Embed Size (px)
Citation preview

59 | P a g e
Chapter 3
Contribution of host heme versus de novo biosynthetic heme in
asexual stage development of malaria parasite

60 | P a g e
3.1 Introduction
Heme biosynthesis is central to the malaria parasite biology. The malaria parasite is capable
of synthesizing heme de novo despite its ability to acquire heme from red blood cell (RBC)
hemoglobin. During the intraerythrocytic stages, the parasite detoxifies hemoglobin-heme by
converting it into hemozoin (Surolia N and Padmanaban G 1992), (Bonday ZQ et.al, 1997).
Detailed studies in Prof G Padamanaban’s lab have completely characterized all the enzymes
in P. falciparum heme-biosynthetic pathway (reviewed in Padmanaban et al., 2013). The
parasite enzymes are unique in terms of their localization and catalytic efficiencies. The first
enzyme, δ-aminolevulinate synthase (PfALAS) (Varadarajan S et.al, 2002), (Sato S et.al,
2004), and the last two enzymes, Protoporphyrinogen IX oxidase (PfPPO) and Ferrochelatase
(PfFC) (Nagaraj VA et.al, 2010), (Nagaraj VA et.al, 2009) localize to the mitochondrion.
The enzymes that catalyze the intermediate steps: ALA dehydratase (PfALAD) (Sato S et.al,
2002), (Dhanasekaran S et.al, 2004), Porphobilinogen deaminase (PfPBGD) (Sato S et.al,
2004), (Nagaraj VA et.al, 2008), and Uroporphyrinogen III Decarboxylase (PfUROD)
(Nagaraj VA et.al, 2009) localize to the apicoplast (a chloroplast relic), whereas, the next
enzyme Coproporphyrinogen III oxidase (PfCPO) is cytosolic (Nagaraj VA et.al, 2010). The
enzymes that localize to the apicoplast have a very low catalytic efficiency compared with
RBC counterparts (Padmanaban G et.al, 2007), (Padmanaban G et.al, 2013).
The apicoplast is involved in the synthesis of heme, fatty acids, iron-sulfur proteins, and
isoprenoids (Lim L et.al, 2010). Yeh and Risi (Yeh E and Risi JL, 2011) showed that a
chemical knockout of apicoplast function could be rescued by isopentenyl pyrophosphate
supplement to P. falciparum cultures in vitro. This suggests that during the intraerythrocytic
stages, the parasite requires apicoplast function for isoprenoid synthesis, but not for heme or
fatty acid synthesis. However, heme as such is essential for parasite survival in the
intraerythrocytic stages, by minimally constituting the cytochrome component of the Electron
Transport Chain (ETC). The ETC is used as a sink for electrons generated in the pyrimidine
pathway (Painter HJ et.al, 2007). Atovaquone inhibits parasite growth by inhibiting
cytochrome bc1 activity of the ETC, most likely by competitively inhibiting the cytochrome
b quinone oxidation site (Hammond DJ et.al, 1985), (Barton V et.al, 2010). Previously, it has
been shown that PfPPO requires the ETC and is likewise inhibited by atovaquone (Nagaraj
VA et.al, 2010). Heme can also serve as a source of iron for the iron-sulfur proteins involved
in isopentenyl pyrophosphate synthesis (Lim L et.al, 2010).

61 | P a g e
The acquisition of heme from RBC hemoglobin and the storage of heme as hemozoin in the
food vacuole of the parasite are reasonably well understood (vanDooren GG et.al, 2012;
Elliott DA et.al, 2008). The question arises whether the parasite depends on de novo heme
biosynthesis or heme from hemoglobin or a combination of both to make mitochondrial
cytochromes. In the present study, the gene knockouts for parasite-encoded heme
biosynthetic enzymes (PbALAS and PbFC) were generated and growth of the parasite in the
intraerythrocytic stage was examined. Radiolabeling of heme was carried out in mouse
reticulocyte cultures infected with wildtype and knockout parasites using [4-14
C] ALA. The
Radiolabeling of parasite hemozoin and the cytochrome c complex was examined. Based on
the radiolabeling of host heme in mouse reticulocytes and the parasite heme in WT but not in
the Knock out parasites, the contribution of host v/s parasite de novo heme in the asexual
stage development was studied.
3.2 Material and Methods
3.2.1 In vitro culture of P. falciparum
In vitro cultures for P. falciparum 3D7 isolate were maintained continuously on human O+
red cells of 5% hematocrit supplemented with 10 % O+ serum or 0.5% Albumax II in RPMI
1640 medium containing L-glutamine (GIBCO) by the candle jar method (Trager W and
Jensen, 1976) or in a CO2 incubator. The reagents for culturing the parasites were procured
from GIBCO Invitrogen and Sigma Aldrich unless otherwise stated. In brief, the red cells
were prepared by collecting human O+ whole blood in anticoagulant and centrifuged at
2000xg for 10 min to remove plasma and Buffy coat and washed thrice with RPMI 1640
medium and adjusted to a final haematocrit of 50% and stored at 4°C till use. O+ serum was
prepared by collecting whole blood without anticoagulant and kept overnight at 4°C followed
by aseptically transferring the separated serum and centrifuged at 10000xg for 20 min to
remove any clotted debris and heat inactivated at 56°C for 30 min and stored at -20°/-80°C
till use. Complete medium was prepared in sterile Milli Q water by adding RPMI 1640 media
(containing L-glutamine, 25mM HEPES), 0.225% Sodium bicarbonte, 2% D-Glucose,
50mg/L hypoxanthine, and 40µg/ml gentamycin with 10% heat inactivated O+ serum or
0.5% Albumax II (Invitrogen) and finally filtered through 0.22µm membrane filter (Nalgene
Inc.). (Note: Hypoxanthine was added to complete medium only when Albumax II was used
as a supplement). The initial parasitaemia was adjusted to 0.5 to 1% by adding unifected red
cells to get 5% cell suspension and maintained at 37°C in CO2 incubator with daily media

