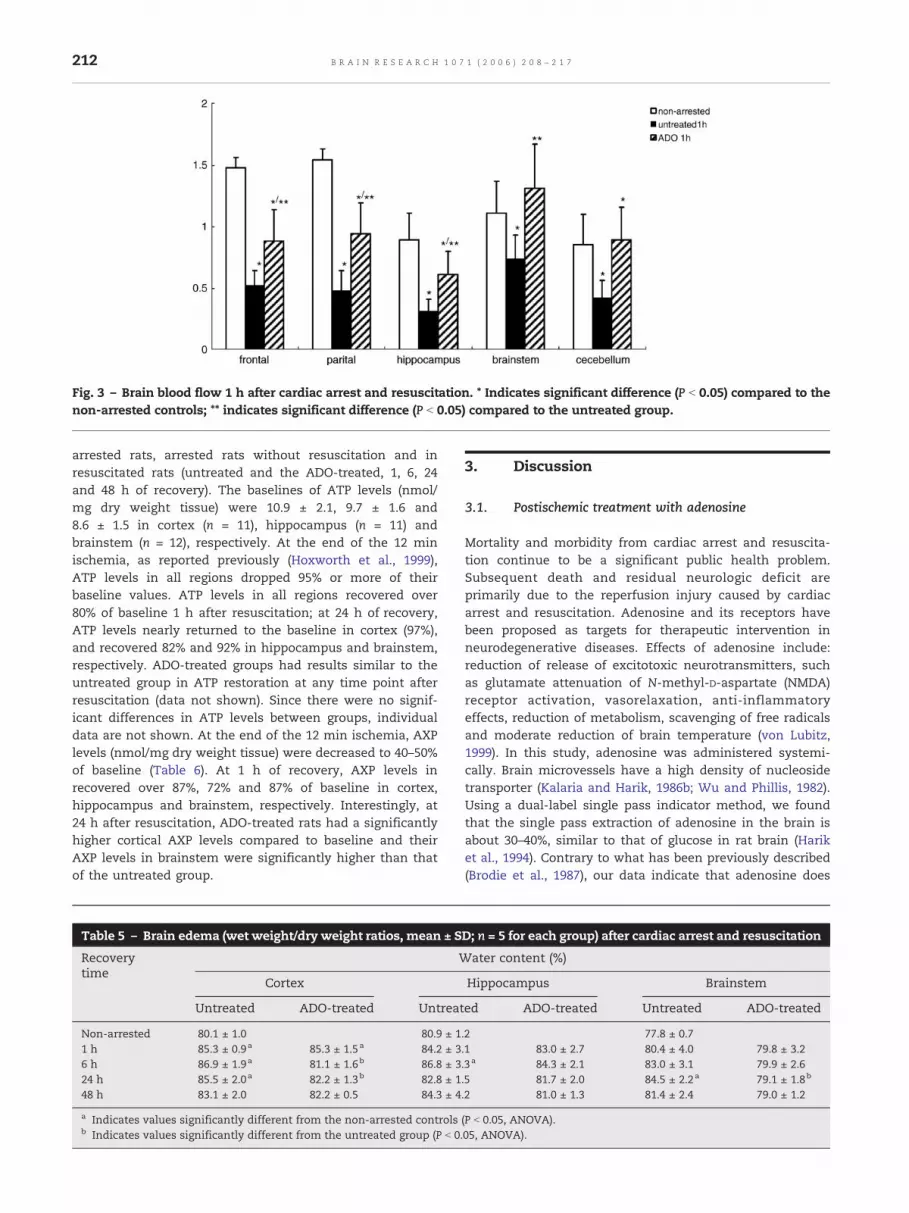
Fig. 3 – Brain blood flow 1 h after cardiac arrest and resuscitation. * Indicates significant difference (P b 0.05) compared to thenon-arrested controls; ** indicates significant difference (P b 0.05) compared to the untreated group.
212 B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
arrested rats, arrested rats without resuscitation and inresuscitated rats (untreated and the ADO-treated, 1, 6, 24and 48 h of recovery). The baselines of ATP levels (nmol/mg dry weight tissue) were 10.9 ± 2.1, 9.7 ± 1.6 and8.6 ± 1.5 in cortex (n = 11), hippocampus (n = 11) andbrainstem (n = 12), respectively. At the end of the 12 minischemia, as reported previously (Hoxworth et al., 1999),ATP levels in all regions dropped 95% or more of theirbaseline values. ATP levels in all regions recovered over80% of baseline 1 h after resuscitation; at 24 h of recovery,ATP levels nearly returned to the baseline in cortex (97%),and recovered 82% and 92% in hippocampus and brainstem,respectively. ADO-treated groups had results similar to theuntreated group in ATP restoration at any time point afterresuscitation (data not shown). Since there were no signif-icant differences in ATP levels between groups, individualdata are not shown. At the end of the 12 min ischemia, AXPlevels (nmol/mg dry weight tissue) were decreased to 40–50%of baseline (Table 6). At 1 h of recovery, AXP levels inrecovered over 87%, 72% and 87% of baseline in cortex,hippocampus and brainstem, respectively. Interestingly, at24 h after resuscitation, ADO-treated rats had a significantlyhigher cortical AXP levels compared to baseline and theirAXP levels in brainstem were significantly higher than thatof the untreated group.
