Embed Size (px)
Citation preview


• La primera publicación sobre hiperbilirrubi-nemia fue realizada en 1473 por Barthomomaeus Mitlinger.
• En 1847, Jacques Hervieux describió las primeras alteraciones patológicas de la hiperbilirrubinemia en el SNC.
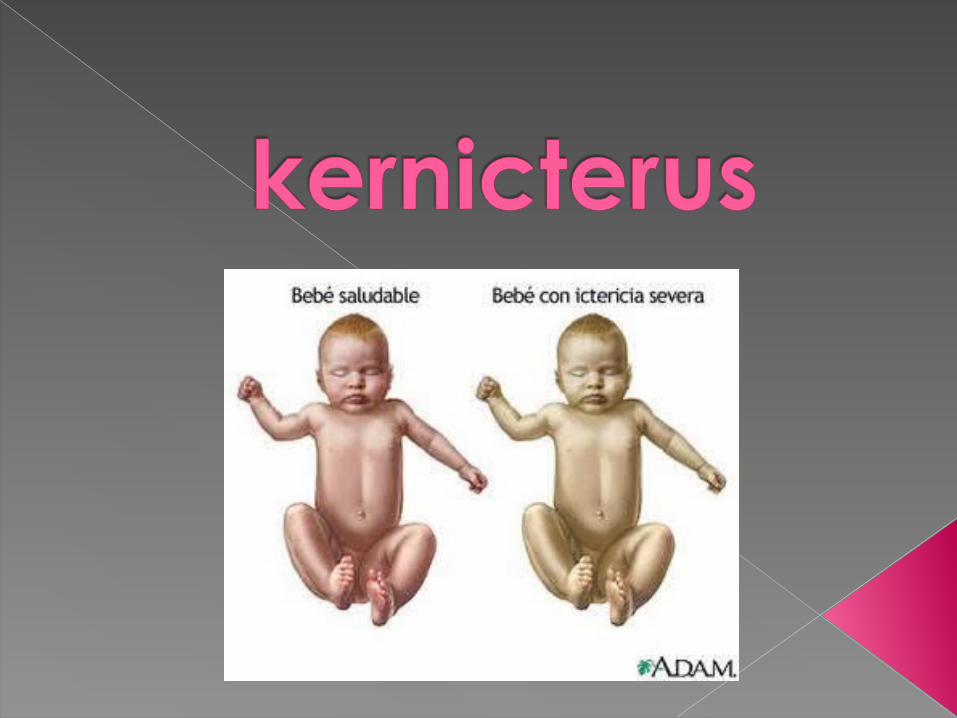
• En 1904, Schmol utilizó el vocablo de Kernic (ictericia nuclear) para describir los hallazgos post mórten en recién nacidos a término, como resultado de ictericia grave neonatal.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

• Wallerstein, en 1946, publicó, en la revista Science, el manejo conexanguinotransfusión; en 1952.
• Hsia y colaboradores reportaron que el 50% de los RN con eritroblastosis fetal y cifras mayores de 30 mg/ dl desarrollaron kernicterus.
• En 1970, Rhogan y colaboradores introducen el uso de fototerapia profiláctica.
• Ostrow, en 1990, publica nuevos conceptos en la fisiopatología de las bilirrubinas.

La encefalopatía por bilirrubinas es un síndrome neurológico que resulta del depósito de bilirrubina no conjugada en el sistema nervioso central (especialmente en los nucleos de la base, núcleos del tallo y cerebelo).
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

En los recién nacidos pretérmino, no hay una cifra que pueda considerarse de peligro, y los datos son variables. Crosse y Yar anotan niveles por encima de 18 mg/dl; Crosse y Obste, niveles mayores de 22 mg/dl; para Koch, niveles mayores de 20 mg/dl, y, según los datos de Hugh-Jones y colaboradores, en 1960, los valores de peligro están por encima de 30 mg/dl.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

Se estima la presencia de kernicterus en un 8% en pacientes con niveles de bilirrubina no conjugada de 19 a 24 mg/dl.
Un 33% de recién nacidos con cifras de bilirrubinas indirectas de 25 a 29 mg/dl.
Un 73% en recién nacidos con niveles de bilirrubinas de 30 a 40 mg/dl.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

Multifactorial. Relacionada con niveles elevados
de bilirrubina no conjugada libre.
Alteración de la barrera hematoencefálica (BHE) por otras enfermedades y susceptibilidad neuronal.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

Se describia que esta entidad era exclusiva del recién nacido a término con enfermedad hemolítica.
En la década de los 50 a los 60, se empieza a publicar su aparición en recién nacidos pretérmino, no solo asociada a hemólisis, sino a otras patologías propias del prematuro, como neumonía, sepsis, y se confirma la presencia de bilirrubina no conjugada en los nucleos de la base.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

No se realiza el diagnóstico temprano porque hay menor tiempo de hospitalización del recién nacido (RN).
Los controles del RN posteriores al alta son tardíos.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

