Embed Size (px)
Citation preview

Molecular Cell, Vol. 18, 25–36, April 1, 2005, Copyright ©2005 by Elsevier Inc. DOI 10.1016/j.molcel.2005.02.029
Structure of the p53 Binding Domain of HAUSP/USP7Bound to Epstein-Barr Nuclear Antigen 1:Implications for EBV-Mediated Immortalization
Vivian Saridakis,1,6 Yi Sheng,2,6 Feroz Sarkari,1
Melissa N. Holowaty,1 Kathy Shire,1 Tin Nguyen,1
Rongguang G. Zhang,5 Jack Liao,2 Weontae Lee,2
Aled M. Edwards,1,3,4 Cheryl H. Arrowsmith,2,3,4
and Lori Frappier1,*1Department of Medical Genetics and Microbiology2Ontario Cancer Institute andDepartment of Medical Biophysics3Banting and Best Department of Medical Research4Structural Genomics ConsortiumUniversity of TorontoToronto, Ontario M5S 1A8Canada5Biosciences DivisionStructural Biology CenterArgonne National Laboratory9700 South Cass AvenueArgonne, Illinois 60439
Summary
USP7/HAUSP is a key regulator of p53 and Mdm2 andis targeted by the Epstein-Barr nuclear antigen 1(EBNA1) protein of Epstein-Barr virus (EBV). We havedetermined the crystal structure of the p53 bindingdomain of USP7 alone and bound to an EBNA1 pep-tide. This domain is an eight-stranded � sandwichsimilar to the TRAF-C domains of TNF-receptor asso-ciated factors, although the mode of peptide bindingdiffers significantly from previously observed TRAF-peptide interactions in the sequence (DPGEGPS) andthe conformation of the bound peptide. NMR chemi-cal shift analyses of USP7 bound by EBNA1 and p53indicated that p53 binds the same pocket as EBNA1but makes less extensive contacts with USP7. Func-tional studies indicated that EBNA1 binding to USP7can protect cells from apoptotic challenge by low-ering p53 levels. The data provide a structural andconceptual framework for understanding how EBNA1might contribute to the survival of Epstein-Barr virus-infected cells.
Introduction
EBV infects more than 90% of people worldwide andefficiently immortalizes infected cells, predisposing thehost to a variety of cancers. Cellular immortalization byEBV occurs as part of its latent infectious cycle andinvolves a few EBV proteins including LMP1, whichmimics an activated tumor necrosis factor receptor,and EBNA2, which activates the transcription of severalcellular and viral genes (Dolcetti and Masucci, 2003).Whereas cellular transformation by other DNA tumorviruses (e.g., adenovirus, SV40, and papillomavirus) hasclearly been shown to involve targeting of the p53 tu-
*Correspondence: [email protected]
6 These authors contributed equally to this work.mor suppressor protein, surprisingly none of the EBVproteins required for immortalization have been shownto act through p53.
EBNA1 is the only EBV protein consistently ex-pressed in all proliferating infected cells and plays sev-eral important roles in EBV latent infection, includingthe initiation of EBV DNA replication, the mitotic segre-gation of the EBV genomes, and transcriptional activa-tion of other EBV latency proteins (Kieff and Rickinson,2001). In addition, several pieces of evidence suggestthat EBNA1 plays a direct role in cellular transformationby EBV. First, EBNA1 is expressed in all EBV-associ-ated tumors and is the only viral protein expressed insome of these tumors. Second, transgenic mice ex-pressing EBNA1 develop malignant B cell lymphomas(Wilson et al., 1996). Third, the expression of EBNA1 inHodgkin cells enhances their ability to form tumors innonobese diabetic-SCID mice (Kube et al., 1999).Fourth, EBV genomes lacking the EBNA1 gene are sev-eral thousand-fold less efficient at B cell immortaliza-tion than EBV genomes expressing EBNA1 (Hume etal., 2003). Fifth, interference with EBNA1 function inBurkitt’s lymphoma cells by overexpression of theEBNA1 DNA binding domain increased cell death, sug-gesting that EBNA1 normally provides a survival func-tion for these cells (Kennedy et al., 2003).
EBNA1 was recently shown to stably interact with theubiquitin-specific protease called USP7 or HAUSP (Her-pes virus Associated USP; [Holowaty et al., 2003b]),which was originally identified as a binding target of theICP0 protein of herpes simplex virus (Everett et al.,1997). EBNA1 sequences mediating this interactionwere mapped to within amino acids 395–450, justN-terminal to the DNA binding domain. An EBNA1 mu-tant lacking this sequence (�395–450) failed to bindUSP7 but continued to bind other known cellular pro-tein targets of EBNA1. Functional studies with �395–450 showed that USP7 binding was not required for thereplication, segregation, or transcriptional activationfunctions of EBNA1 but may inhibit the ability of EBNA1to activate replication (Holowaty et al., 2003b). Thedeletion of the USP7 binding sequence also had no de-tectable effect on EBNA1 turnover or cell surface pre-sentation. The lack of requirement of the USP7 interac-tion for the known EBNA1 functions suggested that thesignificance of this interaction may lie in EBNA1-induced changes to the cell.
A link to the p53 pathway was revealed by Li et al.(2002b), who showed that USP7 bound and deubiquiti-nated p53. Overexpression of USP7 stabilized p53, re-sulting in p53-mediated growth repression and apopto-sis, whereas decreased USP7 levels destabilized p53.However, the role of USP7 in p53 regulation was re-cently shown to be more complicated than originallythought, as ablation of USP7 expression resulted in p53accumulation, as opposed to the expected destabiliza-tion of p53 (Cummins et al., 2004; Li et al., 2004). Thiseffect has been shown to be the result of the ability ofUSP7 to stabilize Mdm2, a ubiquitin ligase that pro-motes the degradation of p53. Therefore USP7 appears
Molecular Cell26
to play multiple roles in regulating the p53-Mdm2 stpathway.
The USP7-p53 interaction occurs between the USP7 lrN-terminal domain (NTD), amino acids 53–208, and res-
idues 357–382 of the C-terminal regulatory region of wsp53 (Hu et al., 2002). This USP7 domain, which is sim-
ilar in sequence to a TRAF domain (Zapata et al., 2001), Tsis also responsible for the interaction with EBNA1 (Ho-
lowaty et al., 2003a). The fact that EBNA1 and p53 bind e1the same domain of USP7 raises the possibility that
EBNA1 affects the regulation of p53 by disrupting the scinteraction of USP7 with p53. Indeed, the 395–450 frag-
ment of EBNA1 binds USP7 with 1 �M affinity, whereas icthe p53 regulatory fragment (with or without the tetra-
merization domain) binds with 10-fold lower affinity Ts(Holowaty et al., 2003a). The EBNA1 peptide 395–450
also displaces the p53 peptide from the p53-USP7 9ucomplex. These results indicate that EBNA1 and p53
bind the same or overlapping sites in the USP7 NTD toand suggest that EBNA1 could sequester USP7 from
p53 in vivo, thereby destabilizing p53. In this paper, we uprovide structural and functional evidence supportinga connection between EBNA1 and p53. M
AsResultsBaStructure of the USP7 NTD
To gain insight into the molecular basis for the EBNA1 qiand p53 interactions with USP7, the structure of the
USP7 NTD (residues 54–204), previously shown to bind bfp53 and EBNA1, was determined; first alone and then
in complex with a peptide from EBNA1 (see Table 1 for u
Table 1. X-Ray Data Collection, Structure Solution, and Refinement Parameters
X-Ray Data Native Peak EBNA1 Complex
Space group P32 P32 P41
Resolution (Å) 1.9 2.0 1.7Unit cell axes (Å3) 102.6 × 102.6 × 45.2 102.5 × 102.5 × 45.0 70.0 × 70.0 × 45.9Molecules/AU 3 3 1Se sites (No.) — 12 —Total observations (No.) 103 005 24 8482 96 636Unique reflections (No.) 40 510 70 663 24 692Intensity (I/σ<I>) 20.8 (2.7) 16.8 (2.7) 13.7 (1.8)Completeness (%) 96.7 (80.6) 98.8 (91.9) 99.7 (97.7)aRsym 0.049 (0.289) 0.070 (0.336) 0.078 (0.534)bFigure of merit (%) — 32.3 —MR RF-function — — 0.099MR monitor 0.48cRwork 0.235 0.191Rfree 0.295 0.225Protein atoms (No.) 2473 1181Water molecules (No.) 90 186Sodium atoms (No.) — 21Rmsd bonds (Å) 0.009 0.006Rmsd angles (°) 1.57 1.40Rmsd dihedrals (°) 27.4 25.4Rmsd improper (°) 0.97 0.94Thermal factor (Å2) 22.8 18.7
Numbers in brackets refer to the highest resolution shell, 1.97–1.90 Å for the native data, 2.07–2.00 Å for the MAD data, and 1.76–1.70 Å forthe EBNA1 complex data. Data were integrated and scaled by using HKL2000.a Rsym = S|I − <I>|/SI where I is the observed intensity and <I> is the average intensity from multiple observations of symmetry-relatedreflections.b Figure of Merit of Phasing = |SP(α)eια|/SP(α) where P(α) is the phase probability distribution and α is the phase angle.c R = S|Fobs − Fcalc|/|Fobs|.
