Embed Size (px)
Citation preview

Brain, Behavior, and Immunity 25 (2011) 1187–1196
Contents lists available at ScienceDirect
Brain, Behavior, and Immunity
journal homepage: www.elsevier .com/locate /ybrbi
Restraint stress and stress hormones significantly impact T lymphocytemigration and function through specific alterations of the actin cytoskeleton q
Melanie S. Flint a,b,⇑, Raluca A. Budiu c, Pang-ning Teng a,b, Mai Sun a,b, Donna B. Stolz d, Megan Lang d,Brian L. Hood b, Anda M. Vlad c, Thomas P. Conrads a,b,⇑a Department of Pharmacology and Chemical Biology, University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USAb Cancer Biomarkers Facility – Mass Spectrometry Platform, University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USAc Department of Obstetrics, Gynecology and Reproductive Sciences, University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USAd Center for Biologic Imaging, University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA
a r t i c l e i n f o a b s t r a c t
Article history:Received 24 November 2010Received in revised form 7 March 2011Accepted 14 March 2011Available online 21 March 2011
Keywords:StressT cellProteomicsCytoskeletonMoesin
0889-1591/$ - see front matter � 2011 Elsevier Inc. Adoi:10.1016/j.bbi.2011.03.009
q This work was supported by an award from the D⇑ Corresponding authors. Address: 204 Craft Ave.
15213, USA. Fax: +1 412 641 2356 (M.S. Flint), PreseIntegrated Research Center at Inova Health System, WWoodburn Rd., Annandale, VA 22003, USA. Fax: +1 41
E-mail addresses: [email protected] (M.S. Flint)Conrads).
Stress triggers complex response mechanisms designed to recognize and adapt to perturbations inhomeostasis. The immune system is highly responsive to stress, although the complete mechanisms link-ing stress and immune mediators including T lymphocytes, are not fully understood. Stress exerts itseffects on immune effectors through two primary pathways: the sympathetic–adrenal–medullary path-way, and the hypothalamic–pituitary–adrenal pathway which modulate adaptive immunity and lympho-cyte migration. In this report we show that stress via release of stress hormones induces early T cellactivation and greatly impacts the cytoskeleton by modulating numerous actin-regulating proteins. Inparticular, proteomic profiling revealed significant decreases in numerous key actin-binding proteinsincluding moesin. Although confocal microscopy showed that moesin and actin were uniformly distrib-uted on the surface of resting T cells, a remarkable polarization and redistribution of moesin and actinwas observed following treatment with stress hormones with moesin localizing at the distal pole com-plex. In addition, the alteration in moesin localization and eventual decrease in expression were accom-panied by a loss of CD43; a receptor involved in negatively regulating T cell activation. In conclusion, wehave defined a novel molecular mechanism whereby stress hormones negatively impact T cell activationand migration through regulation of key cytoskeletal and plasma membrane factors.
� 2011 Elsevier Inc. All rights reserved.
1. Introduction
The stress response is a complex multi-system mechanismdesigned to adapt to perturbations in homeostasis. Stress exertsits effects through two primary pathways: the sympathetic–adre-nal–medullary pathway and the hypothalamic–pituitary–adrenalpathway. The duration of stress is important with respect to thephysiological response. Acute stress (short term) activates theautonomic nervous system resulting in increased levels of gluco-corticoids [such as cortisol (Cort)] and catecholamines [epineph-rine (E) and norepinephrine (NE)]. These neuroendocrine changestypically translate into widely recognized physiological parameterssuch as increased heart and breathing rate and higher blood
ll rights reserved.
avid Scaife Foundation (TPC)., Office B477, Pittsburgh, PAnt address: Women’s Health
oodburn II, Suite 375, 32892 641 2356 (T.P. Conrads).
, [email protected] (T.P.
pressure. The consequences of stress on immunity are robust innature and only partially understood.
The immune system combines multiple players, largely classi-fied as either innate or adaptive (acquired) immune effectors. Itis generally accepted that chronic stress is immunosuppressiveleading to increased susceptibility to infections and diseases(Glaser and Kiecolt-Glaser, 2005; Glaser et al., 2005; Godboutand Glaser, 2006; Kiecolt-Glaser et al., 1996). On the other hand,evidence from several studies suggests that acute stress may haveimmunoenhancing effects on both the innate and adaptive im-mune responses (Dhabhar, 1998, 2000, 2002, 2003; Viswanathanand Dhabhar, 2005). While activation of innate immune responsescurrently dominates the research landscape, further studies onstress-induced changes in adaptive immunity are warranted.
We focus here on T lymphocytes, highly specialized adaptiveimmune effectors and their reaction to stress. A successful T cell-mediated immune response depends on (i) effective antigenpriming, (ii) robust cellular activation and (iii) timely migrationat target anatomical sites. T cells express glucocorticoid and adren-ergic stress hormone receptors (Glass and Saijo, 2010; Warner

1188 M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196
et al., 2010) and are thus highly susceptible to variations in stresshormone levels. Several studies to date have demonstrated thatstress can alter the migration of lymphocyte subsets between cir-culating blood and lymphoid or extra-lymphoid tissues (Engleret al., 2004a; Stefanski, 2000). In particular, acute stress increasesmigration of major leukocyte subpopulations to a site of immuneactivation and can affect localization of different T cell subsets inthe blood (Dhabhar, 2003; Dhabhar et al., 1996; Fauci and Dale,1974, 1975; Viswanathan and Dhabhar, 2005). Investigators havealso examined the effects of individual stress hormones andshowed that glucocorticoids cause decreases in the number of cir-culating lymphocytes, (Fauci and Dale, 1974, 1975; Miller et al.,1994) while catecholamines decrease lymphocyte adhesion andmigration (Carlson et al., 1996; Madden et al., 1994). Furthermore,following acute stress, naïve T cells have been shown to be retainedin the lymphoid tissue prepared for antigen exposure whereaseffector T cells in the periphery are ready to migrate to othertissues (Atanackovic et al., 2006). Altogether, these reports havecontributed to a major transformation in our understanding ofstress-and-immunity and provide the rationale for further mecha-nistic studies deciphering the molecular details of stress-inducedchanges in T cell functionality.
The purpose of our work was to elucidate the intracellularmechanism through which stress impacts T cell function, particu-larly activation and migration. We have previously shown thatacute restraint stress enhances corticosterone levels in femaleBALB/c mice although no consequences on T cell immunity wereassessed (Flint et al., 2005). Using in vivo and in vitro approaches,we examined here the effect of acute restraint stress and stress-produced hormones (glucocorticoids and catecholamines) onCD3+ T cells isolated from control and stressed female BALB/c mice.The expression of CD69, an early activation marker of T cells (San-cho et al., 2005), the T cell actin cytoskeleton and actin binding/regulating proteins such as moesin and Rho GTPase were assessedin both naïve and mitogen stimulated cells.