62 | P a g e
change. The growth of the parasites was monitored by microscopic examination of Giemsa
stained thin smear and parasitaemia was determined as percentage of infected red cells.
3.2.2 Cryopreservation and revival of asexual stage Plasmodium falciparum
The P. falciparum culture selected for cryopreservation should contain more than 50% of the
parasites in ring stages as higher parasitic stages gets lysed in due course. In brief, the
cryopreservative or cryoprotectant is prepared by adding 28ml of glycerol to 72ml of 4.2%
sorbitol in normal saline and filtered through 0.22µm membrane filter. The cultured parasites
predominantly at rings stage are centrifuged at 2000xg to 10 min to remove the spent medium
and the resultant cell pellet is resuspended in an equal volume of cryoprotectant and
distributed in cryotubes and frozen quickly by immersing in liquid nitrogen. To revive, the P.
falciparum frozen stocks was removed from liquid nitrogen and thawed immediately at 37°C.
The culture suspension was transferred to microfuge tube and centrifuged at 1500xg for 10
min to remove the supernatant, and to the pellet an equal volume of 3.5% Sodium Chloride
was added (drop wise) and again centrifuged to remove supernatant followed by wash (twice)
with complete medium supplemented with 15% serum or 1% Albumax II. After washing the
cryopreserved cells, the culture was initiated by adding fresh uninfected red cells at 5%
haematocrit and cultured as described in section 2.2.1.
3.2.3 Synchronization of P. falciparum
Synchronization of P. falciparum in culture was carried out by sorbitol treatment (Lambros C
and Vanderberg JP, 1979). In brief, 5% sorbitol (Sigma Aldrich) was prepared in sterile Milli
Q water and filtered through 0.22µm membrane filter. To 1ml of culture pellet, 9ml of 5%
sorbitol was added and allowed to stand at room temperature for 5 min followed by
centrifugation at 300xg for 5 min to discard supernatant and the pellet was washed twice with
complete media and finally fresh media was added to the pellet and incubated at 37°C as
described in section 2.2.1.
3.2.4 Preparation of free parasites
The free parasites were obtained from infected erythrocytes at the late trophozoite and
schizont stages by treatment with an equal volume of 0.15% (w/v) saponin in PBS (Cowman
AE et.al, 1991). In brief, an equal volume of 0.15% saponin (Sigma Aldrich) prepared in PBS
was added to culture pellet that was washed thrice with PBS and incubated to 37°C for 20
min following by the addition of 3-5 volume of ice cold PBS and centrifuged at 10000x g for

63 | P a g e
10 min at 4°C. After removing the supernatant, the parasite pellet obtained was washed with
ice cold to remove any detectable hemoglobin.
3.2.5 Maintenance and propagation of Plasmodium berghei
The routine propagation of P. berghei ANKA strain (MRA-311, MR4, ATCC Manassas
Virginia) was carried out in 6-8 weeks old Swiss mice. In brief, mice were injected
intraperitoneally (I.P) with 105 P. berghei infected-RBCs/reticulocytes and the parasite
growth was routinely monitored by assessing the percentage of parasitemia in Giemsa stained
thin smears prepared from tail vein blood. On day 8-10 post-infection, mice were
anesthetized with ketamine/xylazine and the infected blood was collected through cardiac
puncture. The blood obtained was diluted with PBS to initiate fresh infections in mice (COX
FEG, 1988), (Helmbby H et.al, 2008). Parasite isolation was carried out as described earlier
(Cowman AE et.al, 1991) under the section 3.2.4.
3.2.6 Cryopreservation and revival of Plasmodium berghei
To cryopreserve P. berghei, freshly collected parasite infected blood in heparinized PBS from
anesthetized mice was mixed with glycerol (final concentration of glycerol is 30%) and
frozen in liquid nitrogen. To initiate fresh infection in mice, P. berghei cryopreserved stock
containing ~ 0.5ml per stock is mixed with equal volume of PBS and thawed at room
temperature. A total volume of 1 ml suspension was then injected intra peritoneal (I.P) into
the flanks of appropriate number of mice (200µl/ mice).
3.2.7 Drugs used during P. berghei growth and maintenance
For various specific experimental purposes in P. berghei, the following drugs were used
during parasite growth in mice:
(a) Phenylhydrazine: P. berghei has a strong preference for reticulocytes as compared to
mature red cells, thus for the specific experimental purpose, reticulocytosis was induced in
mice by injecting 2.5mg of phenylhydrazine (Sigma-Aldrich) in saline per mouse through I.P.
Phenylhydrazine was administered to increase the number of asexual blood stages and
gametocytes and also for the establishment of synchronized parasite infections in mice. Two
days later, fresh infection of P. berghei was initiated in phenylhydrazine treated mice and (or)
mouse reticulocytes were separated for radiolabelling experiments.
(b) Pyrimethamine: The selection of transfected parasites can be carried out using either
pyrimethamine for the PbDHFR/TS selectable marker or WR99210 for both the hDHFR and