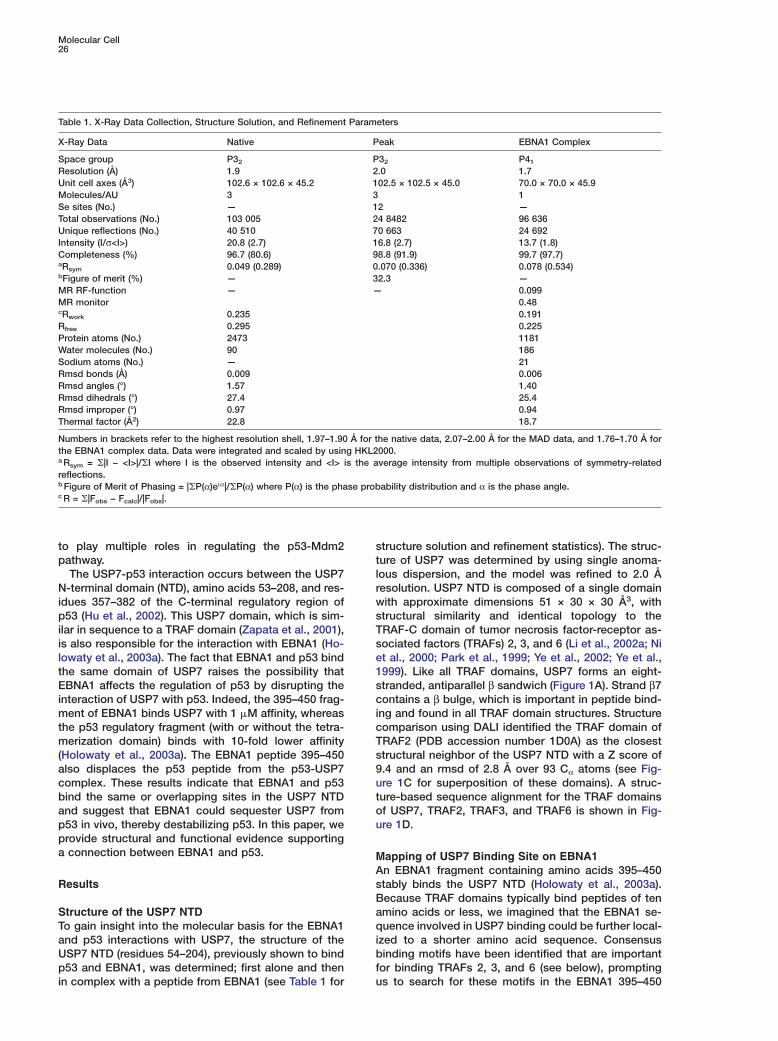
tructure solution and refinement statistics). The struc-ure of USP7 was determined by using single anoma-ous dispersion, and the model was refined to 2.0 Åesolution. USP7 NTD is composed of a single domainith approximate dimensions 51 × 30 × 30 Å3, withtructural similarity and identical topology to theRAF-C domain of tumor necrosis factor-receptor as-ociated factors (TRAFs) 2, 3, and 6 (Li et al., 2002a; Nit al., 2000; Park et al., 1999; Ye et al., 2002; Ye et al.,999). Like all TRAF domains, USP7 forms an eight-tranded, antiparallel β sandwich (Figure 1A). Strand β7ontains a β bulge, which is important in peptide bind-
ng and found in all TRAF domain structures. Structureomparison using DALI identified the TRAF domain ofRAF2 (PDB accession number 1D0A) as the closesttructural neighbor of the USP7 NTD with a Z score of.4 and an rmsd of 2.8 Å over 93 Cα atoms (see Fig-re 1C for superposition of these domains). A struc-ure-based sequence alignment for the TRAF domainsf USP7, TRAF2, TRAF3, and TRAF6 is shown in Fig-re 1D.
apping of USP7 Binding Site on EBNA1n EBNA1 fragment containing amino acids 395–450tably binds the USP7 NTD (Holowaty et al., 2003a).ecause TRAF domains typically bind peptides of tenmino acids or less, we imagined that the EBNA1 se-uence involved in USP7 binding could be further local-
zed to a shorter amino acid sequence. Consensusinding motifs have been identified that are important
or binding TRAFs 2, 3, and 6 (see below), promptings to search for these motifs in the EBNA1 395–450
Structural Basis for EBNA1 and p53 Binding to USP727
Figure 1. Crystal Structure of the USP7 NTDwith EBNA1 Peptide
(A) Ribbon representation of the crystalstructure of the USP7 NTD bound by EBNA1peptide (stick form).(B) Electrostatic surface representation of (A).(C) Superposition of the TRAF domains ofUSP7 (blue) with TRAF2 (silver).(D) Structure based sequence alignment be-tween the TRAF domains of USP7, TRAF2,TRAF3, and TRAF6. Residues that are iden-tical (asterisks) or conserved (dots) in all foursequences are indicated. Residues involvedin EBNA1 binding are in bold.
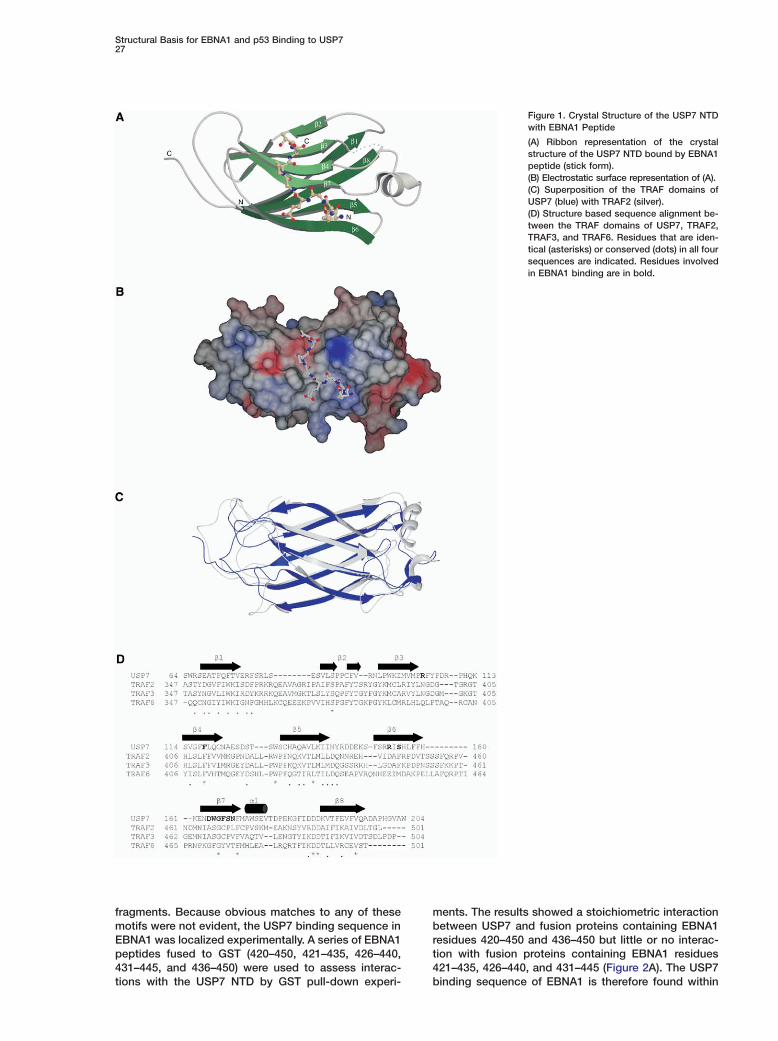
fragments. Because obvious matches to any of thesemotifs were not evident, the USP7 binding sequence inEBNA1 was localized experimentally. A series of EBNA1peptides fused to GST (420–450, 421–435, 426–440,431–445, and 436–450) were used to assess interac-tions with the USP7 NTD by GST pull-down experi-
ments. The results showed a stoichiometric interactionbetween USP7 and fusion proteins containing EBNA1residues 420–450 and 436–450 but little or no interac-tion with fusion proteins containing EBNA1 residues421–435, 426–440, and 431–445 (Figure 2A). The USP7binding sequence of EBNA1 is therefore found within
Molecular Cell28
Figure 2. Interactions of EBNA1 Peptideswith the USP7 NTD
(A) An equimolar mixture of the USP7 NTDand a GST fusion protein containing the indi-cated EBNA1 peptide (L) was mixed withglutathione-sepharose. After washing, pro-tein was eluted with glutathione (E).(B) Increasing amounts of EBNA1 peptides395–450 with wild-type sequence (circles) orE444A (squares), S447A (triangles) or E444A/S447A (diamonds) mutations were incubatedwith the USP7 NTD and binding was quanti-fied by change in tryptophan fluorescence.
