Embed Size (px)
Citation preview

Human heat shock protein (Hsp) 90 interferes withNeisseria meningitidis adhesin A (NadA)-mediatedadhesion and invasion
Paolo Montanari,1† Giuseppe Bozza,1
Barbara Capecchi,1 Elena Caproni,1
Riccardo Barrile,1 Nathalie Norais,1 Mirco Capitani,2
Michele Sallese,2 Paola Cecchini,3,4‡ Laura Ciucchi,1
Zhenai Gao,5 Rino Rappuoli,1 Mariagrazia Pizza,1
Beatrice Aricò1 and Marcello Merola1,6*1Research Center, Novartis Vaccines and Diagnostics,Via Fiorentina 1, 53100 Siena, Italy.2Unit of Genomic Approaches to Membrane Traffic,Consorzio Mario Negri Sud, Via Nazionale 8/A, 66030S. Maria Imbaro (CH), Italy.3Dipartimento di Scienze Biomediche Sperimentali,Università di Padova, Via U. Bassi 58/B, I-35131Padova, Italy.4Centro di Ricerca Interdipartimentale per leBiotecnologie Innovative, Università di Padova, Via U.Bassi 58/B, I-35131 Padova, Italy.5Novartis Institute for Biomedical Research, 4560Horton St. M/S:4.5, Emeryville, CA 94608-2916, USA.6Dipartimento Biologia Strutturale e Funzionale,Università di Napoli ‘Federico II’, Via Cinthia 21, 80126Napoli, Italy.
Summary
NadA (Neisseria meningitidis adhesin A), a menin-gococcal surface protein, mediates adhesion toand invasion of human cells, an activity in whichhost membrane proteins have been implicated.While investigating these host factors in humanepithelial cells by affinity chromatography, we dis-covered an unanticipated interaction of NadA withheat shock protein (Hsp) 90, a molecular chaper-one. The specific in vitro interaction of recombi-nant soluble NadA and Hsp90 was confirmed byco-immunoprecipitations, dot and far-Westernblot. Intriguingly, ADP, but not ATP, was required
for this association, and the Hsp90 inhibitor17-AAG promoted complex formation. Hsp90binding to an Escherichia coli strain used ascarrier to express surface exposed NadA con-firmed these results in live bacteria. We alsoexamined RNA interference, plasmid-driven over-expression, addition of exogenous rHsp90 and17-AAG inhibition in human epithelial cells tofurther elucidate the involvement of Hsp90 inNadA-mediated adhesion and invasion. Together,these data suggest an inverse correlation betweenthe amount of host Hsp90 and the NadA adhesive/invasive phenotype. Confocal microscopy alsodemonstrated that meningococci interact with cel-lular Hsp90, a completely novel finding. Altogetherour results show that variation of host Hsp90expression or activity interferes with adhesive andinvasive events driven by NadA.
Introduction
Neisseria meningitidis causes mortality worldwide due tosepticaemia and meningitis. Host–bacterial interaction forthis organism begins with epithelial colonization, followedby invasion, intracellular persistence and transcytosis.Although some meningococcal vaccines exist againstcommon pathogenic serogroups A, C, W-135 and Y, thesearch for a broadly protective vaccine against serogroupB required new means of antigen identification thatresulted in the discovery of surface-expressed proteins,including potential new virulence factors. The character-ization of these proteins is hoped to provide a betterunderstanding of meningococcal pathogenesis (Pizzaet al., 2000; Giuliani et al., 2006).
One such protein is NadA (Neisseria meningitidisadhesin A), a phase-variable meningococcal surface-exposed protein, present in three of the four known hyper-virulent serogroup B lineages (Comanducci et al., 2002;2004; Metruccio et al., 2009). NadA was included as amajor antigen in the multicomponent vaccine 4CMenB(Giuliani et al., 2006; Bambini et al., 2009). NadA belongsto the ‘Oca’ (Oligomeric coiled-coil adhesin) family, a sub-group of the trimeric autotransporter adhesins (Surana
Received 15 April, 2011; revised 14 October, 2011; accepted 20October, 2011. *For correspondence. E-mail [email protected]; Tel. (+39) 0577 243864; Fax (+39) 0577 243564.Present addresses: †Max Planck Institute for Infection Biology,Charitéplatz 1, D-10117 Berlin, Germany; ‡Babraham ResearchCampus, Babraham, Cambridge CB22 3AT, UK.
Cellular Microbiology (2012) 14(3), 368–385 doi:10.1111/j.1462-5822.2011.01722.xFirst published online 8 December 2011
© 2011 Blackwell Publishing Ltd
cellular microbiology

et al., 2004; Cotter et al., 2006; Linke et al., 2006). Gen-erally, Oca proteins mediate bacterial interaction withhost cells or extracellular matrix (ECM) proteins orinduce invasion into target cells (Yang and Isberg,1993; McMichael et al., 1998; Eitel and Dersch, 2002;Laarmann et al., 2002; Ray et al., 2002; Roggenkampet al., 2003; Li et al., 2004; Riess et al., 2004; Zhanget al., 2004; Girard and Mourez, 2006; Heise and Dersch,2006; Scarselli et al., 2006; Serruto et al., 2009). Likeother Oca proteins, such as YadA of Yersinia spp. (Bliskaet al., 1993; Iriarte and Cornelis, 1996; El Tahir andSkurnik, 2001), UspAs proteins of Moraxella catarrhalis(Lafontaine et al., 2000; Hill and Virji, 2003), Vomp pro-teins of Bartonella quintana (Zhang et al., 2004), BadA ofB. henselae (Riess et al., 2004) and HadA of Haemophi-lus influenzae biogroup aegyptius (Serruto et al., 2009),NadA has a conserved C-terminal membrane anchorthrough which the protein is translocated to the cellsurface, a central alpha helical domain (stalk) with highpropensity to form coiled-coil structures, and anN-terminal globular head that has been associated withbinding to specific cellular receptors.
NadA forms stable trimers on the bacterial surface andthereby contributes to mediate N. meningitidis adhesionto and invasion of epithelial cells. A trimeric protein andproperly folded N-terminal domain are necessary toNadA-cell binding to human cells (Capecchi et al., 2005;Tavano et al., 2011). A protein receptor molecule, whichis differentially expressed by various human epithelialcell lines, appears to mediate the binding of the trimericNadA (Capecchi et al., 2005). The expression of full-length NadA on the surface of Escherichia coli andthe purification of soluble recombinant NadAD351–405
(rNadA) have been described (Comanducci et al., 2002;Capecchi et al., 2005). This soluble rNadA was includedin 4CMenB (Giuliani et al., 2006) and was shown toinduce high levels of bactericidal antibodies in variousmodels (Comanducci et al., 2002; Bowe et al., 2004;Ciabattini et al., 2008). Further, rNadA activates humanmonocyte-derived dendritic cells and monocytes/macrophages (Mazzon et al., 2007; Franzoso et al.,2008) and is recognized by convalescent serum fromchildren (Litt et al., 2004). These studies suggest thatrNadA retains the functional features of native NadA,which is expressed and immunogenic in vivo.
As N. meningitidis has diverse, and likely redundant,virulence factors (Virji, 2009) identifying the precise role ofNadA is difficult; however, E. coli expressing NadA helpsisolate its specific contribution. Surface-exposed NadAbehaves similarly in E. coli and meningococci, firstforming a stable trimer on the bacterial surface and thenmediating adhesion to and invasion of host cells. The hostcell mechanisms behind these activities remain some-what poorly understood. To explore meningococcal infec-
tion on the molecular level, we used transformed E. coliexpressing surface NadA, rNadA and unencapsulatedN. meningitidis strains, to investigate the interaction ofNadA with epithelial cells. We discovered a novel interac-tion between NadA and human Hsp90 that was promotedby ADP and the specific Hsp90 inhibitor 17-AAG (17-N-allylamino-17-demethoxygeldanamycin). Unexpectedly,bacterial adhesion to and invasion of a human epithelialcell line were inversely correlated with the expression ofHsp90 by host cells.
Hsp90 cycles between an apo conformation, in theabsence of associated nucleotides, and two conforma-tionally distinct types that are associated with ADP or ATP(Mayer, 2010). Hsp90 primarily forms complexes in thepresence of ATP (Hutchison et al., 1993; Stancato et al.,1993; Dittmar et al., 1997; Pearl and Prodromou, 2006);thus, inhibitors of ATPase activity, like 17-AAG, a geldana-mycin derivative, cause dissociation and early ubiquitina-tion of client proteins (Stebbins et al., 1997; Jez et al.,2003; Blagg and Kerr, 2006; Powers and Workman,2006). However, ADP-dependent Hsp90 interaction withCHORDC1 has been identified and interactions withsome Hsp90-client proteins that constitute core transcrip-tion machinery have been reinforced by geldanamycin(Gano and Simon, 2010). In fact, Hsp90, compared withother chaperones, has less promiscuous interactions(Picard, 2002; Whitesell and Lindquist, 2005). Extracellu-lar Hsp90 has been described and found to be correlatedwith tumour progression (Eustace and Jay, 2004; Tsut-sumi and Neckers, 2007; Song et al., 2010). Hsp90 canalso function as a receptor component of known bacterialvirulence factors (Jin et al., 2003; Cabanes et al., 2005;Reyes-Del Valle et al., 2005; Rechner et al., 2007; Naet al., 2008). Our finding that cellular Hsp90 interfereswith NadA-mediated adhesion and invasion suggests apreviously unrecognized role for this chaperone duringinfection.
Results
Identification of heat shock protein 90 as aNadA-interacting protein
We performed a NadA affinity column on a membrane-enriched protein fraction from Chang epithelial cells toidentify eukaryotic cell proteins binding to the adhesin. AnrNadA variant with the membrane-anchor region removedwas covalently bound to a CNBr-activated sepharoseresin and used as stationary phase for affinity chromatog-raphy. The sample was adsorbed to the column, part ofthe eluted bound material was separated by SDS-PAGEand blotted. The remaining portion was TCA precipitatedand separated in parallel by SDS-PAGE followed by Coo-massie blue staining.
Hsp90 hampers NadA-mediated infectivity 369
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

Far-Western blot, using rNadA as probe, revealed aspecific band with an apparent molecular weightof ~ 97 kDa in the sample incubated with rNadA(Fig. 1A, left gel) but not the control (Fig. 1A, right gel).The corresponding Coomassie-stained bands wereexcised from the gel then underwent peptide massfingerprint identification using MALDI-TOF TOF massspectrometry analysis (Fig. 1B), which revealed exactmatches with Hsp 90, Alpha (a) and Beta (b) isoforms(Fig. 1C).
Hsp90 binds to NadA in vitro
We confirmed the above results using rNadA and twosources of recombinant Hsp90 (rHsp90): a commerciallyavailable (Stressgen, ADI-SPP-776) product and ananalogous His-tagged product expressed and purified inour laboratory. Results of far-Western and dot blot experi-ments were identical, independent of the source ofrHsp90.