64 | P a g e
PbDHFR/TS. Where possible, it is preferable to use pyrimethamine due to its ease of
application and efficiency in drinking water. Pyrimethamine (Sigma-Aldrich; 7 mg/ml) was
prepared in DMSO and then diluted 100 times with tap water and the pH adjusted between 3
and 5 using 1M HCl. The drug was administered in drinking water to mice and hence is
useful to positively select the genetically modified parasites bearing the dhfr cassette.
3.2.8 Isolation of parasite genomic DNA
Parasite genomic DNA was isolated by SDS/proteinase K method (Lopera-Mesa et al., 2008).
In brief, the parasite pellet (described in section 2.2.4) was resuspended in 500µl of
resuspension buffer containing 50mM Tris pH 8.0, 50mM EDTA, 1% SDS, 100mM NaCl
and 2 µl of RNAase A (1mg/ml) and incubated at 37°C for an hour followed by addition of
20µl of proteinase K (2mg/ml) and incubated for another 2h at 37°C. Next, the suspension
was centrifuged at 13000xg for 10min and the supernatant was carefully transferred into the
fresh microfuge tube and 500µl of buffered phenol: chloroform: isoamylalcohol (25:24:1)
was added and the tube was gently inverted several times and centrifuged at 13000 x g for 5
min. The upper aqueous phase obtained after centrifugation was transferred to new tube and
gently inverted as earlier and centrifuged again at 13000 x g for 5min. The aqueous phase
was again transferred to new tube and equal volume of chloroform was added and gently
mixed by inverting the tube and centrifuged at 13000 x g for 5 min. To the upper aqueous
phase transferred into new tube, 2-2.5 volume of 100% ethanol was added and gently
inverted several times (60-70 times) and DNA was allowed to precipitate at -70°C for 1h then
centrifuged at 13000xg for 15 min at 4°C. The resultant pellet was washed with 70% ethanol
and centrifuged to discard the supernatant and the pellet was dried at 37°C and DNA was re-
suspended in 50µl of sterile Milli Q water.
3.2.9 Generation of P. berghei ALAS and FC knockouts
To generate the knockout parasites, primers were designed and PCR was carried out with P.
berghei genomic DNA to amplify the 670-740 bp fragments that correspond to the 5’- UTR
and 3’-UTR regions of PbALAS/FC genes. Molecular cloning enzymes and restriction
enzymes were procured from New England BioLabs Inc. unless otherwise stated. PCR
amplification for cloning was carried out using Phusion DNA Polymerase (Finnzymes) with
the protocol: initial denaturation at 98°C for 2min, 40 cycles of the following denaturation,
annealing and extension steps: 98°C for 30 seconds, 50-60°C for 30 seconds (depending on
primer melting temperature) and 72°C for 1min, concluding with 10 minutes of final

65 | P a g e
extension step at 72°C. The details of the primers and restriction sites are described in
Table1. The resultant fragments were cloned into the appropriate restriction sites flanking the
human DHFR selection cassette of pL0006 replacement plasmid (MRA-755, MR4, ATCC
Manassas Virginia, deposited by A. P Waters). The plasmid constructs were then digested
with appropriate restriction enzymes, and transfected into P. berghei schizonts that were
purified from intraerythrocytic stage infections initiated by sporozoite injections (Janse et al.,
2006). In brief, parasite infected blood consisting of asynchronous stages of parasites was
collected from phenylhydrazine treated mice and washed twice with RPMI 1640 complete
medium containing L-Glutamine, 25mMHEPE (Gibco) and supplemented with 20% Fetal
bovine serum (FBS) (Invitrogen) and re-suspended in 35 ml of RPMI 1640 complete medium
in an 50ml centrifuge tube followed by underlaying with 10ml of 50% Nycodenz (Sigma)
prepared in PBS and centrifuged at 450 x g for 20 min. The enriched layered of schizont
formed at the interphase was carefully transferred to new tube and washed with RPMI 1640
complete medium and centrifuged at 450 x g for 8 min and finally suspension was kept as
1ml and subjected to nucleofection with appropriate constructs (Nucleofector Kit, Lonza)
followed by pyrimethamine selection. Limiting dilution was carried out for pyrimethamine-
resistant parasites (Janse and Waters, 2002) and the targeted deletion of PbALAS and PbFC
genes in the respective knockout parasites were confirmed by PCR, Southern, Northern and
Western analyses.
Table2. Primers used to generate the knockout parasites and PCR analysis. Restriction sites
are underlined.
Primers Sequence (5’- 3’)
PbALASKO
5’UTR(F)
GCCAGGGCCCCATAAACTTTATTCGATTTGTTTCCGACAAC
ApaI
PbALASKO
5’UTR(R)
GCCCCCGCGGCTTACAACTCTCTCTATATACCCTTATTTATG
SacII
PbALASKO
3’UTR(F)
GCCAGGTACCGAAAGCACTAAGCACATGAATATAATTTTTCC
KpnI
PbALASKO
3’UTR(R)
GCCCGCGGCCGCGAAAATGCATAGACTCCTTGACAAGTCATATAG
NotI
PbFCKO
5’UTR(F)
GCCAGGGCCCGTCTTGAAAATTATCGTTATTATTTTGTTC
ApaI