amino acids 436–450, and residues between 445 and pt450 are essential for the USP7 interaction. This informa-
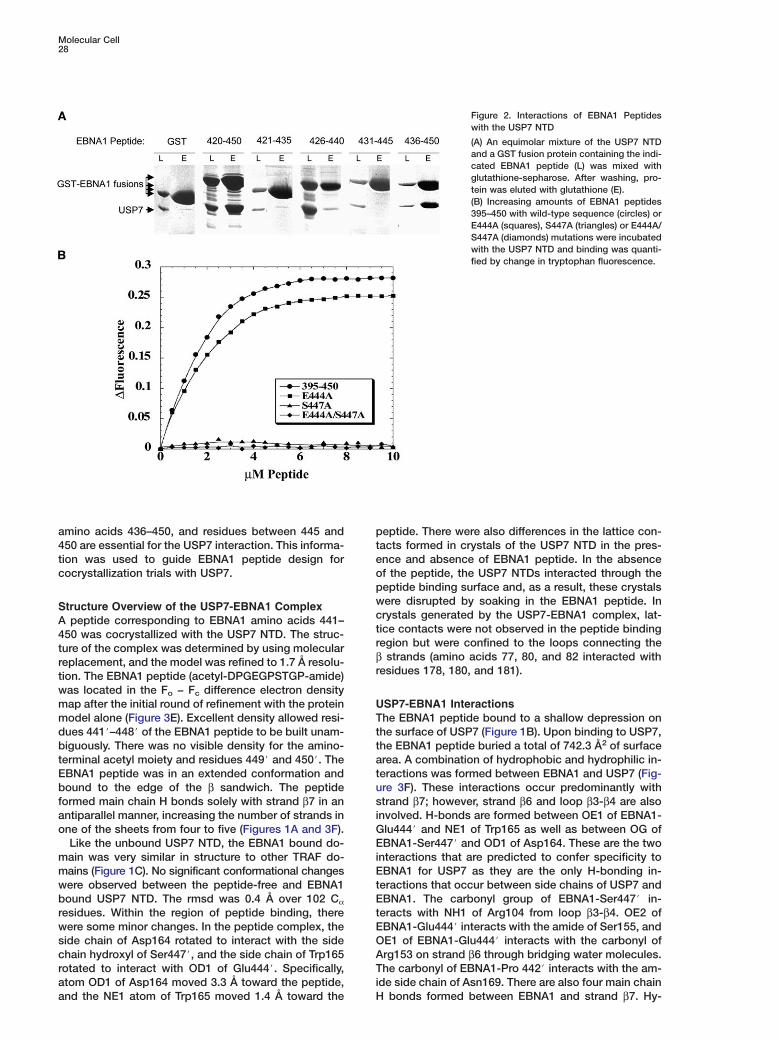
tion was used to guide EBNA1 peptide design for eococrystallization trials with USP7.pwStructure Overview of the USP7-EBNA1 ComplexcA peptide corresponding to EBNA1 amino acids 441–t450 was cocrystallized with the USP7 NTD. The struc-rture of the complex was determined by using molecularβreplacement, and the model was refined to 1.7 Å resolu-rtion. The EBNA1 peptide (acetyl-DPGEGPSTGP-amide)
was located in the Fo − Fc difference electron densitymap after the initial round of refinement with the protein U
Tmodel alone (Figure 3E). Excellent density allowed resi-dues 441#–448# of the EBNA1 peptide to be built unam- t
tbiguously. There was no visible density for the amino-terminal acetyl moiety and residues 449# and 450#. The a
tEBNA1 peptide was in an extended conformation andbound to the edge of the β sandwich. The peptide u
sformed main chain H bonds solely with strand β7 in anantiparallel manner, increasing the number of strands in i
Gone of the sheets from four to five (Figures 1A and 3F).Like the unbound USP7 NTD, the EBNA1 bound do- E
imain was very similar in structure to other TRAF do-mains (Figure 1C). No significant conformational changes E
twere observed between the peptide-free and EBNA1bound USP7 NTD. The rmsd was 0.4 Å over 102 Cα E
tresidues. Within the region of peptide binding, therewere some minor changes. In the peptide complex, the E
Oside chain of Asp164 rotated to interact with the sidechain hydroxyl of Ser447#, and the side chain of Trp165 A
Trotated to interact with OD1 of Glu444#. Specifically,atom OD1 of Asp164 moved 3.3 Å toward the peptide, i
and the NE1 atom of Trp165 moved 1.4 Å toward theeptide. There were also differences in the lattice con-acts formed in crystals of the USP7 NTD in the pres-nce and absence of EBNA1 peptide. In the absencef the peptide, the USP7 NTDs interacted through theeptide binding surface and, as a result, these crystalsere disrupted by soaking in the EBNA1 peptide. Inrystals generated by the USP7-EBNA1 complex, lat-ice contacts were not observed in the peptide bindingegion but were confined to the loops connecting the
strands (amino acids 77, 80, and 82 interacted withesidues 178, 180, and 181).
SP7-EBNA1 Interactionshe EBNA1 peptide bound to a shallow depression onhe surface of USP7 (Figure 1B). Upon binding to USP7,he EBNA1 peptide buried a total of 742.3 Å2 of surfacerea. A combination of hydrophobic and hydrophilic in-eractions was formed between EBNA1 and USP7 (Fig-re 3F). These interactions occur predominantly withtrand β7; however, strand β6 and loop β3-β4 are alsonvolved. H-bonds are formed between OE1 of EBNA1-lu444# and NE1 of Trp165 as well as between OG ofBNA1-Ser447# and OD1 of Asp164. These are the two
nteractions that are predicted to confer specificity toBNA1 for USP7 as they are the only H-bonding in-
eractions that occur between side chains of USP7 andBNA1. The carbonyl group of EBNA1-Ser447# in-
eracts with NH1 of Arg104 from loop β3-β4. OE2 ofBNA1-Glu444# interacts with the amide of Ser155, andE1 of EBNA1-Glu444# interacts with the carbonyl ofrg153 on strand β6 through bridging water molecules.he carbonyl of EBNA1-Pro 442# interacts with the am-
de side chain of Asn169. There are also four main chain
H bonds formed between EBNA1 and strand β7. Hy-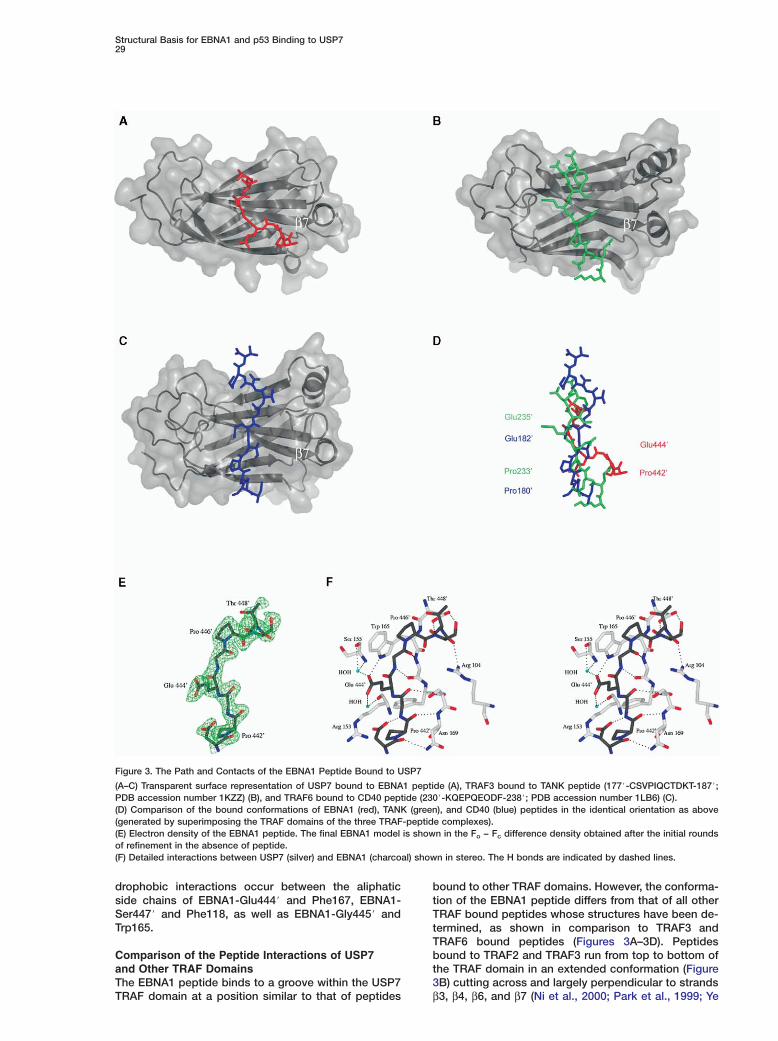
Structural Basis for EBNA1 and p53 Binding to USP729
Figure 3. The Path and Contacts of the EBNA1 Peptide Bound to USP7
(A–C) Transparent surface representation of USP7 bound to EBNA1 peptide (A), TRAF3 bound to TANK peptide (177#-CSVPIQCTDKT-187#;PDB accession number 1KZZ) (B), and TRAF6 bound to CD40 peptide (230#-KQEPQEODF-238#; PDB accession number 1LB6) (C).(D) Comparison of the bound conformations of EBNA1 (red), TANK (green), and CD40 (blue) peptides in the identical orientation as above(generated by superimposing the TRAF domains of the three TRAF-peptide complexes).(E) Electron density of the EBNA1 peptide. The final EBNA1 model is shown in the Fo − Fc difference density obtained after the initial roundsof refinement in the absence of peptide.(F) Detailed interactions between USP7 (silver) and EBNA1 (charcoal) shown in stereo. The H bonds are indicated by dashed lines.
drophobic interactions occur between the aliphaticside chains of EBNA1-Glu444# and Phe167, EBNA1-Ser447# and Phe118, as well as EBNA1-Gly445# andTrp165.
Comparison of the Peptide Interactions of USP7and Other TRAF DomainsThe EBNA1 peptide binds to a groove within the USP7
TRAF domain at a position similar to that of peptidesbound to other TRAF domains. However, the conforma-tion of the EBNA1 peptide differs from that of all otherTRAF bound peptides whose structures have been de-termined, as shown in comparison to TRAF3 andTRAF6 bound peptides (Figures 3A–3D). Peptidesbound to TRAF2 and TRAF3 run from top to bottom ofthe TRAF domain in an extended conformation (Figure3B) cutting across and largely perpendicular to strands
β3, β4, β6, and β7 (Ni et al., 2000; Park et al., 1999; Ye
Molecular Cell30
et al., 1999). The path of TRAF6 bound peptides is sim- Hilar to this, although it deviates from the TRAF2 peptide cpath by 40° and makes more extensive contacts with istrand β7 (Li et al., 2002a; Ye et al., 2002) (Figure 3C). TThe most extensive β7 contacts have been seen with Sthe CD40 peptide, which makes five main-chain H obonds with strand β7. EBNA1 peptide follows strand β7 Seven more extensively than CD40, making seven dif- pferent contacts with six different β7 residues, and, as a cresult, bends relative to the other peptides at the posi- otion of Glu444# (Figures 3A and 3D).