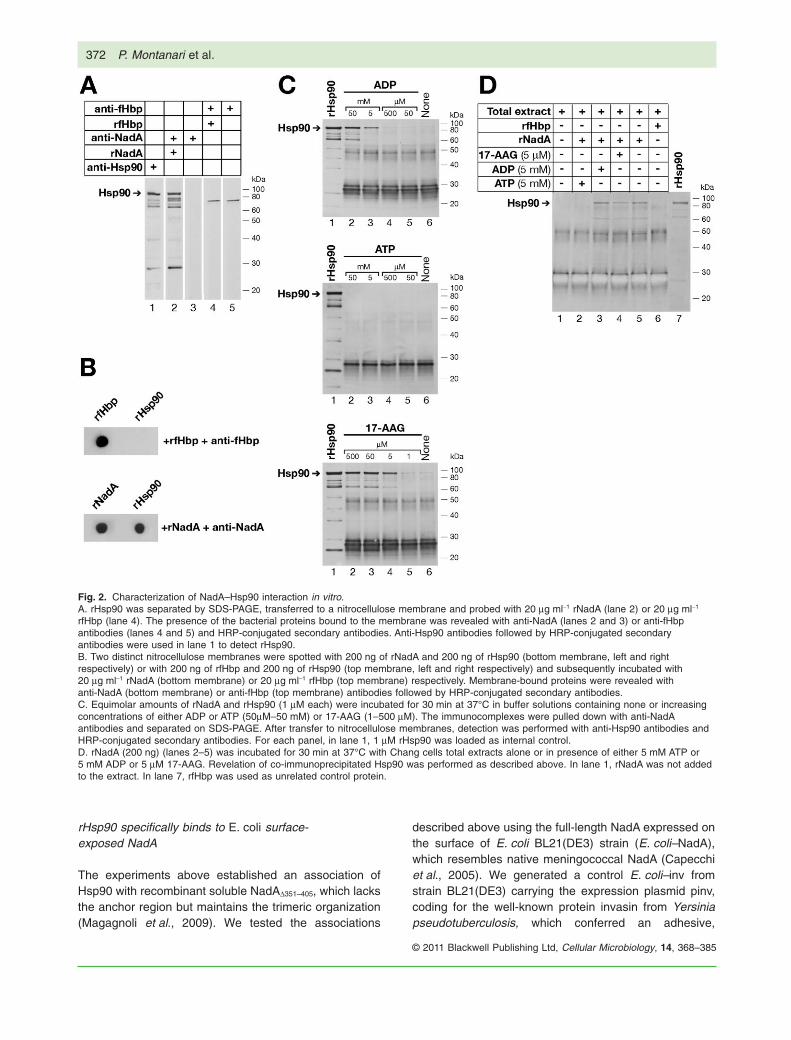
Specific interactions were investigated with SDS-PAGEseparated and blotted rHsp90 and soluble rNadA asprobe (Fig. 2A). Recombinant factor H binding protein(rfHbp), an unrelated meningococcal surface protein(Masignani et al., 2003) was used as an internal control.Binding of bacterial proteins to immobilized rHsp90 wasrevealed by anti-NadA and anti-fHbp antibodies. Furthercontrols included the incubation of membrane blottedrHsp90 with anti-NadA and anti-fHbp antibodies in theabsence of rNadA and rfHbp (Fig. 2A, lanes 3 and 5respectively). The position of rHsp90 on the membranewas assessed by anti-Hsp90 antibodies (Fig. 2A, lane 1).Immobilized rHsp90 was specifically recognized by rNadA(Fig. 2A, lane 2) and not by rfHbp (Fig. 2A, lane 4) or theantibodies against rNadA and rfHbp (Fig. 2A, lanes 3 and5 respectively).
The potential for binding of rNadA was further tested onmembrane-immobilized rHsp90 in a dot blot experiment.As shown in Fig. 2B, a nitrocellulose membrane spottedwith rHsp90 and probed with rNadA (Fig. 2B, bottom right)or rfHbp (Fig. 2B, top right) showed, after extensive wash-ings, the presence of specifically retained proteins withanti-NadA antibodies (Fig. 2B, bottom right) or anti-fHbpantibodies (Fig. 2B, top right). Only rNadA bound to immo-bilized rHsp90. On the left side of the blots, rNadA andrfHbp were spotted on the membrane as controls foranti-NadA (Fig. 2B, bottom left) and anti-fHbp (Fig. 2B,top left) antibodies.
Specific binding between rNadA and rHsp90 wastested in a series of co-immunoprecipitation experimentsusing rNadA and both rHsp90 and cellular extracts assources of Hsp90. The specific recognition of client pro-teins by Hsp90 is strongly regulated by its ATPase activ-ity, and ATP/ADP binding and co-chaperone association
drive structural changes that coordinate its association-release cycle (Pearl and Prodromou, 2006). Inhibition ofHsp90 ATPase activity causes dissociation and earlyubiquitination of the client proteins (Blagg and Kerr,2006); 17-AAG, a known Hsp90 inhibitor, specificallyaffects the ATP binding pocket (Stebbins et al., 1997;Jez et al., 2003; Powers and Workman, 2006). Thus, weanalysed the effect of ATP, ADP and 17-AAG, over arange of concentrations, on the association of NadAand Hsp90 in parallel with the co-immunoprecipitationexperiments.
Following incubation of the two recombinant proteins(see Experimental procedures below), recovered immu-nocomplexes were subjected to Western blot analysisusing anti-Hsp90 antibodies with rfHbp as control(Fig. 2C). The presence of ADP at a 5 mM concentrationwas a crucial step for the formation of NadA–Hsp90complex in solution (Fig. 2C, top membrane, lane 3). Withsub-optimal concentrations (Fig. 2C, top membrane,lanes 4 and 5) or without ADP (Fig. 2C, top membrane,lane 6) no association was detected; no association wasdetected between rNadA and rHsp90 in presence of ATPat any concentration (Fig. 2C, middle membrane). Addi-tion of 17-AAG 5 mM or more allowed formation of theNadA–Hsp90 complex (Fig. 2C, bottom membrane), indi-cating that 17-AAG stabilizes Hsp90 in a conformationsuitable for NadA binding (Grenert et al., 1997; Zhanget al., 2009). No association of fHbp with Hsp90 wasobserved (data not shown).
To verify these findings using Chang cell lysates as asource of Hsp90, Chang cell total extracts were incu-bated with rNadA, and anti-NadA antibodies were usedto pull down immunocomplexes. Western blot analysisrevealed that the association of exogenous rNadA withendogenous Hsp90 was detectable in the presence ofADP or 17-AAG at the lowest concentrations establishedimmediately above (Fig. 2D, lanes 3 and 4 respectively)as well as in the absence of any exogenous compound(Fig. 2D, lane 5). Of note, NadA co-immunoprecipitatedHsp90 in the absence of any added nucleotide or17-AAG (Fig. 2D, lane 5), suggesting that a pool of thischaperone was potentially in an ADP-like conformation.The addition of ADP or 17-AAG did not increase theamount of the complexes detected (Fig. 2D, comparelane 5 with lanes 3 and 4) but ATP reduced Hsp90co-immunoprecipitation to the background level (Fig. 2D,lane 2). rfHbp, shown in absence of exogenously addedcompounds, did not associate with Hsp90 (Fig. 2D, lane6) in this or any other experimental condition studied(data not shown). Thus, we showed a direct specificinteraction between the cellular chaperone Hsp90 andthe bacterial adhesin NadA and found that an ADP or17-AAG associated conformation of Hsp90 was requiredfor this interaction in vitro.
370 P. Montanari et al.
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

Fig. 1. rNadA binds to a 90 kDa protein from Chang membrane extracts.A. A protein membrane extract from Chang epithelial cells was loaded on a rNadA-sepharose affinity column and bound species were elutedwith 100 mM NaCl. Three fractions were collected and analysed (1–3). An aliquot of each fraction was loaded in duplicate (left membrane andright membrane) on SDS-PAGE and transferred to a nitrocellulose membrane. To reveal NadA interacting proteins, the membrane wasoverlaid with 20 mg ml-1 of rNadA, revealed with anti-NadA antibodies and HRP-conjugated secondary antibodies (left membrane). The controlmembrane was treated with anti-NadA antibodies and HRP-conjugated secondary antibodies (right membrane).B. The remaining material of the NaCl eluted fractions was TCA precipitated and loaded on SDS-PAGE. The gel was stained with Coomassieblue and the protein band corresponding to the Western blot positive signal was analysed by peptide mass fingerprint. Only m/z with asignal/noise ratio above 10 were annotated with the m/z-value and the amino acid position of the corresponding peptide (m/z labelled in blackare common to the sequence of Hsp90a and Hsp90b, m/z labelled in red are specific to Hsp90a, and m/z labelled in blue are specific forHsp90b). The asterisks ‘*’ correspond to identified but not annotated signals for Hsp90a (red) and Hsp90b (blue) or both Hsp90a and Hsp90b(black).C. Sequence coverage of the Hsp90a and Hsp90b obtained from the peptide mass fingerprint.
Hsp90 hampers NadA-mediated infectivity 371
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385
rHsp90 specifically binds to E. coli surface-exposed NadA
The experiments above established an association ofHsp90 with recombinant soluble NadAD351–405, which lacksthe anchor region but maintains the trimeric organization(Magagnoli et al., 2009). We tested the associations
described above using the full-length NadA expressed onthe surface of E. coli BL21(DE3) strain (E. coli–NadA),which resembles native meningococcal NadA (Capecchiet al., 2005). We generated a control E. coli–inv fromstrain BL21(DE3) carrying the expression plasmid pinv,coding for the well-known protein invasin from Yersiniapseudotuberculosis, which conferred an adhesive,
Fig. 2. Characterization of NadA–Hsp90 interaction in vitro.A. rHsp90 was separated by SDS-PAGE, transferred to a nitrocellulose membrane and probed with 20 mg ml-1 rNadA (lane 2) or 20 mg ml-1
rfHbp (lane 4). The presence of the bacterial proteins bound to the membrane was revealed with anti-NadA (lanes 2 and 3) or anti-fHbpantibodies (lanes 4 and 5) and HRP-conjugated secondary antibodies. Anti-Hsp90 antibodies followed by HRP-conjugated secondaryantibodies were used in lane 1 to detect rHsp90.B. Two distinct nitrocellulose membranes were spotted with 200 ng of rNadA and 200 ng of rHsp90 (bottom membrane, left and rightrespectively) or with 200 ng of rfHbp and 200 ng of rHsp90 (top membrane, left and right respectively) and subsequently incubated with20 mg ml-1 rNadA (bottom membrane) or 20 mg ml-1 rfHbp (top membrane) respectively. Membrane-bound proteins were revealed withanti-NadA (bottom membrane) or anti-fHbp (top membrane) antibodies followed by HRP-conjugated secondary antibodies.C. Equimolar amounts of rNadA and rHsp90 (1 mM each) were incubated for 30 min at 37°C in buffer solutions containing none or increasingconcentrations of either ADP or ATP (50mM–50 mM) or 17-AAG (1–500 mM). The immunocomplexes were pulled down with anti-NadAantibodies and separated on SDS-PAGE. After transfer to nitrocellulose membranes, detection was performed with anti-Hsp90 antibodies andHRP-conjugated secondary antibodies. For each panel, in lane 1, 1 mM rHsp90 was loaded as internal control.D. rNadA (200 ng) (lanes 2–5) was incubated for 30 min at 37°C with Chang cells total extracts alone or in presence of either 5 mM ATP or5 mM ADP or 5 mM 17-AAG. Revelation of co-immunoprecipitated Hsp90 was performed as described above. In lane 1, rNadA was not addedto the extract. In lane 7, rfHbp was used as unrelated control protein.
372 P. Montanari et al.
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

invasive phenotype (Isberg and Falkow, 1985; Monackand Theriot, 2001).
We obtained outer membrane protein (OMP) prepara-tions from E. coli–NadA (OMP–NadA) and E. coli–inv(OMP–inv) and checked their binding properties forrHsp90 by dot blot. The nitrocellulose membranes inFig. 3A were spotted with OMP–inv (left membrane, leftside) and rHsp90 (left membrane, right side) or OMP–NadA (right membrane, left side) and rHsp90 (right mem-brane, right side), then probed by overlaying OMP–inv(left membrane) or OMP–NadA (right membrane). Spe-cific binding to immobilized rHsp90 was detected by anti-E. coli antibodies. The NadA in OMP–NadA was retainedby the immobilized rHsp90 (Fig. 3A, right membrane, rightside), which did not bind with any protein in the OMP–inv(left membrane, right side). In another experiment usingOMP obtained from E. coli–pET, a BL21(DE3) carryingthe pET expression vector alone, no binding betweenrHsp90 and OMPs was detected (data not shown). Thisfinding is consistent with a specific interaction betweenthe E. coli membrane-associated NadA, as present in theOMP preparation and closer resembling the meningococ-cal native protein, and rHsp90.
