Embed Size (px)
Citation preview

Original article
Impaired SERCA function contributes to cardiomyocyte dysfunction ininsulin resistant rats
Loren E. Wold b,1, Kaushik Dutta a,1, Meredith M. Mason a, Jun Ren b,c, Steven E. Cala d,Mary L. Schwanke e, Amy J. Davidoff a,*
a College of Osteopathic Medicine, University of New England, 11 Hills Beach Road, Biddeford, ME 04005, USAb Department of Pharmacology, Physiology and Therapeutics, University of North Dakota, Grand Forks, ND, USA
c College of Pharmaceutical Sciences, University of Wyoming, Laramie, WY, USAd School of Medicine, Wayne State University, Detroit, MI, USA
e College of Arts and Sciences, University of Maine at Farmington, Farmington, ME, USA
Received 14 December 2004; received in revised form 9 March 2005; accepted 21 March 2005
Available online 04 May 2005
Abstract
Ventricular dysfunction in type 2 diabetic patients is becoming apparent early after diagnosis of diabetes, but the cellular mechanismscontributing to this dysfunction are not well established. Our group has recently identified cardiomyocyte dysfunction in diet-induced insulinresistant rats that have not developed type 2 diabetes. The present investigation was designed to determine cellular mechanisms contributingto slowed cardiomyocyte relaxation in sucrose (SU)-fed rats. SU-feeding was used to induce whole-body insulin resistance. After 9–12 weekson diet, isolated ventricular myocyte shortening/relengthening were slower in SU-fed adult male Wistar rats (42–63%) compared to starch(ST)-fed controls. Cytosolic Ca2+ removal attributable to Na+/Ca2+ exchange (NCX) and to sarco(endo)plasmic reticulum Ca2+-ATPase(SERCA) was evaluated with fluo-3/AM. Caffeine-releasable Ca2+ and cytosolic Ca2+ clearing through NCX were normal, whereas Ca2+
uptake by SERCA was significantly slower in SU myocytes (330 ± 29 ms) compared to ST cells (253 ± 16 ms). Protein levels for SERCA,NCX and phospholamban were not affected by SU-feeding. Manipulating intracellular Ca2+ with various positive inotropic interventions (e.g.post-rest potentiation, isoproterenol) and changes in stimulus frequency demonstrated that mechanical properties can be improved in subsetsof myocytes. Thus, we conclude that impaired SERCA activity (with normal protein content) contributes to cardiomyocyte dysfunction ininsulin resistant animals, whereas NCX function and expression are normal. These results suggest that subtle changes in Ca2+ regulationwhich occur prior to overt ventricular dysfunction/failure, may be common to early stages of a number of disorders involving insulin resis-tance (e.g. diabetes, obesity, syndrome X and hypertension).© 2005 Elsevier Ltd. All rights reserved.
Keywords: Cardiomyopathy; Type 2 diabetes; Abnormal Ca2+ regulation; SERCA; NCX
1. Introduction
The incidence of type 2 diabetes, which accounts for over90% of all new cases of diagnosed diabetes, is becoming apandemic and is especially alarming considering the increasedincidence of insulin resistance and diabetes in young adultsand children [1,2]. Type 2 diabetes is a progressive, multifac-torial disease, which typically involves co-morbidities such
as dyslipidemia, obesity, hypertension and insulin resistance[1]. The high mortality rate in diabetic patients is predomi-nantly due to heart disease. A significant number of diabeticpatients exhibit diabetic cardiomyopathy, a ventricular dys-function which can develop independent of coronary vascu-lar disease or hypertension, with impaired diastolic functiondeveloping prior to systolic abnormalities [3,4].
Diabetic cardiomyopathy is characterized by phenotypicchanges in ventricular myocytes that have been well describedin animal models with long-term type 1 diabetes. This cardi-omyopathy involves abnormal myocyte excitation-contraction(E-C) coupling (e.g. prolonged action potentials, slowed cyto-solic Ca2+ fluxes and slowed myocyte shortening and relength-
* Corresponding author. Tel.: +1 207 283 0170x2824; fax: +1 207 2945931.
E-mail address: [email protected] (A.J. Davidoff).1 These authors contributed equally to this investigation.
Journal of Molecular and Cellular Cardiology 39 (2005) 297–307
www.elsevier.com/locate/yjmcc
0022-2828/$ - see front matter © 2005 Elsevier Ltd. All rights reserved.doi:10.1016/j.yjmcc.2005.03.014

ening [5–8]). Depressed expression and function of sarco(en-do)plasmic reticulum Ca2+-ATPase (SERCA) and Na+/Ca2+
exchange (NCX) are among the cellular changes contribut-ing to the mechanical dysfunctions [9–11].
The consequences of type 2 diabetes on whole heart func-tion are more subtle than those of type 1 diabetes, and oftendo not become apparent until hearts are challenged metaboli-cally or with increased work loads [12–14]. The existence ofcardiomyocyte dysfunction has been shown in some animalmodels of type 2 diabetes [15–17], but is not seen in all mod-els [18]. We have recently described abnormal cardiomyo-cyte E-C coupling in sucrose (SU)-fed, insulin resistant rats[19,20]. SU-fed animals exhibit metabolic abnormalities (e.g.hyperinsulinemia and hypertriglyceridemia, associated witheuglycemia [21,22]), consistent with early stage insulin resis-tance in humans. Unlike most type 2 diabetes models,SU-induced insulin resistance does not present with othercomplicating factors that may alter cardiac physiology (e.g.obesity and hypertension). Thus, we are using a model withwhich to assess the impact of a pre-diabetic metabolic stateon cardiomyocyte function. In this model, myocyte shorten-ing and relengthening are prolonged and cytosolic Ca2+
removal is slowed, similar to that seen in cardiomyocytes fromtype 1 diabetic rats [5]. Based on the observations thatdepressed SERCA and NCX function contribute to type 1 dia-betic cardiomyopathy, we chose to evaluate these prominentmechanisms of cellular Ca2+ regulation in myocytes frominsulin resistant rats.
A variety of physiological interventions are used to evalu-ate contraction and relaxation in heart tissue, including modi-fications in stimulus frequency, extracellular Ca2+ concentra-tion, and adrenergic receptor ligands. These interventions havebeen used extensively to characterize subcellular changes inCa2+ regulation associated with disease, including type 1 dia-betes [5–7,10]. Both b-adrenergic receptor signaling and fre-quency responses have been shown to be depressed in type1 diabetic cardiomyopathy [5,23]. We, therefore, evaluatedmechanical responses to several positive inotropic interven-tions, including isoproteronol and changes in interpulse inter-vals, in a model of insulin resistance.
2. Methods
2.1. Animals and diet
All protocols were approved by the Institutional AnimalCare and Use Committee at the University of New England,and conformed to the Guide for the Care and Use of Labora-tory Animals (NIH Publication No. 85-23, revised 1996).Male Wistar rats (120–140 g) (Charles River Breeding Lab,Wilmington, MA, USA) were in an animal facility under con-trolled conditions (12:12 light:dark cycle). Animals wereallowed water ad libitum and placed on a purified high starchdiet (ST; 68% of kcal from corn starch, 20% protein, 12%fat) for a 1 week baseline period. After the baseline period,
animals were continued either on the ST diet or fed a highsucrose diet (SU; 68% of kcal from sucrose, 20% protein,12% fat) for 9–12 weeks. Both diets were formulated byResearch Diets, Inc., New Brunswick, NJ. It was previouslyshown that body weights are similar in ST- and SU-fed ani-mals, so no attempts to food restrict any of the groups wereemployed [21]. All diets were based on recommendations bythe American Institute of Nutrition.
2.2. Ventricular myocyte isolation, myocyte mechanicsand intracellular Ca2+ transients
All animals were anesthetized by injecting 80 mg/kg ket-amine and 12 mg/kg xylazine ip. Single ventricular myo-cytes were isolated by methods previously described [20].Myocytes were allowed to attach on laminin-coated (10 µg/ml,Collaborative Biochemical Products, Bedford, MA) glass cov-erslips and maintained in control buffer (see below) at 37 °Cin a 100% humidity and 5% CO2 incubator until used (within6 h after isolation).
Mechanical properties and intracellular Ca2+ transients ofventricular myocytes were assessed using a video-based edgedetection coupled to a fluorescent system (IonOptix Co., Mil-ton, MA) as described previously [24]. In brief, myocyteswere electrically stimulated (ES) at 0.5 Hz (unless otherwisestated) and mechanical properties recorded at a sampling rateof 240 Hz. Ca2+ transients were recorded using fluo-3/AM(Molecular Probes Inc., Eugene, OR) while sampling at1000 Hz. Myocytes were incubated in 1 µM fluo-3 sus-pended in control buffer (see below) containing 20% plu-ronic acid for 30 min in the dark, at room temperature. Forboth twitch and Ca2+ transient recordings, the cells weresuperfused with a control buffer (unless otherwise indicated)which was composed of (in mM): 131 NaCl, 4 KCl, 1 CaCl2,1 MgCl2, 10 glucose, 10 HEPES, at pH 7.4.