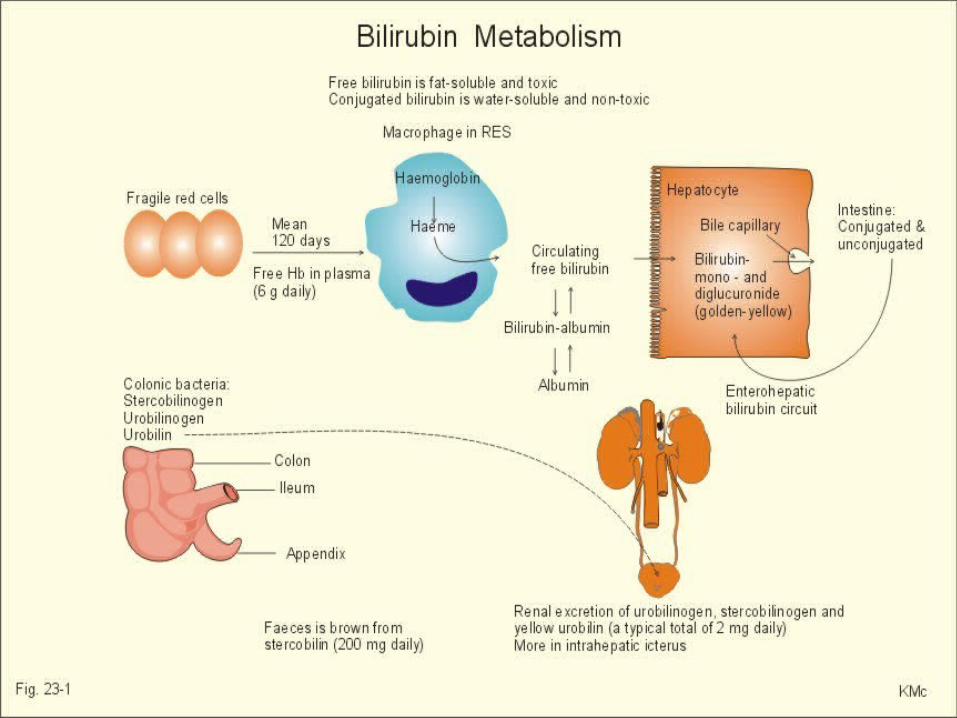
• La bilirrubina proviene de la degradación de la hemoglobina liberada tras la destrucción del eritrocito.
• Luego, se forma el grupo heme por la acción de la hemooxigenasa, la cual produce una molécula de monóxido de carbono por varias moléculas de biliverdina.
• De este producto y por la acción de la biliverdina reductasa, se forma la bilirrubina.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.
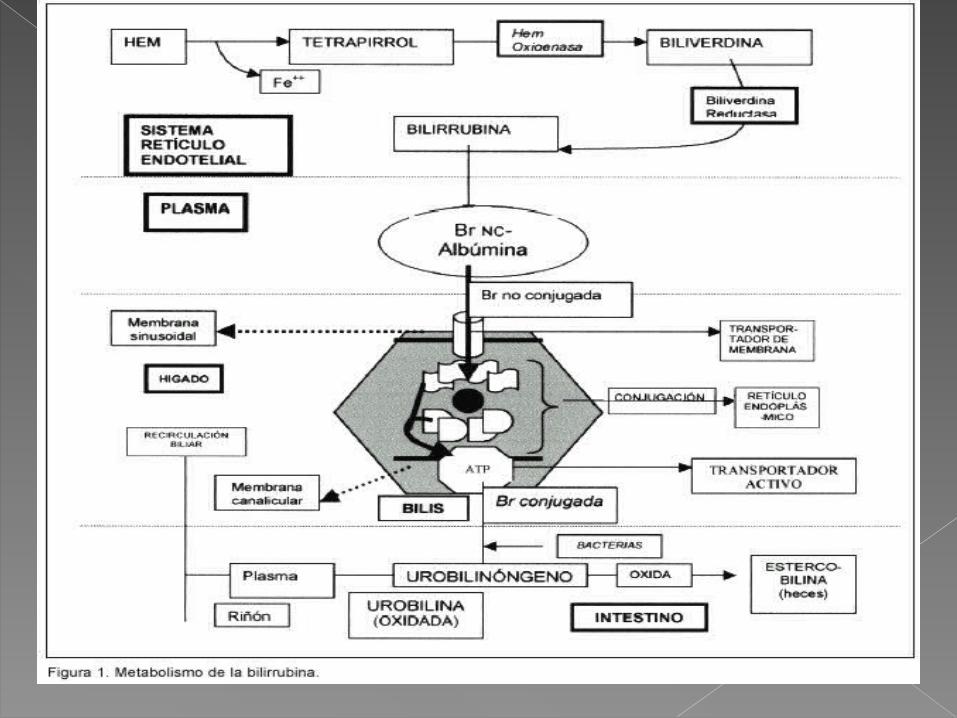

La bilirrubina es conjugada en el hígado por la uridin-difosfato-glucuronil-transferasa (UDPGT).
“ictericia fisiológica del recién nacido”. La bilirrubina no conjugada se une a la
albúmina en la sangre e ingresa al tejido cerebral.
la bilirrubina libre no se une a las proteínas cuando en la sangre la capacidad de unión es excedida o cuando otras sustancias, como las sulfonamidas, compiten en los sitios de unión.Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas
preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

En el SNC, la bilirrubina no conjugada inhibe las enzimas mitocondriales y la síntesis de DNA y proteínas; presenta afinidad por las membranas de fosfolípidos e impide la captación de tirosina, necesaria para la transmisión sináptica, lo que altera la conducción y las señales neuroexcitatorias, especialmente en el VIII par. Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas
preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

La bilirrubina tiene predilección en el SNC por los globos pálidos, núcleos subtalámicos y núcleos del tallo cerebral oculomotores y auditivos, manifestándose clínicamente cuando se presentan estas alteraciones patológicas: parálisis cerebral distónico-coreoatetósica, hipoacusia neurosensorial, neuropatía auditiva y compromiso visual de leve a severo hasta pérdida visual total.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