Table 5 – Brain edema (wet weight/dry weight ratios, mean ± S
Recoverytime
W
Cortex
Untreated ADO-treated Untreat
Non-arrested 80.1 ± 1.0 80.9 ± 11 h 85.3 ± 0.9a 85.3 ± 1.5a 84.2 ± 36 h 86.9 ± 1.9a 81.1 ± 1.6b 86.8 ± 324 h 85.5 ± 2.0a 82.2 ± 1.3b 82.8 ± 148 h 83.1 ± 2.0 82.2 ± 0.5 84.3 ± 4
a Indicates values significantly different from the non-arrested controlsb Indicates values significantly different from the untreated group (P b 0
3. Discussion
3.1. Postischemic treatment with adenosine
Mortality and morbidity from cardiac arrest and resuscita-tion continue to be a significant public health problem.Subsequent death and residual neurologic deficit areprimarily due to the reperfusion injury caused by cardiacarrest and resuscitation. Adenosine and its receptors havebeen proposed as targets for therapeutic intervention inneurodegenerative diseases. Effects of adenosine include:reduction of release of excitotoxic neurotransmitters, suchas glutamate attenuation of N-methyl-D-aspartate (NMDA)receptor activation, vasorelaxation, anti-inflammatoryeffects, reduction of metabolism, scavenging of free radicalsand moderate reduction of brain temperature (von Lubitz,1999). In this study, adenosine was administered systemi-cally. Brain microvessels have a high density of nucleosidetransporter (Kalaria and Harik, 1986b; Wu and Phillis, 1982).Using a dual-label single pass indicator method, we foundthat the single pass extraction of adenosine in the brain isabout 30–40%, similar to that of glucose in rat brain (Hariket al., 1994). Contrary to what has been previously described(Brodie et al., 1987), our data indicate that adenosine does
D; n = 5 for each group) after cardiac arrest and resuscitation
ater content (%)
Hippocampus Brainstem
ed ADO-treated Untreated ADO-treated
.2 77.8 ± 0.7
.1 83.0 ± 2.7 80.4 ± 4.0 79.8 ± 3.2
.3a 84.3 ± 2.1 83.0 ± 3.1 79.9 ± 2.6
.5 81.7 ± 2.0 84.5 ± 2.2a 79.1 ± 1.8b
.2 81.0 ± 1.3 81.4 ± 2.4 79.0 ± 1.2
(P b 0.05, ANOVA)..05, ANOVA).

Table 6 – AXP (nmol/mg dry weight, mean ± SD)a after cardiac arrest and resuscitation
Experimentalgroups
Cortex Hippocampus Brainstem
Untreated ADO Untreated ADO Untreated ADO
Non-arrested 14.4 ± 2.0 15.5 ± 2.1 10.9 ± 2.1(n = 11) (n = 11) (n = 11)
No resuscitation 6.6 ± 1.6b 6.2 ± 2.2b 4.9 ± 1.1b
(n = 6) (n = 7) (n = 6)1 h recovery 12.6 ± 1.6 12.1 ± 2.2 11.1 ± 2.0 11.9 ± 0.9 9.5 ± 1.9 9.8 ± 1.8
(n = 11) (n = 8) (n = 12) (n = 7) (n = 12) (n = 8)6 h recovery 16.2 ± 2.8 16.1 ± 0.9 12.3 ± 2.9 16.6 ± 2.8 11.9 ± 3.8 14.1 ± 3.2
(n = 5) (n = 4) (n = 5) (n = 4) (n = 5) (n = 4)24 h recovery 15.1 ± 1.9 18.2 ± 1.6b 12.5 ± 1.4 12.5 ± 3.6 9.5 ± 0.9 14.0 ± 1.6c
(n = 5) (n = 5) (n = 5) (n = 5) (n = 5) (n = 5)48 h recovery 14.4 ± 2.1 13.6 ± 2.7 10.8 ± 2.5b 11.8 ± 1.5 9.4 ± 1.53 10.8 ± 2.2
(n = 5) (n = 11) (n = 5) (n = 5) (n = 5) (n =5)
a Conversion factors (approximates): nmol/mg wet weight ≈ 20% nmol/mg dry weight; protein content is 11% of nmol/mg wet weight.b Significantly different compared to the value of non-arrested controls (P b 0.05, ANOVA).c Significantly different compared to the value of the untreated group at the same time point (P b 0.05, ANOVA).