Using mass spectrometry proteomic strategies (for molecularanalyses) and key T cell functional studies, our results show thatstress hormones trigger an initial activation of T cells through in-creases in CD69 and a significant re-distribution of the actin cyto-skeleton. Our findings reveal a novel molecular mechanismwhereby stress hormones negatively impact T cell activation andmigration through regulation of key cytoskeletal and plasma mem-brane factors.
2. Methods
2.1. Mice
Young adult female BALB/c mice (20 g average weight) wereused. The animal room was maintained on a 12 h light/dark cycle(lights on at 6:00 a.m.). The mice were housed in a noise-free envi-ronment 1 week prior to the study and handled once a day for2 weeks to allow the mice to acclimatize. Mice were given foodand tap water ad libitum. All animal protocols were approved bythe IACUC at the University of Pittsburgh.
2.2. In vivo restraint stress
The restraint stress was conducted such that each mouse wasplaced in a well-ventilated 50 mL conical plastic tube for 2 h at10:00 a.m. as described previously (Flint et al., 2005). Mice(n = 10) were not physically squeezed and felt no pain. Control,non stressed mice (n = 10) were left in their home cages. Immedi-ately after stress, mice were sacrificed by CO2 inhalation and serawas isolated immediately and stored at �80 �C.
2.3. Analysis of corticosterone by enzyme immunoassay (EIA)
Serum (10 lL) from each mouse was assayed in duplicate forcorticosterone content using a competitive enzyme immunoassayto corticosterone (Assay Designs, Ann Arbor, MI). The samples wererun on one plate to avoid inter assay variability and the intra-assayprecision for high (20,000 pg/mL) and low (160 pg/mL) corticoste-rone in our laboratory was 3.7% and 9.7%, respectively.
2.4. In vitro assessment of stress effects on naïve and activated T cellsub-populations
The spleens and para-aortic lymph nodes were removed andprocessed into single cell suspension (Flint et al., 2005). The cellswere used for either flow cytometry or further processed forCD3+ T cells isolation using the Mouse Biotin Pan T Cell NegativeIsolation Kit 1 (SABiosciences). The cells were placed in RPMI med-ia supplemented with L-glutamine, 1% penicillin/streptomycin and10% FCS (Flint et al., 2005). Cell viability was assessed by trypanblue dye exclusion and cells were 95–98% viable for all experi-ments. In all in vitro experiments, cells were pre-treated with Cort(10�6 M), NE (10�7 M) and E (10�7 M) (all from Sigma) for 2 h.These concentrations mimic the physiological levels of circulatingCort, NE and E generated during acute stress, as reported by others(Rupprecht et al., 1997, 1999). For polyclonal stimulation, the cellswere then incubated with either phytohaemagglutinin (PMA,250 ng/mL)/ionomycin (IONO, 2.5 ng/mL) for 1 h or lipopolysac-charide (LPS, 500 ng/mL) for 24 h. The concentrations for poly-clonal stimulation were selected based on previously optimizedprotocols (data not shown). T cells were assessed for activation,cytotoxicity, and cell migration.
2.5. Analysis of cell activation by flow cytometry
Multicolor-flow cytometry was used to determine T cell sub-populations and activation status by treatment of splenocytes inthe presence of stress hormones for 2 h with and without poly-clonal stimulation by PMA or LPS, as indicated. Cells were stainedwith the following fluorescent antibodies; anti-mouse CD3-FITCconjugated (clone 145-2C11, Miltenyi Biotec), CD4-PacificBlue(clone RM4-5), CD8-APC-Cy7 (clone 53-6.7), CD69 – PE (cloneH1.2F3) and anti-mouse CD43-APC (clone S7) (BD Biosciences).All antibodies were diluted according to manufacturers’ instruc-tions. Gating was set using an isotype-matched control antibody.Stained cells were analyzed on a LSR II flow cytometer using theFACSDiva data analysis software (BD Biosciences).
2.6. Analysis of cell proliferation
Cell proliferation assays were performed by treatment ofsplenocytes or CD3+ sorted cells plated in triplicate in a 96-wellplate with stress hormones for 2 h with or without polyclonalstimulation by PMA or LPS, as indicated. Cell viability was assessedat 96 h using a standard thiazolyl blue tetrazolium bromide (MTT)assay (Sigma). The intra-assay precision for unstimulated cells inour laboratory was 3.3%.
2.7. Assessment of cell migration
Cell migration of freshly isolated CD3+ T cells was determinedusing the Fluorometric Cell Migration Assay kit (5 lm pore size;Cell Biolabs San Diego, CA) as per manufacturers’ instructions. Toassess whether stress hormones resulted in altered chemo-attrac-tant activity, cells were cultured with stress hormones for 2 h withand without polyclonal stimulation by PMA or LPS and placed in-side the insert. Cells were allowed to migrate towards 10% FCS

M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196 1189
(Shynlova et al., 2008) for 4 h at 37 �C. Cells which migratedthrough the membrane and into the medium were lysed and de-tected by the CyQuant GR dye (Invitrogen). Fluorescence measure-ments were made using a Wallace Victor-2 1420 Multilabelcounter (PerkinElmer) with a 490/535 nm filter set. The intra-assayprecision for unstimulated cells was 11.8%.
2.8. Analysis of T cell proteins by mass spectrometry
Proteomic analysis was performed using high resolution massspectrometry to identify quantitative changes in proteins in CD3+
cells either isolated from control and stressed mice, or from T cellstreated in vitro for 2 and 24 h with Cort, NE and E. CD3+ T cells weredissolved in SDS sample buffer, boiled for 5 min and resolved intothe stacking portion of a 4–12% Bis–Tris precast gel. Samples werein-gel digested and analyzed by liquid chromatography tandemmass spectrometry as described (Hood et al., 2010).
Mass spectrometry data were searched against the UniProtmouse proteome database release date 11/09 (http://www.expasy.org) using SEQUEST (Thermo Scientific Inc.). The data werealso searched against a decoy mouse database resulting in anestimated false discovery rate of <1% (Elias and Gygi, 2007). Differ-ences in protein abundance between the samples were derived byspectral counting using identified (e.g. ‘‘proteotypic’’) peptidesequences (Liu et al., 2004).
2.9. Ingenuity pathway analysis
Proteins which were significantly altered in abundance betweenin vivo control vs. stressed groups, and in vitro between stress hor-mones and PMA activated groups were further analyzed using Inge-nuity Pathway Analysis (IPA). Data sets containing the UniProtprotein accession numbers, protein abundance fold changes, and pvalues were uploaded into IPA software (www.ingenuity.com). Thisanalysis package uses evidence from the literature to search theIngenuity Pathway Knowledge Base for protein interactionsbetween our dataset files and other proteins contained in thedatabase, and generates a series of networks generating a statisticalscore for each network identified.