En RN sin hemólisis, el nivel de bilirrubinas no es un buen predictor y va a depender de la cantidad de bilirrubina no conjugada unida a la albúmina, integridad de la BHE por inmadurez del prematuro, infección, acidosis, sepsis y prematurez.
El efecto neurotóxico está determinado por la cantidad de bilirrubina unida a la albúmina.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

Encefalopatía Alteraciones de los reflejos del recién nacido. Postura en opistótonos. Dificultad en la alimentación. Hipotonía. Llanto agudo. Convulsiones. Retardo en el desarrollo psicomotor, hipoacusia
neurosensorial, parálisis cerebral (PCI), alteraciones visuales, trastornos del desarrollo y del aprendizaje, y retardo mental (RM).
Muerte.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

• En el recién nacido a término, la encefalopatía puede iniciarse del segundo al quinto día de nacido, y, en el recién nacido pretérmino, los síntomas se pueden presentar hasta el séptimo día de nacido.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

• Fase 1: (dos primeros días) el recién nacido presenta pobre succión, hipotonía variable y cambiante, alteración del estado de conciencia y, en algunas ocasiones, convulsiones.
• Fase 2: (mitad de la primera semana) aumento del tono muscular en músculos extensores que conllevan a la postura de opistótonos e incremento de la temperatura.Fase 3: (después de la primera semana) hipertonía y convulsiones.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

• A partir de los primeros meses hasta el año de edad, se evidencia hipotonía con hiperreflexia músculo-tendinosa, retardo en el desarrollo psicomotor y reflejos tónicos cervicales presentes.
• Después del primer año, se comprueba trastorno de los movimientos con manifestación de movimientos coreoatetósicos distales, distonía, balismo, temblores distales, oftalmoplejía, ojos en sol poniente, y se puede observar sordera neurosensorial.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

Edad gestacional menor de 38 semanas, hermanos con antecedentes de ictericia neonatal que requirieron manejo con fototerapia, alimentación materna exclusiva e ictericia visible en las primeras 24 horas.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

• El diagnóstico de las alteraciones del neurodesarrollo es clínico, lo mismo que el de parálisis cerebral coreoatetósica.En todo recién nacido con antecedentes de hiperbilirrubinemia, debe realizarse seguimiento, y, ante la presencia de alteraciones en el desarrollo psicomotor, alteraciones del tono muscular y posturas distónicas o movimientos extrapiramidales, independientemente de los niveles de bilirrubinas, se deben efectuar estudios de neuroimágenes y audiológicos. Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas
preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.
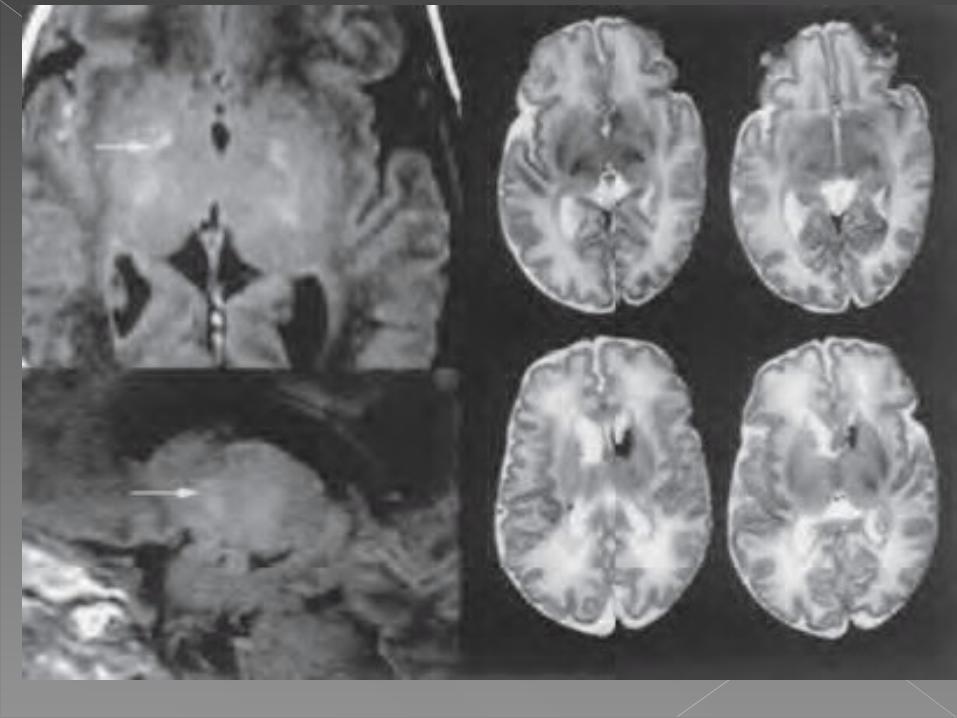

Cuando se diagnostica neuropatía auditiva o hipoacusia neurosensorial, es de importancia el diagnóstico temprano e iniciar el manejo adecuado con audífonos o implante coclear
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

• Pacientes con PC comienzo temprano de los programas de rehabilitación, soportes posturales con manejo integral e interdisciplinario con terapia física, ocupacional, miofuncional, lenguaje, psicología y educación.
• El uso de medicaciones depende del tipo de alteración semiológica, por ejemplo, en caso de coreoatetosis, utilización de haloperidol; en caso de distonía, trihexifenidil, benzodiacepinas y/o baclofeno.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.