213B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
cross the BBB and has a central effect on the brain. Resultsof the present study imply that postresuscitation treatmentwith adenosine following a 12-min cardiac arrest improvedthe overall survival and the preservation of hippocampalCA1 neurons. In addition, adenosine treatment had adepressive effect on brain edema after cardiac arrest andresuscitation. However, the treatment with 2-chloroadeno-sine showed less prominent neuroprotection than adeno-sine itself. Brain microvessels have a lower density ofadenosine A1 receptors than the parenchyma, but docontain A2 receptors (Kalaria and Harik, 1986a; Kalaria andHarik, 1988).
3.2. Adenosine supplementation and total adenylates
During complete ischemia, energy failure is associated with asignificant loss of adenylates (Crumrine and LaManna, 1991;Newman et al., 1998; Phillis et al., 1984), thus limiting thetissue's ability to restore normal levels of ATP during recovery.It has been shown in hippocampal slice preparations thatsupplementing adenosine to the bathing medium increasedtotal tissue adenylates and ATP content in hippocampal slices(Whittingham et al., 1984). Thus, it is possible that one of themechanisms bywhich adenosine acts during reperfusion afterischemia is to replenish the adenylate pool. Consistent withour previous report (Hoxworth et al., 1999), the present studyshows that recovery of ATP and AXP after resuscitation fromcardiac arrest is rapid. ATP and AXP were restored about 80–90% of baseline at 1 h after resuscitation in cortex, hippocam-pus and brainstem. However, supplementing adenosine (7.2mg/kg) did not have significant effects on ATP or AXPrestoration after cardiac arrest and resuscitation at this timepoint.Wealsomeasured theATPandAXPat 6, 24 and48haftercardiac arrest and resuscitation in the ADO-treated and theuntreated rats. There were no significant differences in ATPlevels between the two groups at any time point. However, theADO-treated group had significantly higher (∼30%) AXP levelsthan the non-arrested control baseline in the cortex andbrainstem at 24 h after resuscitation. This result suggests thatadenosine treatment may have a transient effect in enriching
the adenylate pool above baseline; thus, the ADO-treatedgroups had a better overall survival rate and hippocampalneuronal preservation. The increased AXP in this group wouldbe as a result of better tissue preservation. The lower level ofAXP in the hippocampus of the untreated group at 48 hmay bea result of increased delayed neuronal death.
3.3. Adenosine-induced hypothermia
Mild or moderate hypothermia has been shown to protect thebrain from ischemic damage (Busto et al., 1987; Ginsberg et al.,1992; Green et al., 1992; Lanier, 1995; Maher and Hachinski,1993). Hypothermic neuroprotection may involve mechan-isms such as reduction of excitotoxic amino acid release(Busto et al., 1989; Mitani et al., 1991), attenuation of depletionof ATP and reduction of cerebral metabolic rate (Michenfelderand Milde, 1991). It has been found that hypothermia reducespostischemic oxygen radical activity (Globus et al., 1995) andthus results in less consumption of endogenous antioxidants(Karibe et al., 1994) and reduced lipid peroxidation (Lei et al.,1994). This suggests that the neuroprotective effects ofhypothermia are through the inhibition of ametabolic cascadeinvolving free radicals produced during reperfusion. Adeno-sine-induced depression of brain temperature is an importantbut indirect element of its therapeutic properties. Hypother-mia induces supersensitivity of adenosine A1 receptors(Broadley et al., 1985). Neuroprotective impact of adenosineA1 receptor agonists is consequent to their hypothermia-inducing properties (Miller et al., 1980). Our results show thatpostresuscitation infusion of adenosine (7.2 mg/kg) depressedthe brain temperature after resuscitation through the 6-h monitoring period. Preventing adenosine-induced hypo-thermia results in lower survival rate (Table 2) and increasedhippocampal neuronal death (Fig. 1). Thus, one part of theneuroprotective effects of adenosine can be ascribed toadenosine-induced hypothermia. However, CA1 cells are notprotected in long-term recovery even with the hypothermiceffects induced by adenosine. Treatment with 2-chloroadeno-sine, an adenosine A1 receptor agonist, had a more significanthypothermic effect during the period of infusion, but the

214 B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
temperature recoverywasmore rapid, thoughnot complete bythe end of 6 h.
3.4. Postischemic brain blood flow
Reperfusion following global ischemia is often characterizedby an initial hyperemia, followed by a period of hypoperfusion,where cerebral blood flow (CBF) is reduced below preischemicbaseline (Gidday et al., 1996; Kagstrom et al., 1983; Leffler et al.,1989; Miller and Hsu, 1992). Increasing interstitial adenosinelevels secondary to adenosine transport blockade can signif-icantly reduce the magnitude of perfusion deficit followingglobal cerebral ischemia (Gidday et al., 1996). An improvementin functional recovery may derive from strategies aimed atraising CBF. Adenosine may increase CBF through localvasodilation and inhibition of clot formation (Phillis et al.,1995). Our results showed that the treatment with adenosinesignificantly improved brain perfusion at 1 h after resuscita-tion in all regions, especially brainstem. The improvement ofpostresuscitation brain blood flow may be one of themechanisms that results in better overall survival andhippocampal neuronal preservation.