Another difference that distinguishes the EBNA1- TUSP7 interaction from other peptide-TRAF interactions tis the sequence of the bound EBNA1 peptide, which Tdoes not conform to the known TRAF binding se- (quence motifs. TRAF-peptide interactions can be clas- 1sified into two groups based on their peptide specific- tity. TRAF6 binds the consensus sequence, PxExx ((f/acidic) (Ye et al., 2002), whereas TRAFs 1, 2, 3, and 25 all bind the consensus sequence (P/S/A/T) × (Q/E)E tand its variants (fSxEE, QEE, and PxQxxD) (Park et al., �1999; Ye et al., 1999). The TRAF1, TRAF2, TRAF3, and bTRAF5 residues involved in peptide binding are abso- Blutely conserved (including Arg393, Tyr395, Phe447, oSer453, Ser454, Ser455, and Ser467 according to ETRAF2 numbering) despite the fact that these TRAF do- amains share only 52%–64% sequence identity (Wajant net al., 2001). i
The EBNA1 peptide bound by USP7 (PGEGPS) does onot match any of the consensus sequences identified sfor binding other TRAF domains. Although the EBNA1 wsequence contains a PxE motif, which is a found within Usome of the TRAF binding consensus sites, the struc- otural contacts made by these EBNA1 residues are dif- bferent than those of other TRAF bound peptides. In all wpreviously solved TRAF domains, the Pro and Gln/Glu (residues in the bound peptides are largely super impo- bsable. However, the Pro and Glu residues of the EBNA1peptide are out of register in comparison to the TRAF
Cbound TANK and CD40 peptides (Figure 3D). The Cα ofPEBNA1-Glu444# is 7.1 Å away from the Cα of Gln182#Efrom the TANK peptide and 7.4 Å away from the Cα ofcGlu235# from the CD40 peptide. The Cα of Pro442# fromoEBNA1 is 9.4 Å away from the Cα of Pro180# from thetTANK peptide and 6.8 Å away from the Cα of Pro233#Ufrom the CD40 peptide. The functions of the PxE motifsfare also different between EBNA1 and the other TRAFtbinding peptides. For example, in all TRAF2, TRAF3,land TRAF6 interactions, the Pro and Gln/Glu residuesaof the bound peptide make specific interactions with4the TRAF domain and within the peptide itself (Li et al.,n2002a; Li et al., 2003; McWhirter et al., 1999; Ni et al.,u2000; Park et al., 1999; Ye et al., 2002). In EBNA1-USP7Sinteraction, the specific interactions are mediated byfthe Glu and Ser residues. Thus the PxE sequence iniEBNA1 appears to be fortuitous.SConsistent with the different peptide sequencewbound by USP7, the ten USP7 residues that contactpthe EBNA1 peptide are not conserved in other TRAFtdomains (Figure 1D), with the exception of Phe118 andTGly166 (USP7 numbering), which make similar peptide(interactions in all TRAF-peptide structures. Phe118 andaGly166 make aliphatic interactions with the appropri-
ately positioned peptide residues and two main chain S
bonds with the peptide, respectively. USP7 does notontain the residues that mediate peptide interactions
n TRAF2 and TRAF3 and are highly conserved inRAFs 1, 2, 3, and 5 (Arg393, Tyr395, Phe447, Ser453,er454, Ser455, and Ser467) nor are residues at mostf these positions used for EBNA1 peptide binding.imilarly, the residues in TRAF6 that are important foreptide binding (Arg392, Phe471, and Tyr473) are notonserved in USP7. Therefore, USP7 forms a new classf peptide binding TRAF domain.To further investigate the specificity of the USP7
RAF domain, we tested binding to five different pep-ides known to bind TRAF2 or TRAF6; namely, theRAF2 binding peptides QVPFSKEEC from TNF-R2
Park et al., 1999), PQQATDDSS from LMP1 (Ye et al.,999), and PVQETLH from hCD40 (Ye et al., 1999), andhe TRAF6 binding peptides QMPTEDEY from TRANCE-RYe et al., 2002) and KQEPQEIDF from hCD40 (Ye et al.,002). None of these peptides gave detectable bindingo USP7 at any of the concentrations tested (up to 100M), whereas the cocrystallized EBNA1 10 mer peptideound USP7 with a Kd of 0.86 �M in the same assay.ecause EBNA1-USP7 sequence-specific contacts inur structure were mediated by Glu444# and Ser447# ofBNA1, we also generated the EBNA1 395–450 aminocid fragment with these two residues mutated to ala-ines in order to verify their importance for USP7 bind-
ng. In keeping with the structural information, mutationf these residues severely decreased binding to USP7uch that no binding was detected up to 100 �M,hereas the wild-type (wt) 395–450 fragment boundSP7 with a Kd of 0.97 �M (Figure 2B). Point mutationf the Glu444# residue alone only slightly decreasedinding, giving a Kd of 1.36 �M, whereas no bindingas detected when Ser447# was mutated to alanine
Figure 2B). Thus, Ser447 plays a major role in USP7inding.
omparison of the Interaction of p53 and EBNA1eptides with USP7 by NMRBNA1 and p53 bind to the TRAF domain of USP7 andompete for binding. To elucidate the molecular basisf p53 binding, we used NMR chemical shift mappingo compare the binding of EBNA1 and p53 peptides toSP7. 85% of the backbone resonances were assigned
or the NTD of USP7 (amino acids 62–205), and NMRitration experiments were performed on uniformly 15N-abeled USP7 in the presence of unlabeled p53 (aminocids 355–393) and unlabeled EBNA1 (amino acids10–450). Upon binding EBNA1 peptide, the reso-ances in β7 of USP7 showed strong perturbations (Fig-re 4A), especially residues Trp165, Gly166, Phe167,er168, Asn169, Phe170, and Met171. The resonances
or some residues in β3, β4, and β6 were also affected,ncluding Met100, Met102, Phe 117, Phe118, ander155. The results from NMR mapping data agree wellith the crystal structure of the USP7 NTD EBNA1 com-lex (Figure 4C). Phe117 and Phe118 of β4 form con-acts with the backbone amide groups of the peptide.hough the backbone amides of Met100 and Met102
β3) seem far away from the peptide, their side chainsre in close proximity of the C terminus of the peptide.horter EBNA1 peptides (amino acids 420–450 and
Structural Basis for EBNA1 and p53 Binding to USP731
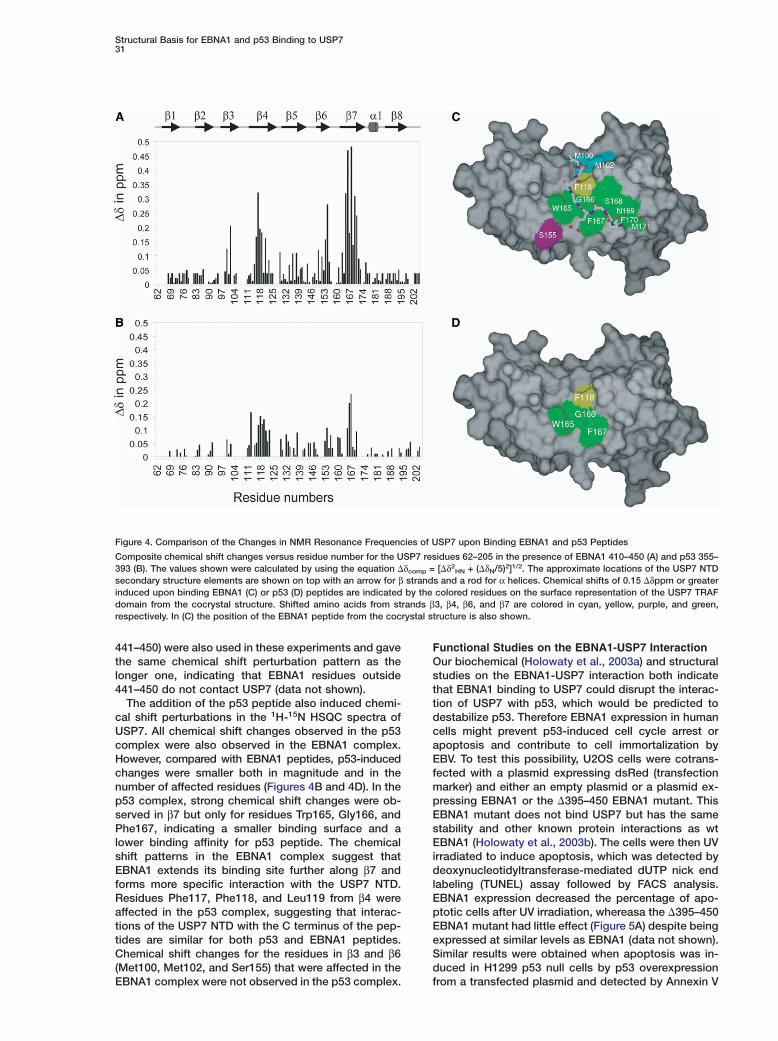
Figure 4. Comparison of the Changes in NMR Resonance Frequencies of USP7 upon Binding EBNA1 and p53 Peptides
Composite chemical shift changes versus residue number for the USP7 residues 62–205 in the presence of EBNA1 410–450 (A) and p53 355–393 (B). The values shown were calculated by using the equation �δcomp = [�δ2
HN + (�δN/5)2]1/2. The approximate locations of the USP7 NTDsecondary structure elements are shown on top with an arrow for β strands and a rod for α helices. Chemical shifts of 0.15 �δppm or greaterinduced upon binding EBNA1 (C) or p53 (D) peptides are indicated by the colored residues on the surface representation of the USP7 TRAFdomain from the cocrystal structure. Shifted amino acids from strands β3, β4, β6, and β7 are colored in cyan, yellow, purple, and green,respectively. In (C) the position of the EBNA1 peptide from the cocrystal structure is also shown.
