Embed Size (px)
Citation preview

Role of Copper Transport Protein Antioxidant 1 inAngiotensin II–Induced Hypertension
A Key Regulator of Extracellular Superoxide Dismutase
Kiyoshi Ozumi, Varadarajan Sudhahar, Ha Won Kim, Gin-Fu Chen, Takashi Kohno, Lydia Finney,Stefan Vogt, Ronald D. McKinney, Masuko Ushio-Fukai, Tohru Fukai
Abstract—Extracellular superoxide dismutase (SOD3) is a secretory copper enzyme involved in protecting angiotensin II(Ang II)–induced hypertension. We found previously that Ang II upregulates SOD3 expression and activity as acounterregulatory mechanism; however, underlying mechanisms are unclear. Antioxidant 1 (Atox1) is shown to act asa copper-dependent transcription factor, as well as a copper chaperone, for SOD3 in vitro, but its role in Ang II–inducedhypertension in vivo is unknown. Here we show that Ang II infusion increases Atox1 expression, as well as SOD3expression and activity, in aortas of wild-type mice, which are inhibited in mice lacking Atox1. Accordingly, Ang IIincreases vascular superoxide production, reduces endothelium-dependent vasodilation, and increases vasoconstrictionin mesenteric arteries to a greater extent in Atox1�/� than in wild-type mice. This contributes to augmented hypertensiveresponse to Ang II in Atox1�/� mice. In cultured vascular smooth muscle cells, Ang II promotes translocation of Atox1to the nucleus, thereby increasing SOD3 transcription by binding to Atox1-responsive element in the SOD3 promoter.Furthermore, Ang II increases Atox1 binding to the copper exporter ATP7A, which obtains copper from Atox1, as wellas translocation of ATP7A to plasma membranes, where it colocalizes with SOD3. As its consequence, Ang IIdecreases vascular copper levels, which is inhibited in Atox1�/� mice. In summary, Atox1 functions to preventAng II–induced endothelial dysfunction and hypercontraction in resistant vessels, as well as hypertension, in vivoby reducing extracellular superoxide levels via increasing vascular SOD3 expression and activity. (Hypertension.2012;60:00-00.) ● Online Data Supplement
Key Words: angiotensin II � hypertension � oxidative stress � antioxidant 1 � SOD3 � SOD1 � copper
Angiotensin II (Ang II), the principal effector peptide ofthe renin-angiotensin system, plays a major role in the
initiation and progression of vascular diseases, such ashypertension, in part through reactive oxygen species.1 AngII–induced increase in reactive oxygen species in particular,superoxide (O2
·�), leads to decreased bioavailability of NO,which impairs endothelium-dependent vasodilatation andpromotes vasoconstriction. A major antioxidant defense sys-tem against O2
·� is the superoxide dismutases (SODs): thecytoplasmic Cu/ZnSOD (SOD1), the mitochondrial MnSOD,and the extracellular Cu/ZnSOD (SOD3).2 SOD3 is a majorextracellular antioxidant enzyme highly expressed in thevasculature, and it plays an important role in regulating bloodpressure and endothelial function by reducing extracellularO2
·� level, thereby preventing oxidative inactivation of NO
released from endothelium.3–5 We and others reported thatAng II–induced increase in blood pressure, vascular O2
·�
levels, and endothelial dysfunction is augmented in SOD3�/�
mice,4,5 which are ameliorated by treatment with recombinantSOD3.4 We have also shown that Ang II increases expressionand activity of SOD3 in vitro and in vivo.6 These findingssuggest that Ang II–induced modulation of SOD3 is animportant protective mechanism that prevents overproductionof O2
·�; however, underlying regulatory mechanisms remainunknown.
Copper is an essential micronutrient by serving as acofactor for key metabolic and redox-related proteins, andSOD1 and SOD3 require catalytic copper for their fullenzymatic activity.2 Under physiological conditions, the in-tracellular level of free copper is extraordinarily restricted.7
Received December 18, 2011; first decision January 5, 2012; revision accepted June 5, 2012.From the Departments of Medicine (Section of Cardiology) and Pharmacology (K.O., V.S., H.W.K., G.-F.C., T.K., R.D.M., T.F.) and Department of
Pharmacology (R.D.M., M.U.-F.), Center for Cardiovascular Research, Center for Lung and Vascular Biology, University of Illinois at Chicago, Chicago,IL; Jesse Brown VA Medical Center, Chicago, IL (T.K., R.D.M., T.F.); Biosciences Division (L.F.) and X-Ray Science Division (L.F., S.V.), ArgonneNational Laboratory, Argonne, IL.