Paralisis cerebral coreoatetósica.
Oftalmoplejía. Hipoacusia neurosensorial. Displasia del esmalte dental
y déficit cognitivo.
Eugenia Espinosa G. . El kernicterus: un viejo diagnóstico y nuevas preocupaciones . CCAP Volumen 10 Número 1 . 2012. Pags 17-23.


La enfermedad de Wilson (EW) o degeneracion hepatolenticular progresiva es un trastorno hereditario del metabolismo del cobre, caracterizado por un defecto de su excrecion biliar que conduce a su acumulacion en el organismo, principalmente en higado y encefalo, con efectos toxicos por dano oxidante y progresion invariable a muerte en ausencia de tratamiento.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

La prevalencia citada en la mayor parte de la bibliografia es de un caso por cada 30.000 habitantes.
Con una frecuencia de portadores de 1/90.
Mas frecuente en cosanguineos.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

• Fue descrita inicialmente en 1912 por Kinnier Wilson, como un trastorno neurodegenerativo asociado con cirrosis hepatica, de presentacion familiar3.
• Su relacion con el cobre fue establecida posteriormente en diversos trabajos que demostraron la presencia de un exceso de este metal en los tejidos de individuos afectados.
• En 1952 se puso de manifiesto la deficiencia de ceruloplasmina en el suero de estos pacientes.
• En 1974 fue documentada la alteracion de la excrecion biliar de cobre.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

El cobre es un elemento traza esencial.
Actua comocofactor de cuproenzimas como la ferroxidasa o ceruloplasmina citocromo-c oxidasa superoxido-dismutas, dopamina -hidroxilasa), tirosinasa. y lisil oxidasa).
Participa asi en procesos vitales como la oxidacion del hierro, respiracion celular, eliminacion de radicales libres, biosintesis de catecolaminas y melanina, asi como en la formacion de tejido conectivo.
Sin embargo, ese mismo potencial oxidoreducto le capacita en situaciones de exceso para generar radicales libres altamente toxicos para el organismo.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Presente en alimentos como semillas, crustaceos, higado y leguminosas, el cobre procedente de la dieta, una vez captado por los enterocitos duodenales, puede quedar unido a proteinas intracelulares (metalotioneinas) o bien ser exportado a la circulacion a traves de la membrana basolateral.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Unido a albumina, transcupreina e histidina, el cobre es transportado por la vena porta al higado, organo de reserva y principal regulador de su homeostasis.
Puesto que la excrecion biliar es la via fundamental para su eliminacion del organismo, no existiendo circulacion enterohepatica significativa.
El hepatocito lo capta gracias a los transportadores de membrana CTR1 y CTR2.
Debido a su potencial toxico las concentraciones de cobre libre dentro de la celula son extremadamente bajas, uniendose a una familia de proteinas llamadas chaperonas de cobre que liberan el metal especificamente a las distintas rutas que sintetizan cuproenzimas.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

El defecto metabolico responsable de la enfermedad de Wilson esta en la disfuncion de la proteina ATP7B que da lugar a la acumulacion de cobre, inicialmente en el higado.
En principio, la metalotion y el glutation del citoplasma celular juegan un papel importante protegiendo a la celula de sus efectos toxicos, pero cuando se sobrepasa su capacidad, el metal libre produce un dano oxidativo en el hepatocito
junto con activacion de la apoptosis, liberandose al torrente circulatorio lo que provoca elevacion de la concentracion serica de cobre libre.

Distribuyebdose el cobre al resto de los
tejidos donde tambien se podra acumular, aumentando asimismo, su excrecion en orina.
La sobrecarga de cobre en los hematies
conduce a hemolisis intravascular con prueba de Coombs negativa.

• Estudios familiares indicaron que la enfermedad mostraba un patron de herencia autosomico recesivo.
• Los progenitores son al menos, portadores heterocigotos de un alelo mutado y los hermanos tienen una probabilidad del 25% de padecer la enfermedad y un 50% de ser portadores heterocigotos, en tanto que el riesgo de
• enfermedad de los hijos es de 1/180.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

En 1985 fue establecida la localizacion del gen responsable ATP7B, en el brazo largo del cromosoma 13 (13q14.3).
En 1993 el gen fue identificado y clonado, demostrandose que se trataba de un locus muy conservado evolutivamente, que codificaba para una ATPasa transportadora de cobre.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Se han descrito mas de 400 mutaciones distintas en el gen ATP7B, tanto a lo largo de sus 21 exones como en supromotor y en regiones intronicas.
Asi, la mutacion mas comun en pacientes del norte, centro y este de Europa es H1069Q, que da lugar a la sustitucion de una histidina por una glutamina en la posicion 1069 de la ATPasa. Sin embargo, esta mutacion parece estar ausente en el este de Asia donde predomina R778L27.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Se han encontrado diferencias tanto en la edad de inicio como en la gravedad y tipo de presentacion.
Lo mas frecuente es que se diagnostique entre los 5 y los 35 anos.
Clinicamente puede presentarse como una enfermedad hepatica o como un trastorno neuropsiquiatrico progresivo en el que la alteracion hepatica puede ser menos aparente u ocasionalmente ausente.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111


Son las mas frecuentes entre los 5 ˜anos y la adolescencia La presentacion hepatica es la manifestacion inicial en e 40-50% de pacientes.
El espectro de enfermedad hepatica abarca desde cuadros asintomaticos con hepatomegalia, esplenomegalia o elevacion de aminotransferasas que se detectan de forma incidental, hasta casos de fallo hepatico fulminante.
La forma de presentacion mas comun es una enfermedad hepatica cronica activa en la que puede haber evidencia de cirrosis bien compensada o descompensada.
.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