441–450) were also used in these experiments and gavethe same chemical shift perturbation pattern as thelonger one, indicating that EBNA1 residues outside441–450 do not contact USP7 (data not shown).
The addition of the p53 peptide also induced chemi-cal shift perturbations in the 1H-15N HSQC spectra ofUSP7. All chemical shift changes observed in the p53complex were also observed in the EBNA1 complex.However, compared with EBNA1 peptides, p53-inducedchanges were smaller both in magnitude and in thenumber of affected residues (Figures 4B and 4D). In thep53 complex, strong chemical shift changes were ob-served in β7 but only for residues Trp165, Gly166, andPhe167, indicating a smaller binding surface and alower binding affinity for p53 peptide. The chemicalshift patterns in the EBNA1 complex suggest thatEBNA1 extends its binding site further along β7 andforms more specific interaction with the USP7 NTD.Residues Phe117, Phe118, and Leu119 from β4 wereaffected in the p53 complex, suggesting that interac-tions of the USP7 NTD with the C terminus of the pep-tides are similar for both p53 and EBNA1 peptides.Chemical shift changes for the residues in β3 and β6(Met100, Met102, and Ser155) that were affected in theEBNA1 complex were not observed in the p53 complex.
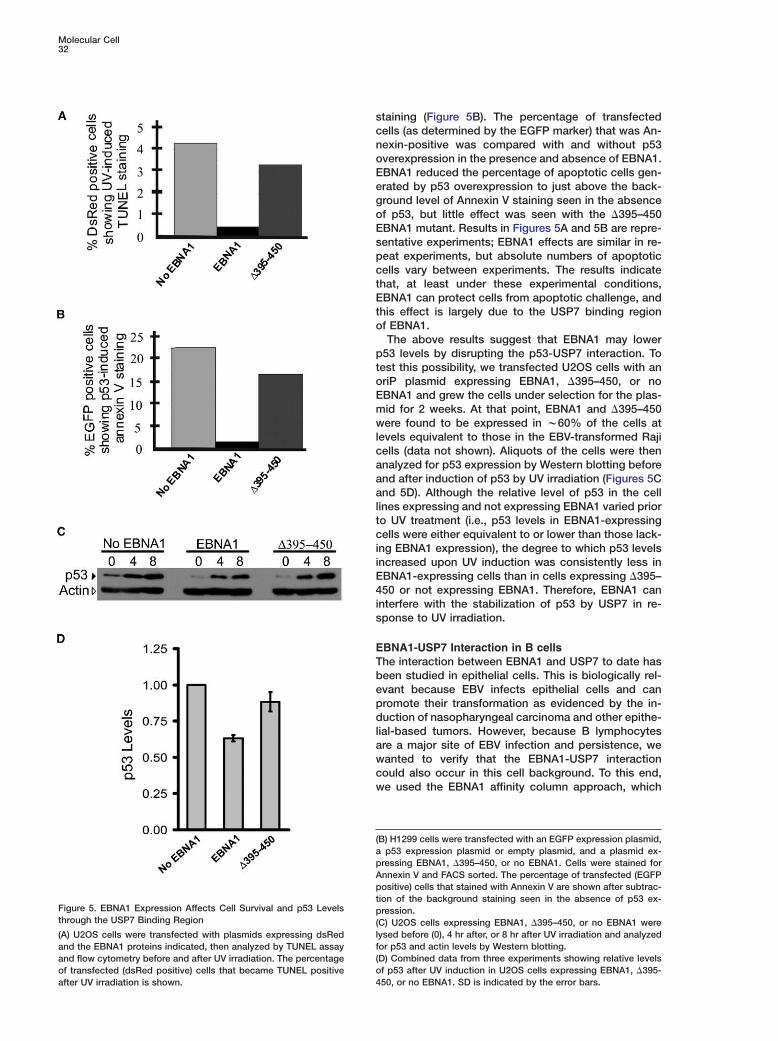
Functional Studies on the EBNA1-USP7 InteractionOur biochemical (Holowaty et al., 2003a) and structuralstudies on the EBNA1-USP7 interaction both indicatethat EBNA1 binding to USP7 could disrupt the interac-tion of USP7 with p53, which would be predicted todestabilize p53. Therefore EBNA1 expression in humancells might prevent p53-induced cell cycle arrest orapoptosis and contribute to cell immortalization byEBV. To test this possibility, U2OS cells were cotrans-fected with a plasmid expressing dsRed (transfectionmarker) and either an empty plasmid or a plasmid ex-pressing EBNA1 or the �395–450 EBNA1 mutant. ThisEBNA1 mutant does not bind USP7 but has the samestability and other known protein interactions as wtEBNA1 (Holowaty et al., 2003b). The cells were then UVirradiated to induce apoptosis, which was detected bydeoxynucleotidyltransferase-mediated dUTP nick endlabeling (TUNEL) assay followed by FACS analysis.EBNA1 expression decreased the percentage of apo-ptotic cells after UV irradiation, whereasa the �395–450EBNA1 mutant had little effect (Figure 5A) despite beingexpressed at similar levels as EBNA1 (data not shown).Similar results were obtained when apoptosis was in-duced in H1299 p53 null cells by p53 overexpressionfrom a transfected plasmid and detected by Annexin V
Molecular Cell32
scnoEegoEspctEto
ptoEmwlcaaaltciiE4is
ETbepdlawcw
(apApt
Figure 5. EBNA1 Expression Affects Cell Survival and p53 Levels pthrough the USP7 Binding Region (
l(A) U2OS cells were transfected with plasmids expressing dsRedfand the EBNA1 proteins indicated, then analyzed by TUNEL assay(and flow cytometry before and after UV irradiation. The percentageoof transfected (dsRed positive) cells that became TUNEL positive4after UV irradiation is shown.
e used the EBNA1 affinity column approach, which
B) H1299 cells were transfected with an EGFP expression plasmid,p53 expression plasmid or empty plasmid, and a plasmid ex-
ressing EBNA1, �395–450, or no EBNA1. Cells were stained fornnexin V and FACS sorted. The percentage of transfected (EGFPositive) cells that stained with Annexin V are shown after subtrac-
ion of the background staining seen in the absence of p53 ex-ression.
C) U2OS cells expressing EBNA1, �395–450, or no EBNA1 wereysed before (0), 4 hr after, or 8 hr after UV irradiation and analyzedor p53 and actin levels by Western blotting.D) Combined data from three experiments showing relative levelsf p53 after UV induction in U2OS cells expressing EBNA1, �395-50, or no EBNA1. SD is indicated by the error bars.
taining (Figure 5B). The percentage of transfectedells (as determined by the EGFP marker) that was An-exin-positive was compared with and without p53verexpression in the presence and absence of EBNA1.BNA1 reduced the percentage of apoptotic cells gen-rated by p53 overexpression to just above the back-round level of Annexin V staining seen in the absencef p53, but little effect was seen with the �395–450BNA1 mutant. Results in Figures 5A and 5B are repre-entative experiments; EBNA1 effects are similar in re-eat experiments, but absolute numbers of apoptoticells vary between experiments. The results indicatehat, at least under these experimental conditions,BNA1 can protect cells from apoptotic challenge, and
his effect is largely due to the USP7 binding regionf EBNA1.The above results suggest that EBNA1 may lower
53 levels by disrupting the p53-USP7 interaction. Toest this possibility, we transfected U2OS cells with anriP plasmid expressing EBNA1, �395–450, or noBNA1 and grew the cells under selection for the plas-id for 2 weeks. At that point, EBNA1 and �395–450ere found to be expressed in w60% of the cells at
evels equivalent to those in the EBV-transformed Rajiells (data not shown). Aliquots of the cells were thennalyzed for p53 expression by Western blotting beforend after induction of p53 by UV irradiation (Figures 5Cnd 5D). Although the relative level of p53 in the cell
ines expressing and not expressing EBNA1 varied prioro UV treatment (i.e., p53 levels in EBNA1-expressingells were either equivalent to or lower than those lack-
ng EBNA1 expression), the degree to which p53 levelsncreased upon UV induction was consistently less inBNA1-expressing cells than in cells expressing �395–50 or not expressing EBNA1. Therefore, EBNA1 can
nterfere with the stabilization of p53 by USP7 in re-ponse to UV irradiation.