The indices used to describe isotonic shortening have beenpreviously reported [24,25] and include peak fractional short-ening (PS; peak shortening amplitude normalized to restingcell length), and area under the shortening (contraction) phasenormalized to peak shortening amplitude (AC/PK). The indexused to describe isotonic relengthening is area under therelengthening (relaxation) phase normalized to peak ampli-tude (AR/PK). All indices of shortening/relengthening weredetermined after averaging ~10 steady-state twitches for eachmyocyte, and analyzed off-line using Clampfit (Axon Instru-ments, Foster City, CA). Myocytes were chosen randomlyfrom those meeting the inclusion criteria of sharp, regularstriations and the ability to achieve steady-state twitches (orCa2+ transients) during electrical stimulation. Steady-state wasdefined as consistent peak amplitudes during the recordingperiod for each cell.
2.3. Rapid switching of superfusate
Pharmacological interventions were used to evaluate cyto-solic Ca2+ clearing attributable to SERCA, NCX, or slow pro-
298 L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307

cesses (e.g. mitochondrial Ca2+ uptake), using a rapid switch-ing device as previously described [24]. This system consistsof a multi-barreled pipette that was positioned close to themyocyte being recorded (within ~300 µm). The pipette wasmade of four polyethylene tubes converging to a single efflu-ent tube (dead space was estimated to be less than 10 µl).Electronically controlled valves allowed for rapid switchingof the gravity feed effluent, with a single stream superfusingthe cell. All baseline recordings were made while superfus-ing myocytes with control buffer (see recipe above). Thesuperfusate was then switched to isolate either SR Ca2+ uptakeor NCX (see Section 3 for details).
2.4. Immunoblot assays and antibodies
Myocytes isolated from ST or SU animals were plated on60 mm diameter laminin-coated culture dishes (~105 cellsper dish) and rinsed to minimize cellular debris and non-myocyte cells. For immunoblotting, cells were then homog-enized in 1 ml of a 1% sodium dodecyl sulfate (SDS) solu-tion and heated for 3 min at 70°–80 °C. Cell homogenates inSDS were collected and assayed for protein using the methodof Lowry. SDS-polyacrylamide gel electrophoresis and immu-noblotting were performed as previously described [24]. Pro-teins (60 µg per lane) separated by SDS-PAGE were trans-ferred electrophoretically onto nitrocellulose membranes(0.45 µm; BioRad) in 50 mM sodium phosphate (pH 7.2).The antibodies for NCX were purchased from Swant (Bell-inzona, Switzerland), anti-[17Thr-P]-PLB from Badrilla Ltd.(Leeds, UK) and anti-[16Ser-P]-PLB from Upstate Biotech-nology (Lake Placid, NY). Monoclonal antibodies toSERCA2a (A7R5) and PLB (2D12) were provided by Dr.Larry Jones (Indiana University School of Medicine).Antigen-antibody complexes were detected using [125I] pro-tein A (NEN Life Science Products, Boston, MA) and auto-radiography using Kodak BioMax MS film. To avoid compli-cations associated with non-linearity of X-ray film,quantification of radioactivity was done by excising bandsfrom nitrocellulose and gamma counting. For detecting bandsusing the anti-[17Thr-P]-PLB, enhanced chemiluminescencewas also used because of the weak signal.
2.5. Manipulation of intracellular Ca2+
In a variety of animal models with cardiomyopathies(including diabetes and congestive heart failure), the heartand myocyte dysfunctions are exacerbated by increased stimu-lus frequencies and elevated extracellular Ca2+, and showimpaired responsiveness to b-adrenergic receptor (b-AR)stimulation [6,10,26]. Therefore, we chose several protocolsdesigned to manipulate cellular Ca2+: increasing perfusateCa2+ concentration (1 vs. 2 mM), adding ISO (10–9 M) tocontrol buffer (containing 1 mM Ca2+), and manipulatingstimulus frequency (0.5–5 Hz). A post-rest potentiation pro-tocol was used which involved electrically stimulating myo-cytes at a baseline frequency of 0.5 Hz until steady-state
twitches were attained, then recording the initial twitch fol-lowing a 15 s rest period.
2.6. Statistical analyses
Data are presented as mean ± S.E. Statistical significancebetween control (ST-fed) and insulin resistant (SU-fed) groupswas determined for myocyte mechanics and Ca2+ transientsby a Student’s t-test for all analyses except for the frequencyprotocols. Each index was analyzed separately. A two wayANOVA with repeated measures was used to analyze ST andSU group differences when each myocyte was stimulated tocontract at 1, 2, 3 and 5 Hz. When the ANOVA was found tobe significant, a Bonferroni post hoc test for repeated mea-sures was used for multiple comparisons (SYSTAT, Rich-mond, CA, USA).
3. Results
3.1. Characterization of cellular mechanisms underlyingcardiomyocyte dysfunction
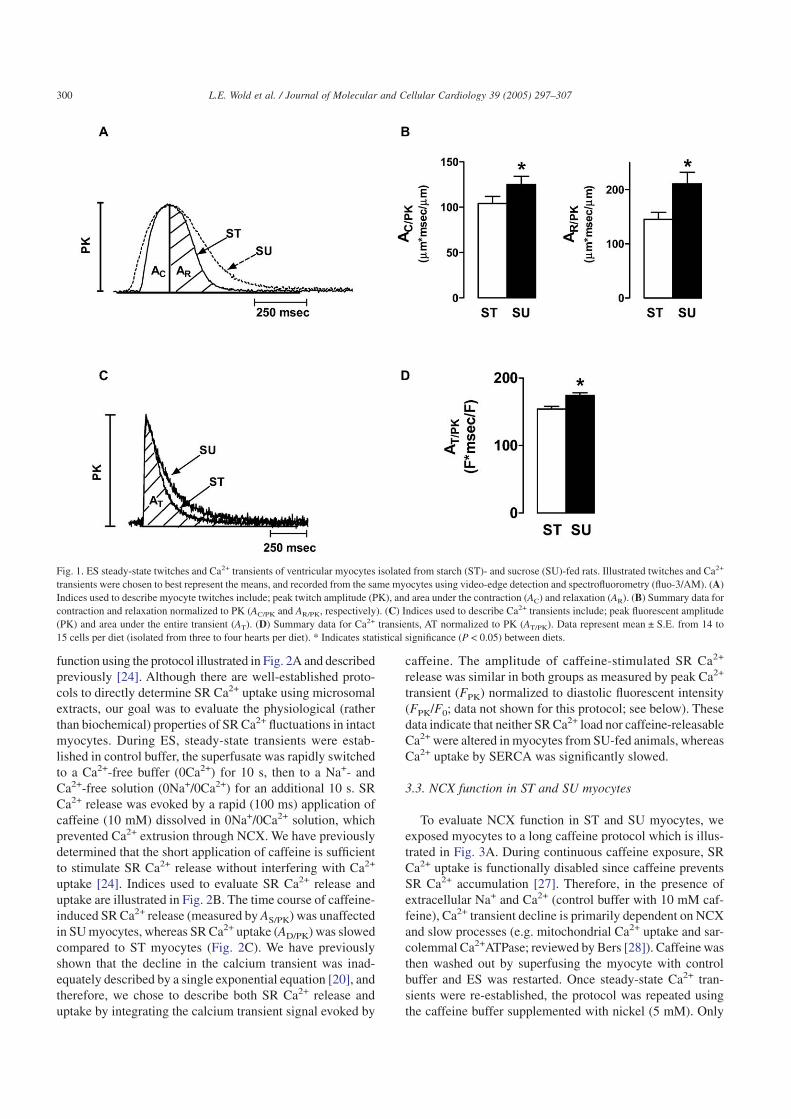
It has been well-established that animals fed a diet high insucrose (SU) develop whole-body insulin resistance [20–22].We have most recently shown that SU-feeding results in ametabolic condition characterized by euglycemia, hyperin-sulinemia, and hypertriglyceridemia without obesity [19,20].As we have previously shown [19,20] and repeated here-within, cardiomyocyte dysfunction develops in SU-fed ani-mals compared to starch (ST)-fed controls (Fig. 1). Indicesused to evaluate myocyte twitches and Ca2+ transients areillustrated in Figs. 1A, C, respectively. The area indices ofshortening and relengthening phases (each normalized to peakshortening amplitude) account for both the time course andshape of the contractile cycle. Larger areas describing short-ening and relengthening indicate slower contraction and relax-ation, respectively. Isolated ventricular myocytes from SU-fedanimals had prolonged shortening (AC/PK) and relengthening(AR/PK) when compared to myocytes from ST-fed animals(Fig. 1B).