En el 40-45% de pacientes con EW, los primeros sintomas son neurologicos y neuropsiquiatricos.
Suelen presentarse durante la 2.a y 3.a decadas de la vida. Estos son principalmente trastornos del movimiento como distonia, incordinacion y temblores.
Tambien pueden aparecer disartria y disfagia, trastornos del sistema nervioso autonomo,
asi como perdida de memoria, cefalalgias y convulsiones.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Alrededor del 50% de los pacientes con dano neurologico posee algun antecedente de trastornos de la conducta.
Otras manifestaciones son la depresion, ansiedad y psicosis.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

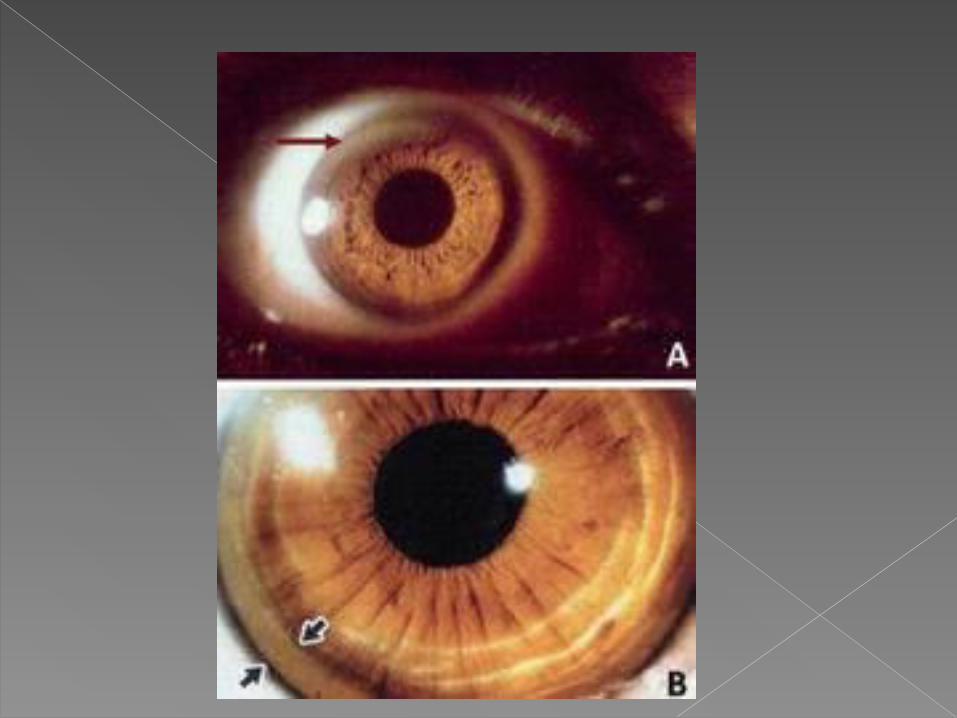
Mediante exploracion oftalmologica con lampara de hendidura se detectan anillos de Kayser-Fleischer (KF) por deposito del cobre en la cornea, en mas del 95% de enfermos con manifestaciones neurologicas o psiquiatricas pero solo en alrededor del 50% con clinica hepatica.
Tras el tratamiento suele observarse su regresion.
Tambien puede aparecer una catarata central «en girasol» por acumulacion del cobre en el cristalino.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Otras manifestaciones mas raras incluyen lesiones renales con sindrome tubular, desmineralizacion osea por la hipercalciuria y la hiperfosfaturia que provoca la disfuncion tubular, miocardiopatia, pancreatitis, hipoparatiroidismo, abortos espontaneos repetidos e infertilidad.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

La EW debe ser considerada en el diagnostico diferencialde individuos con anormalidades de la funcion hepatica y/otrastornos neurologicos inexplicables por otras causas masfrecuentes, fundamentalmente en ni˜nos o adultos jovenes
ocuando aparece hemolisis29.Puesto que ninguna prueba de forma aislada tiene
suficientesensibilidad y especificidad para el diagnostico dela EW, este requiere la utilizacion combinada de diversosdatos clinicos, bioquimicos y geneticos, siendo inicialmenteesencial la sospecha clinica de estos casos.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111



La ceruloplasmina es una glicoproteina sintetizada principalmente
en el higado, que contiene de seis a ocho sitios de fijacion de cobre por molecula. Posee actividad ferroxidasa, estando implicada en el metabolismo del hierro.
Asi su ausencia, en la rara enfermedad hereditaria de la aceruloplasminemia, no produce acumulo de cobre, mientras que
estos pacientes pueden presentar hemosiderosis.
Una concentracion extremadamente baja (< 0,05 g/L) deberia ser considerada una fuerte evidencia para el diagnostico de EW, especialmente en asociacion con la presencia de anillos de Kayser-Fleischer.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