66 | P a g e
PbFCKO
5’UTR(R)
GCCCAGATCTTATTAAATATAAAAGTATATCAAACTATAAATTCG
BglII
PbFCKO
3’UTR(F)
GCCAGGTACCAATTATTATAAAATTCTTTAACAAGAATAAATC
KpnI
PbFCKO
3’UTR(R)
GCCCGCGGCCGCGTAATAATGTAGTATTATATTTATTGGATCAAC
NotI
PbALASKO(F) ATGAGAAAGAAAAAAGCATTAAAGGTGAGTC
PbALASKO(R) TTAAAGCTTCATTTCGATTTTGTTTTTTTTGTTG
PbPBGD(F) ATGCATCTATTAACTTTCATTATATTAAATATATATATAAC
PbPBGD(R) TTATTTGTAATATAAAGCTGCCTCATCCTTTATTTTATTAAAC
PbFCKO(F) ATGGATATAGACGATTTCTTAAAATGTAACAATTTAAAC
PbFCKO(R) TTACCAGCCACTTAGATTTTTTTCAATAATATTC
3.2.10 In vitro radiolabeling experiments
In vitro radiolabeling of heme in mouse reticulocytes was carried out at 37°C in a CO2
incubator, for a period of 9 h in RPMI-1640 medium containing 10% FBS, by adding 1 µCi
of [4-14
C]ALA to a total volume of 5 ml containing 109 reticulocytes. In brief, reticulocytosis
was induced in mice by injecting a single dose of phenylhydrazine (2.5 mg in saline/mouse)
intraperitoneally. Two days later, reticulocytes from the mice blood were separated by
performing the density-gradient centrifugation on isotonic percoll (Liu et al., 2010), washed
thrice with the medium and used for labelling. Labelling studies in human RBCs were also
carried out in a similar fashion with 109 human RBCs in RPMI-medium containing 10%
human serum. To perform in vitro labelling for the intraerythrocytic stages of P. berghei
wild-type and knockout parasites, the respective blood-stage infections were initiated in
phenylhydrazine-treated mice by intraperitoneal injection of 105
infected erythrocytes. The
blood was collected when the parasitemia reached around 5-8% with parasites predominantly
in early trophozoites. After washing thrice with RPMI-1640 containing FBS, the cells were
resuspended in 10 ml of the medium to a final hematocrit of 5% and labelling was carried out
for 9 h as described for reticulocytes by adding 3 µCi of [4-14
C]ALA. To study the in vitro
effect of Succinyl Acetone (SA) on heme labelling, cultures were treated for 3 h with 50 µM
SA prior to the addition of [4-14
C]ALA, and the labelling was carried out for 9 h in the
presence of SA. For Chloroquine (CQ) treatment, PbFCKO-infected mice were injected
intraperitoneally with two doses of 0.5 mg CQ dissolved in water at 6 h time interval, when

67 | P a g e
the blood stage parasites were predominantly in early rings. The blood was collected 1 h after
the second dosage and the cells were washed with medium, followed by in vitro labelling for
9 h with 3 µCi of [4-14
C]ALA. In vitro labelling for P. falciparum in the presence and
absence of SA was carried out with synchronized cultures harbouring 5-8% early
trophozoites maintained in RPMI-1640 containing 10 % O+ serum or 0.5% Albumax II .
3.2.11 Preparation of parasite mitochondria and food vacuole
Mitochondria isolation was carried out as described (Chavalitshewinkoon-Petmitr et al.,
2000) by homogenizing the parasite pellet in 10 volumes of buffer pH 7.4 containing 5mM
Hepes-KOH, 75mM sucrose, 225 mM mannitol, 5 mM MgCl2, 5mM KH2PO4 and 1 mM
EGTA with protease inhibitors. The homogenate was then centrifuged at 4500 x g for 5 min
at 4°C and the supernatant obtained was subjected to 44,700 x g for 7 min at 4°C to pellet
mitochondria. Labelling of hemoproteins in the parasite mitochondria was examined by
solubilizing the pellet in 20 mM Tris buffer pH 7.5 containing 5% Triton X-100 and protease
inhibitors, and centrifuging at 20,000 x g to remove membrane debris, followed by loading
the supernatant on to a 5% Native-PAGE. The radiolabelled sharp band seen at the top of the
gel in silver staining was subjected to MALDI analysis. To measure the intensity of
radiolabeling, the gel was dried and exposed to phosphorimager screen for 24 h. For food
vacuole preparation, 4500xg pellet was processed as described (Saliba et al., 1998; Lamarque
et al., 2008). After lysis in ice cold water pH 4.2 and DNaseI treatment in uptake buffer (25
mM HEPES, 25 mM NaHCO3, 100 mM KCl, 10 mM NaCl, 2 mM MgSO4, and 5 mM
sodium phosphate, pH 7.4), food vacuoles were purified by titurating the pellet in 42%
percoll containing 0.25 M sucrose and 1.5 mM MgSO4, and centrifuging at 16,000 x g for 10
min at 4°C. The food vacuole pellet obtained was washed with 1 ml of uptake buffer to
remove percoll.
3.2.12 Extraction of total heme and hemozoin-heme
Extraction of free and protein-bound heme (total heme) was carried out as described earlier
(Nagaraj et al., 2010). Briefly, the parasite pellet was extracted with 10 volumes of ethyl
acetate: glacial acetic acid (4:1) for 30 min at 4°C and centrifuged at 16,000 x g for 10 min.
The organic phase containing heme and porphyrins was separated and washed thrice with 1.5
N HCl of one-third total volume, and twice with water to remove porphyrins and any ALA
present. The extracted organic phase containing heme was dried under a stream of nitrogen