2.10. Moesin detection by Western blotting
CD3+ T cells were isolated from the spleen, lysates were pre-pared from above, and western analyses performed as previouslydescribed (Bateman et al., 2010) using a primary mouse monoclo-nal antibody directed against moesin (Abcam) and GAPDH (SantaCruz) at 4 �C, and a secondary HRP-labeled anti-mouse monoclonalantibody at 1:50000 dilution (R&D Systems).
2.11. Analysis of F-actin and moesin distribution by confocalmicroscopy
CD3+ T cells were cultured in the presence or absence of stresshormones at various times and the distribution of moesin andF-actin were assessed by confocal microscopy. Cells were attachedto glass coverslips coated with BD Cell-Tak (BD Biosciences) andfixed with 2% paraformaldehyde in PBS. For intracellular stainingthe cells were permeabilized in 0.1% Triton� X-100 for 20 min.Cells were first stained with a rabbit monoclonal anti-moesin[EP1863Y] (Abcam) at a 1:100 dilution and stained with the sec-ondary antibody Cy3-conjugated goat anti-rabbit at a 1:1000 dilu-tion (Jackson Immunoresearch). All cells were counterstained withAlexa Fluor� 647-conjugated phalloidin and 1% Hoescht dye at a1:250 concentration to label T-cell F-actin and nuclei, respectively.Optical sections (0.4 lm thick) were generated using OlympusFluoView 1000 scanning confocal microscope, images were taken
with a 100� optical lens (Olympus UPLSAPO; NA = 1.40) with a2.0� digital zoom.
2.12. Statistical analyses
For the corticosterone assay, results are expressed as mean ± SD.Based on our sample size (n = 10) it was difficult to estimate thepopulation distribution for the data. Therefore, we elected to use anon parametric test which does not make assumptions about thepopulation distribution. The Wilcoxon rank-sum test was used todetermine significant difference in corticosterone between controland stressed mice. For the proteomics studies, the variance inspectral count among the observation groups was studied usingthe Mann–Whitney rank sum test. The significance level was set to0.05. The p value was computed using Fisher’s exact test.
For the in vitro studies, results are expressed as mean ± SD ofthree independent experiments. Statistically significant differencesin viable cells between two experimental conditions were deter-mined by the Student’s two sample t-test.
3. Results
3.1. Effects of stress hormones on T cell activation, proliferation andmigration
We first examined the effects of in vitro exposure to stress hor-mones on activation and proliferation of T cells isolated fromhealthy mice. Exposure of sorted spleen CD3+ T cells to stress hor-mones resulted in a subtle increase of the early activation markerCD69 at 3 h (Fig. 1A), and a 2-fold increase (p = 0.043) at 24 h(Fig. 1B). As expected, exposure of T cells to PMA/IONO (polyclonalstimuli) and LPS (an activation-inducer) also increased expressionof CD69 in CD3+ cells at 3 h (PMA/IONO; p = 0.01) and at 24 h (LPS;p = 0.001). This effect was further enhanced, although not signifi-cantly by stress hormones (Fig. 1A and B).
To determine if stress hormones alter T cell proliferation, cellswere either left unstimulated or exposed to stress hormones foreither 3 (Fig. 2A) or 24 h (Fig. 2B). Cell proliferation was assessedat 96 h by the MTT assay. Incubation with stress hormones alonefor 24 h, significantly decreased cell proliferation (p = 0.01,Fig. 2B). As expected, both mitogenic stimuli PMA/IONO and LPSsignificantly increased proliferation at both time points (p = 0.01;Fig. 2A and p = 0.01; Fig. 2B). However, pre-exposure of cells toSH followed by PMA/IONO or LPS significantly abrogated the cells’capacity to proliferate in response to both stimulation protocols.
Having established the effects of stress hormones on activationand proliferation, we next examined the effects of stress hormoneson T cell migration via fluorescent cell migration assays. We ob-served that stress hormones significantly impacted cell migrationat 3 h (p = 0.03) with 40% of cells failing to migrate (Fig. 3A). Fur-thermore, treatment with PMA/IONO for 1 h had no significant ef-fect on cell migration. To further dissect the outcome of stress, weexamined the effects of stress hormones (Cort, NE and E) individu-ally. Twenty-four hour exposure to combined stress hormones re-sulted in significantly decreased cell migration by 45% (p = 0.0005).Exposure of T cells to Cort and NE separately resulted in a signifi-cant decrease in cell migration by either hormones (p = 0.007 and0.009, respectively). As previously reported in the literature(Zanin-Zhorov et al., 2007), exposure to LPS also significantlyaffected T cell migration (p = 0.003; Fig. 3B) and the suppressiveresponse was further enhanced by stress hormones (p = 0.006).
Taken together, these results demonstrate that stress hormonestrigger apparently divergent effects on T cells: they can increase Tcell activation (as demonstrated by CD69 up-regulation) on onehand and diminish T cell proliferation and migration on the other.

2540 25
35
40
p = 0 001
20
30
35
A B
p = 0.001
25
30p = 0.043
15
3CD
69
20
25
3CD
69
10%C
D3
15
20
%C
D3
p = 0.01
10
15
5
5
00Unstim SH LPS SH/LPSUnstim SH PMA/IONO SH/PMA/IONO
Fig. 1. Effect of stress hormones on T cell activation in vitro. Percentages of CD3+CD69+ cells were determined by flow cytometry in splenocytes isolated from healthy mice.The cells were either unstimulated, treated with stress hormones (SH = Cort; 10�6 M, NE; 10�7 M and E; 10�7 M) for 2 h or treated with stress hormones for 2 h followed bytreatment with PMA (250 ng/mL)/ionomycin (IONO, 2.5 ng/mL) for 1 h (A) or LPS (500 ng/mL) for 24 h (B). The numbers represent percent cells from the parent gate. All gateswere set based on isotype controls. Data are expressed as mean ± SD of three independent experiments. p Values indicate significant differences between the two groups.
p = 0 0049p = 0 010.5 0.5
p = 0.05
p = 0.0049
p = 0.01
p = 0.01
0 4 0 4
A B
0.4 0.4
p = 0.01
0.3
nM 0.3
nM
D a
t 490
n
D a
t 490
n
0.2OD
0.2OD
0.1 0.1
0
Unstim SH PMA/IONO SH/PMA/IONO0
Unstim SH LPS SH/LPS
Fig. 2. Effect of stress hormones on T cell proliferation in vitro. Cell proliferation was assessed by the MTT assay in CD3+ cells isolated from the spleen of healthy mice. Thecells were either unstimulated, treated with stress hormones (SH = Cort; 10�6 M, NE; 10�7 M and E; 10�7 M) for 2 h, or treated with stress hormones for 2 h followed bytreatment with PMA (250 ng/mL)/ionomycin (IONO, 2.5 ng/mL) for 1 h (A) or LPS (500 ng/mL) for 24 h (B). Cell proliferation was assessed in triplicate wells of a 96 well plateat 96 h. Data shown are expressed as mean ± SD of three independent experiments. p Values indicate significant differences between the two groups.