BNA1-USP7 Interaction in B cellshe interaction between EBNA1 and USP7 to date haseen studied in epithelial cells. This is biologically rel-vant because EBV infects epithelial cells and canromote their transformation as evidenced by the in-uction of nasopharyngeal carcinoma and other epithe-
ial-based tumors. However, because B lymphocytesre a major site of EBV infection and persistence, weanted to verify that the EBNA1-USP7 interactionould also occur in this cell background. To this end,
Structural Basis for EBNA1 and p53 Binding to USP733
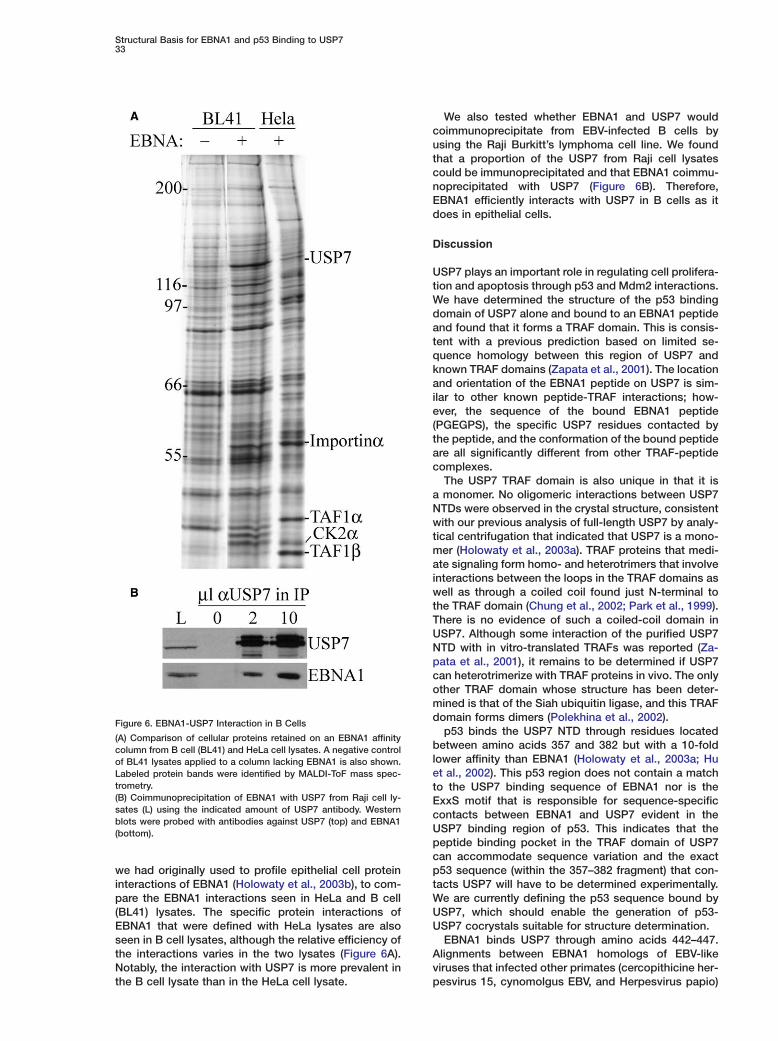
Figure 6. EBNA1-USP7 Interaction in B Cells
(A) Comparison of cellular proteins retained on an EBNA1 affinitycolumn from B cell (BL41) and HeLa cell lysates. A negative controlof BL41 lysates applied to a column lacking EBNA1 is also shown.Labeled protein bands were identified by MALDI-ToF mass spec-trometry.(B) Coimmunoprecipitation of EBNA1 with USP7 from Raji cell ly-sates (L) using the indicated amount of USP7 antibody. Westernblots were probed with antibodies against USP7 (top) and EBNA1(bottom).
we had originally used to profile epithelial cell proteininteractions of EBNA1 (Holowaty et al., 2003b), to com-pare the EBNA1 interactions seen in HeLa and B cell(BL41) lysates. The specific protein interactions ofEBNA1 that were defined with HeLa lysates are alsoseen in B cell lysates, although the relative efficiency ofthe interactions varies in the two lysates (Figure 6A).Notably, the interaction with USP7 is more prevalent inthe B cell lysate than in the HeLa cell lysate.
We also tested whether EBNA1 and USP7 wouldcoimmunoprecipitate from EBV-infected B cells byusing the Raji Burkitt’s lymphoma cell line. We foundthat a proportion of the USP7 from Raji cell lysatescould be immunoprecipitated and that EBNA1 coimmu-noprecipitated with USP7 (Figure 6B). Therefore,EBNA1 efficiently interacts with USP7 in B cells as itdoes in epithelial cells.
Discussion
USP7 plays an important role in regulating cell prolifera-tion and apoptosis through p53 and Mdm2 interactions.We have determined the structure of the p53 bindingdomain of USP7 alone and bound to an EBNA1 peptideand found that it forms a TRAF domain. This is consis-tent with a previous prediction based on limited se-quence homology between this region of USP7 andknown TRAF domains (Zapata et al., 2001). The locationand orientation of the EBNA1 peptide on USP7 is sim-ilar to other known peptide-TRAF interactions; how-ever, the sequence of the bound EBNA1 peptide(PGEGPS), the specific USP7 residues contacted bythe peptide, and the conformation of the bound peptideare all significantly different from other TRAF-peptidecomplexes.
The USP7 TRAF domain is also unique in that it isa monomer. No oligomeric interactions between USP7NTDs were observed in the crystal structure, consistentwith our previous analysis of full-length USP7 by analy-tical centrifugation that indicated that USP7 is a mono-mer (Holowaty et al., 2003a). TRAF proteins that medi-ate signaling form homo- and heterotrimers that involveinteractions between the loops in the TRAF domains aswell as through a coiled coil found just N-terminal tothe TRAF domain (Chung et al., 2002; Park et al., 1999).There is no evidence of such a coiled-coil domain inUSP7. Although some interaction of the purified USP7NTD with in vitro-translated TRAFs was reported (Za-pata et al., 2001), it remains to be determined if USP7can heterotrimerize with TRAF proteins in vivo. The onlyother TRAF domain whose structure has been deter-mined is that of the Siah ubiquitin ligase, and this TRAFdomain forms dimers (Polekhina et al., 2002).
p53 binds the USP7 NTD through residues locatedbetween amino acids 357 and 382 but with a 10-foldlower affinity than EBNA1 (Holowaty et al., 2003a; Huet al., 2002). This p53 region does not contain a matchto the USP7 binding sequence of EBNA1 nor is theExxS motif that is responsible for sequence-specificcontacts between EBNA1 and USP7 evident in theUSP7 binding region of p53. This indicates that thepeptide binding pocket in the TRAF domain of USP7can accommodate sequence variation and the exactp53 sequence (within the 357–382 fragment) that con-tacts USP7 will have to be determined experimentally.We are currently defining the p53 sequence bound byUSP7, which should enable the generation of p53-USP7 cocrystals suitable for structure determination.
EBNA1 binds USP7 through amino acids 442–447.Alignments between EBNA1 homologs of EBV-likeviruses that infected other primates (cercopithicine her-pesvirus 15, cynomolgus EBV, and Herpesvirus papio)

Molecular Cell34
mshowed that, this USP7 binding sequence (PGEGPS) issabsolutely conserved in these viruses, whereas se-aquences in the 395–430 region are highly divergent.q
This suggests that EBNA1 residues 442–447 are func-tionally important for the virus and that USP7 interac- Ctions likely also occur with EBV-related viruses. D
CBecause USP7 binding to p53 results in the stabiliza-otion of p53 (Li et al., 2002b), our structural data on theCEBNA1 and p53 interactions with USP7 predict thatlEBNA1 would interfere with the stabilization of p53 byt
blocking the p53-USP7 interaction. In keeping with the iprediction, EBNA1, but not an EBNA1 mutant deficient w
Min USP7 binding, was found to increase the survival oflcells that were induced to undergo apoptosis either bycDNA damage or p53 overexpression. Similar antiapo-nptotic effects of EBNA1 have been reported by Ken-(
nedy et al. (2003). In keeping with this antiapoptotic ef- afect, EBNA1 was found to decrease the stabilization of 2
pp53 that occurs in response to UV-induced DNA dam-8age, and this effect required the USP7 binding regionfof EBNA1. Not surprisingly, we have not seen reproduc-rible effects of EBNA1 expression on p53 levels in1
rapidly growing cells where p53 is unstable and ex- 1pressed at low levels. The apoptotic protection experi- 1
aments presented here were performed in the presencerof EBNA1 that was expressed at levels approximatelyt8-fold higher than in Raji cells, and it remains to beodetermined whether similar protection is conferred by
EBNA1 in EBV-infected cells. However, this possibility Cis supported by the findings that (1) EBNA1 binds USP7 Din EBV-infected cells and (2) EBNA1 interferes with the U
pUV-induced stabilization of p53 when expressed atmlevels similar to those in EBV-infected cells. Overall, thewdata indicate that EBNA1 can indirectly destabilize p534
by binding USP7, which could be important for initial ccell immortalization by EBV, continued proliferation and msurvival of latently infected cells, and/or malignant S
atransformation.irExperimental ProceduresadExpression and Purification of USP7
USP7 fragments coding for amino acids 54–205 and 62–205 wereGexpressed in E. coli from pET15b plasmids and purified as de-ascribed (Holowaty et al., 2003a). Expression of selenomethionineO(Se-Met)-containing USP7 54–205 was conducted in BL21(DE3)4Gold cells according to Doublie (1997) and purified as for native2protein. For NMR measurements, uniformly labeled 15N, 13C/15N,aand 13C/15N/2H USP7 (amino acids 62–205) were produced in M9(media with 15N ammonium chloride (0.8 g/l) and 13C glucose (2 g/l)sas the sole nitrogen and/or carbon sources, respectively, and usingDdeuterated water (90%) for 2H-labeled samples. Labeled USP7 62–a205 for NMR was prepared as in Holowaty et al. (2003a) except thatwbuffers containing 20 mM sodium phosphate (pH 7.5), 250 mMmNaCl, 2 mM DTT, and 90% H2O/10%D2O were used. The concen-gtration of the purified proteins for NMR experiments ranged be-btween 0.5 and 0.8 mM.a
Peptide PreparationAThe EBNA1 peptide crystallized with USP7 consisted of aminoEacids 441–450 (DPGEGPSTGP). This peptide was synthesized byUDalton Chemicals (Toronto, Canada) with both amino terminal acet-mylation and carboxy-terminal amidation to mimic the native pep-
tides. Peptides containing known TRAF binding sequences used inUSP7 binding assays were also synthesized by Dalton Chemicals. N
NHuman p53 (355–393) and EBNA1 (410–450) peptides used in NMRstudies and the EBNA1 395–450 fragment (with or without point 6
utations in Glu444, Ser447, or both) were generated by expres-ion of relevant sequences from pET15b (Novagen) and purifications described in Holowaty et al. (2003a). Resulting clones were se-uence verified.
rystallization, Data Collection, and Structureetermination of Unbound USP7 NTDrystals of native or Se-Met-enriched USP7 NTD (30 mg/ml) werebtained in 35% MPD, 0.2 M MgOAc, and 0.1 M MES, (pH 6.5).omplete native and MAD data sets from frozen crystals were col-
ected at beamline 19ID at the Advanced Photon Source by usinghe SBC-3 CCD detector. Data collection statistics are presentedn Table 1. The native and MAD data were merohedrally twinnedith a twinning factor of 0.36 for the native data and 0.40 for theAD data. The structure was determined by using single anoma-
ous dispersion with the peak data. There were three USP7 mole-ules in the asymmetric unit and each was monomeric. The sele-ium substructure was located as described in Saridakis et al.