We then performed immunofluorescence analysis toinvestigate whether recombinant Hsp90 could binddirectly to the surface of E. coli carrying surface exposedmembrane-anchored NadA. E. coli–NadA and E. coli–invwere pre-incubated with rHsp90 in PBS buffer for 30 minat 37°C alone or with: ADP (5 mM), ATP (5 mM) or17-AAG (5 mM). Following extensive washings, sampleswere stained with anti-Hsp90 and revealed with second-ary antibodies Alexa Fluor 488-conjugated. The bacterialchromosome was stained with DAPI (4′,6-diamidino-2-phenylindole). Immunofluorescence microscopy analysisrevealed a significant level of fluorescence, consistentwith binding, associated with E. coli–NadA when incu-bated with rHsp90 in the presence of ADP and 17-AAG(Fig. 3B, second and fourth panel from the top), but not inthe presence of ATP or alone (Fig. 3B, first and third panelfrom the top). Binding with rHsp90 was specific to E. coliexpressing surface NadA; no rHsp90 associated withE. coli–inv in any condition tested (see Fig. 3B for incu-bation with ADP). These results indicate that directbinding of Hsp90 to NadA occurs with the membraneanchored protein on the bacterial surface and the speci-ficity of such an interaction relies on ADP or 17-AAG.
Low levels of Hsp90 strengthen the adhesion andincrease the entry of E. coli–NadA in Changepithelial cells
Although it is normally a cytosolic chaperone, Hsp90 hasbeen reported extracellularly (Tsutsumi and Neckers,2007), and it can function as a receptor component of
bacterial virulence factors (Jin et al., 2003; Cabaneset al., 2005; Reyes-Del Valle et al., 2005; Rechner et al.,2007; Na et al., 2008). To investigate the biological sig-nificance of the results presented above and to explorethe functional implications of NadA–Hsp90, we investi-gated the possibility that Hsp90 acts as a NadA receptorusing E. coli expressing NadA and Chang cells in whichHsp90 was efficiently reduced by RNA silencing. Changcells were transfected either with Hsp90-targeted siRNAor control siRNA (scrambled); Hsp90 reduction at 48 hwas checked by Western blot analysis of total cell lysates(Fig. 4A). We infected the Hsp90-depleted cells withE. coli–NadA and E. coli–inv to determine the rate ofadhesion and invasion mediated by NadA and to comparethe effect of decreased Hsp90 levels on a control invasin.Standard adhesion and invasion values were calculatedby infecting control cells with E. coli–NadA or E. coli–inv.Adhesion and invasion levels in all tested conditions areshown in Fig. 4B. Chang cells, transfected with siRNA forHsp90 were more susceptible to adhesion (1.8-foldincrease) and invasion (3.8-fold increase) by E. coli–NadA compared with the control cells transfected withscrambled siRNA. E. coli–inv adhesive and invasive phe-notypes were not influenced by Hsp90 silencing. Theseresults suggest that Hsp90 is unlikely to play a receptor-like role in the NadA-mediated adhesion and invasionprocess, but that total amount of Hsp90 seems to be animportant parameter specifically influencing NadA-mediated infectivity.
High levels of Hsp90 impairs the infection propertiesmediated by NadA
To assess the consequences of E. coli–NadA infection inChang epithelial cells under conditions of increased quan-tity of the chaperone, we transfected Chang cells with apQE-TriSystem vector containing hsp90a cDNA (His-tagged) to overexpress Hsp90 and tested this expressionlevel by Western blot of total cell lysates (Fig. 5A), com-pared with control cells transfected with a pQE-TriSystemempty vector. Hsp90 overexpressing cells and controlcells were infected with E. coli–NadA or E. coli–inv; adhe-sion or invasion was quantified respectively. The Hsp90overexpressing cells showed a 30% reduction in E. coli–NadA adhesion and a 50% reduction in invasion com-pared with control cells (Fig. 5B). No effects on E. coli–invinteractions were observed in Hsp90 overexpressingcells.
Following detection of direct Hsp90 binding to E. coli–NadA, the effect of exogenously added rHsp90 on NadA-mediated adhesion and invasion was investigated.E. coli–NadA and E. coli–inv were pre-incubated withrHsp90 then added to Chang epithelial cells for adhesionand invasion assays. The same concentration of
Hsp90 hampers NadA-mediated infectivity 373
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385
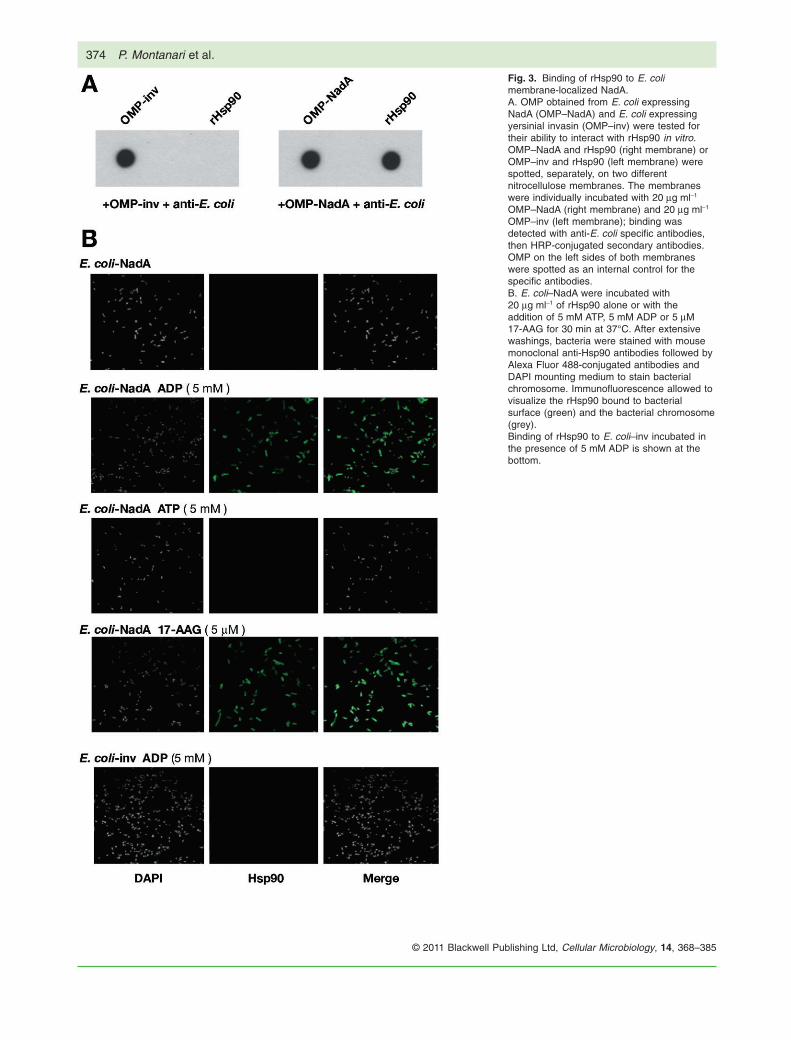
Fig. 3. Binding of rHsp90 to E. colimembrane-localized NadA.A. OMP obtained from E. coli expressingNadA (OMP–NadA) and E. coli expressingyersinial invasin (OMP–inv) were tested fortheir ability to interact with rHsp90 in vitro.OMP–NadA and rHsp90 (right membrane) orOMP–inv and rHsp90 (left membrane) werespotted, separately, on two differentnitrocellulose membranes. The membraneswere individually incubated with 20 mg ml-1
OMP–NadA (right membrane) and 20 mg ml-1
OMP–inv (left membrane); binding wasdetected with anti-E. coli specific antibodies,then HRP-conjugated secondary antibodies.OMP on the left sides of both membraneswere spotted as an internal control for thespecific antibodies.B. E. coli–NadA were incubated with20 mg ml-1 of rHsp90 alone or with theaddition of 5 mM ATP, 5 mM ADP or 5 mM17-AAG for 30 min at 37°C. After extensivewashings, bacteria were stained with mousemonoclonal anti-Hsp90 antibodies followed byAlexa Fluor 488-conjugated antibodies andDAPI mounting medium to stain bacterialchromosome. Immunofluorescence allowed tovisualize the rHsp90 bound to bacterialsurface (green) and the bacterial chromosome(grey).Binding of rHsp90 to E. coli–inv incubated inthe presence of 5 mM ADP is shown at thebottom.
374 P. Montanari et al.
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385
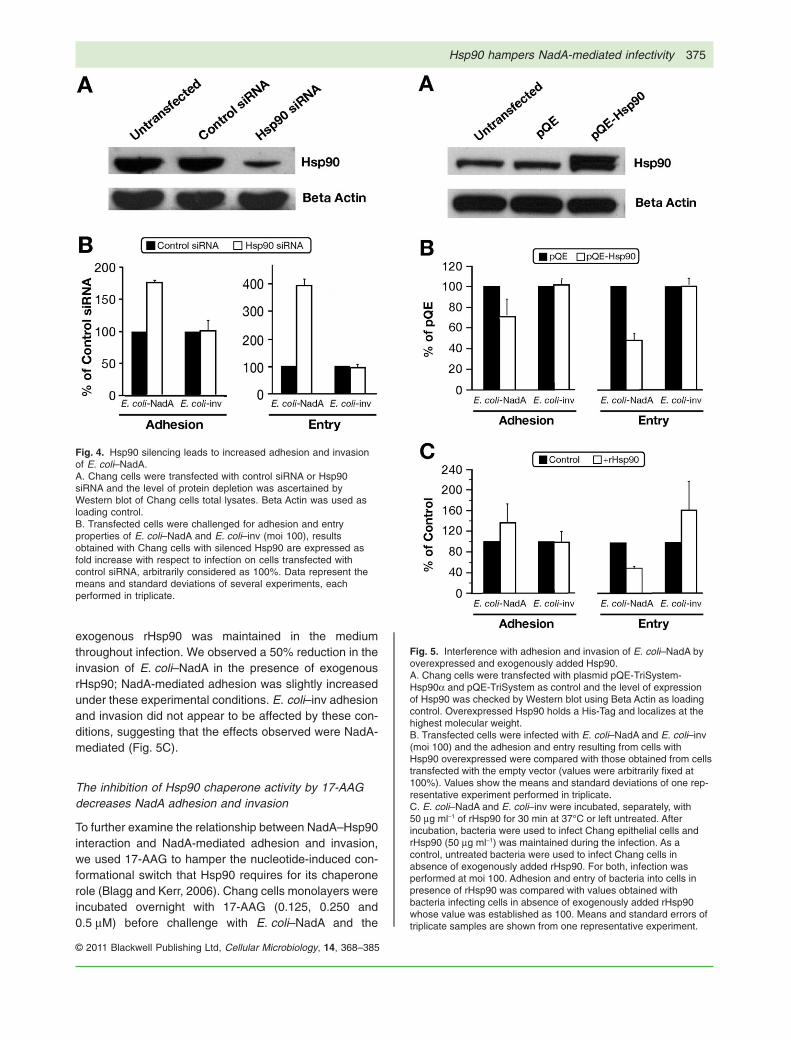
exogenous rHsp90 was maintained in the mediumthroughout infection. We observed a 50% reduction in theinvasion of E. coli–NadA in the presence of exogenousrHsp90; NadA-mediated adhesion was slightly increasedunder these experimental conditions. E. coli–inv adhesionand invasion did not appear to be affected by these con-ditions, suggesting that the effects observed were NadA-mediated (Fig. 5C).