3.5. Postresuscitation brain edema
Brain edema raises intracranial pressure and leads to furthertissuedamagethroughsecondary ischemia.Theextentofbrainedema is critical in determining postresuscitation survival;especially in regions such as brainstem which containscardiovascular and respiratory centers. In our study, most oftheratsthatdidnotsurvivefor4daysdiedwithinthefirst2daysof recovery. High mortality in the early recovery days may bedue to brain edema, which peaks at 1, 6 and 24 h after resus-citation in the cortex and hippocampus, and 24 h in the brain-stem. Vascular endothelial growth factor (VEGF) is thought toinduce transient vascular leakageunder the conditions suchashypoxia or ischemia. VEGF expression has been reported toincrease significantly between 12 and 48 h after cardiac arrestand resuscitation (Pichiule et al., 1999). Induction of VEGF aftercardiac arrest and resuscitation may result in increasedvascular permeability and alteration of BBB characteristicsfollowing global ischemia (Pluta et al., 1994) and further lead tovasogenic edema. Mild hypothermia has been suggested toreduce brain edema (Belanger et al., 2005; Nito et al., 2004). Thetreatmentwithadenosineattenuatedtheextentofbrainedemaafter resuscitation from cardiac arrest; possibly attributablemechanistically to adenosine-induced hypothermia.
In conclusion, these results demonstrate that adenosine treat-ment protects the brain from reperfusion injury induced bycardiac arrest and resuscitation. Additionally, adenosine-inducedhypothermia and improvement of postresuscitation blood flowmay be the two major contributors to its neuroprotection.
4. Experimental procedures
4.1. Animal preparation
The experimental protocol employed by this study was approvedby the Animal Care and Use Committee at Case Medical School.
Male Wistar rats (250–300 g) purchased from Charles River werehoused in our animal resources facility for at least 5 days on a 12h day–night diurnal cycle.
Rats were randomly assigned to the following groups: (1)non-arrested group; (2) untreated group; (3) ADO-treated group(ADO 4.8 mg/kg, 80μg/kg/min for 60 min); (4) ADO-treated group(ADO 7.2 mg/kg, 80μg/kg/min for 90 min); (5) ADO-Temp group(7.2 mg/kg, 80μg/kg/min for 90 min) with correction of ADO-induced hypothermia; (6) 2-chloroadenosine-treated group(2CADO). On the day of experiment, anesthesia was inducedby halothane (2.5% halothane, 70% N2O in O2) and maintainedwith 1–2% halothane and 70% nitrous oxide in oxygen througha nasal cone. A Silastic catheter (0.64 mm i.d., 1.19 mm o.d.;Dow Corning) was inserted through the external jugular veininto the right atrium. The ventral tail artery or femoral artery(for the blood flow study) was cannulated with Polyethylenetubing (Intramedic PE-50, 0.058 mm i.d., 0.965 mm o.d.; BectonDickson) for monitoring systemic arterial blood pressure andobtaining blood samples for the measurement of blood gases,pH (ABL5, Radiometer Medical; Copenhagen, Denmark), hemat-ocrit, plasma glucose and plasma lactate. The rats were allowedto recover from anesthesia for at least 1 h in plastic rodentrestraints. For blood flow and extraction fraction measure-ments, rats were then orotracheally intubated and ventilatedwith a gas mixture (1–2% halothane, 30% oxygen in 70% nitrousoxide) at a rate of ∼70 times/min, tidal volume of 1 ml/100 gbody weight. Body temperature during each experiment wasmaintained at 37 °C by an infrared heat lamp (250 W, 45 cmabove the body) regulated by feedback from a rectal tempera-ture probe.
4.2. Induction of reversible total cerebral ischemia
Reversible total cerebral ischemia was achieved as previouslyreported (Crumrine et al., 1991; Hoxworth et al., 1999). Cardiacarrest was induced in the conscious rat by the rapid sequentialintra-atrial injection of D-tubocurare (0.3 mg) and ice-cold KClsolution (0.5 M; 0.12 ml/100 g of body weight). Resuscitated ratswere orotracheally intubated with a 14-guage catheter (Angiocath)and the tracheal tube was attached to a rodent ventilator.Resuscitation was initiated at 7 min after arrest, and ventilation(100%O2, tidal volume: 10 cm3/kg; respiratory rate: 80 breaths/min)was begun simultaneously with chest compressions and theadministration of intravenous fluid (0.9% sodium chloride) at arate of approximately 0.5 ml/min. Once a spontaneous heart beatreturned, epinephrine (4–10 μg) was administered intravenously tobring the mean blood pressure above 80% of prearrest value, thepoint at which animal was considered to be resuscitated. Theduration of arrest in this study was about 12 min. Afterresuscitation, the oxygen content was decreased gradually to 21–30% balanced with N2O. Ventilation was adjusted, on the basis ofblood gases, to normal ranges until the rats regained theirspontaneous respiration.