ES Ca2+ transients declined more slowly in myocytes iso-lated from SU-fed rats than from ST-fed animals (Fig. 1C,D), indicating that cytosolic Ca2+ removal is slower in myo-cytes from insulin resistant rats. The time course of the fluo-rescence signal decay was described by the area under thetransient normalized to the peak fluorescent intensity (AT/PK).We chose this method of analysis because it describes changesin cytosolic Ca2+ during the entire contractile cycle withoutmaking the assumption that cytosolic Ca2+ clearing follows aspecific time course (e.g. a single exponential decay [20]). Alarger AT/PK indicates slower cytosolic Ca2+ clearing. Repre-sentative transients are shown in Fig. 1C and summary datain Fig. 1D.
3.2. SERCA function in ST and SU myocytes
To determine whether impaired SR Ca2+ uptake contrib-utes to slowed cytosolic Ca2+ removal, we evaluated SERCA
299L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307
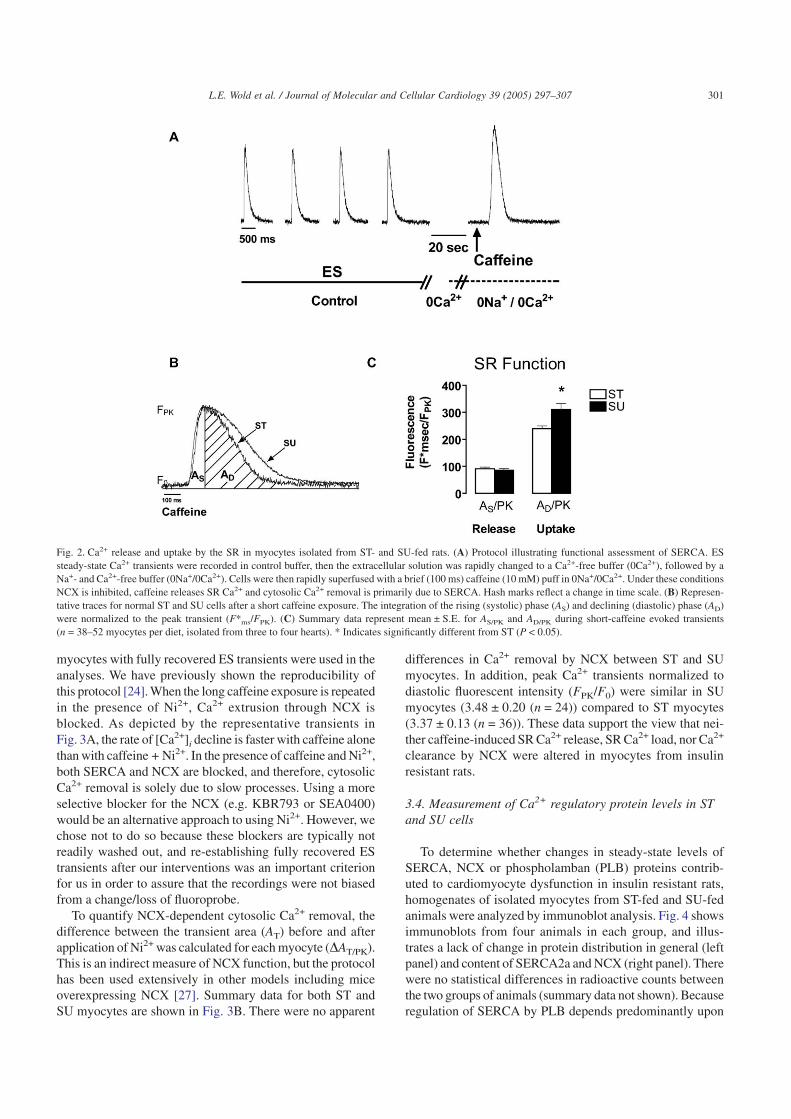
function using the protocol illustrated in Fig. 2A and describedpreviously [24]. Although there are well-established proto-cols to directly determine SR Ca2+ uptake using microsomalextracts, our goal was to evaluate the physiological (ratherthan biochemical) properties of SR Ca2+ fluctuations in intactmyocytes. During ES, steady-state transients were estab-lished in control buffer, the superfusate was rapidly switchedto a Ca2+-free buffer (0Ca2+) for 10 s, then to a Na+- andCa2+-free solution (0Na+/0Ca2+) for an additional 10 s. SRCa2+ release was evoked by a rapid (100 ms) application ofcaffeine (10 mM) dissolved in 0Na+/0Ca2+ solution, whichprevented Ca2+ extrusion through NCX. We have previouslydetermined that the short application of caffeine is sufficientto stimulate SR Ca2+ release without interfering with Ca2+
uptake [24]. Indices used to evaluate SR Ca2+ release anduptake are illustrated in Fig. 2B. The time course of caffeine-induced SR Ca2+ release (measured by AS/PK) was unaffectedin SU myocytes, whereas SR Ca2+ uptake (AD/PK) was slowedcompared to ST myocytes (Fig. 2C). We have previouslyshown that the decline in the calcium transient was inad-equately described by a single exponential equation [20], andtherefore, we chose to describe both SR Ca2+ release anduptake by integrating the calcium transient signal evoked by
caffeine. The amplitude of caffeine-stimulated SR Ca2+
release was similar in both groups as measured by peak Ca2+
transient (FPK) normalized to diastolic fluorescent intensity(FPK/F0; data not shown for this protocol; see below). Thesedata indicate that neither SR Ca2+ load nor caffeine-releasableCa2+ were altered in myocytes from SU-fed animals, whereasCa2+ uptake by SERCA was significantly slowed.
3.3. NCX function in ST and SU myocytes
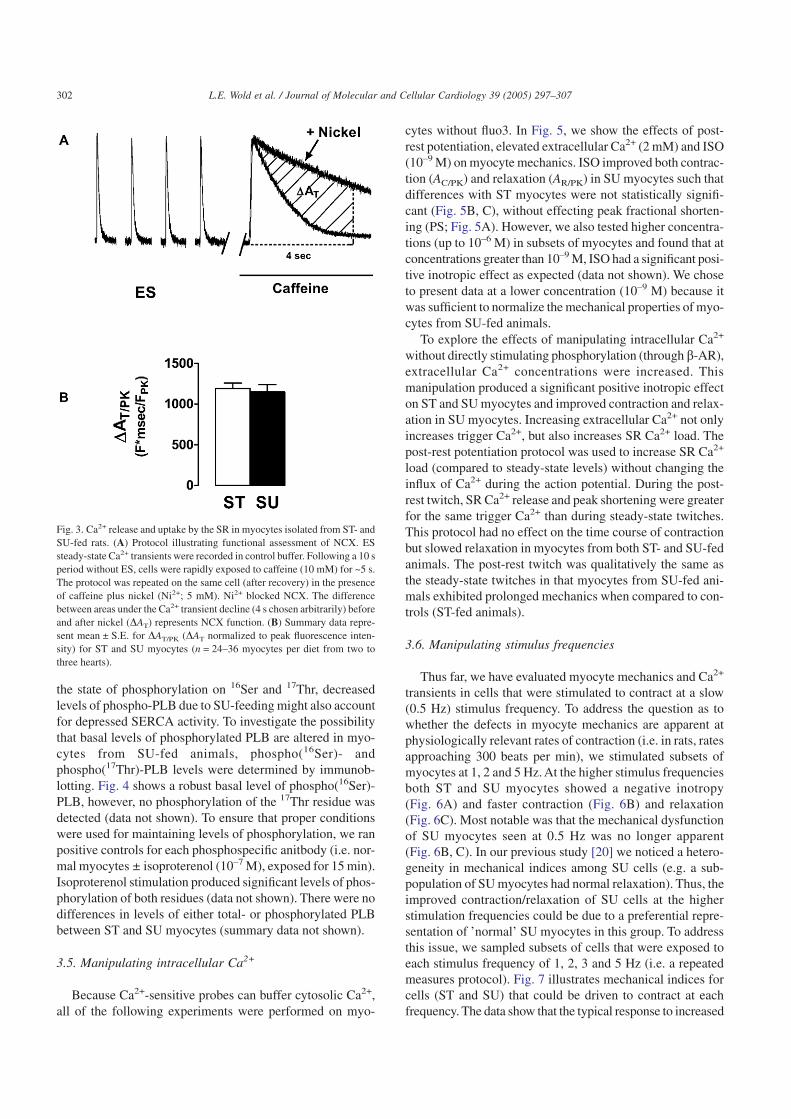
To evaluate NCX function in ST and SU myocytes, weexposed myocytes to a long caffeine protocol which is illus-trated in Fig. 3A. During continuous caffeine exposure, SRCa2+ uptake is functionally disabled since caffeine preventsSR Ca2+ accumulation [27]. Therefore, in the presence ofextracellular Na+ and Ca2+ (control buffer with 10 mM caf-feine), Ca2+ transient decline is primarily dependent on NCXand slow processes (e.g. mitochondrial Ca2+ uptake and sar-colemmal Ca2+ATPase; reviewed by Bers [28]). Caffeine wasthen washed out by superfusing the myocyte with controlbuffer and ES was restarted. Once steady-state Ca2+ tran-sients were re-established, the protocol was repeated usingthe caffeine buffer supplemented with nickel (5 mM). Only
Fig. 1. ES steady-state twitches and Ca2+ transients of ventricular myocytes isolated from starch (ST)- and sucrose (SU)-fed rats. Illustrated twitches and Ca2+
transients were chosen to best represent the means, and recorded from the same myocytes using video-edge detection and spectrofluorometry (fluo-3/AM). (A)Indices used to describe myocyte twitches include; peak twitch amplitude (PK), and area under the contraction (AC) and relaxation (AR). (B) Summary data forcontraction and relaxation normalized to PK (AC/PK and AR/PK, respectively). (C) Indices used to describe Ca2+ transients include; peak fluorescent amplitude(PK) and area under the entire transient (AT). (D) Summary data for Ca2+ transients, AT normalized to PK (AT/PK). Data represent mean ± S.E. from 14 to15 cells per diet (isolated from three to four hearts per diet). * Indicates statistical significance (P < 0.05) between diets.