The inhibition of Hsp90 chaperone activity by 17-AAGdecreases NadA adhesion and invasion
To further examine the relationship between NadA–Hsp90interaction and NadA-mediated adhesion and invasion,we used 17-AAG to hamper the nucleotide-induced con-formational switch that Hsp90 requires for its chaperonerole (Blagg and Kerr, 2006). Chang cells monolayers wereincubated overnight with 17-AAG (0.125, 0.250 and0.5 mM) before challenge with E. coli–NadA and the
Fig. 4. Hsp90 silencing leads to increased adhesion and invasionof E. coli–NadA.A. Chang cells were transfected with control siRNA or Hsp90siRNA and the level of protein depletion was ascertained byWestern blot of Chang cells total lysates. Beta Actin was used asloading control.B. Transfected cells were challenged for adhesion and entryproperties of E. coli–NadA and E. coli–inv (moi 100), resultsobtained with Chang cells with silenced Hsp90 are expressed asfold increase with respect to infection on cells transfected withcontrol siRNA, arbitrarily considered as 100%. Data represent themeans and standard deviations of several experiments, eachperformed in triplicate.
Fig. 5. Interference with adhesion and invasion of E. coli–NadA byoverexpressed and exogenously added Hsp90.A. Chang cells were transfected with plasmid pQE-TriSystem-Hsp90a and pQE-TriSystem as control and the level of expressionof Hsp90 was checked by Western blot using Beta Actin as loadingcontrol. Overexpressed Hsp90 holds a His-Tag and localizes at thehighest molecular weight.B. Transfected cells were infected with E. coli–NadA and E. coli–inv(moi 100) and the adhesion and entry resulting from cells withHsp90 overexpressed were compared with those obtained from cellstransfected with the empty vector (values were arbitrarily fixed at100%). Values show the means and standard deviations of one rep-resentative experiment performed in triplicate.C. E. coli–NadA and E. coli–inv were incubated, separately, with50 mg ml-1 of rHsp90 for 30 min at 37°C or left untreated. Afterincubation, bacteria were used to infect Chang epithelial cells andrHsp90 (50 mg ml-1) was maintained during the infection. As acontrol, untreated bacteria were used to infect Chang cells inabsence of exogenously added rHsp90. For both, infection wasperformed at moi 100. Adhesion and entry of bacteria into cells inpresence of rHsp90 was compared with values obtained withbacteria infecting cells in absence of exogenously added rHsp90whose value was established as 100. Means and standard errors oftriplicate samples are shown from one representative experiment.
Hsp90 hampers NadA-mediated infectivity 375
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385
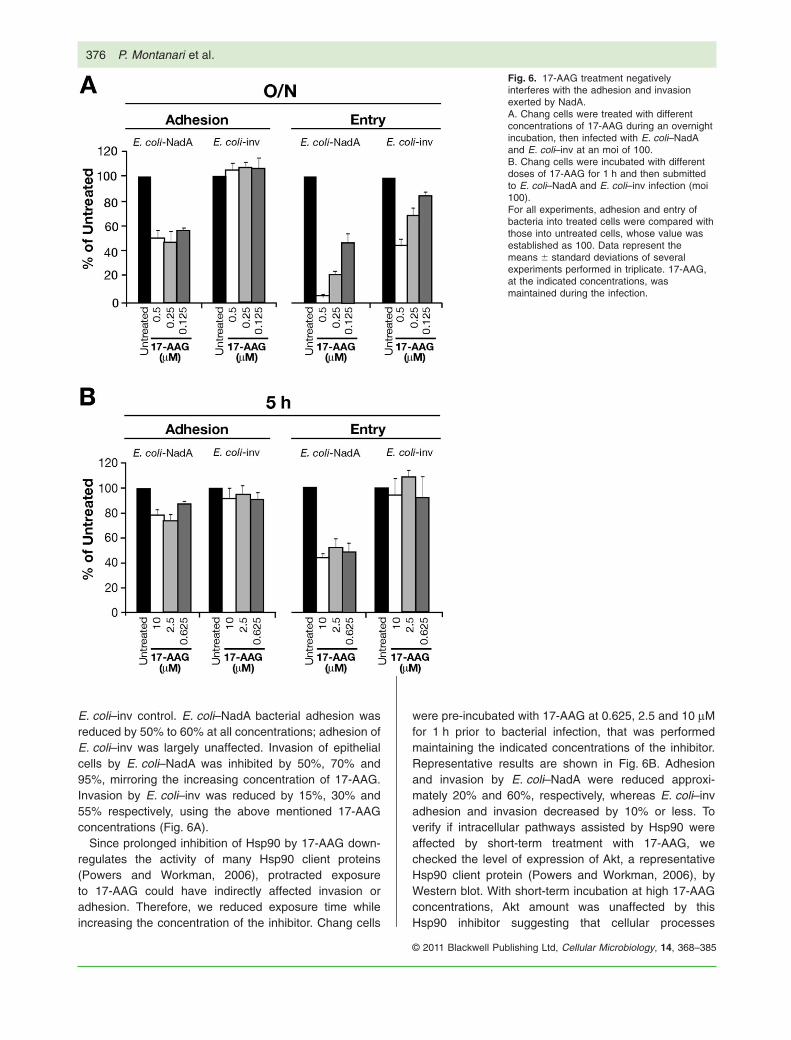
E. coli–inv control. E. coli–NadA bacterial adhesion wasreduced by 50% to 60% at all concentrations; adhesion ofE. coli–inv was largely unaffected. Invasion of epithelialcells by E. coli–NadA was inhibited by 50%, 70% and95%, mirroring the increasing concentration of 17-AAG.Invasion by E. coli–inv was reduced by 15%, 30% and55% respectively, using the above mentioned 17-AAGconcentrations (Fig. 6A).
Since prolonged inhibition of Hsp90 by 17-AAG down-regulates the activity of many Hsp90 client proteins(Powers and Workman, 2006), protracted exposureto 17-AAG could have indirectly affected invasion oradhesion. Therefore, we reduced exposure time whileincreasing the concentration of the inhibitor. Chang cells
were pre-incubated with 17-AAG at 0.625, 2.5 and 10 mMfor 1 h prior to bacterial infection, that was performedmaintaining the indicated concentrations of the inhibitor.Representative results are shown in Fig. 6B. Adhesionand invasion by E. coli–NadA were reduced approxi-mately 20% and 60%, respectively, whereas E. coli–invadhesion and invasion decreased by 10% or less. Toverify if intracellular pathways assisted by Hsp90 wereaffected by short-term treatment with 17-AAG, wechecked the level of expression of Akt, a representativeHsp90 client protein (Powers and Workman, 2006), byWestern blot. With short-term incubation at high 17-AAGconcentrations, Akt amount was unaffected by thisHsp90 inhibitor suggesting that cellular processes
Fig. 6. 17-AAG treatment negativelyinterferes with the adhesion and invasionexerted by NadA.A. Chang cells were treated with differentconcentrations of 17-AAG during an overnightincubation, then infected with E. coli–NadAand E. coli–inv at an moi of 100.B. Chang cells were incubated with differentdoses of 17-AAG for 1 h and then submittedto E. coli–NadA and E. coli–inv infection (moi100).For all experiments, adhesion and entry ofbacteria into treated cells were compared withthose into untreated cells, whose value wasestablished as 100. Data represent themeans � standard deviations of severalexperiments performed in triplicate. 17-AAG,at the indicated concentrations, wasmaintained during the infection.
376 P. Montanari et al.
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

indirectly assisted by this chaperone were not hampered(data not shown).
While our results suggest that the first line of Hsp90interference with NadA-mediated adhesion and invasionis independent of any action on client proteins, this con-clusion would not rule out a requirement for indirectmechanisms, which could be shared by other pathogensand could confirm the reduced invasion of E. coli–inv afterprolonged periods of inhibitor treatment.
Hsp90 inhibitors strongly induce Hsp90 expression byactivating Hsf1 (heat shock factor 1), an essential hsp90transcription factor (Whitesell et al., 2003). We usedWestern blot analysis to estimate the amount of Hsp90 inChang cells lysates following 17-AAG treatment over-night at concentrations of 0.125, 0.25 and 0.5 mM or for5 h at concentrations of 0.625, 2.5 and 10 mM; thesewere the same exposure times used to generate the datain Fig. 6. A representative result is in Fig. 7. In Changcells, Hsp90 levels increased after 17-AAG treatment(Fig. 7, compare with lane 1), and Hsp90 induction overthe basal level (Fig. 7, lane 1) was observed following17-AAG exposure, both overnight (Fig. 7, lanes 2–4) andfor 5 h (Fig. 7, lanes 5–7). Western blot indicated that theconcentration of 17-AAG did not affect Hsp90 levels inthese time frames, although longer incubation periodsled to stronger expression.
Hsp90 colocalizes with unencapsulated meningococcalstrains during infection of Chang cells
Escherichia coli expressing NadA allows the analysisof the specific contribution of this adhesin/invasin inmeningococcal host interaction because the diverseN. meningitidis virulence factors have likely redundantfunctions (Virji, 2009). To assess possible interactionsbetween cellular Hsp90 and meningococci after infec-tion, we conducted confocal analysis of Chang cellsinfected with two unencapsulated meningococcalstrains: a nadA knockout mutant, MC58 SiaD-/NadA-
that carries a truncated form of nadA gene and lacks theexpression of the protein (Capecchi et al., 2005) andstrain MC58 SiaD-/NadA-/cNadA that overexpressesNadA by mean of genetic complementation as described
in Experimental procedures. In the MC58 SiaD-/NadA-/cNadA strain, nadA is controlled by a constitutive pro-moter (Ptac) and NadA is produced at a higher extentcompared with the wild-type strain, which allowed us toovercome the low basal expression of NadA and poten-tial phase-variability.
Chang cells were infected for 6 h with the two menin-gococcal strains then underwent fixation, permeabiliza-tion and staining. A representative confocal analysis isshown in Fig. 8. Bacteria penetrated into cells (Fig. 8,bottom panels), generally in close proximity to Hsp90;the overexpressing strain is shown in Fig. 8A and theknockout strain in Fig. 8B. Hsp90 appeared to form intra-cellular cluster-like structures that extended around themeningococci; however, the apical surface of infectedcells revealed marked differences between the strains.Cells infected with the nadA knockout strain showedmany clustered bacteria with very limited colocalizationwith Hsp90 (Fig. 8B, top panels), while the NadA over-expressing strain was rarely found on the cell surface.Clusters of the NadA overexpressing strain were com-pletely surrounded by Hsp90 (Fig. 8A, top panels). Thus,NadA specifically affected interaction of Hsp90 withadhered meningococci. The relevance of this findingdeserves further investigation.
Discussion
In the course of investigations of cellular interactions withNadA, a meningococcal adhesin and invasin, we identi-fied a new, non-canonical role for Hsp90, as a line ofinterference with NadA-mediated activity. This roleappeared independent of any action on Hsp90 client pro-teins. We provided robust evidence of in vitro associationsbetween NadA and Hsp90 by ligand overlay assay, dotblots, co-immunoprecipitation and immunofluorescence.The similarity of the specific Hsp90 binding properties forboth soluble and anchored NadA was also demonstrated.Further, the association between Hsp90 and NadA wasfound to be finely tuned by chaperone-binding small mol-ecules in vitro. While NadA associated with the ADP-bound conformer of Hsp90 and 17-AAG-bound Hsp90, inthe absence of nucleotides or in the presence of ATP, no
Fig. 7. 17-AAG induces Hsp90 expression.Chang cells lysates were tested for the levelof expression of Hsp90 by Western blot afterboth an overnight (O/N) and a 5 h treatmentwith different concentrations of 17-AAG. BetaActin was used as loading control.