68 | P a g e
and dissolved in methanol, followed by thin-layer chromatography (TLC) on silica gel using
the mobile phase 2,6-lutidine and water (5:3) in ammonia atmosphere (Marks, 1969). The
intensity of radiolabelling was quantified by exposing the TLC sheets to phosphorimager
screen for 8 h. To extract heme from hemozoin, the food vacuole pellet was resuspended in
10 volumes of cold acetone containing 0.1 N HCl, vortexed for 30 min at 4°C and
centrifuged at 16,000 x g for 10 min. The supernatant obtained was dried, dissolved in
methanol and analyzed by TLC as described for total heme. The complete extraction of heme
from hemozoin can be easily visualized by the color change of the pellet from dark brown to
pale and if necessary, the extraction was carried out twice.
3.2.13 Other procedures
Total RNA from the parasite was prepared using Trizol reagent (Invitrogen) according to the
manufacturer’s protocol. PCR, Western, Southern and Northern analyses were carried out
using standard procedures. Polyclonal antibodies for ALAS and FC, cross-reacting with the
proteins of both human and mouse origin, were procured from Santa Cruz Biotechnology,
Inc. To detect P. berghei ALAS and FC, polyclonal antibodies raised against P. falciparum
ALAS and FC, cross-reacting with P. berghei proteins were used. All these antibodies were
used in 1:1000 dilution for Western blotting. Hemoglobin was purified from mouse
reticulocytes and human RBCs by resuspending the cells in hypotonic lysis buffer containing
20 mM Tris pH 7.5 and protease inhibitors. The lysate was incubated in ice for 30 min,
followed by centrifugation at 20,000 x g for 20 min and the supernatant obtained was loaded
on to a UNOsphereQ column (Bio-Rad). After washing the column with 10 mM NaCl,
haemoglobin was eluted with lysis buffer containing 50 mM NaCl. To perform MALDI
analysis, the protein complex was eluted from 5% Native-PAGE and resolved in 12% SDS-
PAGE, followed by in-gel trypsin digestion. Proteins were identified by searching the
National Center for Biotechnology Information (NCBI) nr protein database using MASCOT
peptide mass fingerprint with cysteine carbamidomethylation and methionine oxidation as
fixed and variable modifications, respectively, and taking into account of one missed
cleavage and 0.5 Da peptide mass tolerance. MALDI analysis was carried out at Proteomics
Facility, Molecular Biophysics Unit, Indian Institute of Science.
3.2.14 Statistical analysis
Statistical analysis was performed using unpaired t-test of Excel software with two-tailed
distribution and unequal sample variance. P values of < 0.05 were considered as significant.

69 | P a g e
Graphs were prepared using Sigmaplot 10.0. Error bars given in the figures represent the
standard deviations. The band intensities were quantified using Fujifilm Multi guage V3.0
software.
3.3 Results
3.3.1 The role of parasite-synthesized heme during the intraerythrocytic
stages of P. berghei
In vivo animal model of parasite infection was used to determine the role of heme
biosynthesis during all the stages of parasite development. Results pertaining to asexual
stages of parasite development have been presented here, whereas the role of heme
biosynthesis during the sexual and liver stage of parasite development has been presented in
chapter 4. Figure 3.1 depicts the double crossover recombination strategy followed to obtain
PbALAS and PbFC KOs. Table 2 shows the primers used to amplify the 5’ upstream and 3’
downstream regions of PbALAS and PbFC. Figure 3.2–3.6 shows the detailed
characterization of the KOs based on genomic DNA-PCR, RT-PCR, Southern, Northern, and
Western analyses. The intraerythrocytic-stage infection was initiated by injecting 105
parasites intraperitoneally into mice by bypassing the liver stage infection cycle. There was
no significant difference in the growth of the PbKO parasites compared with the PbWT
parasites (Figure 3.7). These results indicate that the parasite may be acquiring host heme
during the intraerythrocytic stages.
Figure 3.1: Double cross-over recombination strategy to generate PbALAS and PbFC KOs.