La medicion cuantitativa de cobre hepatico sigue siendo considerada la prueba bioquimica mas importante para el diagnostico de la enfermedad, pero debido a su carácter invasivo se reserva para aquellos casos con sospecha de EW en los que otras pruebas no muestran resultados definitivos.
En individuos no tratados, valores inferiores a 0,63-0,79 mmol/kg de tejido seco (40-50 g/g), casi siempre excluyen el diagnostico de EW.
Mientras que resultados iguales o superiores a 3,9 mmol/kg de tejido seco (≥ 250g/g) son habitualmente considerados diagnosticos de EW en ausencia de otras patologias en las que tambien se observan estaselevaciones como la enfermedad hepatica colestatica o la cirrosis infantil de la India
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Valores inferiores a 3,9 mmol/kg de tejido seco (250g/g), sin embargo, no excluyen la enfermedad.
El valor de 1,2 mmol/kg de tejido seco (75 g/g) ha mostrado aumentar considerablemente la SD de la prueba (83,3% vs. 96,5%) aunque se pierde algo de ED (98,6% vs. 95,4%). Por ello, en pacientes con resultados entre 1,2 y 3,9 mmol/kg de tejido seco (75 y 250 g/g), se recomienda realizar otros estudios, fundamentalmente geneticos.
rnandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111 He

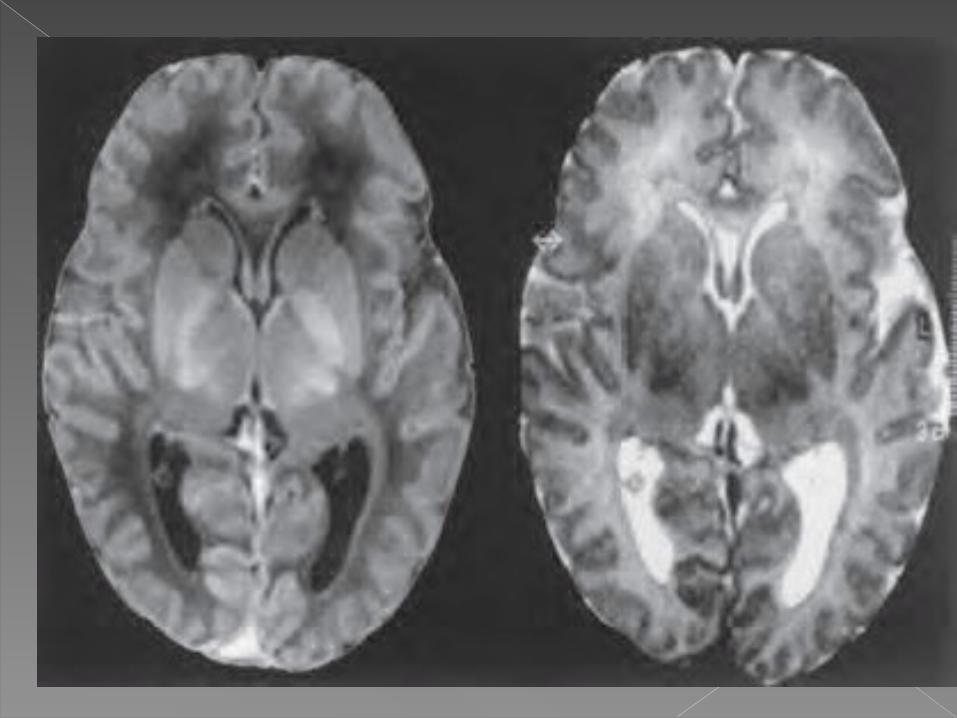
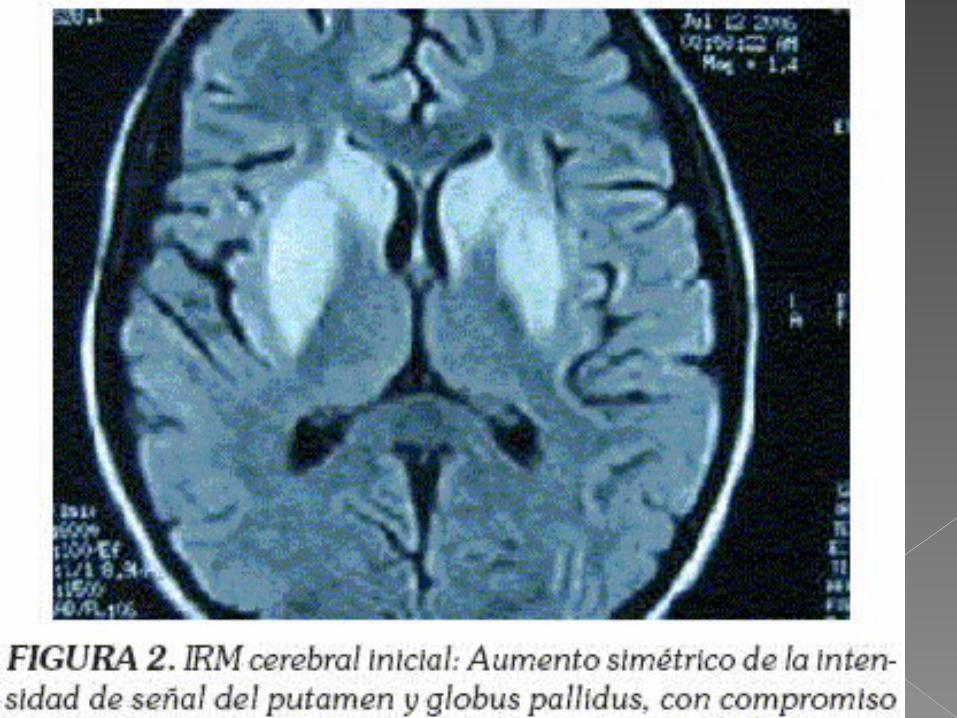
La tomografia computarizada
La resonancia magnetica revelan en presencia de enfermedad neurologica, dano en ganglios basales y en ocasiones en otras zonas del encefalo.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111
Actualmente el estudio genetico resulta todavia complicado
y caro debido al elevado numero de mutaciones.
Sin embargo, su importancia en el diagnostico es cadavez mayor fundamentalmente en ciertas poblaciones en
lasque se observa un predominio de determinadas
mutacionesespecificas, lo que permite realizar un cribado inicial de laenfermedad estudiando solamente determinadas regionesde gen ATP7B.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Quelantes de cobre que promueven la excrecion del metal por orina, o de sales de cinc que reducen su absorcionintestinal,o bien de una combinacion de ambos.
El primer tratamiento farmacologico, el dimercaptopropanol (BAL) i.m., que fue rapidamente reemplazado por la D-penicilamina (1956), otro agente quelante de cobre que, administrado via oral, induce ademas la sintesis de metalotioneina reduciendo su fraccion libre intracelular.
Actualmente esta siendo desplazado por otros quelantes con menos efectos secundarios como la Trientina y el tetratiomolibdato amonico (este todavia no
disponible).
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