300 L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307
myocytes with fully recovered ES transients were used in theanalyses. We have previously shown the reproducibility ofthis protocol [24]. When the long caffeine exposure is repeatedin the presence of Ni2+, Ca2+ extrusion through NCX isblocked. As depicted by the representative transients inFig. 3A, the rate of [Ca2+]i decline is faster with caffeine alonethan with caffeine + Ni2+. In the presence of caffeine and Ni2+,both SERCA and NCX are blocked, and therefore, cytosolicCa2+ removal is solely due to slow processes. Using a moreselective blocker for the NCX (e.g. KBR793 or SEA0400)would be an alternative approach to using Ni2+. However, wechose not to do so because these blockers are typically notreadily washed out, and re-establishing fully recovered EStransients after our interventions was an important criterionfor us in order to assure that the recordings were not biasedfrom a change/loss of fluoroprobe.
To quantify NCX-dependent cytosolic Ca2+ removal, thedifference between the transient area (AT) before and afterapplication of Ni2+ was calculated for each myocyte (DAT/PK).This is an indirect measure of NCX function, but the protocolhas been used extensively in other models including miceoverexpressing NCX [27]. Summary data for both ST andSU myocytes are shown in Fig. 3B. There were no apparent
differences in Ca2+ removal by NCX between ST and SUmyocytes. In addition, peak Ca2+ transients normalized todiastolic fluorescent intensity (FPK/F0) were similar in SUmyocytes (3.48 ± 0.20 (n = 24)) compared to ST myocytes(3.37 ± 0.13 (n = 36)). These data support the view that nei-ther caffeine-induced SR Ca2+ release, SR Ca2+ load, nor Ca2+
clearance by NCX were altered in myocytes from insulinresistant rats.
3.4. Measurement of Ca2+ regulatory protein levels in STand SU cells
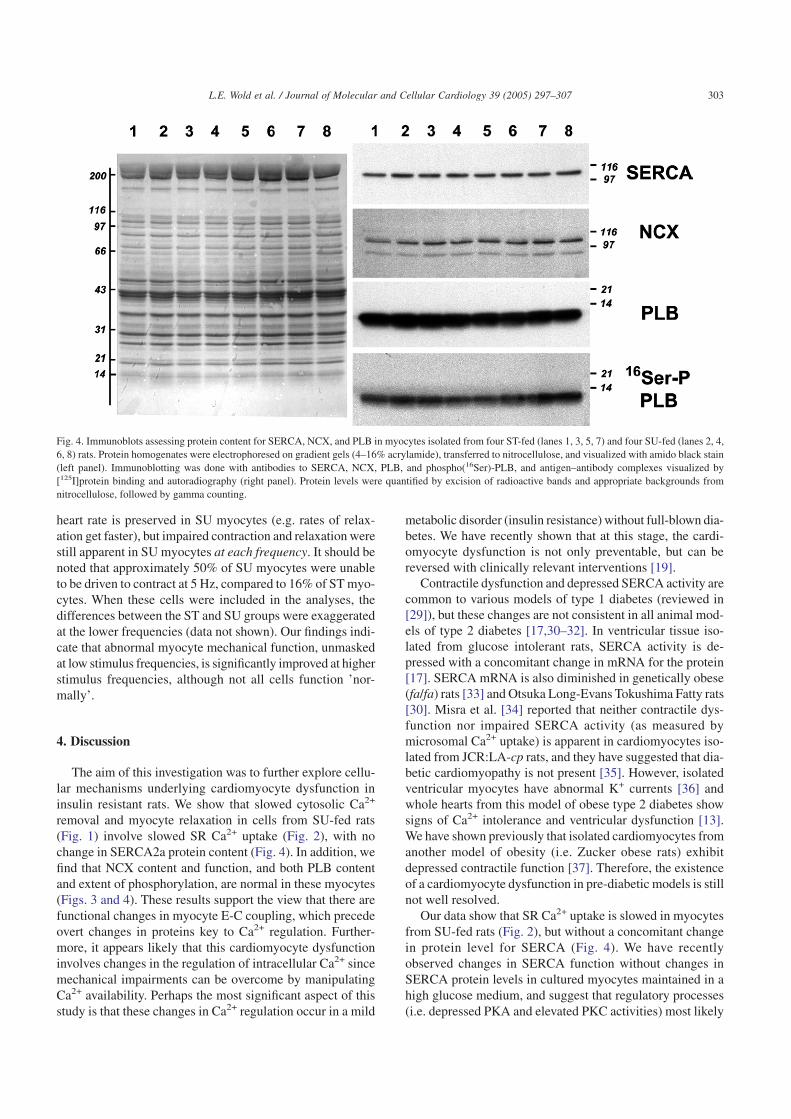
To determine whether changes in steady-state levels ofSERCA, NCX or phospholamban (PLB) proteins contrib-uted to cardiomyocyte dysfunction in insulin resistant rats,homogenates of isolated myocytes from ST-fed and SU-fedanimals were analyzed by immunoblot analysis. Fig. 4 showsimmunoblots from four animals in each group, and illus-trates a lack of change in protein distribution in general (leftpanel) and content of SERCA2a and NCX (right panel). Therewere no statistical differences in radioactive counts betweenthe two groups of animals (summary data not shown). Becauseregulation of SERCA by PLB depends predominantly upon
Fig. 2. Ca2+ release and uptake by the SR in myocytes isolated from ST- and SU-fed rats. (A) Protocol illustrating functional assessment of SERCA. ESsteady-state Ca2+ transients were recorded in control buffer, then the extracellular solution was rapidly changed to a Ca2+-free buffer (0Ca2+), followed by aNa+- and Ca2+-free buffer (0Na+/0Ca2+). Cells were then rapidly superfused with a brief (100 ms) caffeine (10 mM) puff in 0Na+/0Ca2+. Under these conditionsNCX is inhibited, caffeine releases SR Ca2+ and cytosolic Ca2+ removal is primarily due to SERCA. Hash marks reflect a change in time scale. (B) Represen-tative traces for normal ST and SU cells after a short caffeine exposure. The integration of the rising (systolic) phase (AS) and declining (diastolic) phase (AD)were normalized to the peak transient (F*ms/FPK). (C) Summary data represent mean ± S.E. for AS/PK and AD/PK during short-caffeine evoked transients(n = 38–52 myocytes per diet, isolated from three to four hearts). * Indicates significantly different from ST (P < 0.05).
301L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307
the state of phosphorylation on 16Ser and 17Thr, decreasedlevels of phospho-PLB due to SU-feeding might also accountfor depressed SERCA activity. To investigate the possibilitythat basal levels of phosphorylated PLB are altered in myo-cytes from SU-fed animals, phospho(16Ser)- andphospho(17Thr)-PLB levels were determined by immunob-lotting. Fig. 4 shows a robust basal level of phospho(16Ser)-PLB, however, no phosphorylation of the 17Thr residue wasdetected (data not shown). To ensure that proper conditionswere used for maintaining levels of phosphorylation, we ranpositive controls for each phosphospecific anitbody (i.e. nor-mal myocytes ± isoproterenol (10–7 M), exposed for 15 min).Isoproterenol stimulation produced significant levels of phos-phorylation of both residues (data not shown). There were nodifferences in levels of either total- or phosphorylated PLBbetween ST and SU myocytes (summary data not shown).