70 | P a g e
Figure 3.2: Genomic DNA-PCR analysis indicating the targeted deletion of (A) ALAS and
(B) FC sequences in the KOs.
Figure 3.3: RT-PCR analysis indicating absence of mRNAs for (A) ALAS and (B) FC in the
KOs.
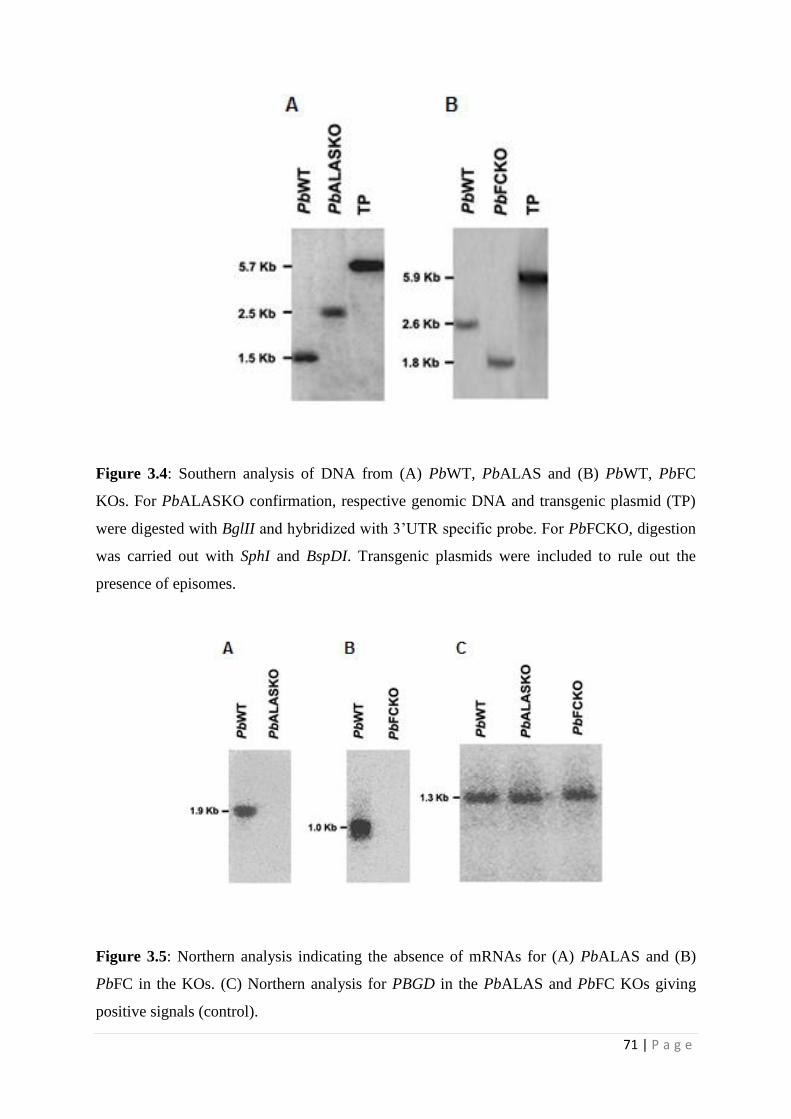
71 | P a g e
Figure 3.4: Southern analysis of DNA from (A) PbWT, PbALAS and (B) PbWT, PbFC
KOs. For PbALASKO confirmation, respective genomic DNA and transgenic plasmid (TP)
were digested with BglII and hybridized with 3’UTR specific probe. For PbFCKO, digestion
was carried out with SphI and BspDI. Transgenic plasmids were included to rule out the
presence of episomes.
Figure 3.5: Northern analysis indicating the absence of mRNAs for (A) PbALAS and (B)
PbFC in the KOs. (C) Northern analysis for PBGD in the PbALAS and PbFC KOs giving
positive signals (control).

72 | P a g e
Figure 3.6: Western analysis indicating the absence of (A) PbALAS and (B) PbFC proteins
in the KOs. (C) Western analysis for hsp60 in the PbWT and PbKOs giving positive signal
(control).
Figure 3.7: Growth curves for intraerythrocytic stages of P. berghei WT and KO parasites in
mice. The Mice were injected intraperitoneally with 105 P. berghei infected-
RBCs/reticulocytes and the parasite growth was routinely monitored as described in
Materials and Methods. Multiple fields were used to quantify the parasite infected cells. The
data provided represent the mean + S.D. obtained from 6 animals.
3.3.2 Radiolabeling of hemoglobin-heme and tracing its path in the parasite
The potential of human RBCs and mouse reticulocytes to synthesize heme was explored in
this study. The ALAS and FC proteins were detected by Western analysis in mouse
reticulocytes but not in human RBCs (Figure 3.8). Unlike in human RBCs, it was possible to
radiolabel the total heme and hemoglobin-heme in short-term mouse reticulocyte cultures
incubated with [4-14
C]ALA (Figure 3.9–3.10). Because P. berghei prefers reticulocytes, the

73 | P a g e
experimental system made it feasible to study the availability of hemoglobin-heme not only
for hemozoin formation but also for parasite cytochromes. Furthermore, the heme labeling in
the mouse reticulocyte cultures was blocked using succinyl acetone (SA), a specific inhibitor
of ALAD (Figure 3.11).
Figure 3.8: Western analysis for (A) ALAS and (B) FC. 1, mouse reticulocyte lysate; 2,
human RBC lysate.
Figure 3.9: (A) Total heme and hemoglobin-heme from mouse reticulocyte loaded on TLC
(B) Radiolabeling of bands depicted in A. Labeling was carried out with [4-14
C]ALA for 9 h
in short-term cultures. (C) Total heme and hemoglobin-heme from human RBC loaded on
TLC. (D) Radiolabeling of the bands depicted in C.

74 | P a g e
Figure 3.10: Quantification of radioactivity in total and hemoglobin-heme from mouse
reticulocytes and human RBC. The data represent the radioactive counts obtained from three
independent experiments. MRet, mouse reticulocytes; HRBC, human RBCs; TH, total heme;
HbH, hemoglobin-heme.
Figure 3.11: Effect of SA (50 µM) on heme synthesis in mouse reticulocyte cultures labeled
with [4-14
C] ALA. (A) Amount of total heme loaded on TLC. (B) Radiolabeling of bands
depicted in A. (C) Quantification of radioactivity in the heme bands. The data represent the
radioactive counts obtained from three independent experiments; P < 0.005.
Since P. berghei can only grow, but poorly infect fresh reticulocytes in vitro, reticulocytes
infected in vivo with PbWT and PbKO parasites were used to perform short-term
radiolabeling experiments in the presence of [4-14
C]ALA. The incorporation of [4-14
C] ALA
into total heme and hemozoin-heme was found in PbWT parasites and both of the PbKO
parasites. SA inhibited the radiolabeling (Figure 3.12). Radiolabeled heme appearing in the
PbWT and PbALASKO parasites could come from host hemoglobin as well as from parasite
heme biosynthesis. But, we would not expect to find [4-14
C]ALA incorporated into the heme
synthesized by the PbFCKO parasites. The ethyl acetate: acetic acid mixture used to extract