Hsp90 hampers NadA-mediated infectivity 377
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385
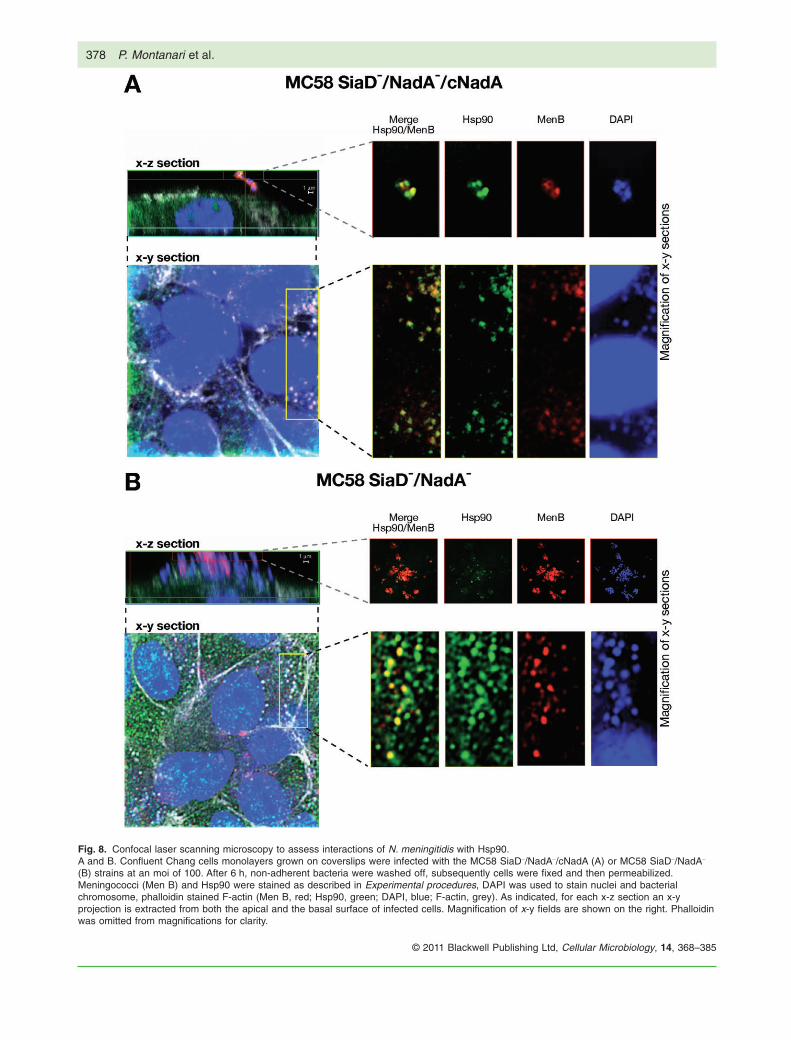
Fig. 8. Confocal laser scanning microscopy to assess interactions of N. meningitidis with Hsp90.A and B. Confluent Chang cells monolayers grown on coverslips were infected with the MC58 SiaD-/NadA-/cNadA (A) or MC58 SiaD-/NadA-
(B) strains at an moi of 100. After 6 h, non-adherent bacteria were washed off, subsequently cells were fixed and then permeabilized.Meningococci (Men B) and Hsp90 were stained as described in Experimental procedures, DAPI was used to stain nuclei and bacterialchromosome, phalloidin stained F-actin (Men B, red; Hsp90, green; DAPI, blue; F-actin, grey). As indicated, for each x-z section an x-yprojection is extracted from both the apical and the basal surface of infected cells. Magnification of x-y fields are shown on the right. Phalloidinwas omitted from magnifications for clarity.
378 P. Montanari et al.
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

association between NadA and Hsp90 was detected.Either ADP or 17-AAG was also necessary for specificbinding of Hsp90 to E. coli expressing surface NadA.
Previous identification of surface Hsp90 and Gp96, theendoplasmic reticulum homologue of Hsp90, as patho-gen protein binding factors was made by ligand overlay(Jin et al., 2003; Cabanes et al., 2005) or immunoblotting/pull down assay (Reyes-Del Valle et al., 2005; Rechneret al., 2007; Na et al., 2008). We used an analogousapproach to identify the association of NadA to Hsp90.Interactions of Hsp90 with client proteins are less promis-cuous than those of other chaperones because of itsinvolvement with proteins that maintain the structuralintegrity of essential regulators of cellular homeostasis(Picard, 2002; Whitesell and Lindquist, 2005). Associa-tion of Hsp90 to client proteins requires ATP as co-factorwhereas hydrolysis to ADP induces dissociation of thecomplexes (Hutchison et al., 1993). Surprisingly, usingrecombinant proteins in vitro, we discovered that Hsp90binding to NadA required ADP or 17-AAG and was inhib-ited by ATP. These results were inconsistent with theprevious understanding of Hsp90. Further, NadA wasable to co-immunoprecipitate Hsp90 from a crude cellextract, with the unanticipated result that Hsp90 bindingwith rNadA was then independent of any added nucle-otide. Co-factors and co-chaperones in the cell lysatecould have played a role in this result, counteracting thehigh concentration of cellular ATP relative to ADP, whichwas shown to be necessary in other experiments. Alter-natively, an adequate amount of cellular Hsp90 couldhave been bound to ADP to produce this result. In alltested conditions, however, the addition of ATP causedHsp90–NadA complex dissociation while exogenous ADPor 17-AAG allowed complex formation. NadA binding tothe ADP or 17-AAG form of Hsp90 suggests a function forthis chaperone outside of its classical ATP-dependentbinding and ATP-hydrolysis release of client proteins.Reassuringly, our finding is supported indirectly by recentwork that identifies ADP-dependent Hsp90 interactionwith CHORDC1 and several additional client protein inter-actions that are reinforced by geldanamycin (Gano andSimon, 2010).
To isolate the role of NadA as a single meningococcalfactor, we exploited a heterologous well-defined bacterialcarrier, E. coli, which is able to correctly fold and carrysurface NadA (Capecchi et al., 2005). This systemallowed us to perform adhesion/invasion assays whoseoutcome could be directly ascribed to NadA. In infectionassays with Chang epithelial cells, we found evidence ofa role of Hsp90 interfering against NadA-driven infection.Lower cellular levels of Hsp90 allowed greater NadAattachment and massive invasion of E. coli bearingsurface-exposed NadA. Conversely, plasmid-driven over-expression of Hsp90 reduced E. coli–NadA adhesion and
entry. The addition of exogenous rHsp90 into the infectionsystem led to a substantial decrease in the invasion medi-ated by NadA supporting the hypothesis of an interferenceeffect by the chaperone.
To evaluate the interaction between Hsp90 and NadAin the meningococcus we carried out a confocal micros-copy analysis of Chang epithelial cells infected witheither a nadA knockout strain or a NadA overexpressingstrain. Results indicated that Hsp90 colocalized withintracellular bacteria for both strains. However, differ-ences between extracellular and intracellular bacteriawere pronounced. Specifically, many fewer NadA over-expressing meningococci were identified extracellularlywhen compared with nadA null mutants; moreover, theformer were completely surrounded by Hsp90 whereasthe latter were merely colocalized. This observationcould be explained by two paradoxical mechanisms:accelerated intracellular localization of the NadA overex-pressing strain or a faster kinetics of association/dissociation from the cell surface for Hsp90 coatedbacteria. Although the full significance of this observationremains to be demonstrated, our analysis suggests thatHsp90 binding could represent a redundant functionshared by other meningococcal factors. Nevertheless,the presence of NadA could fine-tune the process ofinfection so that Hsp90 becomes crucial. Studies areongoing to clarify our findings.
One important consideration for our work is that theinvasion and adhesion mechanisms of NadA remainlargely unelucidated, a circumstance which in factprompted the initial experiments reported here. An inter-esting finding was that the inhibition of NadA, a surfaceprotein belonging to an obligate human pathogen, byHsp90 appeared to be dependent on the amount of eachprotein present, which raises the question whether men-ingococcal strains without NadA will bind to Hsp90. Ourresults suggest diverse associations with a nadA knock-out strain; however, further experimentation using strainsthat lack the nadA gene would help clarify this relation-ship. Interestingly, NadA is present on relatively few men-ingococcal strains as compared with other surfaceproteins like PorA or fHbp. Evolutionary pressure couldgradually have eliminated NadA-harbouring strainsbecause human cell factors evolved to recognize NadAand mount immune responses against it, yet an associa-tion of NadA with pathogenic isolates has been noted(Comanducci et al., 2002; 2004). Another area requiringfurther investigation is the phase variation of NadA, whichhas been indirectly suggested in other contexts (Metruc-cio et al., 2009). As a matter of fact, we ignore if NadA isselectively expressed during critical steps of meningococ-cal pathogenesis.
The Hsp90 inhibitor 17-AAG is another co-factor thatallowed binding of Hsp90 to NadA. Members of this
Hsp90 hampers NadA-mediated infectivity 379
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

drug-derivative family mimic nucleotides and hamperADP/ATP exchange as well as intrinsic Hsp90 ATPaseactivity preventing the chaperone from assisting itsclient proteins (Powers and Workman, 2006; Workmanet al., 2007). We showed that short-term exposuresto 17-AAG specifically interfered with NadA-mediatedinfection, while long-term Hsp90 inhibition affectedinvasion processes mediated by yersinial inv, our controlinvasin. We speculate that chemical inhibition byHsp90 hinders NadA invasion in two additive ways: abackground, 17-AAG-dose-dependent inhibition towardsputative common mechanisms relevant for bacterialinvasion, such as endocytosis or cytoskeleton remodel-ling (Yang et al., 2004; Amiri et al., 2007), and a specificNadA–Hsp90 interaction-related process relying onthe availability of Hsp90 species appropriate to bindNadA. Increased protein expression of Hsp90 after17-AAG treatment appeared homogeneous and dose-independent within a single incubation time frame. Wehypothesize that a physical association between menin-gococcal NadA and Hsp90 interferes with bacterialattachment to, and invasion of, Chang cells, in a mannerdependent on the relative amounts of these proteins.Thus, the NadA adhesive phenotype appears unaffectedby background side-effects. Further, nucleotides orco-chaperones might participate in association/dissociation cycles between NadA and Hsp90 regulatingsuch phenomena, should they be found to occur. Sinceextracellular Hsp90 has been identified (Eustace andJay, 2004; Tsutsumi and Neckers, 2007; Song et al.,2010), this would support different roles for this chaper-one in intra- and extracellular contexts and could there-fore be of use in further interpretation of the results wepresented above. Yet, while the confocal microscopyanalysis of Chang epithelial cells suggests that extracel-lular Hsp90 may bind NadA overexpressing meningo-cocci and showed that intracellular Hsp90 colocalizedwith both nadA knockout and overexpressing meningo-coccal strains, confirmatory data are required. Ourresults also suggest that Hsp90 binding could representa redundant function shared by other meningococcalfactors and that examining the interactions of Hsp90with NadA could help identify areas in the infectiousprocess where Hsp90 represents a crucial component.Cellular Hsp90 might play a role in the context of innateimmune mechanisms by preventing the meningococcalNadA from promoting cell adhesion and invasion.
Our results demonstrated NadA–Hsp90 interaction thatrelied on specific cofactors such as ADP or 17-AAG, andthe displacement of such interaction was driven by ATP.Based on these observations, it would be of interest tofurther characterize the NadA infection process underconditions of impaired ADP/ATP ratio, as during alteredenergy balance.
Experimental procedures
All procedures were performed following appropriate ethicalguidelines for the treatment of laboratory animals wheneverapplicable and good laboratory practice.
Cell cultures
Chang epithelial cells (Wong-Kilbourne derivative, clone 1-5c-4,human conjunctiva, ATCC CCL-20.2) were maintained in Dulbec-co’s modified Eagle’s medium (DMEM) supplemented with15 mM L-glutamine, antibiotics and 10% heat-inactivated FBS(FBSi). Cells were grown at 37°C in 5% CO2.