1190 M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196
3.2. The effect of restraint stress on cytoskeletal proteins in T cells
Given the central role of the cytoskeleton rearrangement in cellmigration, we postulated next that stress hormones may affect Tcell motility via changes in actin binding proteins. To address this,we used T cells from stress-exposed mice and employed a massspectrometry-based approach. Mice (n = 10) were either restrainedfor 2 h or left in their home cages as described previously by us(Flint et al., 2005). This 2 h restraint stress model triggers a 5-foldincrease in serum corticosterone in restrained mice compared tonon-restrained controls. The corticosterone levels were
955 ± 1152 pg/mL in controls and 5311 ± 3215 pg/mL in stressedmice, p = 0.001 (S1).
Immediately upon completion of the stress protocol, the micewere euthanized and the spleens and lymph nodes were removedand subjected to T cell isolation protocols. T cell proteomes wereanalyzed by mass spectrometry, resulting in the identification ofan average of 1153 ± 156 proteins per splenic mouse T cell sampleand 466 ± 171 proteins per lymph node T cell sample (S2).
Significant changes in the abundance of 79 proteins (p < 0.05)across all 10 experimental mice were observed as a result ofrestraint stress in splenic T cells, (Fig. 4A) and significant changes

p = 0.003
A Bp = 0.0009
p = 0.0005
160350 p = 0.007
p
p = 0.03
140300
120250
its
p = 0.006
100
nt U
nit
s
200
cenc
e U
ni
80
uou
resc
en
150Flu
ores
c
60
ativ
e F
lu
100
150
Rel
ativ
e F
40
Rel
a
100R
2050
0
Unstim CORT NE SH LPS SH/LPS
0
Unstim SH PMA/IONO SH/PMA/IONO
Fig. 3. Effect of stress hormones on T cell migration in vitro. Cell migration was assessed using the Fluorometric Cell Migration Assay kit with FBS as the chemo-attractant.CD3+ cells were isolated from the spleen of healthy mice and cells were unstimulated or treated for 2 h with stress hormones (Cort; 10�6 M, NE; 10�7 M and E; 10�7 M) orstress hormones followed by treatment with PMA/IONO for 1 h (A) or LPS for 24 h (B). CD3+ cells were seeded at 500,000 cells per well of a 24-well plate in triplicate andallowed to migrate toward 10% FBS for 4 h. Migratory cells on the bottom of the polycarbonate membrane quantified in a fluorescence plate reader. Data are expressed asmean ± SD of two independent experiments. p Values indicate significant differences between the two groups.
M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196 1191
in 15 proteins were found in lymph node isolated T cells (S2). Fiveof the proteins were found in both splenic and lymph node T cells.The biological functions of the significantly altered proteins(p < 0.05) between control and restrained mice were examinedby IPA. The analysis determined that the most significantly alteredcanonical pathways were actin cytoskeleton signaling and thepathway associated with cardiovascular disease, cellular assemblyand organization, and cell morphology. These pathways are perti-nent to cell migration and statistically significant decreases(p < 0.05) in a number of actin-regulating cytoskeleton proteinsincluding talin, ezrin, profilin-1, annexin A2 and annexin A6 in Tcells isolated from the spleen were observed (Fig. 4B). In addition,significant decreases were found for key proteins involved in regu-lating cell migration such as Rho GDP dissociation inhibitor (RhoGDI) 2 (Ishizaki et al., 2006). Although RhoA CDC42 and Rac-2which are key cytoskeleton regulators were identified, their abun-dances were not significantly altered at the 2 h time-point. How-ever, significant decreases in ezrin, profilin-1, vimentin, Rho GDI1 in T cells isolated from the lymph nodes were observed (S3A).Pathway analysis of T cells isolated from the lymph nodes betweenstressed and control mice revealed that the most dysregulated net-works to be cell morphology, cellular development and cellularmovement (S3B).
3.3. In vitro analysis of stress hormones on cytoskeletal pathways
We confirmed in vitro the effects of stress hormones on cyto-skeletal pathways using freshly isolated spleen T cells from controlmice incubated in vitro with stress hormones for 2 and 24 h. The Tlymphocyte proteome was analyzed by mass spectrometry, en-abling the identification of 734 ± 134 proteins per mouse T cellsample (S3). Similar to the in vivo mass spectrometry data, signif-icant decreases in the following actin-regulating proteins were ob-served: moesin (linker protein), talin (actin binding), and profilin
(actin polymerization) (Table 1 and S3). In addition, there were de-creases in key proteins influencing migration, such as cofilin-1 andRho GDI 1 and 2 and Rho/Cdc42/Rac GTP-ase-activating proteinand the transforming protein RhoA (summarized in Table 1).
Interestingly, when cells were activated by PMA followingstress hormones exposure, there was an increase in the actin-reg-ulating proteins moesin, profilin-1, cofilin-1 and talin. We furtherexamined by IPA the proteins altered at least 4-fold and the topnetwork was found to be cellular assembly and organization,cellular function and maintenance, cell to cell signaling and inter-action (Fig. 5A). Amongst our findings, the linker and dominant Tcell ezrin, radixin and moesin protein, moesin was found to besignificantly decreased. Because moesin plays a key role in activa-tion/migration, we further verified the down regulatory effects ofstress on moesin using western blotting. Moesin was indeed de-creased in CD3+ cells exposed to stress hormones and increasedin PMA activated T cells and these data correlated with the spectralcount data obtained by mass spectrometry (Fig. 5B).