For the untreated rats, normal saline (10 ml/kg) wasadministrated intravenously for 90 min after resuscitation. Forthe two dosage groups (4.8 mg/kg and 7.2 mg/kg) of adenosine-treated rats, intravenous administration of adenosine (Sigma)was begun immediately after resuscitation at a constant rate of80μg/kg/min for 60 or 90 min, respectively. In ADO-Temp group,adenosine was given at a constant rate of 80μg/kg/min for 90min; temperature maintained to match the untreated group for6 h with an infrared heat lamp (250 W, 45 cm above the rathead) and regulated by feedback from a temperature probe inthe temporalis muscle. For the 2-chloroadenosine-treated group,2-chloroadenosine (0.03 mg/kg, Sigma) was given intravenouslyover 90 min after the rats were resuscitated.
For metabolite assay and brain edema evaluation, rats werefrozen in situ by funnel freezing with liquid nitrogen (Hoxworth etal., 1999; Ponten et al., 1973) at 1, 6, 24 and 48 h after resuscitation.

215B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
Non-arrested control rats underwent all surgical proceduresexcept cardiac arrest and were frozen in situ as above. Ratsundergoing arrest without recovery were submersed in liquidnitrogen completely after 12min of arrest and the frozen rats werestored at −80 °C.
For the long-term recovery rats (4 days and 30 days), overallsurvival rates were determined.
4.3. Measurement of temporalis muscle temperature
The temperature probe was inserted into the temporalis musclebefore cardiac arrest. The temperature was monitored (YSI tele-thermometer) in resuscitated rats up to 6 h of recovery. For theADO-Temp group, the temporalis muscle temperature was main-tained at 37 ± 0.5 °C during the 6-h period by an infrared heat lamp(250 W, 45 cm above the head).
4.4. Hippocampal neuronal counts
For hippocampal neuronal survival analysis, the rats that survivedfor 4 days, 30 days and non-arrested controls were deeplyanesthetized with halothane, perfused through the heart withabout 200 ml 0.1 M phosphate sodium buffer and perfusion-fixedwith 4% paraformaldehyde. The brains were removed andembedded in paraffin. Serial 5 μm coronal sections were made atthe level of anterior hippocampus (atlas plate 30; Palkovits, 1988)and H&E stained. The entire length of the hippocampal pyramidalcell layer in the hippocampus was viewed under high power lightmicroscopy (400×). Neurons with rounded cell bodies and clearlyvisible nucleoli were counted. The number of neurons survivingwas evaluated in CA1 region and other regions (CA2–CA3) ofpyramidal cells in hippocampus.
4.5. Extraction fraction of adenosine and regional brain bloodflow
Extraction fraction of adenosine was measured by the singlepass dual-label indicator fractionation method (LaManna andHarik, 1985; LaManna and Harik, 1986; Puchowicz et al., 2003),which also allowed simultaneous blood flow measurement. Thearterial cannula was connected to a withdrawal pump calibratedto 1.6 ml/min. A bolus (0.15 ml) containing 10 μCi n-[14C]butanol(reference isotope) and 40 μCi [3H]adenosine (1.0 mCi/ml,American Radiolabeled Chemicals, St. Louis, MO) plus normalsaline was injected in the jugular vein catheter 3 s after thewithdrawal pump was started. The animal was decapitated andthe pump was stopped simultaneously 10 s after bolus injection.After collecting a venous sample from the foramen magnumusing capillary tubes, the brain was removed rapidly andbilateral samples (frontal and parietal cortex, hippocampus,brainstem and cerebellum) were weighed and solubilized inpreweighed scintillation vials. The withdrawn arterial blood wasreweighed and collected quantitatively into weighed vials.Aliquots of arterial blood, venous plasma and brain samples,as previously described (LaManna et al., 1993; Puchowicz et al.,2003), were measured for their radioactive contents on a β-scintillation counter (1600 TR Liquid Scintillation Analyzer,Packard). From measured radioisotope contents in each of thetissue and blood samples, the following calculations wereperformed:
E ¼ ½ð3H=14CÞtissue�=½ð3H=14C Þart�BF ¼ Fs � ½14C �tissue=ð½14C �art � tissue wtÞ
Where E is the extraction fraction; BF is blood flow, ml/min/g; Fsis the withdrawal rate of the syringe pump; DPMs in brainsamples were corrected for intravascular compartment volumes
(Vpl) by: DPMtissue correct = dpmtissue − (dpmvenous / μl × Vpl × tissuewt), as previously described (LaManna et al., 1993).