2004), and 73 out of 151 amino acids of molecule A were builtutomatically (residues 70–77, 85–100, 116–121, 128–158, and 191–02). Models of molecules B and C were built manually. The finalrotein model of molecule A consists of 97 residues from 65–78,5–104, 115–142, 150–173, and 188–203. The amino terminus andour loops are completely disordered. In molecule B, the followingesidues were modeled: 67–78, 85–104, 115–142, 150–175, and89–204 and in molecule C, 67–74, 85–102, 115–142, 150–178, and88–204. The rmsd between the different molecules ranges from.0 Å for molecules B and C to 1.3 Å for molecules A and C or Bnd C over the same number of Cα residues. The final models wereefined to 2.0 Å with an Rcryst of 23.5 and an Rfree of 29.5 and con-ain 62 water molecules. All of the residues are in the best regionsf the Ramachandran plot.
rystallization, Data Collection, and Structureetermination of EBNA1 Bound USP7 NTDSP7 NTD (100 mg/ml) was cocrystallized with a 10 mer EBNA1eptide corresponding to EBNA1 amino acids 441–450 at 1.5-foldolar excess of peptide. Large clusters of rods appeared after 4eeks at 4°C in the dark in three conditions containing 30% PEG000, 0.1 M Tris (pH 8.5), and either lithium sulfate, magnesiumhloride, or sodium acetate. The structure was determined by usingolecular replacement and was refined and rebuilt as described inaridakis et al. (2004). The final model refined to an Rwork of 0.21nd an Rfree of 0.25. There are 186 water molecules and 21 sodium
ons. All residues are in the most favored and additionally allowedegions of the Ramachandran plot. Residues 54–62 and 107–111re disordered in the final model of the complex. A summary ofata collection and refinement statistics is presented in Table 1.
eneration of GST-EBNA1 Fusion Proteinsnd Use in USP7 Binding Assaysligonucleotides encoding EBNA1 amino acids 421–435, 426–440,31–445, and 436–450 were expressed as GST-fusions from GST-TK plasmid (Amersham) in BL21-CodonPlus E. coli (Stratagene)t 37°C for 3 hr. Proteins were purified on glutathione-sepharose
Amersham) by using standard methods then dialyzed against as-ay buffer (20 mM sodium phosphate [pH 7.5], 250 mM NaCl, 2 mMTT, 1 mM Benzamidine, and 0.5 mM PMSF). For USP7 bindingssays, purified USP7 NTD (amino acids 62–205) was incubatedith GST or GST-EBNA1 fusion proteins in the assay buffer in a 1:1olar ratio at 4°C for 1 hr. The mixture was passed through a 0.2 mllutathione-sepharose column. After extensive washing with assayuffer, bound proteins were eluted with 20 mM reduced glutathionend detected by SDS-PAGE and Coomassie staining.
ssays of USP7 Binding by Tryptophan FluorescenceBNA1 and TRAF binding peptides were titrated with the purifiedSP7 NTD (62–205), and change in tryptophan fluorescence waseasured as described in Holowaty et al. (2003a).
MR MeasurementsMR spectra were acquired at 30°C on a Varian Inova-500 and00 MHz spectrometers equipped with pulsed field gradient triple-

Structural Basis for EBNA1 and p53 Binding to USP735
resonance probes or a Bruker 600 MHz spectrometer equippedwith a triple resonance cryo probe. The backbone 1H, 15N, and 13Cresonances were assigned by using TROSY-HNCACB, TROSY-CBCA(CO)NH, HNCACB, HNCO, and 15N-NOESY experiments(Salzmann et al., 1998; Kay, 2001). Greater than 80% of the back-bone atoms were assigned. The interaction of USP7 with EBNA1and p53 peptides was monitored by using the 1H-15N HSQC experi-ment at 30°C in a buffer containing 20 mM sodium phosphate (pH7.5), 250 mM NaCl. Briefly, 0.7 mM 15N-labeled USP7 (amino acids62–205) was titrated with unlabeled p53 (residues 355–393) orEBNA1 (residues 395–450, 410–450, or 441–450) peptides up to a10:1 peptide:USP7 molar ratio. The data shown were collected byusing a 3:1 and a 5:1 molar ratio of EBNA1:USP7 and p53:USP7,respectively, and no further changes in chemical shifts were de-tected in the 1H-15N HSQC spectra with higher peptide ratios.
Apoptosis AssaysThe effect of EBNA1 expression on cell death was determined inthe U2OS osteosarcoma line by TUNEL assay upon induction ofp53 by UV irradiation. Cells were seeded at 50% confluency on 60mm2 dishes in duplicate 18 hr prior to transfection with 3 �g ofpc3oriP plasmid expressing either EBNA1, EBNA1�395–450, or noEBNA1 (Holowaty et al., 2003b) and with 250 ng of pDsRed1-N1(BD Bioscience) as a transfection marker. 24 hr later, one set oftransfections was subjected to UV irradiation in a Stratagene 1800ultraviolet crosslinker at 50 × 100 �J/cm2. Cells were grown another24 hr, then stained by using the In Situ Cell Death Detection Kit,Fluorescein (Roche Applied Sciences) according to the manufac-turer. Cells were filtered through a 0.7 �m strainer cap (BD Biosci-ence) prior to analysis on a Beckman-Coulter EPICS Elite (FlowCytometry Facility, University of Toronto).
The effect of EBNA1 on p53-mediated apoptosis was determinedin the H1299 p53 null osteosarcoma line by Annexin V staining.Cells were seeded as above, 18 hr prior to transfection with 2 �gof pc3oriP plasmid expressing either EBNA1, EBNA1�395–450, orno EBNA1, 1 �g of pcDNA3p53 (Leng et al., 2003), and 500 ng ofpEGFP-C1 (BD Biosciences) as a transfection marker. 24 hr later,cells were harvested and stained by using Annexin V-APC (BD Bio-sciences) according to the manufacturer. Cells were fixed in 2%paraformaldehyde/PBS overnight, washed in PBS, then filtered andanalyzed by flow cytometry as described above.
Effect of EBNA1 on p53 LevelsU2OS cells in 60 mm dishes were transfected with 2 �g of pc3oriPplasmid expressing either EBNA1, �395–450, or no EBNA1 by usingLipofectamine 2000 (Invitrogen). 5 hr later, cells were replated in 15cm dishes and grown in the presence of 0.4 mg/ml G418 to selectfor cells containing the pc3oriP plasmids. After 2 weeks, cells wereeither harvested or UV irradiated (as described above) and har-vested 4 or 8 hr post-UV treatment. Cells were lysed in 9 M urea, 5mM Tris-HCl (pH 6.8), and sonicated. 30 �g of total protein wasanalyzed by Western blotting using antibodies PAb1801 for p53(Banks et al., 1986), Ab-1 for actin (Oncogene research products)and R4 serum for EBNA1 (Holowaty et al., 2003b). Blots were devel-oped by using the ECL plus system (Amersham), and enhancedchemifluorescence was quantified by using a Typhoon 9400 scan-ner (Amersham) and ImageQuant 5.0 software. p53 levels were de-termined in relationship to the actin loading control.
Assays of EBNA1-USP7 Interactions in B CellsEBNA1 affinity column assays were conducted by using whole-cellextracts from BL41 (at 10 mg/ml) and HeLa (at 14 mg/ml) cells,which were generated as described in Holowaty et al. (2003b). 400�l of each lysate was applied under physiological salt conditionsto a 40 �l column containing EBNA1�61–83 (Wu et al., 2002) at 1mg/ml and, after washing, bound proteins were eluted in high saltand identified by MALDI-ToF mass spectrometry as previously de-scribed (Holowaty et al., 2003b). For coimmunoprecipitation experi-ments, 2.5 × 108 Raji (EBV-positive Burkitt’s lymphoma) cells werelysed by dounce homogenization in 10 mM HEPES (pH 7.9), 1.5mM MgCl2, 10 mM KCl, 0.1% Triton X-1001, and Roche proteaseinhibitors using 1.33 mls buffer per mg cell pellet. After addition ofan equal volume of extraction buffer (50 mM HEPES [pH 7.5], 1.5
mM MgCl2, 1.26 M potassium acetate, and 75% glycerol), cellswere further dounce homogenized and extracted on ice for 30 min.The lysate was clarified by centrifugation, dialysed overnightagainst 10 mM HEPES (pH 7.9), 150 mM NaCl then precleared byincubation with 100 �l bed volume of Protein A Sepharose for 15min. USP7 was immunoprecipitated from 500 �l (2.4 mg) lysate with0 (negative control), 2, and 10 �l BL851 USP7 antibody (BethylLaboratories) and 20 �l bed volume Protein A Sepharose. After 5hr of mixing at 4°C, beads were washed three times in 10 mMHEPES (pH 7.9), 150 mM NaCl and eluted with 40 �l 1% SDS. 20�l of lysate and eluates were analyzed by Western blot usingEBNA1 OT1X monoclonal antibody. The blots were then strippedand reprobed with BL851 USP7 antibody.