3.4. Effect of stress hormones on the cellular distribution of moesin andF-actin in T cells
To further analyze the role of each stress hormone on the distri-bution of moesin and F-actin, CD3+ cells were incubated in thepresence of stress hormones for several time points and the F-actinand moesin were assessed by confocal microscopy. At all timepoints, unstimulated T cells show an even distribution of moesinand actin; however, when stress hormones were administered incombination for 30 min there was a striking polarization of moesinand a redistribution of actin (Fig. 6). This effect was also observedfollowing treatment with Cort or catecholamines separately. Inhi-bition of the GC receptor using mifepristone (RU-486) reversed thiseffect. Similarly, inhibition of the b-adrenergic receptor by pro-pranolol also impacted the polarization, albeit to a lesser extent

A
B
Fig. 4. Role of in vivo stress on regulatory proteins of the cytoskeleton. Mice (n = 10) were restrained for 2 h or left alone (n = 10) in home cages and spleens were removed andCD3+ cells were isolated and analyzed using label-free mass spectrometry. Supervised hierarchal cluster analyses of significant differentially abundant proteins (Mann–Whitney rank-sum, p < 0.05) in splenic T cells between control and restrained mice (A). Ingenuity Pathway Analyses was used to identify pathways of significant differentiallyabundant proteins between control and restrained mice CD3+ cells (B). The red–green color scheme indicates the rank normalized abundance of a protein relative to itsaverage value across all ten mice. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
1192 M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196

Table 1Proteomic analysis of selected actin binding and regulatory proteins using mass spectrometry. CD3+ cells were isolated from healthy mouse spleens and cytoskeletal proteinswere analyzed using label-free mass spectrometry. CD3+ cells were unstimulated or treated with stress hormones (Cort; 10�6 M, NE; 10�6 M and E; 10�6 M) or treated with stresshormones for 2 h followed by treatment with (250 ng/mL)/ionomycin (IONO, 2.5 ng/mL) for 1 h PMA/IONO. Data shown are spectral count data for cytoskeletal proteins identifiedin unstimulated, stress hormones (Cort, NE and E) treated and PMA treated CD3+ cells. Spectral counts correlate linearly with protein abundance.
Name Accession UNSTIM SH 3 SH 24 PMA 3 SH/PMA 3
Moesin P26041 42 43 8 26 57Profilin-1 P62962 30 29 0 28 43Cofilin-1 P18760 20 21 3 14 37Rho GDP-dissociation inhibitor 2 Q61599 10 16 0 5 21Talin-1 P26039 43 30 17 20 56F-actin-capping protein subunit a-1 P47753 4 2 2 5 9Rho GDP-dissociation inhibitor 1 Q99PT1 5 4 0 5 19F-actin-capping protein subunit b P47757 1 1 1 2 7Vasodilator-stimulated phosphoprotein P70460 4 6 0 2 9Rho/Cdc42/RacGTPase-activating protein RICS Q811P8 1 0 0 0 0Transforming protein RhoA Q9QUI0 5 0 0 1 2
Fig. 5. Effect of in vitro stress hormones on regulatory proteins of the cytoskeleton.The ingenuity pathway analysis program was used to identify pathways ofsignificant differentially abundant proteins between CD3+ cells isolated from thespleen and treated with stress hormones (Cort; 10�6 M, NE; 10�6 M and E; 10�6 M)for 24 h vs. unstimulated cells (A). Moesin protein in CD3+ cells was verified at 3 and24 h by Western blot analyses. The corresponding spectral count data for moesinare shown at the top and approximate molecular masses (kDa) are shown on theside.
M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196 1193
than RU-486, suggesting that Cort may exert a more dramatic ef-fect on cytoskeletal rearrangements than catecholamines. A kineticstudy also showed that the polarization of moesin was persistentand this effect was observed at 30, 60 and 120 min (S5).
3.5. Analysis of stress hormones on CD43 expression on T cells
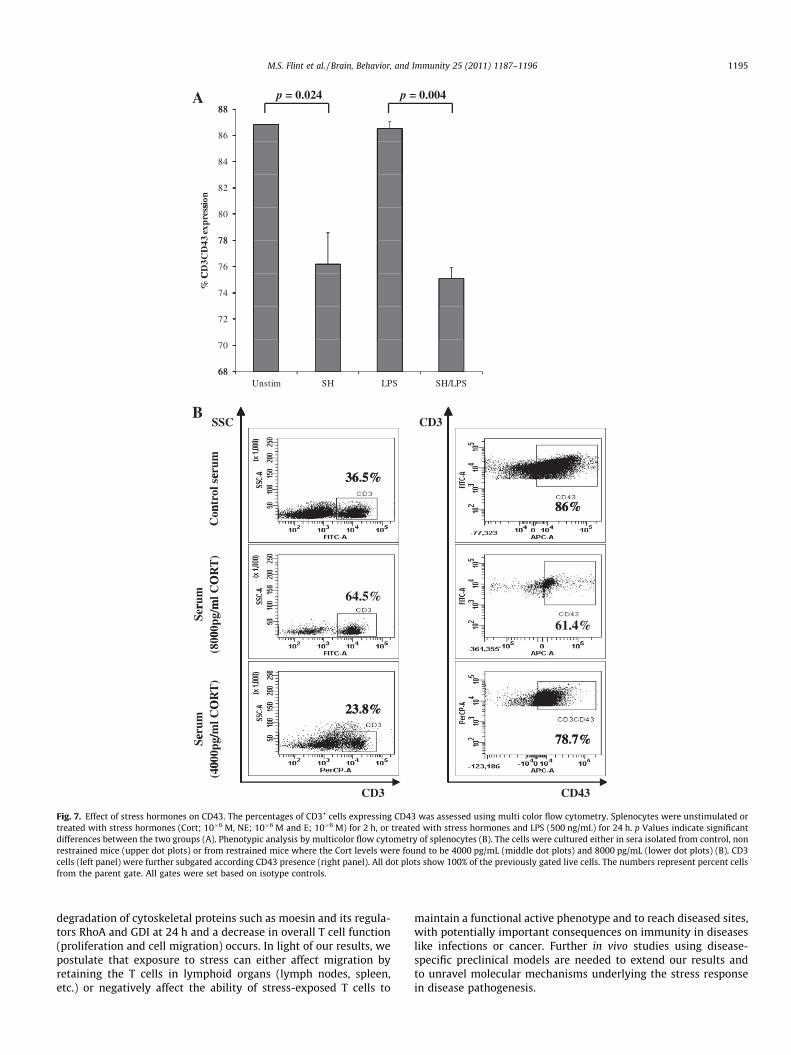
To elucidate whether stress hormones affect the moesin bindingmembrane protein, CD43, splenocytes were treated with stresshormones in the presence/absence of LPS and analyzed by flowcytometry. Stress hormones significantly decreased CD43 expres-sion on resting (p = 0.024) and LPS-treated cells (p = 0.004)(Fig. 7A). As expected, LPS alone did not alter CD43 expression.To further analyze the effects of cort on CD43, splenocytes weretreated with sera isolated from control and stressed mice. Spleno-cytes incubated in the presence of sera isolated from stressed micealso presented with a decrease in CD43 (Fig. 7B). There were corre-lations between the amount of corticosterone measured in individ-ual mouse serum and CD43, such that higher levels of Cort (andtherefore the mouse response to stress) resulted in lower CD43 lev-els. However, serum containing less than 2500 pg/mL Cort resultedin only subtle decreases in CD43.