In another set of rats, the time course of regional brain bloodflow was determined by [14C]-iodantipyrine (IAP, New EnglandNuclear) autoradiography (Lauro et al., 1999) in the untreatedrats (1, 6, 24 and 48 h of recovery) and in the ADO-treated group(1 h of recovery). For each determination, the femoral arterycatheter was attached to a withdrawal syringe pump set to awithdraw rate at 1.6 ml/min. A bolus of 25 μCi of [14C] IAP in 150μl of normal saline was injected intra-arterial 3 s after the pumpwas started. The rat was decapitated and the pump stoppedsimultaneously 10 s after the bolus injection. The brain wasquickly removed, frozen on dry ice and stored at −80 °C. Thereference arterial blood was collected and its radioactive contentwas determined (see above). For autoradiography, each frozenbrain was sectioned (20 μm) at the levels of atlas plate 13, 30 and69 (Palkovits, 1988) in a cryomicrotome (−20 °C). Brain sectionsand Amersham [14C]-Micro-scale standards were placed on glassslides, covered with an Amersham Hyperfilm β-Max autographicfilm and exposed for 21 days. The images were digitized andbackground corrected using a BIOQUANT image analysis system(R&M Biometries, INC). Optical densities were converted tonanoCuries per gram using standard curves generated from[14C]-Micro-scale standards. The blood flow was calculated bythe equation below:
Blood flowðml=g=minÞ¼ ðTissue nCi=g � Pump rateðml=minÞÞ=ðReference Blood nCiÞ
4.6. Tissue water content
The brains of frozen rats were removed in a glove box (−30 °C) andtissue samples from representative frozen brains were taken in acryomicrotome (−30 °C) by a small scoop from one side (the otherside was used for metabolite assays) of cortical, hippocampal andbrainstem regions. Tissue samples were quickly placed into tared,dried, capped Eppendorf tubes and reweighed to determine tissuewet weights. The tissues were then dried to constant weight at100 °C to determine tissue dry weights. Water content (%) wascalculated as (wet weight − dry weight) / (wet weight) × 100.
4.7. Metabolite assays
The frozen brainswere sectioned (20 μm) at the levels of atlas plate13, 30 and 69 (Palkovits, 1988) in a cryomicrotome (−20 °C). Thesamples of the cortex, brain stem and hippocampus weredissected from frozen lyophilized sections. Levels of ATP andtotal adenylates (AXP = ATP + ADP + AMP) were analyzed in tissuesamples by microquantitative histochemistry assay methodsdescribed previously (Lust et al., 1981). Metabolite levels werereported as nanomoles per milligram dry tissue weight.
4.8. Statistical analysis
Statistical analyses were performed using SPSS v13.0 forWindows.Group comparisons were made by one-way analysis of variance(ANOVA) using Tukey's statistic. The comparison between any twogroupswasanalyzedwith a t test for paired sample, two-tailed. Thesurvival analysiswasperformedusing aWilcoxon (Gehan) survivalanalysis. Significance was considered at the level of P b 0.05.
Acknowledgments
This work was supported by NIH grants GM066309, NS38632and NS46074, and a grant from the Kenneth HaasMedical Care

216 B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
Trust. We would like to especially thank Sue Foss and AndreIvy for their assistance in preparation of the manuscript.
R E F E R E N C E S
Andine, P., Rudolphi, K.A., Fredholm, B.B., Hagberg, H., 1990. Effectof propentofylline (HWA 285) on extracellular purines andexcitatory amino acids in CA1 of rat hippocampus duringtransient ischaemia. Br. J. Pharmacol. 100, 814–818.
Belanger, M., Desjardins, P., Chatauret, N., Rose, C., Butterworth,R.F., 2005. Mild hypothermia prevents brain edema andattenuates up-regulation of the astrocytic benzodiazepinereceptor in experimental acute liver failure. J. Hepatol. 42,694–699.
Broadley, K.J., Broome, S., Paton, D.M., 1985. Hypothermia-induced supersensitivity to adenosine for responsesmediated via A1-receptors but not A2-receptors. Br. J.Pharmacol. 84, 407–415.
Brodie, M.S., Lee, K., Fredholm, B.B., Stahle, L., Dunwiddie, T.V.,1987. Central versus peripheral mediation of responses toadenosine receptor agonists: evidence against a central modeof action. Brain Res. 415, 323–330.
Busto, R., Dietrich, W.D., Globus, M.Y., Valdes, I., Scheinberg, P.,Ginsberg, M.D., 1987. Small differences in intraischemic braintemperature critically determine the extent of ischemicneuronal injury. J. Cereb. Blood Flow Metab. 7, 729–738.
Busto, R., Dietrich, W.D., Globus, M.Y., Ginsberg, M.D., 1989.Postischemic moderate hypothermia inhibits CA1hippocampal ischemic neuronal injury. Neurosci. Lett. 101,299–304.
Crumrine, R.C., LaManna, J.C., 1991. Regional cerebral metabolites,blood flow, plasma volume, and mean transit time in totalcerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 11,272–282.
Crumrine, R.C., LaManna, J.C., Lust, W.D., 1991. Regional changesin intracellular pH determined by neutral redhistophotometry and high energy metabolites during cardiacarrest and following resuscitation in the rat. Metab. Brain Dis.6, 145–155.