También son utiles las sales de cinc que bloquean la absorcion intestinal del cobre induciendo la sintesis de metalotioneina intestinal que lo secuestra favoreciendo su eliminacion en heces.
El tratamiento debe ser iniciado lo mas precozmente posible, incluyendo a los individuos presintomaticos y mantenido durante toda la vida.
Se debe recomendar una dieta pobre en cobre y tambien se ha sugerido que la adicion de antioxidantes, principalmente vitamina E mejora la sintomatologia.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

El trasplante hepatico esta indicado en pacientes con cirrosis hepatica terminal que no responden al tratamiento con quelantes y en caso de fallo hepatico fulminante.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Enfermedad de Fahr

Ferrocalcinosis cerebrovascular o calcicosis de los núcleos del cerebro de origen idiopático, siendo una enfermedad neurológica rara, caracterizada por calcificaciones bilaterales y simétricas limitadas a los núcleos grises centrales , preferentemente en los ganglios basales, o extendidas a otras áreas cerebrales,asociada a trastornos neurológicos.
Carrillo R et al. Enfermedad de Fahr y gestaciónClin Invest Gin Obst. 2006;33(4):152-3

En honor a Karl Theodor Fahr, quien en 1930 describió la calcificación de los nucleos de la base en un paciente con demencia asociada a rigidez
Afecta por igual a ambos sexos y es más frecuente por encima de los 50 años. Su prevalencia aumenta con la edad. Su máxima frecuencia se da entre los 30 y
los 60 años de edad y se desarrolla de forma variable dentro de los miembros de una misma familia.
Prevalencia de <1/1,000,000.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Idiopatica y secundaria Se asocia frecuentemente con
hipoparatiroidismo. Otras causas relacionadas con ella son
infecciones por el virus de Epstein-Barr, virus de la inmunodeficia humana.
Lupus eritematoso sistémico. Hipoxia perinatal. Radioterapia y quimioterapia. Intoxicaciones por CO2 o plomo. Secundario al uso prolongado de
anticonvulsionantes.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Presenta un patrón hereditario que sugiere una transmisión autosómica dominante en la mayoría de las familias estudiadas.
Recientemente se ha identificado la localización primaria en el cromosoma 14q2,3.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111

Suele presentarse con una sintomatología extrapiramidal similar a la de la enfermedad de Parkinson (alteración del tono muscular, distonías, discinesias, con parálisis espástica, atetosis, crisis epilépticas.
Datos cerebelosos (ataxia) Deterioro de las funciones mentales
(cambios de personalidad y trastornos obsesivo-compulsivos).
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111


De sospecha es clínico. Su confirmación se realiza por estudios
por imagen (tomografía computarizada y resonancia magnética).
La TAC es la técnica más fiable para su detección.
La resonancia magnética, la mejor prueba para realizar el diagnóstico diferencial.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111