3.5. Manipulating intracellular Ca2+
Because Ca2+-sensitive probes can buffer cytosolic Ca2+,all of the following experiments were performed on myo-
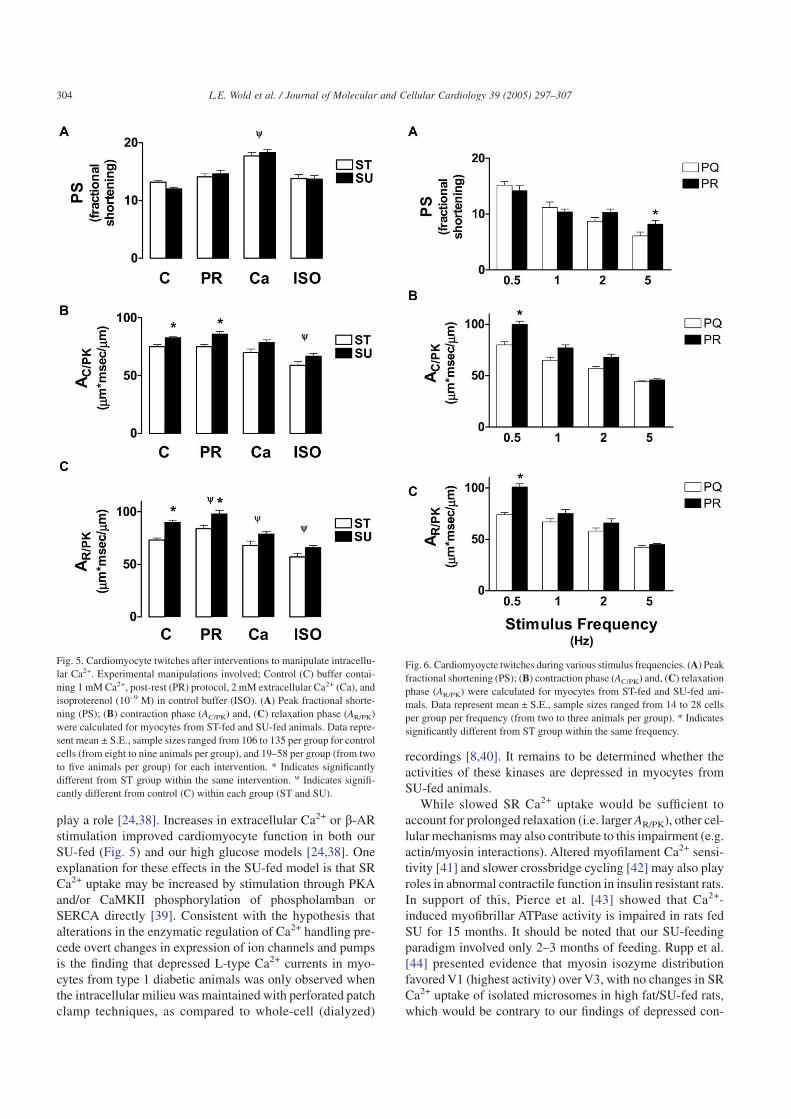
cytes without fluo3. In Fig. 5, we show the effects of post-rest potentiation, elevated extracellular Ca2+ (2 mM) and ISO(10–9 M) on myocyte mechanics. ISO improved both contrac-tion (AC/PK) and relaxation (AR/PK) in SU myocytes such thatdifferences with ST myocytes were not statistically signifi-cant (Fig. 5B, C), without effecting peak fractional shorten-ing (PS; Fig. 5A). However, we also tested higher concentra-tions (up to 10–6 M) in subsets of myocytes and found that atconcentrations greater than 10–9 M, ISO had a significant posi-tive inotropic effect as expected (data not shown). We choseto present data at a lower concentration (10–9 M) because itwas sufficient to normalize the mechanical properties of myo-cytes from SU-fed animals.
To explore the effects of manipulating intracellular Ca2+
without directly stimulating phosphorylation (through b-AR),extracellular Ca2+ concentrations were increased. Thismanipulation produced a significant positive inotropic effecton ST and SU myocytes and improved contraction and relax-ation in SU myocytes. Increasing extracellular Ca2+ not onlyincreases trigger Ca2+, but also increases SR Ca2+ load. Thepost-rest potentiation protocol was used to increase SR Ca2+
load (compared to steady-state levels) without changing theinflux of Ca2+ during the action potential. During the post-rest twitch, SR Ca2+ release and peak shortening were greaterfor the same trigger Ca2+ than during steady-state twitches.This protocol had no effect on the time course of contractionbut slowed relaxation in myocytes from both ST- and SU-fedanimals. The post-rest twitch was qualitatively the same asthe steady-state twitches in that myocytes from SU-fed ani-mals exhibited prolonged mechanics when compared to con-trols (ST-fed animals).
3.6. Manipulating stimulus frequencies
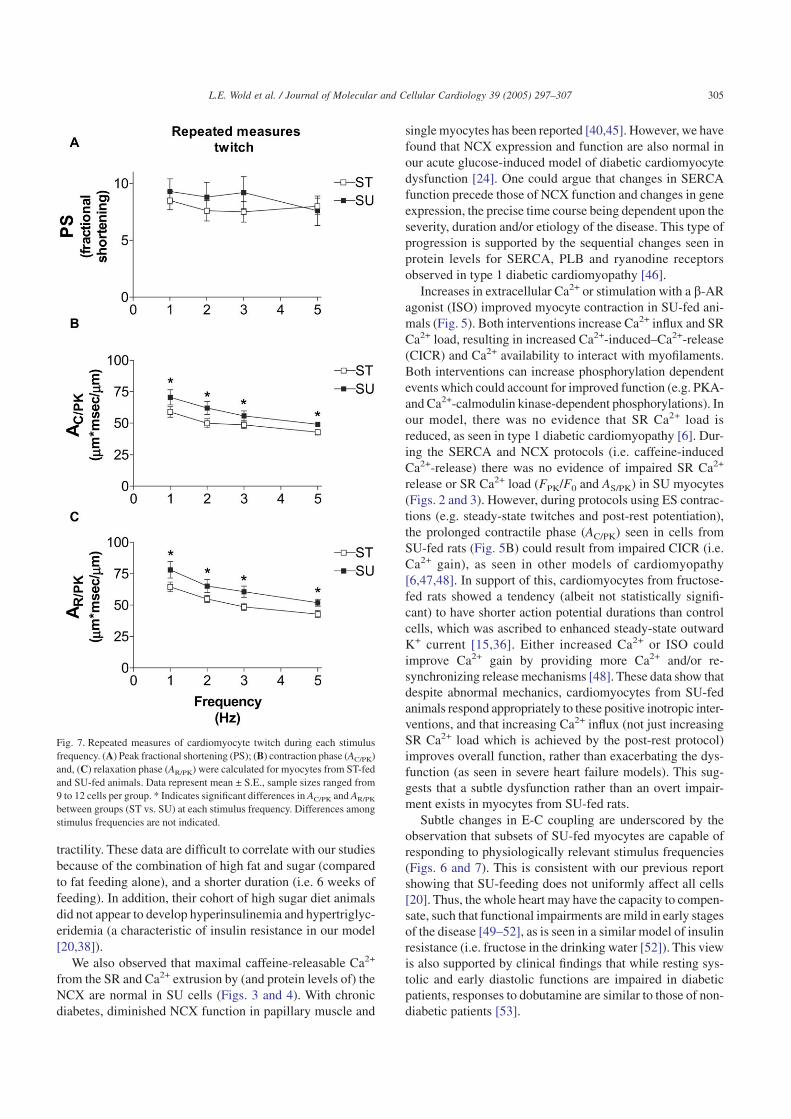
Thus far, we have evaluated myocyte mechanics and Ca2+
transients in cells that were stimulated to contract at a slow(0.5 Hz) stimulus frequency. To address the question as towhether the defects in myocyte mechanics are apparent atphysiologically relevant rates of contraction (i.e. in rats, ratesapproaching 300 beats per min), we stimulated subsets ofmyocytes at 1, 2 and 5 Hz. At the higher stimulus frequenciesboth ST and SU myocytes showed a negative inotropy(Fig. 6A) and faster contraction (Fig. 6B) and relaxation(Fig. 6C). Most notable was that the mechanical dysfunctionof SU myocytes seen at 0.5 Hz was no longer apparent(Fig. 6B, C). In our previous study [20] we noticed a hetero-geneity in mechanical indices among SU cells (e.g. a sub-population of SU myocytes had normal relaxation). Thus, theimproved contraction/relaxation of SU cells at the higherstimulation frequencies could be due to a preferential repre-sentation of ’normal’ SU myocytes in this group. To addressthis issue, we sampled subsets of cells that were exposed toeach stimulus frequency of 1, 2, 3 and 5 Hz (i.e. a repeatedmeasures protocol). Fig. 7 illustrates mechanical indices forcells (ST and SU) that could be driven to contract at eachfrequency. The data show that the typical response to increased
Fig. 3. Ca2+ release and uptake by the SR in myocytes isolated from ST- andSU-fed rats. (A) Protocol illustrating functional assessment of NCX. ESsteady-state Ca2+ transients were recorded in control buffer. Following a 10 speriod without ES, cells were rapidly exposed to caffeine (10 mM) for ~5 s.The protocol was repeated on the same cell (after recovery) in the presenceof caffeine plus nickel (Ni2+; 5 mM). Ni2+ blocked NCX. The differencebetween areas under the Ca2+ transient decline (4 s chosen arbitrarily) beforeand after nickel (DAT) represents NCX function. (B) Summary data repre-sent mean ± S.E. for DAT/PK (DAT normalized to peak fluorescence inten-sity) for ST and SU myocytes (n = 24–36 myocytes per diet from two tothree hearts).