Bacterial strains, growth conditions andOMP preparation
Escherichia coli BL21(DE3) (Novagen) was used to expressgenes coding for full-length NadA and NadAD351–405 as previouslydescribed (Capecchi et al., 2005).
The expression of Y. pseudotuberculosis invasin in E. coli wasobtained by transforming the plasmid pinv (Monack and Theriot,2001), a generous gift from Professor Monack (Stanford Univer-sity School of Medicine, Stanford, CA, USA), into E. coliBL21(DE3).
Escherichia coli was cultured at 37°C in Luria–Bertani (LB)broth supplemented with 100 mg ml-1 ampicillin (E. coli–NadA) or30 mg ml-1 chloramphenicol (E. coli–inv). Protein expression forfull-length NadA and yersinial invasin was achieved without addi-tion of IPTG (uninduced conditions), exploiting expression due toleakage of the induction system.
Meningococcal MC58 SiaD-/NadA- and MC58 SiaD-/NadA-/cNadA were serogroup B strains. Unencapsulated MC58SiaD-/NadA- as previously described (Capecchi et al., 2005)lacks NadA expression. To achieve complementation ofNadA a copy of the nadA gene was inserted under thecontrol of the Ptac promoter in the non-coding region of theMC58 SiaD-/NadA- chromosome between the convergingopen reading frames NMB1428 and NMB1429. The plasmidfor complementation of the nadA null mutant, pCOMnadA, waspreviously described (Tavano et al., 2009) and was used totransform the MC58 SiaD-/NadA- strain to generate theMC58 SiaD-/NadA-/cNadA strain. The restoration of NadAexpression on the surface of the complemented strain wasconfirmed by Western blot and FACS analysis. NadA was pro-duced in a trimeric form and expressed on the surface of MC58SiaD-/NadA-/cNadA at a higher extent compared with MC58SiaD-, the isogenic unencapsulate wild-type strain (data notshown).
Outer membrane proteins (OMPs) were recovered fromE. coli strains on the basis of Sarkosyl insolubility followingthe rapid procedure described by Carlone et al. (1986). Briefly,bacteria were harvested, suspended in 1 ml of 10 mM Hepesbuffer (pH 7.4) and sonicated on ice. Cell membranes wererecovered by successive centrifugations at 15 600 g at 4°Cin a microcentrifuge. Cytoplasmic membranes were solubilizedby addition of an equal volume of 2% Sarkosyl in 10 mMHepes (pH 7.4). The outer membranes were then recovered bycentrifugation and resuspending the pellet in 10 mM Hepesbuffer.
380 P. Montanari et al.
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

Purified proteins, antibodies and reagents
Recombinant NadAD351–405 was purified according to previouslydescribed procedures (Capecchi et al., 2005). Recombinant fHbp(variant 1) was obtained as previously described (Masignaniet al., 2003). Recombinant Hsp90 was purchased from Stress-gen (ADI-SPP-776). Recombinant His-tagged Hsp90 wasexpressed by an E. coli BL21(DE3) strain transformed with theplasmid pDEST14Hsp90, provided by the Protein Science Groupat Novartis Institute for Biomedical Research (Emeryville, CA,USA). Recombinant His-tagged Hsp90 was purified as follows:one single colony of E. coli BL21(DE3) strain expressing Hsp90-His was inoculated in LB + ampicillin and grown overnight at37°C, diluted in fresh LB medium and grown at 37°C to an OD of0.6–0.8. The protein overexpression was induced by the additionof 1 mM isopropyl-1-thio-b-D-galactopyranoside (IPTG; Sigma)for 3 h. Recombinant Hsp90 6¥ His fusion protein was purified byaffinity chromatography on Ni2+-conjugated chelating fast-flowSepharose (Pharmacia). The purity was checked by SDS-PAGEelectrophoresis staining with Coomassie blue. The proteincontent was quantified by Bradford reagent (Bio-Rad).
The mAb (9F11) recognizing NadA was produced by immuniz-ing 4- to 6-week-old female CD1 mice with 20 mg of NadA recom-binant protein (allele 3) administered intraperitoneally togetherwith complete Freund’s adjuvant (except for the third dose,which was administered without adjuvant). Three days later,the mice were sacrificed and their spleen cells were fusedwith myeloma cells (P3 ¥ 63-Ag8.653) at a ratio of five spleencells to one myeloma cell. After a 2-week incubationin hypoxanthine-aminopterin-thymidine selective medium, thehybridoma supernatants were screened for antibody bindingactivity by enzyme-linked immunosorbent assays (ELISAs).
The mouse polyclonal anti-serum against fHbp had beenobtained previously (Masignani et al., 2003). The mAb (AC88)against Hsp90 was purchased from Stressgen (SPA-830).
The rabbit polyclonal anti-serum against Hsp90 was obtainedby immunizing a New Zealand White rabbit with 25 mg of recom-binant His-tagged Hsp90. The recombinant protein was givensubcutaneously with Freund’s incomplete adjuvant for the firstdose and with Freund’s complete adjuvant for the second (day21) and the third (day 35) doses. A blood sample was taken onday 49. Finally, the serum was purified by affinity chromatographyon CNBr activated Sepharose 4B resin (Pharmacia) according tothe manufacturer’s instructions. Rabbit polyclonal anti-E. coliserum (DAKO) was used to recognize E. coli OMPs. The mAbs(AC74) against Beta Actin was purchased from Sigma (A2228,Sigma). Rabbit polyclonal anti-Men B OMVs antibodies wereobtained as previously described (Giuliani et al., 2006). Poly-clonal Goat Anti-Mouse or Anti-Rabbit Immunoglobulins/HRPwere purchased from DAKO. Alexa Fluor 488 goat anti-mouseIgG, Alexa Fluor 568 goat anti-mouse IgG, Alexa Fluor 647 goatanti-rabbit IgG and Alexa Fluor 488-conjugated phalloidin werefrom Molecular Probes.
17-AAG (17-N-allylamino-17-demethoxygeldanamycin), ADPand ATP were resuspended, stored and implied in accordancewith the manufacturer’s specifications (Sigma).
Identification of Hsp90 by affinity chromatography
About 2 ¥ 108 monolayered Chang cells were detached usingCDS solution (Sigma), washed with PBS and lysed using hypo-
tonic solution (10 mM NaCl, 10 mM Tris-base, 0.2 mM CaCl2,1.5 mM MgCl2) supplemented with complete protease inhibitor(Roche) for 40 min at 4°C in rotation. A pellet was collected bycentrifugation at low speed 3000 g for 5 min. Membrane proteinswere extracted from the pellet in 2% Brij 96 in 50 mM HepespH 7.4, 150 mM NaCl and the complete protease inhibitor andsample were submitted for two subsequent centrifugations at21 000 g for 10 min and at 190 000 g for 20 min. Supernatantwas recovered, diluted 1:2.5 with 50 mM Hepes pH 7.4 contain-ing complete protease inhibitor to equilibrate in 0.8% Brij 96 and60 mM NaCl. The sample was pre-cleared on a deactivatedCNBr-activated Sepharose 4-Fast Flow resin, pre-equilibrated inthe same buffer, for 1 h at 4°C. The pre-cleared material was thenloaded on CNBr-activated Sepharose 4-Fast Flow resin coupledwith rNadA pre-equilibrated in 0.8% Brij 96 in 50 mM HepespH 7.4, 60 mM NaCl and complete protease inhibitor. The boundmaterial was eluted with 100 mM NaCl in 50 mM Hepes, and0.8% Brij 96 collecting three fractions. For each fraction, about1/50 of the total volume was checked in ligand overlay assay(far-Western blot) for the presence of specific bands recognizedby rNadA protein. The remaining material of each fraction wasTCA precipitated and loaded on a parallel identical gel stainedwith Coomassie blue.
In-gel protein digestion and MALDI-TOF TOF massspectrometry analysis
Protein spots were excised from the gels, washed with 50 mMammonium bicarbonate, acetonitrile (50:50, v/v), washed oncewith pure acetonitrile, and air-dried. Dried spots were digested for2 h at 37°C in 12 ml of 0.012 mg ml-1 sequencing grade modifiedtrypsin (Promega) in 5 mM ammonium bicarbonate. After diges-tion, 0.6 ml was loaded on a matrix PAC target (PrespottedAnchorChip 96, set for proteomics, Bruker Daltonics) and air-dried. Spots were washed with 0.6 ml of a solution of 70%ethanol, 0.1% trifluoroacetic acid. Mass spectra were acquired onan Ultraflex MALDI-TOF TOF mass spectrometer (Bruker Dalton-ics) in reflectron, positive mode in the mass range of 900–3500 Da. Spectra were externally calibrated by using acombination of standards prespotted on the target (Bruker Dal-tonics). MS spectra were analysed with flexAnalysis (flexAnalysisversion 2.4, Bruker Daltonics). Monoisotopic peaks were anno-tated with flexAnalysis default parameters and manually revised.Protein identification was carried from the generated peak listusing the Mascot program (Mascot server version 2.2.01, MatrixScience). Mascot was run on a public database, National Centerfor Biotechnology Information non-redundant (NCBInr).
Detergent lysis of Chang cells
For immunoprecipitation using Chang cells total extracts and foranalysis of protein level expression, cells were lysed on ice inRIPA buffer (Sigma) supplemented with complete protease inhibi-tor (Roche) for 30 min. Cell debris was removed by centrifugationat 14 000 g for 15 min. In some cases, sonication was performedto increase yields.
SDS-PAGE, Western blotting and ligand overlay assay
All SDS-PAGE reagents were purchased from Invitrogen. Equalamounts of proteins were prepared in 4¥ NuPAGE LDS Sample
Hsp90 hampers NadA-mediated infectivity 381
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

Preparation Buffer and 10¥ NuPAGE Sample Reducing Agentand separated on NuPAGE polyacrylamide gels using NuPAGESDS Running Buffers. Proteins were transferred to nitrocellulosemembranes for Western blot analysis. Membranes transferred(for Western blot) or spotted (for dot blot) proteins were blockedwith PBS containing 0.05% Tween 20 (PBST) + 5% dried skimmilk at room temperature for 1 h. For dot blot and far-Westernblot analysis, prior to primary antibodies incubation, membraneswere overlaid with PBS containing 25 mg ml-1 of recombinantoverlaying proteins or 25 mg ml-1 of OMPs at 4°C for 4 h.After extensive washings in PBST, proteins bound on nitro-cellulose membranes were detected with specific primaryantibodies followed by the corresponding HRP-conjugated sec-ondary antibodies.
Co-immunoprecipitations
Dynabeads® protein G (Invitrogen) were incubated with anti-NadA monoclonal mouse antibodies (9F11) in PBS + 0.1%Tween 20 for 40 min at room temperature. Extensive washingswith PBS + 0.1% Tween 20 were performed to eliminateexcess antibodies. Equimolar amounts (1 mM) of rNadA andrHsp90 were incubated for 30 min at 37°C in immunoprecipita-tion buffer (10 mM MgCl2, 300 mM KCl, 2.5% Triton X-100 inPBS) with 1 mM DTT. Nucleotides or 17-AAG were added tothe incubation mixture at the concentration specified in eachlegend, ranging from 50 mM to 50 mM for ADP and ATP and1 mM to 500 mM for 17-AAG. Samples were incubated withanti-NadA antibodies pre-loaded magnetic beads in immuno-precipitation buffer in presence of 1 mM DTT and gently rotatedfor 30 min at room temperature. After removal of the superna-tant, beads were washed three times with PBS + 1 mM DTT.Protein complexes were recovered by adding SDS-PAGEsample buffer including reducing agent and boiling for 5 min at100°C. Samples underwent SDS-PAGE and Western blotanalysis. The presence of co-immunoprecipitated Hsp90 onthe membrane was revealed with rabbit polyclonal anti-Hsp90 antibodies followed by specific HRP-conjugatedantibodies.