Es una enfermedad degenerativa para la que no existe tratamiento curativo.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111
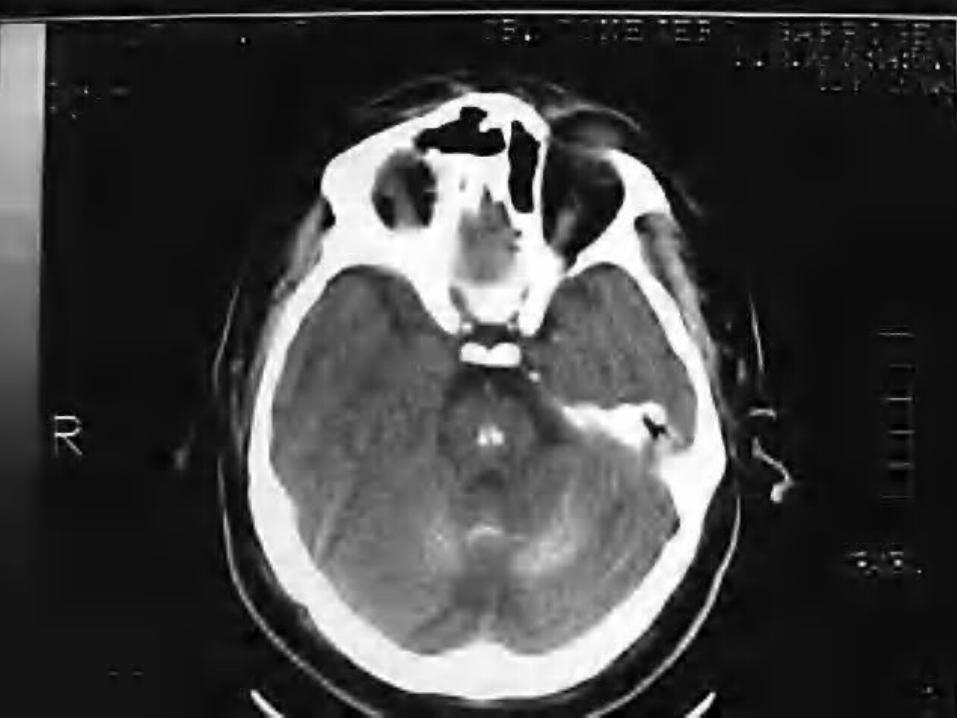
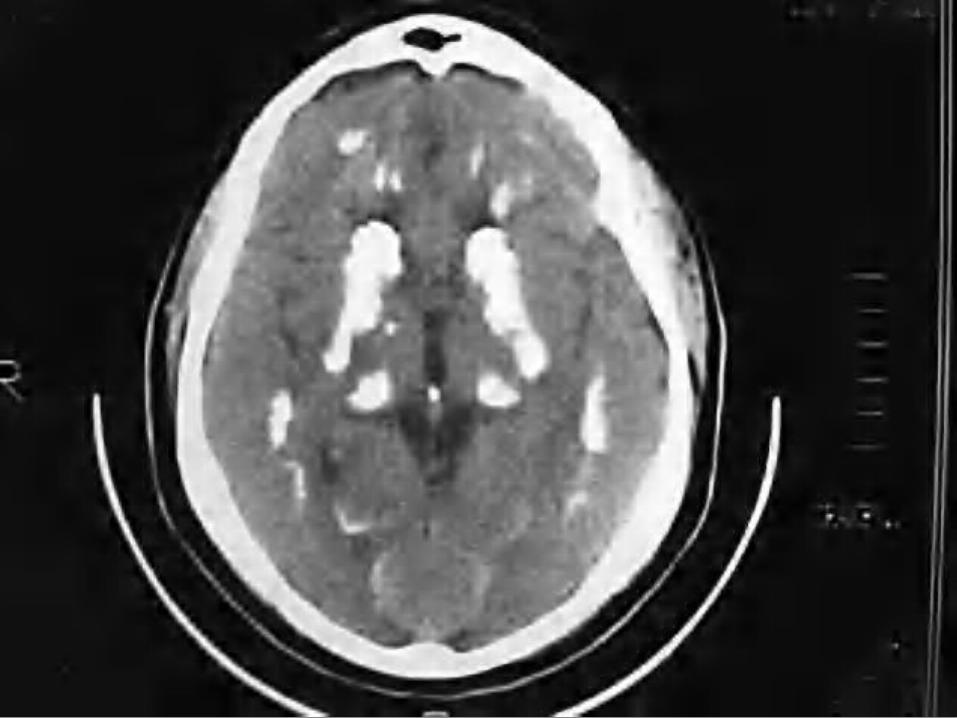
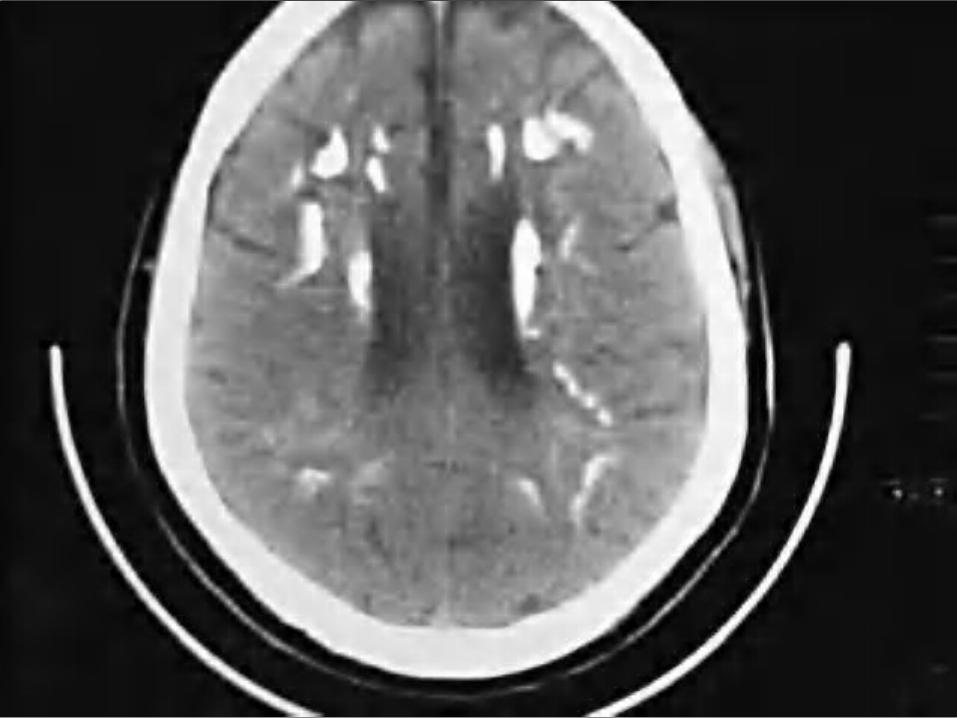

Es grave debido al deterioro neurológico progresivo, deterioro general y discapacidad progresiva, sin tratamiento específico.
Hernandez Villen, S. Lopez Martinez. Enfermedad de Wilson. Rev Lab Clin. 2011;4(2):102—111





