302 L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307
heart rate is preserved in SU myocytes (e.g. rates of relax-ation get faster), but impaired contraction and relaxation werestill apparent in SU myocytes at each frequency. It should benoted that approximately 50% of SU myocytes were unableto be driven to contract at 5 Hz, compared to 16% of ST myo-cytes. When these cells were included in the analyses, thedifferences between the ST and SU groups were exaggeratedat the lower frequencies (data not shown). Our findings indi-cate that abnormal myocyte mechanical function, unmaskedat low stimulus frequencies, is significantly improved at higherstimulus frequencies, although not all cells function ’nor-mally’.
4. Discussion
The aim of this investigation was to further explore cellu-lar mechanisms underlying cardiomyocyte dysfunction ininsulin resistant rats. We show that slowed cytosolic Ca2+
removal and myocyte relaxation in cells from SU-fed rats(Fig. 1) involve slowed SR Ca2+ uptake (Fig. 2), with nochange in SERCA2a protein content (Fig. 4). In addition, wefind that NCX content and function, and both PLB contentand extent of phosphorylation, are normal in these myocytes(Figs. 3 and 4). These results support the view that there arefunctional changes in myocyte E-C coupling, which precedeovert changes in proteins key to Ca2+ regulation. Further-more, it appears likely that this cardiomyocyte dysfunctioninvolves changes in the regulation of intracellular Ca2+ sincemechanical impairments can be overcome by manipulatingCa2+ availability. Perhaps the most significant aspect of thisstudy is that these changes in Ca2+ regulation occur in a mild
metabolic disorder (insulin resistance) without full-blown dia-betes. We have recently shown that at this stage, the cardi-omyocyte dysfunction is not only preventable, but can bereversed with clinically relevant interventions [19].
Contractile dysfunction and depressed SERCA activity arecommon to various models of type 1 diabetes (reviewed in[29]), but these changes are not consistent in all animal mod-els of type 2 diabetes [17,30–32]. In ventricular tissue iso-lated from glucose intolerant rats, SERCA activity is de-pressed with a concomitant change in mRNA for the protein[17]. SERCA mRNA is also diminished in genetically obese(fa/fa) rats [33] and Otsuka Long-Evans Tokushima Fatty rats[30]. Misra et al. [34] reported that neither contractile dys-function nor impaired SERCA activity (as measured bymicrosomal Ca2+ uptake) is apparent in cardiomyocytes iso-lated from JCR:LA-cp rats, and they have suggested that dia-betic cardiomyopathy is not present [35]. However, isolatedventricular myocytes have abnormal K+ currents [36] andwhole hearts from this model of obese type 2 diabetes showsigns of Ca2+ intolerance and ventricular dysfunction [13].We have shown previously that isolated cardiomyocytes fromanother model of obesity (i.e. Zucker obese rats) exhibitdepressed contractile function [37]. Therefore, the existenceof a cardiomyocyte dysfunction in pre-diabetic models is stillnot well resolved.
Our data show that SR Ca2+ uptake is slowed in myocytesfrom SU-fed rats (Fig. 2), but without a concomitant changein protein level for SERCA (Fig. 4). We have recentlyobserved changes in SERCA function without changes inSERCA protein levels in cultured myocytes maintained in ahigh glucose medium, and suggest that regulatory processes(i.e. depressed PKA and elevated PKC activities) most likely
Fig. 4. Immunoblots assessing protein content for SERCA, NCX, and PLB in myocytes isolated from four ST-fed (lanes 1, 3, 5, 7) and four SU-fed (lanes 2, 4,6, 8) rats. Protein homogenates were electrophoresed on gradient gels (4–16% acrylamide), transferred to nitrocellulose, and visualized with amido black stain(left panel). Immunoblotting was done with antibodies to SERCA, NCX, PLB, and phospho(16Ser)-PLB, and antigen–antibody complexes visualized by[125I]protein binding and autoradiography (right panel). Protein levels were quantified by excision of radioactive bands and appropriate backgrounds fromnitrocellulose, followed by gamma counting.
303L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307
play a role [24,38]. Increases in extracellular Ca2+ or b-ARstimulation improved cardiomyocyte function in both ourSU-fed (Fig. 5) and our high glucose models [24,38]. Oneexplanation for these effects in the SU-fed model is that SRCa2+ uptake may be increased by stimulation through PKAand/or CaMKII phosphorylation of phospholamban orSERCA directly [39]. Consistent with the hypothesis thatalterations in the enzymatic regulation of Ca2+ handling pre-cede overt changes in expression of ion channels and pumpsis the finding that depressed L-type Ca2+ currents in myo-cytes from type 1 diabetic animals was only observed whenthe intracellular milieu was maintained with perforated patchclamp techniques, as compared to whole-cell (dialyzed)
recordings [8,40]. It remains to be determined whether theactivities of these kinases are depressed in myocytes fromSU-fed animals.
While slowed SR Ca2+ uptake would be sufficient toaccount for prolonged relaxation (i.e. larger AR/PK), other cel-lular mechanisms may also contribute to this impairment (e.g.actin/myosin interactions). Altered myofilament Ca2+ sensi-tivity [41] and slower crossbridge cycling [42] may also playroles in abnormal contractile function in insulin resistant rats.In support of this, Pierce et al. [43] showed that Ca2+-induced myofibrillar ATPase activity is impaired in rats fedSU for 15 months. It should be noted that our SU-feedingparadigm involved only 2–3 months of feeding. Rupp et al.[44] presented evidence that myosin isozyme distributionfavored V1 (highest activity) over V3, with no changes in SRCa2+ uptake of isolated microsomes in high fat/SU-fed rats,which would be contrary to our findings of depressed con-
Fig. 5. Cardiomyocyte twitches after interventions to manipulate intracellu-lar Ca2+. Experimental manipulations involved; Control (C) buffer contai-ning 1 mM Ca2+, post-rest (PR) protocol, 2 mM extracellular Ca2+ (Ca), andisoproterenol (10–9 M) in control buffer (ISO). (A) Peak fractional shorte-ning (PS); (B) contraction phase (AC/PK) and, (C) relaxation phase (AR/PK)were calculated for myocytes from ST-fed and SU-fed animals. Data repre-sent mean ± S.E., sample sizes ranged from 106 to 135 per group for controlcells (from eight to nine animals per group), and 19–58 per group (from twoto five animals per group) for each intervention. * Indicates significantlydifferent from ST group within the same intervention. w Indicates signifi-cantly different from control (C) within each group (ST and SU).
Fig. 6. Cardiomyoycte twitches during various stimulus frequencies. (A) Peakfractional shortening (PS); (B) contraction phase (AC/PK) and, (C) relaxationphase (AR/PK) were calculated for myocytes from ST-fed and SU-fed ani-mals. Data represent mean ± S.E., sample sizes ranged from 14 to 28 cellsper group per frequency (from two to three animals per group). * Indicatessignificantly different from ST group within the same frequency.
304 L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307
tractility. These data are difficult to correlate with our studiesbecause of the combination of high fat and sugar (comparedto fat feeding alone), and a shorter duration (i.e. 6 weeks offeeding). In addition, their cohort of high sugar diet animalsdid not appear to develop hyperinsulinemia and hypertriglyc-eridemia (a characteristic of insulin resistance in our model[20,38]).