75 | P a g e
heme did not extract hemozoin. Therefore, the hemozoin was extracted using acid-acetone
solvent.
Figure 3.12: (A) Radiolabeling of total parasite heme. (B) Radiolabeling of hemozoin-heme.
The labelling of mitochondrial proteins was analysed by non-denaturing PAGE. A sharp band
at the top of the gel was observed after silver staining. The band was radiolabeled in PbWT
parasites and in both of the PbKO parasites. The radiolabeling was almost completely
inhibited by SA (Figure 3.13). SDS-PAGE analysis of the band excised and eluted from non-
denaturing PAGE showed five separate protein bands and MALDI analysis revealed the
presence of two cytochrome oxidase subunits. The sharp silver-stained band in non-
denaturing PAGE thus appeared to represent a complex of proteins and needs to be further
characterized in detail (Figure 3.14–3.15). For now, it is clear that the PbWT parasites and
both of the PbKO parasites incorporated hemoglobin-heme into mitochondrial hemoproteins
and into hemozoin.
Figure 3.13: Radiolabeling of parasite mitochondrial cytochrome complex from (A) PbWT,
(B) PbALASKO and (C) PbFCKO

76 | P a g e
Figure 3.14: MALDI analysis of the cytochrome complex from P. berghei. (A) Coomassie
staining of the gel after resolving the mitochondrial proteins in non-denaturing PAGE (B)
SDS-PAGE analysis of the band from A.
Figure 3.15: Mass spectra and the protein sequences derived from the two prominent bands
(A) Cytochrome C Oxidase subunit and (B) Cytochrome C Oxidase subunit2.
Next, the ability of parasite to use hemozoin-heme to make mitochondrial cytochromes was
examined. The effect of CQ was tested, which is known to block hemozoin formation (Tilley
et.al, 2001), on P. berghei-infected short-term reticulocyte cultures. PbFCKO parasite was

77 | P a g e
used to avoid any contribution from parasite-synthesized heme. CQ was injected into
PbFCKO-infected mice as described in the Materials and Methods. After 7 h, the infected
reticulocytes were incubated in short-term cultures and the incorporation of [4-14
C]ALA into
hemozoin and mitochondrial cytochromes over a period of 9 h was measured. The drug
treatment was given in vivo to the animals, since the direct addition of the drug to
reticulocytes culture failed to inhibit the hemozoin formation under the conditions used, even
at high concentrations. Figures 3.16 show that the CQ treatment inhibited hemozoin labeling
by 70% but did not affect the labeling of mitochondrial cytochromes. These results suggest
that host hemoglobin may provide heme to mitochondrial cytochromes and hemozoin
through independent pathways.
Figures 3.16: Radiolabeling of (A) hemozoin-heme and (B) mitochondrial cytochrome
complex after chloroquine (CQ) treatment. Equal numbers of infected reticulocytes were used
to perform the radiolabeling of PbFCKO parasites and the data obtained for CQ treatment
were compared with untreated control.
3.3.3 The role of parasite biosynthetic heme in P. falciparum cultures
The radiolabeling of hemoglobin-heme made it impossible to assess the contribution of
parasite-synthesized heme using [4-14
C]ALA in P. berghei-infected reticulocytes. However,
the contribution of parasite-synthesized heme could be assessed in P. falciparum cultures. In
those cultures, all of the radiolabeled heme was synthesized by the parasite. The hemoglobin-
heme was not radiolabeled in the P. falciparum cultures because the human RBCs used in the
in vitro cultures lacked the mitochondrial enzymes required to synthesize heme (Figure 3.8).

78 | P a g e
Although not radiolabeled, the preformed hemoglobin in the RBCs could act as a heme
source for the parasite. The incorporation of [4-14
C]ALA into the total heme, hemozoin-hem
and mitochondrial hemoproteins was found in P. falciparum cultures. SA (50 µM) inhibited
the radiolabeling (Figure 3.17). Earlier studies used SA at a fixed concentration ranging from
1 to 2 mM to inhibit heme synthesis and parasite growth (Surolia N and Padmanaban G,
1992). The present study showed that while the 50% growth inhibitory concentration was
around 1 to 2 mM (Figure 3.18 A), concentrations as low as 50 µM inhibited heme synthesis
(Figure 3.17). Similar results were observed in short-term P. berghei cultures (Figure 3.18
B). The P. falciparum mitochondrial cytochromes also formed a complex in non-denaturing
PAGE and need to be further characterized in detail.
Figure 3.17: Radiolabeling of (A) total heme, (B) hemozoin-heme and (C) mitochondrial
cytochrome complex in P.falciparum.
Figure 3.18: Effect of SA on the in vitro growth of P. falciparum (A) and P. berghei (B).
Experiments were carried out in triplicates and growth was measured based on 3H-
hypoxanthine uptake.