4. Discussion
Acute psychological stress has been reported to affect severalaspects of immunity including lymphocyte activation and migra-tion (Dhabhar, 2000, 2003; Dhabhar et al., 1996; Engler et al.,2004a,b; Glaser and Kiecolt-Glaser, 2005). Little is known howeverabout the intracellular mechanisms which govern communicationbetween stress and T cells. Herein, we investigated the role ofacute restraint stress (in vivo) and exposure to stress hormones(in vitro) on the intracellular mechanism through which stress im-pacts T cell activation and migration. We found significant in-creases in the percentages of CD3+ T cells expressing CD69 afteracute stress hormone exposure, equivalent to polyclonal stimula-tion indicating an early activated status of the stress hormone-exposed T cells. This is consistent with evidence that stress maybe immune enhancing (Dhabhar, 2000, 2002, 2009; Viswanathanand Dhabhar, 2005). However, despite this activation, T cellsexposed to stress hormones were unable to proliferate in responseto mitogens and exhibited impaired migratory properties, in linewith theories on stress-induced impaired redistribution of effectorT lymphocytes between blood, lymphoid organs and peripheraltissue (Engler et al., 2004a; Stefanski, 2000).
To investigate these migratory defects further, we examinedproteomic changes in CD3+ T cells exposed to stress in vivo andin vitro. Actin-dependent cellular processes are associated withmembrane dynamics, and in T cells, the highly regulated polymer-ization of actin filaments against cellular membranes provides theenergy for processes such as cell migration and activation. Ourextensive proteomic profiling revealed significant changes in

Fig. 6. Effect of stress hormones on in vitro polarization of moesin and actin. The localization of moesin and F-actin in CD3+ cells was assessed by confocal microscopy. CD3+
cells were isolated from the spleen of healthy mice. Cells were left unstimulated or treated for 30 min with stress hormones (combination of Cort; 10�6 M, E; 10�7 M and NE;10�7 M) or with Cort and catecholamines separately. CD3+ cells were also pre-treated for 30 min to the GC antagonist, RU-486 and the b-adrenergic antagonist, propranolol(PRO). Moesin (red) was detected with a rabbit monoclonal anti-moesin antibody [EP1863Y] while actin (blue) was identified by Alexa Fluor� 647-conjugated phalloidin.Arrows indicate examples of polarization of moesin. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
1194 M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196
several proteins important for cytoskeletal rearrangement and cellmigration: actin binding and regulating proteins, including moesin,Rho GTP ases and Rho GDI proteins. We focused here on moesin, anactin-regulatory protein and a member of the ERM complex pro-teins. On T cells, moesin co-localizes with the cell surface moleculeCD43 and together play key roles in T cell activation and migration(Belkina et al., 2009; Burkhardt et al., 2008; Charrin and Alcover,2006; del Pozo et al., 1998; Lee et al., 2004; Li et al., 2007; Niggliand Rossy, 2008; Ramoni et al., 2002; Romero et al., 2002).
Our confocal microscopy analysis revealed an initial rapidpolarization of moesin followed by a significant decrease in proteinlevels with resultant defective migration capabilities in response tostress hormones. Reportedly, unstimulated T cells contain activeezrin–radixin–moesin proteins in a random distribution and T cellreceptor dependent reorganization of the cytoskeleton is essentialfor formation of the immunological synapse at the T cell and anti-gen-presenting cell interface. Upon T cell activation, ezrin–radixin–moesin proteins rapidly undergo dephosphorylation followed byre-phosphorylation driving ezrin–radixin–moesin protein redistri-bution and their binding partners to the distal pole (Lee et al.,2004; Li et al., 2007). Our confocal results showed moesin and actindistribution on opposing T cell sites suggesting that stresshormones do indeed initiate cell activation; however, due to thesubsequent robust decreases in key cytoskeletal proteins, thestress-exposed T cell seem to be prevented from migrating andproliferating. Others have shown that ezrin- and moesin- deficientcells show early intact T cell receptor (TCR) signaling but decreasedlevels of IL-2 (Shaffer et al., 2009) suggesting that although theymay not necessarily impact early TCR signaling, both proteins arerequired for robust T cell activation and subsequent activation offunctional genes. This evidence is in line with the decreased migra-tion we observe in vitro, and lends support to the idea that acutestress retains lymphocytes in the lymphoid organs with potentiallyimportant consequences for first line of immunity in stressful‘‘fight or flight’’ situations (Atanackovic et al., 2006).
We also demonstrated that stress hormones significantlydecreased CD43 expression. CD43 interacts with moesin and
functions as a signaling molecule to regulate T cell migration(Mody et al., 2007) where for efficient migration and activationCD43 must be lost from the surface (Takai et al., 2008). Immunoflu-oresence studies have shown that moesin, but not ezrin, selectivelyco-localizes with CD43 in both resting and activated T cells andthat loss of CD43 occurs when moesin is de-phosphorylated (Ilaniet al., 2007). It was interesting that we observed a decrease in CD43again demonstrating an activated phenotype, however these cellswere still unable to migrate, indicating that the intracellularmolecular events play a dominant role in migration.
In some of our experiments we tested only the global effect ofcombined stress hormones and did not dissect the individual contri-butions of E/NE or Cort. Although Cort, NE and E exerted similar ef-fects with respect to migration and cytoskeletal activation, it wouldbe desirable to test the hormones individually for their impact on thecytoskeleton. Also, our experiments were mainly focused on the ef-fects of acute (2 h) stress in vivo and (2 h) stress hormones followedby 1 and 24 h exposure to mitogens in the presence of hormones, wedid not examine longer exposures equivalent to chronic stress.Acute stress is experienced in response to an immediate perceivedthreat also known as the fight-or-flight response, and is immunoen-hancing, whereas chronic stress (weeks to months duration) isimmunosuppressive (Dhabhar and McEwen, 1997). The molecularmechanisms of immunosuppression warrant further verification.Finally, although we report here that stress suppresses cell prolifer-ation by the MTT assay, and also we observe decreases in the numberof viable cells treated with serum containing high concentrations ofCort by flow cytometry, we have not performed additional experi-ments to see if this finding is due to apoptosis or other types of celldeath. Further work is ongoing in our laboratory to further dissectout the role of each stress hormone and to identify chronic-stressinduced changes in cytoskeleton rearrangements.