We also observed that maximal caffeine-releasable Ca2+
from the SR and Ca2+ extrusion by (and protein levels of) theNCX are normal in SU cells (Figs. 3 and 4). With chronicdiabetes, diminished NCX function in papillary muscle and
single myocytes has been reported [40,45]. However, we havefound that NCX expression and function are also normal inour acute glucose-induced model of diabetic cardiomyocytedysfunction [24]. One could argue that changes in SERCAfunction precede those of NCX function and changes in geneexpression, the precise time course being dependent upon theseverity, duration and/or etiology of the disease. This type ofprogression is supported by the sequential changes seen inprotein levels for SERCA, PLB and ryanodine receptorsobserved in type 1 diabetic cardiomyopathy [46].
Increases in extracellular Ca2+ or stimulation with a b-ARagonist (ISO) improved myocyte contraction in SU-fed ani-mals (Fig. 5). Both interventions increase Ca2+ influx and SRCa2+ load, resulting in increased Ca2+-induced–Ca2+-release(CICR) and Ca2+ availability to interact with myofilaments.Both interventions can increase phosphorylation dependentevents which could account for improved function (e.g. PKA-and Ca2+-calmodulin kinase-dependent phosphorylations). Inour model, there was no evidence that SR Ca2+ load isreduced, as seen in type 1 diabetic cardiomyopathy [6]. Dur-ing the SERCA and NCX protocols (i.e. caffeine-inducedCa2+-release) there was no evidence of impaired SR Ca2+
release or SR Ca2+ load (FPK/F0 and AS/PK) in SU myocytes(Figs. 2 and 3). However, during protocols using ES contrac-tions (e.g. steady-state twitches and post-rest potentiation),the prolonged contractile phase (AC/PK) seen in cells fromSU-fed rats (Fig. 5B) could result from impaired CICR (i.e.Ca2+ gain), as seen in other models of cardiomyopathy[6,47,48]. In support of this, cardiomyocytes from fructose-fed rats showed a tendency (albeit not statistically signifi-cant) to have shorter action potential durations than controlcells, which was ascribed to enhanced steady-state outwardK+ current [15,36]. Either increased Ca2+ or ISO couldimprove Ca2+ gain by providing more Ca2+ and/or re-synchronizing release mechanisms [48]. These data show thatdespite abnormal mechanics, cardiomyocytes from SU-fedanimals respond appropriately to these positive inotropic inter-ventions, and that increasing Ca2+ influx (not just increasingSR Ca2+ load which is achieved by the post-rest protocol)improves overall function, rather than exacerbating the dys-function (as seen in severe heart failure models). This sug-gests that a subtle dysfunction rather than an overt impair-ment exists in myocytes from SU-fed rats.
Subtle changes in E-C coupling are underscored by theobservation that subsets of SU-fed myocytes are capable ofresponding to physiologically relevant stimulus frequencies(Figs. 6 and 7). This is consistent with our previous reportshowing that SU-feeding does not uniformly affect all cells[20]. Thus, the whole heart may have the capacity to compen-sate, such that functional impairments are mild in early stagesof the disease [49–52], as is seen in a similar model of insulinresistance (i.e. fructose in the drinking water [52]). This viewis also supported by clinical findings that while resting sys-tolic and early diastolic functions are impaired in diabeticpatients, responses to dobutamine are similar to those of non-diabetic patients [53].
Fig. 7. Repeated measures of cardiomyocyte twitch during each stimulusfrequency. (A) Peak fractional shortening (PS); (B) contraction phase (AC/PK)and, (C) relaxation phase (AR/PK) were calculated for myocytes from ST-fedand SU-fed animals. Data represent mean ± S.E., sample sizes ranged from9 to 12 cells per group. * Indicates significant differences in AC/PK and AR/PK
between groups (ST vs. SU) at each stimulus frequency. Differences amongstimulus frequencies are not indicated.
305L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307

The pathogenic factors which contribute to these subtlechanges in E-C coupling have yet to be resolved. However, inthe SU-fed model insulin resistance involves elevated lipids(including free fatty acids) as well as hyperinsulinemia. Wehave preliminary data supporting the role of high fatty acids(e.g. palmitate and oleate) in producing abnormal E-C cou-pling (consistent with that shown for SU-fed animals) andblunted response to insulin (as measured by glucose uptakeassays). In addition, we have recently demonstrated that bothabnormal E-C coupling and impaired response to insulin occurin early stages of type 1 diabetes [38]. An intriguing studyhas shown that insulin receptor substrates (IRS1 and IRS2)co-immunoprecipitate with SERCA2a in cardiac muscle, andthat this association is decreased in type 1 diabetes [54]. Toour knowledge, a physiological link between insulin signal-ing and SERCA function has not been established. However,this relationship is worth pursuing given the increased inci-dence of insulin resistance and its association with cardiovas-cular disease.
In summary, the cardiomyocyte dysfunction in diet-induced insulin resistant rats involves depressed SERCAactivity contributing to impaired relaxation. At this stage ofthe disease there is no impact on NCX function, nor are therechanges in protein content of SERCA2a, NCX, or PLB. Wehave yet to resolve the underlying mechanisms associated withmechanical dysfunction. In light of the fact that there are noapparent changes in expression of these Ca2+ regulating pro-teins, and that cardiomyocyte mechanics are improved athigher (physiologically relevant) frequencies, we refer to acardiomyocyte dysfunction rather than a cardiomyopathy. Thedata presented here provide insight into the underlying cellu-lar mechanisms involved in the transition from normal cardi-omyocyte function to the pathophysiological consequencesassociated with type 2 diabetes mellitus. These data under-score the point that subtle cellular changes occur very earlyin the development of metabolic insulin resistance, which pre-cedes overt type 2 diabetes and obesity.
Acknowledgements
This research was supported by grants from the NationalInstitutes of Health, Bethesda, MD (NIH/NHIBL 60303 and66895 to AJD), and American Heart Association, NewEngland Affiliate Undergraduate Research Fellowship (toMMM).
References
[1] Zimmet P, Alberti MM, Shaw J. Global and societal implications ofthe diabetes epidemic. Nature 2001;414:782–7.
[2] Steinberger J, Daniels SR. Obesity, insulin resistance, diabetes, andcardiovascular risk in children. Circulation 2003;107:1448–53.
[3] Gargiulo P, Jacobellis G, Vaccari V, Andreani D. Diabeticcardiomyopathy: pathophysiological and clinical aspects. Diab NutrMetab 1998;11:336–46.
[4] Zarich SW, Nesto RW. Diabetic cardiomyopathy. Curriculum Cardiol1989;118:1000–12.
[5] Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctionsin isolated ventricular myocytes. Am J Physiol Heart Circ Physiol1997;272:H148–H158.
[6] Lagadic-Gossmann DL, Buckler KJ, Le Prigent K, Feuvray D.Altered Ca2+ handling in ventricular myocytes isolated from diabeticrats. Am J Physiol Heart Circ Physiol 1996;270:H1529–H1537.
[7] Okayama H, Hamada M, Hiwada K. Contractile dysfunction in thediabetic-rat heart is an intrinsic abnormality of the cardiac myocyte.Clin Sci 1994;86:257–62.
[8] Jourdon P, Feuvray D. Calcium and potassium currents in ventricularmyocytes isolated from diabetic rats. J Physiol 1993;470:411–29.
[9] Ganguly PK, Pierce GN, Dhalla KS, Dhalla NS. Defective sarcoplas-mic reticular calcium transport in diabetic cardiomyopathy. Am JPhysiol Endocrinol Metab 1983;244:E528–E535.
[10] Bouchard RA, Bose D. Influence of experimental diabetes on sarco-plasmic reticulum function in rat ventricular muscle. Am J PhysiolHeart Circ Physiol 1991;260:H341–H354.
[11] Kim HW, Ch YS, Lee HR, Park SY, Kim YH. Diabetic alterations incardiac sarcoplasmic reticulum Ca2+-ATPase and phospholambanprotein expression. Life Sci 2001;70:367–79.
[12] Maddaford TG, Russell JC, Pierce GN. Postischemic cardiac perfor-mance in the insulin-resistant JCR:LA-cp rat. Am J Physiol Heart CircPhysiol 1997;273:H1187–H1192.
[13] Lopaschuk GD, Russell JC. Myocardial function and energy substratemetabolism in the insulin-resistant JCR:LA corpulent rat. J ApplPhysiol 1991;71:1302–8.
[14] Rösen P, Herberg L, Reinauer H. Different types of postinsulin recep-tor defects contribute to insulin resistance in hearts of obese Zuckerrats. Endocrinology 1986;119:1285–91.
[15] Shimoni Y, Ewart HS, Severson D. Type I and II models of diabetesproduce different modifications of K+ currents in rat heart: role ofinsulin. J Physiol 1998;507:485–96.
[16] Schaffer SW, Ballard-Croft C, Boerth S, Allo SN. Mechanisms under-lying depressed Na+/Ca2+ exchanger activity in the diabetic heart.Cardiovasc Res 1997;34:129–36.