Total extracts from Chang cells, used as source of Hsp90for co-immunoprecipitation experiments, were prepared asdescribed above. Cellular extracts from 106 cells were incubatedwith 200 ng of rNadA for 30 min at 37°C under the conditionsdescribed above. Co-immunoprecipitated Hsp90 was revealedas detailed above.
Immunofluorescence analysis
For demonstration of specific coating of rHsp90 onto E. coliexpressing NadA, E. coli–NadA and E. coli–inv strains were sus-pended in PBS and rHsp90 added to a final concentration of20 mg ml-1 in the presence or absence of either 5 mM ATP or5 mM ADP or 5 mM 17-AAG. After incubation with gentle mixingfor 30 min at 37°C, bacteria were washed extensively with PBSand spread on polylysine-coated plates. Samples were then fixedin 3.7% paraformaldehyde, washed and blocked with PBS + 3%Bovine Serum Albumin (BSA) (Sigma) +10% Normal Goat Serum(Invitrogen) for 1 h at room temperature. After multiple washings,samples were incubated with mouse monoclonal anti-Hsp90 anti-bodies (1:100) for 1 h at room temperature. Washings to remove
unbound primary antibodies were followed by incubation withAlexa Fluor 488 goat anti-mouse IgG (1:400). Labelled prepara-tions were mounted with ProLong® Gold antifade reagent withDAPI (Molecular Probes) and analysed with a Zeiss LSM-710confocal microscope.
Measurement of bacterial association and invasion byviable counting
Chang cells were seeded on 24-well tissue culture plates (1 ¥ 105
cells per well), and after 24 h of incubation in an antibiotic-freemedium, approximately 3 ¥ 107 [multiplicity of infection (moi) of100:1] bacteria were added per well in DMEM supplemented with1% FBSi and incubated for 4 h at 37°C in 5% CO2. After removalof non-adherent bacteria by washing, cells were lysed with 1%saponin (Sigma), DMEM + 1% FBSi was used to harvest bacte-ria, and serial dilutions of the suspension were plated onto LBagar to calculate the number of colony-forming units. To deter-mine the number of intracellular bacteria, infected Chang mono-layers were treated with gentamicin (200 mg ml-1) for 1 h at 37°C.After washing, cells were lysed and the bacteria recovered andplated.
To test the effect of exogenously added rHsp90, E. coli–NadAand E. coli–inv were suspended in DMEM + 1% FBSi containing50 mg ml-1 of rHsp90 and incubated with gentle mixing for 30 minat 37°C. Bacteria were then used to infect Chang epithelial cells;rHsp90 was maintained in the medium throughout the infectionperiod.
To test the effect of Hsp90 chaperone activity inhibition, cellswere pre-incubated either for an overnight period (approximately15 h) or for 1 h at 37°C before infection with different concentra-tions of 17-AAG, which remained constant throughout the infec-tion period. No effect on bacterial or cell viability at theconcentrations and times used was observed (data not shown).
Cell transfections: Hsp90 siRNA and overexpression
To silence gene expression by siRNA, Chang cells were trans-fected either with a mixture containing four independent siRNAconstructs (2.5 nM each, sc-35608, Santa Cruz Biotechnology)directed to both hsp90a and hsp90b or with a scrambled con-struct siRNA (10 nM, sc-44230, Santa Cruz Biotechnology), asnegative control, using HiPerfect Transfection Reagent (Qiagen)according to the manufacturer’s instructions. Chang cells wereinfected with E. coli–NadA or E. coli–inv 48 h after transfection.
For overexpression experiments cells were transfected eitherwith 0.5 mg of pQE-TriSystem vector containing hsp90a cDNAand encoding for an additional 10X His-Tag, or with 0.5 mg ofpQE-TriSystem empty vector, as a negative control (both fromQIAgenes Expression Kit, Qiagen) using FuGene 6 reagent(Roche) as recommended by the manufacturer. Infection assayswere performed 24 h after transfection. All plasmid DNA wasprepared using the EndoFree Plasmid Maxi Kit (endotoxin free;Qiagen).
Confocal microscopy
Dual labelling of Hsp90 and meningococci was performed in aseries of steps. Chang epithelial cells were seeded on coverslips
382 P. Montanari et al.
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

(1 ¥ 105 cells per coverslip) and grown to confluency. The dayof infection the culture medium was removed and freshDMEM + 1% FBSi added. Bacteria grown on GC agar plateswere washed in PBS once, resuspended in DMEM + 1% FBSiand then added (~3 ¥ 107 bacteria per coverslip, moi of 100:1) tomonolayers. Cells and bacteria were incubated for 6 h at 37°C in5% CO2. After removal of non-adherent bacteria by washing,samples were fixed in 2% paraformaldehyde, washed and thenpermeabilized using PBS + 0.l% Triton X-100 + 1% saponin for20 min at room temperature. Monolayers were washed andblocked with PBS + 0.1% Triton X-100 + 3% BSA + 10% NormalGoat Serum for 1 h at room temperature, then washed andincubated with: (i) rabbit polyclonal anti-Men B OMVs antibodies(1:500) followed by Alexa Fluor 647 goat anti-rabbit IgG (1:400)to detect meningococci and (ii) mouse monoclonal anti-Hsp90antibodies (1:1000) followed by Alexa Fluor 568 goat anti-mouseIgG (1:400) to detect cellular Hsp90. Each antibody was diluted inPBS + 0.1% Triton X-100 + 1% BSA, and incubations took placeat room temperature. Alexa Fluor 488-conjugated phalloidin(1:200) was used together with secondary antibodies to stainF-actin. Glass coverslips were mounted with ProLong® Gold anti-fade reagent with DAPI and analysed with a Zeiss LSM-710confocal microscope.
Acknowledgements
We thank Marialina Bernardini (Università La Sapienza, Roma)and Marco Soriani (Novartis Vaccines and Diagnostics) for veryhelpful discussions. Professor Monack of Stanford Universitygenerously provided a plasmid, as described above. Lisa DeTora(Novartis Vaccines and Diagnostics) is gratefully acknowledgedfor providing editorial guidance and support. We are grateful toGiorgio Corsi for artwork and Mirko Cortese for technical help.
References
Amiri, A., Noei, F., Feroz, T., and Lee, J.M. (2007) Geldana-mycin anisimycins activate Rho and stimulate Rho- andRock-dependent actin stress fiber formation. Mol CancerRes 5: 933–942.
Bambini, S., Muzzi, A., Olcen, P., Rappuoli, R., Pizza, M., andComanducci, M. (2009) Distribution and genetic variabilityof three vaccine components in a panel of strains repre-sentative of the diversity of serogroup B meningococcus.Vaccine 27: 2794–2803.
Blagg, B.S., and Kerr, T.D. (2006) Hsp90 inhibitors: smallmolecules that transform the Hsp90 protein folding machin-ery into a catalyst for protein degradation. Med Res Rev26: 310–338.
Bliska, J.B., Copass, M.C., and Falkow, S. (1993) The Yers-inia pseudotuberculosis adhesin YadA mediates intimatebacterial attachment to and entry into HEp-2 cells. InfectImmun 61: 3914–3921.
Bowe, F., Lavelle, E.C., McNeela, E.A., Hale, C., Clare, S.,Arico, B., et al. (2004) Mucosal vaccination against sero-group B meningococci: induction of bactericidal antibodiesand cellular immunity following intranasal immunizationwith NadA of Neisseria meningitidis and mutants ofEscherichia coli heat-labile enterotoxin. Infect Immun 72:4052–4060.
Cabanes, D., Sousa, S., Cebriá, A., Lecuit, M., García-delPortillo, F., and Cossart, P. (2005) Gp96 is a receptor for anovel Listeria monocytogenes virulence factor, Vip, asurface protein. EMBO J 24: 2827–2838.
Capecchi, B., Adu-Bobie, J., Di Marcello, F., Ciucchi, L.,Masignani, V., Taddei, A., et al. (2005) Neisseria meningiti-dis NadA is a new invasin which promotes bacterial adhe-sion to and penetration into human epithelial cells. MolMicrobiol 55: 687–698.
Carlone, G.M., Thomas, M.L., Rumschlag, H.S., and Sottnek,F.O. (1986) Rapid microprocedure for isolating detergent-insoluble outer membrane proteins from Haemophilusspecies. J Clin Microbiol 24: 330–332.
Ciabattini, A., Giomarelli, B., Parigi, R., Chiavolini, D., Pettini,E., Aricò, B., et al. (2008) Intranasal immunization of micewith recombinant Streptococcus gordonii expressing NadAof Neisseria meningitidis induces systemic bactericidalantibodies and local IgA. Vaccine 26: 4244–4250.
Comanducci, M., Bambini, S., Brunelli, B., Adu-Bobie, J.,Aricò, B., Capecchi, B., et al. (2002) NadA, a novel vaccinecandidate of Neisseria meningitidis. J Exp Med 195: 1445–1454.
Comanducci, M., Bambini, S., Caugant, D.A., Mora, M.,Brunelli, B., Capecchi, B., et al. (2004) NadA diversity andcarriage in Neisseria meningitidis. Infect Immun 72: 4217–4223.
Cotter, S.E., Surana, N.K., Grass, S., and St Geme, J.W. 3rd(2006) Trimeric autotransporters require trimerization ofthe passenger domain for stability and adhesive activity.J Bacteriol 188: 5400–5407.
Dittmar, K.D., Demady, D.R., Stancato, L.F., Krishna, P., andPratt, W.B. (1997) Folding of the glucocorticoid receptor bythe heat shock protein (hsp) 90-based chaperone machin-ery. The role of p23 is to stabilize receptor.hsp90 hetero-complexes formed by hsp90.p60.hsp70. J Biol Chem 272:21213–21220.
Eitel, J., and Dersch, P. (2002) The YadA protein of Yersiniapseudotuberculosis mediates high-efficiency uptake intohuman cells under environmental conditions in which inva-sion is repressed. Infect Immun 70: 4880–4891.
El Tahir, Y., and Skurnik, M. (2001) YadA, the multifacetedYersinia adhesin. Int J Med Microbiol 291: 209–218.
Eustace, B.K., and Jay, D.G. (2004) Extracellular roles for themolecular chaperone, hsp90. Cell Cycle 3: 1098–1100.
Franzoso, S., Mazzon, C., Sztukowska, M., Cecchini, P.,Kasic, T., Capecchi, B., et al. (2008) Human monocytes/macrophages are a target of Neisseria meningitidisAdhesin A (NadA). J Leukoc Biol 83: 1100–1110.
Gano, J.J., and Simon, J.A. (2010) A proteomic investigationof ligand-dependent HSP90 complexes reveals CHORDC1as a novel ADP-dependent HSP90-interacting protein. MolCell Proteomics 9: 255–270.
Girard, V., and Mourez, M. (2006) Adhesion mediated byautotransporters of Gram-negative bacteria: structural andfunctional features. Res Microbiol 157: 407–416.
Giuliani, M.M., Adu-Bobie, J., Comanducci, M., Aricò, B.,Savino, S., Santini, L., et al. (2006) A universal vaccine forserogroup B meningococcus. Proc Natl Acad Sci USA 103:10834–10839.
Grenert, J.P., Sullivan, W.P., Fadden, P., Haystead, T.A.,Clark, J., Mimnaugh, E., et al. (1997) The amino-terminal
Hsp90 hampers NadA-mediated infectivity 383
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

domain of heat shock protein 90 (hsp90) that bindsgeldanamycin is an ATP/ADP switch domain that regulateshsp90 conformation. J Biol Chem 272: 23843–23850.