Evans, M.C., Swan, J.H., Meldrum, B.S., 1987. An adenosineanalogue, 2-chloroadenosine, protects against long termdevelopment of ischaemic cell loss in the rat hippocampus.Neurosci. Lett. 83, 287–292.
Gidday, J.M., Kim, Y.B., Shah, A.R., Gonzales, E.R., Park, T.S., 1996.Adenosine transport inhibition ameliorates postischemichypoperfusion in pigs. Brain Res. 734, 261–268.
Ginsberg, M.D., Sternau, L.L., Globus, M.Y., Dietrich,W.D., Busto, R.,1992. Therapeutic modulation of brain temperature: relevanceto ischemic brain injury. Cerebrovasc. Brain Metab. Rev. 4,189–225.
Globus, M.Y., Alonso, O., Dietrich, W.D., Busto, R., Ginsberg, M.D.,1995. Glutamate release and free radical production followingbrain injury: effects of posttraumatic hypothermia.J. Neurochem. 65, 1704–1711.
Green, E.J., Dietrich, W.D., van Dijk, F., Busto, R., Markgraf, C.G.,McCabe, P.M., Ginsberg, M.D., Schneiderman, N., 1992.Protective effects of brain hypothermia on behavior andhistopathology following global cerebral ischemia in rats. BrainRes. 580, 197–204.
Harik, S.I., Behmand, R.A., LaManna, J.C., 1994. Hypoxia increasesglucose transport at blood–brain barrier in rats. J. Appl. Physiol.77, 896–901.
Hoxworth, J.M., Xu, K., Zhou, Y., Lust, W.D., LaManna, J.C., 1999.Cerebral metabolic profile, selective neuron loss, and survivalof acute and chronic hyperglycemic rats following cardiacarrest and resuscitation. Brain Res. 821, 467–479.
Kagstrom, E., Smith, M.L., Siesjo, B.K., 1983. Cerebral circulatory
responses to hypercapnia and hypoxia in the recovery periodfollowing complete and incomplete cerebral ischemia in therat. Acta Physiol. Scand. 118, 281–291.
Kalaria, R.N., Harik, S.I., 1986a. Adenosine receptors of cerebralmicrovessels and choroid plexus. J. Cereb. Blood Flow Metab. 6,463–470.
Kalaria, R.N., Harik, S.I., 1986b. Nucleoside transporter of cerebralmicrovessels and choroid plexus. J. Neurochem. 47, 1849–1856.
Kalaria, R.N., Harik, S.I., 1988. Adenosine receptors and thenucleoside transporter in human brain vasculature. J. Cereb.Blood Flow Metab. 8, 32–39.
Karibe, H., Chen, S.F., Zarow, G.J., Gafni, J., Graham, S.H.,Chan, P.H., Weinstein, P.R., 1994. Mild intraischemichypothermia suppresses consumption of endogenousantioxidants after temporary focal ischemia in rats. BrainRes. 649, 12–18.
LaManna, J.C., Harik, S.I., 1985. Regional comparisons of brainglucose influx. Brain Res. 326, 299–305.
LaManna, J.C., Harik, S.I., 1986. Regional studies of blood–brainbarrier transport of glucose and leucine in awake andanesthetized rats. J. Cereb. Blood Flow Metab. 6, 717–723.
LaManna, J.C., Harrington, J.F., Vendel, L.M., Abi-Saleh, K., Lust, W.D., Harik, S.I., 1993. Regional blood–brain lactate influx. BrainRes. 614, 164–170.
LaManna, J.C., Griffith, J.K., Cordisco, B.R., Bell, H.E., Lin, C.W.,Pundik, S., Lust, W.D., 1995. Rapid recovery of rat brainintracellular pH after cardiac arrest and resuscitation. BrainRes. 687, 175–181.
Lanier, W.L., 1995. Cerebral metabolic rate and hypothermia: theirrelationship with ischemic neurologic injury. J. Neurosurg.Anesthesiol. 7, 216–221.
Lauro, K.L., Kabert, H., LaManna, J.C., 1999. Methyl isobutylamiloride alters regional brain reperfusion afterresuscitation from cardiac arrest in rats. Brain Res. 831,64–71.
Leffler, C.W., Busija, D.W., Mirro, R., Armstead,W.M., Beasley, D.G.,1989. Effects of ischemia on brain blood flow and oxygenconsumption of newborn pigs. Am. J. Physiol. 257,H1917–H1926.
Lei, B., Tan, X., Cai, H., Xu, Q., Guo, Q., 1994. Effect of moderatehypothermia on lipid peroxidation in canine brain tissue aftercardiac arrest and resuscitation. Stroke 25, 147–152.
Lust, W.D., Feussner, G.K., Barbehenn, E.K., Passonneau, J.V.,1981. The enzymatic measurement of adenine nucleotidesand P-creatine in picomole amounts. Anal. Biochem. 110,258–266.
Maher, J., Hachinski, V., 1993. Hypothermia as a potentialtreatment for cerebral ischemia. Cerebrovasc. Brain Metab.Rev. 5, 277–300.