Acknowledgments
We thank the Oxford Protein Production Facility, University of Ox-ford for performing initial crystal screens with USP7 and Dr. DineshChristendat for help with X-ray data collection. We also thank Dr.J. Lukin for assistance with NMR analysis, Cheryl Smith for FACSanalysis, Dr. Sam Benchimol for cell lines and antibodies, Dr. JaapMiddeldorp for EBNA1 antibody, and Dr. Alan Davidson for use ofhis spectrofluorometer. This work was funded by the CanadianCancer Society through grants to L.F. and C.H.A. from the NationalCancer Institute of Canada (NCIC). V.S. was supported by a NaturalSciences and Engineering Council of Canada postdoctoral fellow-ship and Y.S. was supported by a fellowship from the NCIC.
Received: August 26, 2004Revised: November 23, 2004Accepted: February 23, 2005Published: March 31, 2005
References
Banks, L., Matlashewski, G., and Crawford, L. (1986). Isolation ofhuman-p53-specific monoclonal antibodies and their use in thestudies of human p53 expression. Eur. J. Biochem. 159, 529–534.
Chung, J.Y., Park, Y.C., Ye, H., and Wu, H. (2002). All TRAFs arenot created equal: common and distinct molecular mechanisms ofTRAF-mediated signal transduction. J. Cell Sci. 115, 679–688.
Cummins, J.M., Rago, C., Kohli, M., Kinzler, K.W., Lengauer, C., andVogelstein, B. (2004). Tumour suppression: disruption of HAUSPgene stabilizes p53. Nature 428, 486–487.
Dolcetti, R., and Masucci, M.G. (2003). Epstein-Barr virus: inductionand control of cell transformation. J. Cell. Physiol. 196, 207–218.
Doublie, S. (1997). Preparation of selenomethionyl proteins forphase determination. Methods Enzymol. 276, 523–530.
Everett, R., Meredith, M., Orr, A., Cross, A., Kathoria, M., and Par-kinson, J. (1997). A novel ubiquitin-specific protease is dyamicallyassocited with the PML nuclear domain and binds to a herpesvirusregulatory protein. EMBO J. 16, 1519–1530.
Holowaty, M.N., Sheng, Y., Nguyen, T., Arrowsmith, C., and Frap-pier, L. (2003a). Protein interaction domains of the ubiqutin specificprotease, USP7/HAUSP. J. Biol. Chem. 278, 47753–47761.
Holowaty, M.N., Zeghouf, M., Wu, H., Tellam, J., Athanasopoulos,V., Greenblatt, J., and Frappier, L. (2003b). Protein profiling withEpstein-Barr nuclear antigen 1 reveals an interaction with the her-pesvirus-associated ubiquitin-specific protease HAUSP/USP7. J.Biol. Chem. 278, 29987–29994.
Hu, M., Li, P., Li, M., Li, W., Yao, T., Wu, J.-W., Gu, W., Cohen, R.E.,and Shi, Y. (2002). Crystal structure of a UBP-family deubiquitinat-ing enzyme in isolation and in complex with ubiquitin aldehyde.Cell 111, 1041–1054.
Hume, S., Reisbach, G., Feederle, R., Delecluse, H.-J., Bousset,K., Hammerschmidt, W., and Schepers, A. (2003). The EBV nuclearantigen 1 (EBNA1) enhances B cell immortalization several thou-sanfold. Proc. Natl. Acad. Sci. USA 100, 10989–10994.
Kay, L.E. (2001). Nuclear magnetic resonance methods for high mo-lecular weight proteins: a study involving a complex of maltose

Molecular Cell36
binding protein and beta-cyclodextrin. Methods Enzymol. 339, (l174–203.
ZKennedy, G., Komano, J., and Sugden, B. (2003). Epstein-Barr virusRprovides a survival factor to Burkitt’s lymphomas. Proc. Natl. Acad.nSci. USA 100, 14269–14274.2Kieff, E., and Rickinson, A.B. (2001). Epstein-Barr virus and its repli-
cation. In Fields Virology, D.M. Knipe and P.M. Howley, eds. (Phila-Adelphia: Lippincott Williams and Wilkins), pp. 2511–2573.
Kube, D., Vockerodt, M., Weber, O., Hell, K., Wolf, J., Haier, B., TGrasser, F.A., Muller-Lantzsch, N., Kieff, E., Diehl, V., and Tesch, H. t(1999). Expression of Epstein-Barr virus nuclear antigen 1 is associ- wated with enhanced expression of CD25 in the Hodgkin cell lineL428. J. Virol. 73, 1630–1636.
Leng, R.P., Lin, Y., Ma, W., Wu, H., Lemmers, B., Chung, S., Parant,J.M., Lozano, G., Hakem, R., and Benchimol, S. (2003). Pirh2, ap53-induced ubiquitin-protein ligase, promotes p53 degradation.Cell 112, 779–791.
Li, C., Ni, C.Z., Havert, M.L., Cabezas, E., He, J., Kaiser, D., Reed,J.C., Satteerthwait, A.C., Cheng, G., and Ely, K.R. (2002a). Down-stream regulator TANK binds to the CD40 recognition site onTRAF3. Structure 10, 403–411.
Li, C., Norris, P.S., Ni, C.Z., Havert, M.L., Chiong, E.M., Tran, B.R.,Cabezas, E., Reed, J.C., Satterthwait, A.C., Ware, C.F., and Ely, K.R.(2003). Structurally distinct recognition motifs in lymphotoxin-betareceptor and CD40 for tumor necrosis factor receptor-associatedfactor (TRAF)-mediated signaling. J. Biol. Chem. 278, 50523–50529.
Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev, A.Y., Qin, J., and Gu,W. (2002b). Deubiquitination of p53 by HAUSP is an important path-way for p53 stabilization. Nature 416, 648–653.
Li, M., Brooks, C.L., Kon, N., and Gu, W. (2004). A dynamic role ofHAUSP in the p53-Mdm2 pathway. Mol. Cell 13, 879–886.
McWhirter, S.M., Pullen, S.S., Holton, J.M., Crute, J.J., Kehry, M.R.,and Alber, T. (1999). Crystallographic analysis of CD40 recognitionand signaling by human TRAF2. Proc. Natl. Acad. Sci. USA 96,8408–8413.
Ni, C.Z., Welsh, K., Leo, E., Chiou, C.K., Wu, H., Reed, J.C., and Ely,K.R. (2000). Molecular basis for CD40 signaling mediated byTRAF3. Proc. Natl. Acad. Sci. USA 97, 10395–10399.
Park, Y.C., Burkitt, V., Villa, A.R., Tong, L., and Wu, H. (1999). Struc-tural basis for self-association and receptor recognition of humanTRAF2. Nature 398, 533–538.
Polekhina, G., House, C.M., Traficante, N., Mackay, J.P., Relaix, F.,Sassoon, D.A., Parker, M.W., and Bowtell, D.D.L. (2002). Siah ubiqu-itin ligase is structurally related to TRAF and modulates TNF-α signal-ling. Nat. Struct. Biol. 9, 68–75.
Salzmann, M., Pervushin, K., Wider, G., Senn, H., and Wuthrich, K.(1998). TROSY in triple-resonance experiments: new perspectivesfor sequential NMR assignment of large proteins. Proc. Natl. Acad.Sci. USA 95, 13585–13590.
Saridakis, V., Yakunin, A., Xu, X., Anandakumar, P., Pennycooke, M.,Gu, J., Cheung, F., Lew, J.M., Sanishvili, R., Joachimiak, A., et al.(2004). The structural basis for methylmalonic aciduria. The crystalstructure of archaeal ATP:cobalamin adenosyltransferase. J. Biol.Chem. 279, 23646–23653.
Wajant, H., Henkler, F., and Scheurich, P. (2001). The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors,kinases and their regulators. Cell. Signal. 13, 389–400.
Wilson, J.B., Bell, J.L., and Levine, A.J. (1996). Expression of Ep-stein-Barr virus nuclear antigen-1 induces B cell neoplasia in trans-genic mice. EMBO J. 15, 3117–3126.
Wu, H., Kapoor, P., and Frappier, L. (2002). Separation of the DNAreplication, segregation and transcriptional activation functions ofEpstein-Barr nuclear antigen 1. J. Virol. 76, 2480–2490.
Ye, H., Park, Y.C., Kreishman, M., Kieff, E., and Wu, H. (1999). Thestructural basis for the recognition of diverse receptor sequencesby TRAF2. Mol. Cell 4, 321–330.
Ye, H., Arron, J.R., Lamothe, B., Cirilli, M., Kobayashi, T., Shevde,N.K., Segal, D., Dzivenu, O.K., Vologodskaia, M., Yim, M., et al.
2002). Distinct molecular mechanism for initiating TRAF6 signal-ing. Nature 418, 443–446.
apata, J.M., Pawlowski, K., Haas, E., Ware, C.F., Godzik, A., andeed, J.C. (2001). A diverse family of proteins containing tumorecrosis factor receptor-associated factor domains. J. Biol. Chem.76, 24242–24252.
ccession Numbers
he atomic coordinates for USP7 NTD and USP7 NTD bound tohe EBNA1 peptide have been deposited in the Protein Data Bankith the accession numbers 1YZE and 1YY6, respectively.





