Heise, T., and Dersch, P. (2006) Identification of a domain inYersinia virulence factor YadA that is crucial for extracellu-lar matrix-specific cell adhesion and uptake. Proc NatlAcad Sci USA 103: 3375–3380.
Hill, D.J., and Virji, M. (2003) A novel cell-binding mechanismof Moraxella catharralis ubiquitous surface protein UspA:specific targeting of the N-domain of carcinoembryonicantigen-related cell adhesion molecules by UspA1. MolMicrobiol 48: 117–129.
Hutchison, K.A., Scherrer, L.C., Czar, M.J., Stancato, L.F.,Chow, Y.H., Jove, R., and Pratt, W.P. (1993) Regulation ofglucocorticoid receptor function through assembly of areceptor-heat shock protein complex. Ann N Y Acad Sci684: 35–48.
Iriarte, M., and Cornelis, G.M. (1996) Molecular determinantsof Yersinia pathogenesis. Microbiologia 12: 267–280.
Isberg, R.R., and Falkow, S. (1985) A single genetic locusencoded by Yersinia pseudotuberculosis permits invasionof cultured animal cells by Escherichia coli K-12. Nature317: 262–264.
Jez, J.M., Chen, J.C., Rastelli, G., Stroud, R.M., and Santi,D.V. (2003) Crystal structure and molecular modeling of17-DMAG in complex with human Hsp90. Chem Biol 10:361–368.
Jin, S., Song, Y.C., Emili, A., Sherman, P.M., and Chan, V.L.(2003) JlpA of Campylobacter jejuni interacts with surface-exposed heat shock protein 90alpha and triggers signallingpathways leading to the activation of NF-kappaB and p38MAP kinase in epithelial cells. Cell Microbiol 5: 165–174.
Laarmann, S., Cutter, D., Juehne, T., Barenkamp, S.J., andSt Geme, J.W. (2002) The Haemophilus influenzae Hiaautotransporter harbours two adhesive pockets that residein the passenger domain and recognize the same host cellreceptor. Mol Microbiol 46: 731–743.
Lafontaine, E.R., Cope, L.D., Aebi, C., Latimer, J.L.,McCracken, J.H. Jr, and Hansen, E.J. (2000) The UspA1protein and a second type of UspA2 protein mediate adher-ence of Moraxella catarrhalis to human epithelial cells invitro. J Bacteriol 182: 1364–1373.
Li, L., Matevski, D., Aspiras, M., Ellen, R.P., and Lépine, G.(2004) Two epithelial cell invasion-related loci of the oralpathogen Actinobacillus actinomycetemcomitans. OralMicrobiol Immunol 19: 16–25.
Linke, D.T., Riess, T., Autenrieth, I.B., Lupas, A., and Kempf,V.A. (2006) Trimeric autotransporter adhesins: variablestructure, common function. Trends Microbiol 14: 264–270.
Litt, D.J., Savino, S., Beddek, A., Comanducci, M., Sandiford,C., Stevens, J., et al. (2004) Putative vaccine antigensfrom Neisseria meningitidis recognized by serum antibod-ies of young children convalescing after meningococcaldisease. J Infect Dis 190: 1488–1497.
McMichael, J.C., Fiske, M.J., Fredenburg, R.A., Chakravarti,D.N., VanDerMeid, K.R., Barniak, V., et al. (1998) Isolationand characterization of two proteins from Moraxellacatarrhalis that bear a common epitope. Infect Immun 66:4374–4381.
Magagnoli, C., Bardotti, A., De Conciliis, G., Galasso, R.,Tomei, M., Campa, C., et al. (2009) Structural organization
of NadADelta(351-405), a recombinant MenB vaccinecomponent, by its physico-chemical characterization atdrug substance level. Vaccine 27: 2156–2170.
Masignani, V., Comanducci, M., Giuliani, M.M., Bambini, S.,Adu-Bobie, J., Arico, B., et al. (2003) Vaccination againstNeisseria meningitidis using three variants of the lipopro-tein GNA1870. J Exp Med 197: 789–799.
Mayer, M.P. (2010) Gymnastic of molecular chaperones. MolCell 39: 321–331.
Mazzon, C., Baldani-Guerra, B., Cecchini, P., Kasic, T., Viola,A., de Bernard, M., et al. (2007) IFN-gamma and R-848dependent activation of human monocyte-derived dendriticcells by Neisseria meningitidis Adhesin A. J Immunol 179:3904–3916.
Metruccio, M.M., Pigozzi, E., Roncarati, D., Berlanda Scorza,F., Norais, N., Hill, S.A., et al. (2009) A novel phase varia-tion mechanism in the meningococcus driven by a ligand-responsive repressor and differential spacing of distalpromoter elements. PLoS Pathog 5: e1000710.
Monack, D.M., and Theriot, J.A. (2001) Actin-based motility issufficient for bacterial membrane protrusion formation andhost cell uptake. Cell Microbiol 3: 633–647.
Na, X., Kim, H., Moyer, M.P., Pothoulakis, C., and LaMont,J.T. (2008) gp96 is a human colonocyte plasma membranebinding protein for Clostridium difficile toxin A. Infect Immun76: 2862–2871.
Pearl, L.H., and Prodromou, C. (2006) Structure and mecha-nism of the Hsp90 molecular chaperone machinery. AnnuRev Biochem 75: 271–294.
Picard, D. (2002) Heat-shock protein 90, a chaperone forfolding and regulation. Cell Mol Life Sci 59: 1640–1648.
Pizza, M., Scarlato, V., Masignani, V., Giuliani, M.M., Aricò,B., Comanducci, M., et al. (2000) Identification of vaccinecandidates against serogroup B meningococcus by whole-.genome sequencing. Science 287: 1816–1820.
Powers, M.V., and Workman, P. (2006) Targeting of multiplesignalling pathways by heat shock protein 90 molecularchaperone inhibitors. Endocr Relat Cancer 13 (Suppl. 1):S125–S135.
Ray, S.K., Rajeshwari, R., Sharma, Y., and Sonti, R.V. (2002)A high-molecular weight outer membrane protein of Xan-thomonas oryzae pv. oryzae exhibits similarity to nonfim-brial adhesins of animal pathogenic bacteria and isrequired for optimum virulence. Mol Microbiol 46: 637–647.
Rechner, C., Kühlevein, C., Müller, A., Schild, H., and Rudel,T. (2007) Host glycoprotein Gp96 and scavenger receptorSREC interact with PorB of disseminating Neisseria gon-orrhoeae in an epithelial invasion pathway. Cell HostMicrobe 2: 393–403.
Reyes-Del Valle, J., Chavéz-Salinas, S., Medina, F., and DelAngel, R.M. (2005) Heat shock protein 90 and heat shockprotein 70 are components of dengue virus receptorcomplex in human cells. J Virol 79: 4557–4567.
Riess, T., Andersson, S.G., Lupas, A., Schaller, M., Schäfer,A., Kyme, P., et al. (2004) Bartonella adhesin a mediates aproangiogenic host cell response. J Exp Med 200: 1267–1278.
Roggenkamp, A., Ackermann, N., Jacobi, C.A., Truelzsch, K.,Hoffmann, H., and Heesemann, J. (2003) Molecular analy-sis of transport and oligomerization of the Yersinia entero-colitica adhesin YadA. J Bacteriol 185: 3735–3744.
384 P. Montanari et al.
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385

Scarselli, M., Serruto, D., Montanari, P., Capecchi, B.,Adu-Bobie, J., Veggi, D., et al. (2006) Neisseria meningiti-dis NhhA is a multifunctional trimeric aurotransporter adhe-sion. Mol Microbiol 61: 631–644.
Serruto, D., Spadafina, T., Scarselli, M., Bambini, S.,Comanducci, M., Höhle, S., et al. (2009) HadA is an atypi-cal new multifunctional trimeric coiled-coil adhesion ofHaemophilus influenzae biogroup aegyptius, which pro-motes entry into host cells. Cell Microbiol 11: 1044–1063.
Song, X., Wang, X., Zhuo, W., Shi, H., Feng, D., Sun, Y., et al.(2010) The regulatory mechanism of extracellular Hsp90on matrix metalloproteinase-2 processing and tumor angio-genesis. J Biol Chem 285: 40039–40049.
Stancato, L.F., Chow, Y.H., Hutchison, K.A., Perdew, G.H.,Jove, R., and Pratt, W.B. (1993) Raf exists in a nativeheterocomplex with hsp90 and p50 that can be reconsti-tuted in a cell-free system. J Biol Chem 268: 21711–21716.
Stebbins, C.E., Russo, A.A., Schneider, C., Rosen, N., Hartl,F.U., and Pavletich, N.P. (1997) Crystal structure of anHsp90-geldanamycin complex: targeting of a protein chap-erone by an antitumor agent. Cell 89: 239–250.
Surana, N.K., Cutter, D., Barenkamp, S.J., and St Geme,J.W., 3rd (2004) The Haemophilus influenzae Hiaautotransporter contains an unusually short trimeric trans-locator domain. J Biol Chem 279: 14679–14685.
Tavano, R., Franzoso, S., Cecchini, P., Cartocci, E., Oriente,F., Aricò, B., and Papini, E. (2009) The membrane expres-sion of Neisseria meningitidis adhesin A (NadA) increasesthe proimmune effects of MenB OMVs on human macroph-ages, compared with NadA- OMVs, without further stimu-lating their proinflammatory activity on circulatingmonocytes. J Leukoc Biol 86: 143–153.
Tavano, R., Capecchi, B., Montanari, P., Franzoso, S., Marin,O., Sztukowska, M., et al. (2011) Mapping of the Neisseria
meningitidis NadA cell-binding site: relevance of predicted-helices in the NH2-terminal and dimeric coiled-coilregions. J Bacteriol 193: 107–115.
Tsutsumi, S., and Neckers, L. (2007) Extracellular heat shockprotein 90: a role for a molecular chaperone in cell motilityand cancer metastasis. Cancer Sci 98: 1536–1539.
Virji, M. (2009) Pathogenic neisseriae: surface modulation,pathogenesis and infection control. Nat Rev Microbiol 7:274–286.
Whitesell, L., and Lindquist, S.L. (2005) HSP90 and the chap-eroning of cancer. Nat Rev Cancer 5: 761–772.
Whitesell, L., Bagatell, R., and Falsey, R. (2003) The stressresponse: implications for the clinical developmentof hsp90 inhibitors. Curr Cancer Drug Targets 3: 349–358.
Workman, P., Burrows, F., Neckers, L., and Rosen, N. (2007)Drugging the cancer chaperone HSP90: combinatorialtherapeutic exploitation of oncogene addiction and tumorstress. Ann N Y Acad Sci 1113: 202–216.
Yang, W., Jansen, J.M., Lin, Q., Canova, S., Cerione, R.A.,and Childress, C. (2004) Interaction of activated Cdc42-associated tyrosine kinase ACK2 with HSP90. Biochem J382: 199–204.
Yang, Y., and Isberg, R.R. (1993) Cellular internalization inthe absence on invasion expression is promoted by theYersinia pseudotuberculosis yadA product. Infect Immun61: 3907–3913.
Zhang, P., Chomel, B.B., Schau, M.K., Goo, J.S., Droz, S.,Kelminson, K.L., et al. (2004) A family of variablyexpressed outer-membrane proteins (Vomp) mediatesadhesion and autoaggregation in Bartonella quintana. ProcNatl Acad Sci USA 101: 13630–13635.
Zhang, T., Li, Y., Yu, Y., Zou, P., Jiang, Y., and Sun, D. (2009)Characterization of celastrol to inhibit hsp90 and cdc37interaction. J Biol Chem 284: 35381–35389.
Hsp90 hampers NadA-mediated infectivity 385
© 2011 Blackwell Publishing Ltd, Cellular Microbiology, 14, 368–385