In conclusion, our data demonstrates that restraint stress via re-lease of Cort and catecholamines can initially activate the T cell asevidenced by increases in CD69, early rearrangement of the cyto-skeleton (polarization of moesin and actin) and a loss of CD43 fromthe surface. However, this early activation is not sustained and a
A
B
p = 0.024 p = 0.00488
86
88
84
82
sion
78
803
exp
ress
76
78
CD
3CD
4
74
% C
72
68
70
68Unstim SH LPS SH/LPS
CD3SSC
erum
36 5%
ntro
l se 36.5%
86%
Con 86%
OR
T)
64.5%
rum
m
l CO
61.4%
Ser
00pg
/m(8
00
23 8%OR
T)
78 7%
23.8%
erum
/m
l CO
78.7%Se00
0pg/
(40
CD3 CD43
Fig. 7. Effect of stress hormones on CD43. The percentages of CD3+ cells expressing CD43 was assessed using multi color flow cytometry. Splenocytes were unstimulated ortreated with stress hormones (Cort; 10�6 M, NE; 10�6 M and E; 10�6 M) for 2 h, or treated with stress hormones and LPS (500 ng/mL) for 24 h. p Values indicate significantdifferences between the two groups (A). Phenotypic analysis by multicolor flow cytometry of splenocytes (B). The cells were cultured either in sera isolated from control, nonrestrained mice (upper dot plots) or from restrained mice where the Cort levels were found to be 4000 pg/mL (middle dot plots) and 8000 pg/mL (lower dot plots) (B). CD3cells (left panel) were further subgated according CD43 presence (right panel). All dot plots show 100% of the previously gated live cells. The numbers represent percent cellsfrom the parent gate. All gates were set based on isotype controls.
M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196 1195
degradation of cytoskeletal proteins such as moesin and its regula-tors RhoA and GDI at 24 h and a decrease in overall T cell function(proliferation and cell migration) occurs. In light of our results, wepostulate that exposure to stress can either affect migration byretaining the T cells in lymphoid organs (lymph nodes, spleen,etc.) or negatively affect the ability of stress-exposed T cells to
maintain a functional active phenotype and to reach diseased sites,with potentially important consequences on immunity in diseaseslike infections or cancer. Further in vivo studies using disease-specific preclinical models are needed to extend our results andto unravel molecular mechanisms underlying the stress responsein disease pathogenesis.

1196 M.S. Flint et al. / Brain, Behavior, and Immunity 25 (2011) 1187–1196
Acknowledgment
The authors wish to thank Ms. Nadine Ryan for grammaticalassistance.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.bbi.2011.03.009.
References
Atanackovic, D., Schnee, B., Schuch, G., Faltz, C., Schulze, J., Weber, C.S., Schafhausen,P., Bartels, K., Bokemeyer, C., Brunner-Weinzierl, M.C., Deter, H.C., 2006. Acutepsychological stress alerts the adaptive immune response: stress-inducedmobilization of effector T cells. J. Neuroimmunol. 176, 141–152.
Bateman, N.W., Sun, M., Hood, B.L., Flint, M.S., Conrads, T.P., 2010. Defining centralthemes in breast cancer biology by differential proteomics: conservedregulation of cell spreading and focal adhesion kinase. J. Proteome Res. 9,5311–5324.
Belkina, N.V., Liu, Y., Hao, J.J., Karasuyama, H., Shaw, S., 2009. LOK is a major ERMkinase in resting lymphocytes and regulates cytoskeletal rearrangementthrough ERM phosphorylation. Proc. Nat. Acad. Sci. USA 106, 4707–4712.
Burkhardt, J.K., Carrizosa, E., Shaffer, M.H., 2008. The actin cytoskeleton in T cellactivation. Annu. Rev. Immunol. 26, 233–259.
Carlson, S.L., Beiting, D.J., Kiani, C.A., Abell, K.M., McGillis, J.P., 1996. Catecholaminesdecrease lymphocyte adhesion to cytokine-activated endothelial cells. BrainBehav. Immun. 10, 55–67.
Charrin, S., Alcover, A., 2006. Role of ERM (ezrin-radixin-moesin) proteins in Tlymphocyte polarization, immune synapse formation and in T cell receptor-mediated signaling. Front. Biosci. 11, 1987–1997.
del Pozo, M.A., Nieto, M., Serrador, J.M., Sancho, D., Vicente-Manzanares, M.,Martinez, C., Sanchez-Madrid, F., 1998. The two poles of the lymphocyte:specialized cell compartments for migration and recruitment. Cell Adhes.Commun. 6, 125–133.
Dhabhar, F.S., 1998. Stress-induced enhancement of cell-mediated immunity. Ann.NY Acad. Sci. 840, 359–372.
Dhabhar, F.S., 2000. Acute stress enhances while chronic stress suppresses skinimmunity. The role of stress hormones and leukocyte trafficking. Ann. NY Acad.Sci. 917, 876–893.
Dhabhar, F.S., 2002. Stress-induced augmentation of immune function – the role ofstress hormones, leukocyte trafficking, and cytokines. Brain Behav. Immun. 16,785–798.
Dhabhar, F.S., 2003. Stress, leukocyte trafficking, and the augmentation of skinimmune function. Ann. NY Acad. Sci. 992, 205–217.
Dhabhar, F.S., 2009. Enhancing versus suppressive effects of stress on immunefunction: implications for immunoprotection and immunopathology.Neuroimmunomodulation 16, 300–317.
Dhabhar, F.S., McEwen, B.S., 1997. Acute stress enhances while chronic stresssuppresses cell-mediated immunity in vivo: a potential role for leukocytetrafficking. Brain Behav. Immun. 11, 286–306.
Dhabhar, F.S., Miller, A.H., McEwen, B.S., Spencer, R.L., 1996. Stress-induced changesin blood leukocyte distribution. Role of adrenal steroid hormones. J. Immunol.157, 1638–1644.
Elias, J.E., Gygi, S.P., 2007. Target-decoy search strategy for increased confidence inlarge-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214.
Engler, H., Bailey, M.T., Engler, A., Sheridan, J.F., 2004a. Effects of repeated socialstress on leukocyte distribution in bone marrow, peripheral blood and spleen. J.Neuroimmunol. 148, 106–115.
Engler, H., Dawils, L., Hoves, S., Kurth, S., Stevenson, J.R., Schauenstein, K., Stefanski,V., 2004b. Effects of social stress on blood leukocyte distribution: the role ofalpha- and beta-adrenergic mechanisms. J. Neuroimmunol. 156, 153–162.
Fauci, A.S., Dale, D.C., 1974. The effect of in vivo hydrocortisone on subpopulationsof human lymphocytes. J. Clin. Invest. 53, 240–246.
Fauci, A.S., Dale, D.C., 1975. The effect of Hydrocortisone on the kinetics of normalhuman lymphocytes. Blood 46, 235–243.
Flint, M.S., Carroll, J.E., Jenkins, F.J., Chambers, W.H., Han, M.L., Baum, A., 2005.Genomic profiling of restraint stress-induced alterations in mouse Tlymphocytes. J. Neuroimmunol., 34–44.
Glaser, R., Kiecolt-Glaser, J.K., 2005. Stress-induced immune dysfunction:implications for health. Nat. Rev. Immunol. 5, 243–251.