79 | P a g e
Thus, the present study has shown that both hemoglobin-heme and parasite-synthesized heme
could be incorporated into hemozoin in the food vacuole and into mitochondrial
cytochromes. Hemozoin formation from host hemoglobin in P. falciparum is well
characterized (Elliott et.al, 2008). Hemozoin formation from heme synthesized in the parasite
mitochondrion, however, needs to be studied further. The relative contributions of
hemoglobin-heme and parasite-synthesized heme to parasite cytochrome biosynthesis during
the intraerythrocytic stages need to be assessed under different environmental conditions.
3.4 Discussion
In this study, the role of parasite-synthesized heme in all stages of malaria parasite growth
has been assessed. P. berghei ALAS and FC gene knockout parasites were generated. The
Knockout parasites were used to track the parasite-synthetized heme and host haemoglobin
heme during the intraerythrocytic stages of the parasite development. The knockout parasites
injected intraperitoneally to mice showed no growth defects in the intraerythrocytic stages
and all the infected animals died within 10 to 12 days, when parasitemia reached around
60%. The synthesis of mitochondrial cytochromes is essential for parasite survival, so the
results would mean that the PbKO parasites used hemoglobin-heme to synthesize
cytochromes during the intraerythrocytic stages. This was demonstrated by radiolabeling
hemoglobin-heme with [4-14
C]ALA in short-term mouse reticulocyte cultures. Radiolabelled
hemozoin and mitochondrial cytochromes were found in reticulocytes infected with PbWT,
PbALASKO, and PbFCKO parasites. It was not possible to assess the contribution of
hemoglobin-heme and parasite synthesized-heme in short term in vitro cultures, because the
use of [4-14
C]ALA to radiolabel heme would bypass the potential ALASKO block. At the
same time, the PbFCKO parasites would not be able to incorporate [4-14
C]ALA into heme.
Previous study has shown that P. berghei imports host ALAD as well as host FC (Bonday et
al., 1997). Therefore, the possibility that the parasite used FC imported from the host to
synthesize heme cannot be ruled out. This was addressed using P. falciparum cultured in
human RBC, as the western analysis indicated that the human RBCs used to culture P.
falciparum did not contain detectable levels of ALAS and FC. Further, the human RBCs did
not incorporate [4-14
C]ALA into heme. So, all the radiolabeled heme in P. falciparum was
synthesized de novo by the parasite.
Earlier studies have shown that high concentration of Succinyl Acetone (1-2 mM) (Surolia
and Padmanaban, 1992) inhibited both heme synthesis and parasite growth. However, 50 µM

80 | P a g e
SA was found to completely inhibit heme synthesis in P. falciparum, the concentration much
lower than required to inhibit parasite growth, which means that P. falciparum can use
haemoglobin-heme to sustain growth under these conditions indicating that de novo heme
synthesis is not essential for P. falciparum growth in culture. Similarly, 50 µM SA
completely inhibited heme synthesis in P. berghei-infected reticulocytes as well but, did not
affect P. berghei growth in short-term cultures. The use of specific gene knockouts in the
pathway has re-evaluated the earlier reports correlating the growth of the parasite with
inhibition of heme synthesis or host enzyme import (Surolia and Padmanaban, 1992),
(Bonday et al., 1997; Padmanaban et al., 2007). Since the parasite can survive in the absence
of de novo heme synthesis, it may appear that the parasite heme-biosynthetic pathway has no
role in the intraerythrocytic stages. However, it has been shown for the first time that P.
falciparum growing in human RBCs incorporated parasite-synthesized heme radiolabeled
with [4-14
C]ALA into hemozoin as well as into mitochondrial cytochromes. Hemoglobin-
heme in the RBCs was not radiolabeled; so the heme in the parasite hemozoin and
mitochondrial cytochromes was synthesized de novo by the parasite. It has long been
assumed that only hemoglobin-heme is converted into hemozoin in the parasite food vacuole.
But now, it is clear that parasite-synthesized heme can also give rise to hemozoin in the food
vacuole. It is possible that haemoglobin-heme and parasite synthesized heme serve as back up
mechanisms for each other and the relative contributions of each has to be assessed under
different environmental conditions. Since hemoglobin transport into the food vacuole
involves cytostomes and other vesicle-mediated transformations (Elliott et al., 2008), it is not
clear at this stage how the parasite-synthesized heme made in the mitochondrion finds its way
to the food vacuole. The results from the study also emphasize the fact that hemozoin is,
perhaps, the only mechanism for heme detoxification in the parasite.
A recent study showed that the malaria parasite lacks the canonical heme oxygenase pathway
for heme degradation and relies on hemozoin formation to detoxify heme (Sigala et al, 2012),
although an earlier study suggested the possible presence of heme oxygenase in the
apicoplast (van Dooren et al., 2012; Okada, 2009). It appears that the parasite mitochondrion
would need a two-way transporter for heme: one to incorporate hemoglobin-heme into the
mitochondrion and another to transport mitochondrial heme into the pathway leading to
hemozoin formation in the food vacuole. Free heme was also detected in the erythrocyte at a
concentration around 1 µM (Liu et al., 1988) and the parasite may be able to scavenge this
heme directly (van Dooren et al., 2012). It was also suggested that ferriprotoporphyrin could

81 | P a g e
leach from the food vacuole into the parasite cytosol (Campanale et al., 2003). We found that
SA inhibited the radiolabeling of hemozoin and of mitochondrial cytochromes in PbFCKO
parasites. But, CQ inhibited the radiolabeling of hemozoin but not of mitochondrial
cytochromes. These results suggest that hemoglobin-heme may be incorporated into
mitochondrial cytochromes and into hemozoin through independent processes.







![Millennials abandonment of linear televisiondadun.unav.edu/bitstream/10171/53248/1/Guerrero...Enrique Guerrero [CV] [ ORCID] [ GS] Full Professor. Universidad de Navarra (Spain) -](https://img.pdfslide.net/doc/110x75/5fd797fe7cee8d22154ac53a/millennials-abandonment-of-linear-enrique-guerrero-cv-orcid-gs-full.jpg)





