Marangos, P.J., 1990. Adenosinergic approaches to stroketherapeutics. Med. Hypotheses 32, 45–49.
Michenfelder, J.D., Milde, J.H., 1991. The relationship amongcanine brain temperature, metabolism, and function duringhypothermia. Anesthesiology 75, 130–136.
Miller, L.P., Hsu, C., 1992. Therapeutic potential for adenosinereceptor activation in ischemic brain injury. J. Neurotrauma 9(Suppl 2), S563–S577.
Miller, C.L., Lampard, D.G., Alexander, K., Brown, W.A., 1980. Localcerebral blood flow following transient cerebral ischemia: I.Onset of impaired reperfusion within the first hour followingglobal ischemia. Stroke 11, 534–541.
Mitani, A., Kataoka, K., 1991. Critical levels of extracellularglutamate mediating gerbil hippocampal delayed neuronaldeath during hypothermia: brain microdialysis study.Neuroscience 42, 661–670.
Newman, G.C., Hospod, F.E., Trowbridge, S.D., Motwani, S., Liu,Y., 1998. Restoring adenine nucleotides in a brain slice modelof cerebral reperfusion. J. Cereb. Blood Flow Metab. 18,675–685.

217B R A I N R E S E A R C H 1 0 7 1 ( 2 0 0 6 ) 2 0 8 – 2 1 7
Nito, C., Kamiya, T., Ueda, M., Arii, T., Katayama, Y., 2004. Mildhypothermia enhances the neuroprotective effects of FK506and expands its therapeutic window following transient focalischemia in rats. Brain Res. 1008, 179–185.
Palkovits, M., 1988. In: Brownstein, M.J. (Ed.), Maps and Guide toMicrodissection of Rat Brain.
Phillis, J.W., 1989. Adenosine in the control of the cerebralcirculation. Cerebrovasc. Brain Metab. Rev. 1, 26–54.
Phillis, J.W., Preston, G., Delong, R.E., 1984. Effects of anoxia oncerebral blood flow in the rat brain: evidence for a role ofadenosine in autoregulation. J. Cereb. Blood Flow Metab. 4,586–592.
Phillis, J.W., Perkins, L.M., Smith-Barbour, M., O'Regan, M.H., 1995.Oxypurinol-enhanced postischemic recovery of the rat braininvolves preservation of adenine nucleotides. J. Neurochem.64, 2177–2184.
Pichiule, P., Chavez, J.C., Xu, K., LaManna, J.C., 1999. Vascularendothelial growth factor upregulation in transient globalischemia induced by cardiac arrest and resuscitation in ratbrain. Brain Res. Mol. Brain Res. 74, 83–90.
Pluta, R., Lossinsky, A.S., Wisniewski, H.M., Mossakowski, M.J.,1994. Early blood–brain barrier changes in the rat followingtransient complete cerebral ischemia induced by cardiacarrest. Brain Res. 633, 41–52.
Ponten, U., Ratcheson, R.A., Siesjo, B.K., 1973. Metabolic changes inthe brains of mice frozen in liquid nitrogen. J. Neurochem. 21,1211–1216.
Puchowicz, M.A., Xu, K., LaManna, J.C., 2003. Single-pass dual-labelindicator method. Blood-to-brain transport of glucose andshort-chain monocarboxylic acids. Methods Mol. Med. 89,265–276.
Rudolphi, K.A., Schubert, P., Parkinson, F.E., Fredholm, B.B., 1992.Neuroprotective role of adenosine in cerebral ischaemia.Trends Pharmacol. Sci. 13, 439–445.
Schubert, P., Ogata, T., Marchini, C., Ferroni, S., Rudolphi, K., 1997.Protective mechanisms of adenosine in neurons and glial cells.Ann. N. Y. Acad. Sci. 825, 1–10.
Sweeney, M.I., 1997. Neuroprotective effects of adenosine incerebral ischemia: window of opportunity. Neurosci. Biobehav.Rev. 21, 207–217.
von Lubitz, D.K., 1999. Adenosine and cerebral ischemia:therapeutic future or death of a brave concept? Eur. J.Pharmacol. 371, 85–102.
von Lubitz, D.K., Carter, M.F., Beenhakker, M., Lin, R.C.,Jacobson, K.A., 1995. Adenosine: a prototherapeuticconcept in neurodegeneration. Ann. N. Y. Acad. Sci. 765,163–178.
Whittingham, T.S., Lust, W.D., Passonneau, J.V., 1984. An in vitromodel of ischemia: metabolic and electrical alterations in thehippocampal slice. J. Neurosci. 4, 793–802.
William, M., 1989. Adenosine: the prototypic neuromodulator.Neurochem. Int. 14, 249–264.
Wu, P.H., Phillis, J.W., 1982. Uptake of adenosine by isolated ratbrain capillaries. J. Neurochem. 38, 687–690.





