Glaser, R., Padgett, D.A., Litsky, M.L., Baiocchi, R.A., Yang, E.V., Chen, M., Yeh, P.E.,Klimas, N.G., Marshall, G.D., Whiteside, T., Herberman, R., Kiecolt-Glaser, J.,Williams, M.V., 2005. Stress-associated changes in the steady-state expression
of latent Epstein-Barr virus: implications for chronic fatigue syndrome andcancer. Brain Behav. Immun. 19, 91–103.
Glass, C.K., Saijo, K., 2010. Nuclear receptor transrepression pathways that regulateinflammation in macrophages and T cells. Nat. Rev. Immunol. 10, 365–376.
Godbout, J.P., Glaser, R., 2006. Stress-induced immune dysregulation: implicationsfor wound healing, infectious disease and cancer. J. Neuroimmunol. Pharmacol.1, 421–427.
Hood, B.L., Grahovac, J., Flint, M.S., Sun, M., Charro, N., Becker, D., Wells, A., Conrads,T.P., 2010. Proteomic analysis of laser microdissected melanoma cells from skinorgan cultures. J. Proteome Res. 9, 3656–3663.
Ilani, T., Khanna, C., Zhou, M., Veenstra, T.D., Bretscher, A., 2007. Immune synapseformation requires ZAP-70 recruitment by ezrin and CD43 removal by moesin. J.Cell Biol. 179, 733–746.
Ishizaki, H., Togawa, A., Tanaka-Okamoto, M., Hori, K., Nishimura, M., Hamaguchi,A., Imai, T., Takai, Y., Miyoshi, J., 2006. Defective chemokine-directedlymphocyte migration and development in the absence of Rho guanosinediphosphate-dissociation inhibitors alpha and beta. J. Immunol. 177, 8512–8521.
Kiecolt-Glaser, J.K., Glaser, R., Gravenstein, S., Malarkey, W.B., Sheridan, J., 1996.Chronic stress alters the immune response to influenza virus vaccine in olderadults. Proc. Nat. Acad. Sci. USA 93, 3043–3047.
Lee, J.H., Katakai, T., Hara, T., Gonda, H., Sugai, M., Shimizu, A., 2004. Roles of p-ERMand Rho-ROCK signaling in lymphocyte polarity and uropod formation. J. CellBiol. 167, 327–337.
Li, Y., Harada, T., Juang, Y.T., Kyttaris, V.C., Wang, Y., Zidanic, M., Tung, K., Tsokos,G.C., 2007. Phosphorylated ERM is responsible for increased T cell polarization,adhesion, and migration in patients with systemic lupus erythematosus. J.Immunol. 178, 1938–1947.
Liu, H., Sadygov, R.G., Yates 3rd, J.R., 2004. A model for random sampling andestimation of relative protein abundance in shotgun proteomics. Anal. Chem.76, 4193–4201.
Madden, K.S., Felten, S.Y., Felten, D.L., Hardy, C.A., Livnat, S., 1994. Sympatheticnervous system modulation of the immune system. II. Induction of lymphocyteproliferation and migration in vivo by chemical sympathectomy. J.Neuroimmunol. 49, 67–75.
Miller, A.H., Spencer, R.L., Hassett, J., Kim, C., Rhee, R., Ciurea, D., Dhabhar, F.,McEwen, B., Stein, M., 1994. Effects of selective type I and II adrenal steroidagonists on immune cell distribution. Endocrinology 135, 1934–1944.
Mody, P.D., Cannon, J.L., Bandukwala, H.S., Blaine, K.M., Schilling, A.B., Swier, K.,Sperling, A.I., 2007. Signaling through CD43 regulates CD4 T-cell trafficking.Blood 110, 2974–2982.
Niggli, V., Rossy, J., 2008. Ezrin/radixin/moesin: versatile controllers of signalingmolecules and of the cortical cytoskeleton. Int. J. Biochem. Cell Biol. 40, 344–349.
Ramoni, C., Luciani, F., Spadaro, F., Lugini, L., Lozupone, F., Fais, S., 2002. Differentialexpression and distribution of ezrin, radixin and moesin in human natural killercells. Eur. J. Immunol. 32, 3059–3065.
Romero, I.A., Amos, C.L., Greenwood, J., Adamson, P., 2002. Ezrin and moesin co-localise with ICAM-1 in brain endothelial cells but are not directly associated.Mol. Brain Res. 105, 47–59.
Rupprecht, M., Salzer, B., Raum, B., Hornstein, O.P., Koch, H.U., Riederer, P., Sofic, E.,Rupprecht, R., 1997. Physical stress-induced secretion of adrenal and pituitaryhormones in patients with atopic eczema compared with normal controls. Exp.Clin. Endocrinol. Diabetes 105, 39–45.
Rupprecht, R., Koch, M., Montkowski, A., Lancel, M., Faulhaber, J., Harting, J.,Spanagel, R., 1999. Assessment of neuroleptic-like properties of progesterone.Psychopharmacology (Berl) 143, 29–38.
Sancho, D., Gomez, M., Sanchez-Madrid, F., 2005. CD69 is an immunoregulatorymolecule induced following activation. Trends Immunol. 26, 136–140.
Shaffer, M.H., Dupree, R.S., Zhu, P., Saotome, I., Schmidt, R.F., McClatchey, A.I.,Freedman, B.D., Burkhardt, J.K., 2009. Ezrin and moesin function together topromote T cell activation. J. Immunol. 182, 1021–1032.
Shynlova, O., Tsui, P., Dorogin, A., Lye, S.J., 2008. Monocyte chemoattractant protein-1 (CCL-2) integrates mechanical and endocrine signals that mediate term andpreterm labor. J. Immunol. 181, 1470–1479.
Stefanski, V., 2000. Social stress in laboratory rats: hormonal responses andimmune cell distribution. Psychoneuroendocrinology 25, 389–406.
Takai, Y., Kitano, K., Terawaki, S., Maesaki, R., Hakoshima, T., 2008. Structural basisof the cytoplasmic tail of adhesion molecule CD43 and its binding to ERMproteins. J. Mol. Biol. 381, 634–644.
Viswanathan, K., Dhabhar, F.S., 2005. Stress-induced enhancement of leukocytetrafficking into sites of surgery or immune activation. Proc. Nat. Acad. Sci. USA102, 5808–5813.
Warner, A., Ovadia, H., Tarcic, N., Weidenfeld, J., 2010. The effect of restraint stresson glucocorticoid receptors in mouse spleen lymphocytes: involvement of thesympathetic nervous system. Neuroimmunomodulation 17, 298–304.
Zanin-Zhorov, A., Tal-Lapidot, G., Cahalon, L., Cohen-Sfady, M., Pevsner-Fischer, M.,Lider, O., Cohen, I.R., 2007. Cutting edge: T cells respond to lipopolysaccharideinnately via TLR4 signaling. J. Immunol. 179, 41–44.





