[17] Schaffer SW, Mozafferi M. Abnormal mechanical function indiabetes: relation to myocardial calcium handling. Coron Artery Dis1996;7:109–15.
[18] Clark TA, Pierce GN. Cardiovascular complications of non-insulin-dependent diabetes. The JCR:LA-cp rat. J Pharm Toxic Meth 2000;43:1–10.
[19] Davidoff AJ, Mason MM, Davidson MB, Carmody MW, Hintz K,Wold LE, et al. Sucrose-induced cardiomyocyte dysfunction is bothpreventable and reversible with clinically relevant treatments. Am JPhysiol Endocrinol Metab 2004;286:E718–E724.
[20] Dutta K, Podolin DA, Davidson MB, Davidoff AJ. Cardiomyocytedysfunction in sucrose-fed rats is associated with insulin resistance.Diabetes 2001;50:1186–92.
[21] Podolin DA, Gayles EC, Wei Y, Thresher JS, Pagliassotti MJ. Menha-den oil prevents but not reverse sucrose-induced insulin resistance inrats. Am J Physiol Regul Integr Comp Physiol 1998;274:R840–R848.
[22] Pagliassotti MJ, Prach PA, Koppenhafer TA, Pan DA. Changes ininsulin action, triglycerides, and lipid composition during sucrosefeeding in rats. Am J Physiol Regul Integr Comp Physiol 1996;271:R1319–R1326.
[23] Yu Z, Quamme GA, McNeill JH. Depressed [Ca2+]i responses toisoproterenol and cAMP in isolated cardiomyocytes from experimen-tal diabetic rats. Am J Physiol Heart Circ Physiol 1994;266:H2334–H2342.
[24] Dutta K, Carmody MW, Cala SE, Davidoff AJ. Depressed PKAactivity contributes to impaired SERCA function and is linked to thepathogenesis of glucose-induced cardiomyopathy. J Mol Cell Cardiol2002;34:985–96.
[25] Ren J, Gintant GA, Miller RE, Davidoff AJ. High extracellular glu-cose impairs cardiac E-C coupling in a glycosylation-dependent man-ner. Am J Physiol Heart Circ Physiol 1997;273:H2876–H2883.
306 L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307

[26] Dinçer UD, Bidasee KR, Güner S, Tay A, Özçelikay AT, Altan VM.The effect of diabetes on expression of ß1-, ß2-, and ß3-adreno-receptors in rat hearts. Diabetes 2001;50:455–61.
[27] Yao A, Su Z, Noaka A, Zubair I, Lu L, Philipson KD, et al. Effects ofoverexpression of the Na+–Ca2+ exchanger on [Ca2+]i transients inmurine ventricular myocytes. Circ Res 1998;82:657–65.
[28] Bers DM. Calcium fluxes involved in control of cardiac myocytecontraction. Circ Res 2000;87:275–81.
[29] Pierce GN, Russell JC. Regulation of intracellular Ca2+ in the heartduring diabetes. Cardiovasc Res 1997;34:41–7.
[30] Abe T, Ohga Y, Tabayashi N, Kobayashi S, Sakata S, Misawa H, et al.Left ventricular diastolic dysfunction in type 2 diabetes mellitusmodel rats. Am J Physiol Heart Circ Physiol 2002;282:H138–H148.
[31] Chatham JC, Seymour A-M. Cardiac carbohydrate metabolism inZucker diabetic fatty rats. Cardiovasc Res 2002;55:104–12.
[32] Pierce GN, Maddaford TG, Russell JC. Cardiovascular dysfunction ininsulin-dependent and non-insulin-dependent animal models of dia-betes mellitus. Can J Physiol Pharmacol 1997;75:343–50.
[33] Russ M, Reinauer H, Eckel J. Diabetes-induced decrease in themRNA coding for sarcoplasmic reticulum Ca2+-ATPase in adult ratcardiomyocytes. Biochem Biophys Res Commun 1991;178:906–12.
[34] Misra T, Gilchrist JSC, Russell JC, Pierce GN. Cardiac myofibrillarand sarcoplasmic reticulum function are not depressed in insulin-resistant JCR:LA-cp rats. Am J Physiol Heart Circ Physiol 1999;45:H1811–H1817.
[35] Misra T, Russell JC, Clark TA, Pierce GN. Mg2+-dependent ATPaseactivity in cardiac myofibrils from the insulin-resistant JCR:LA-cprat. Adv Exp Med Biol 2001;498:247–52.
[36] Shimoni Y, Severson D, Ewart HS. Insulin resistance and the modula-tion of rat cardiac K+ currents. Am J Physiol Heart Circ Physiol2000;279:H639–H649.
[37] Ren J, Sowers JR, Walsh MF, Brown RA. Reduced contractileresponse to insulin and IGF-1 in ventricular myocytes from geneti-cally obese Zucker rats. Am J Physiol Heart Circ Physiol 2000;279:H1708–H1714.
[38] Davidoff AJ, Davidson MB, Carmody MW, Davis M-E, Ren J. Dia-betic cardiomyocyte dysfunction and myocyte insulin resistance: roleof glucose-induced PKC activity. Mol Cell Biochem 2004;262:155–63.
[39] Frank KF, Bolck B, Erdmann E, Schwinger RHG. Sarcoplasmicreticulum Ca2+-ATPase modulates cardiac contraction and relaxation.Cardiovasc Res 2003;57:20–7.
[40] Chattou S, Diacono J, Feuvray D. Decrease in sodium-calciumexchange and calcium currents in diabetic rat ventricular myocytes.Acta Physiol Scanda 1999;166:137–44.
[41] Hofmann PA, Menon V, Gannaway KF. Effects of diabetes on isomet-ric tension as a function of [Ca2+] and pH in rat skinned cardiacmyocytes. Am J Physiol Heart Circ Physiol 1995;269:H1656–H1663.
[42] Ishikawa T, Kajiwara H, Kurihara S. Alterations in contractile proper-ties and Ca2+ handling in streptozotocin-induced diabetic rat myocar-dium. Am J Physiol Heart Circ Physiol 1999;277:H2185–H2194.
[43] Pierce GN, Lockwood MK, Eckert CD. Cardiac contractile proteinATPase activity in a diet induced model of noninsulin dependentdiabetes mellitus. Can J Cardiol 1989;5:117–20.
[44] Rupp H, Wahl R, Maisch B, Hansen M. Characterization of sucrose-induced changes in cardiac phenotype. Pflüg Arch, Eur J Physiol2002;445:32–9.
[45] Hattori Y, Matsuda N, Kimura J, Ishitani T, Tamada A, Gando SKO,et al. Diminished function and expression of the cardiac Na+–Ca2+
exchanger in diabetic rats: implication in Ca2+ overload. J Physiol2000;527:85–94.
[46] ZhongY, Ahmed S, Grupp IL, Matlib MA. Altered SR protein expres-sion associated with contractile dysfunction in diabetic rat hearts. AmJ Physiol Heart Circ Physiol 2001;281:H1137–H1147.
[47] Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C,et al. Regulation of cardiac excitation-contraction coupling by actionpotential repolarization: role of the transient outward potassium cur-rent (Ito). J Physiol 2003;546:5–18.
[48] Litwin SE, Zhang D, Bridge JHB. Dyssynchronous Ca2+ sparks inmyocytes from infarcted hearts. Circ Res 2000;87:1040–7.
[49] Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, et al.Streptozotocin-induced changes in cardiac gene expression in theabsence of severe contractile dysfunction. J Mol Cell Cardiol 2000;32:985–96.
[50] Chatham JC, Gao Z-P, Forder JR. Impact of 1 week of diabetes on theregulation of myocardial carbohydrate and fatty acid oxidation. Am JPhysiol Endocrinol Metab 1999;277(40):E342–E351.
[51] Tahiliani AG, McNeill JH. Effects of insulin perfusion and alteredglucose concentrations on heart function in 3-day and 6-week diabeticrats. Can J Physiol Pharmacol 1986;64:188–92.
[52] Dai S, McNeill JH. Effects of fructose loading in streptozotocin-diabetic and nondiabetic rats. Can J Physiol Pharmacol 1992;70:1583–9.
[53] Fang ZY, Najos-Valencia O, Leano R, Marwick TH. Patients withearly diabetic hearts disease demonstrate a normal myocardialresponse to dobutamine. J Am Cell Cardiol 2003;42:446–53.
[54] Algenstaedt P, Antonetti DA, Yaffe MB, Kahn CR. Insulin receptorsubstrate proteins create a link between the tyrosine phosphorylationcascade and the Ca2+-ATPase in muscle and heart. J Biol Chem1997;272:23696–702.
307L.E. Wold et al. / Journal of Molecular and Cellular Cardiology 39 (2005) 297–307





