K.O. and V.S. contributed equally to the article.The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.
111.189571/-/DC1.Correspondence to Tohru Fukai, Departments of Medicine and Pharmacology, University of Illinois at Chicago, 835 S Wolcott, M/C868, E403MSB,
Chicago, IL 60612. E-mail [email protected]© 2012 American Heart Association, Inc.
Hypertension is available at http://hyper.ahajournals.org DOI: 10.1161/HYPERTENSIONAHA.111.189571
1
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Thus, soluble copper carrier proteins, termed “copper chap-erones,” are required to directly transfer copper to specificcellular target proteins. SOD1 obtains copper through inter-action with the cytosolic copper chaperone for SOD1.8 Usingcultured fibroblasts, we demonstrated that exogenous copper-induced full activation of SOD3 requires copper chaperoneantioxidant 1 (Atox1), which delivers copper to the secretorycopper enzymes, likely via interaction with the copper ex-porter ATP7A (Menkes ATPase).9–11 Of note, the Atox1-ATP7A pathway is involved not only to activate secretorycopper enzyme but also to control intracellular copper levelsby regulating secretion of excess copper. Recently, we haveshown that Atox1 also serves as a transcription factor forcopper-induced increase in SOD3 and cyclin D1 expressionin vitro.9,12 The association of copper homeostasis withhypertension has been reported.13 Tissue levels of copper areeither decreased or increased in hypertensive rats, which havebeen proposed to be affected by severity of hypertension.14,15
Serum copper concentration is increased in humans charac-terized by either essential or pulmonary hypertension,16,17 aswell as various rodent hypertension models.14 Copper-deficient diet alters blood pressure in an age-dependentmanner.18,19 However, the role of Atox1 in hypertension invivo has not been investigated.
We performed the present study to test the hypothesis thatAtox1 may play a critical role in modulating blood pressureand vascular function in resistance vessels via regulatingvascular SOD3 during Ang II–induced hypertension. Here wedemonstrate that Ang II infusion markedly increases Atox1protein expression, as well as SOD3 expression, and specificactivity in aortas from wild-type (WT) mice, which areinhibited in Atox1�/� mice. Ang II increases vascular O2
·�
production and promotes endothelial dysfunction in mesen-teric arteries to a greater extent in Atox1�/� than in WT mice.This contributes to the exaggerated hypertensive response toAng II in Atox1�/� mice. Mechanistically, Ang II increasesAtox1 binding to the SOD3 promoter to increase SOD3
transcription, as well as Atox1 binding to copper exporterATP7A, to increase specific activity of SOD3 in culturedvascular smooth muscle cells (VSMCs). Thus, Ang II–induced upregulation of Atox1 may represent an importantcounterregulatory mechanism that blunts overproduction ofO2
·� or elevating blood pressure through regulating SOD3expression and activity in the vasculature.
Materials and MethodsAnimal and Cell Culture StudiesAtox1�/� mice (backcrossed 8 times to C57Bl/6) were obtainedfrom Mutant Mouse Regional Resource Centers. Atox1�/� micewere originally reported to have phenotypes that show perinataldeath (45% of pups) or survive �1 month.20 Our laboratory furtherbackcrossed Atox1�/� mice to C57Bl/6 mice �10 times and, thus,used C57Bl/6 mice as a control. “Survivor” mice were intercrossed�10 times, and Atox1�/� mice used in the present study survived�6 months (90%). For in vivo experiments, we performed Ang IIinfusion by osmotic minipumps, blood pressure measurement by thetail-cuff method, immunohistochemical and Western analysis, real-time PCR, SOD activity assay, vascular O2
·� production, vascularreactivity studies by wire myograph, copper measurements byinductively coupled plasma mass spectrometry, and synchrotronx-ray fluorescence microscopy analysis. All of the studies wereapproved by the animal care and use committee of the University ofIllinois-Chicago. For in vitro experiments, rat VSMCs (betweenpassage 7 and 15) were maintained in DMEM supplemented with10% bovine serum. Using them, we performed nuclear/cytoplasmicfractionation, immunofluorescence, plasmid constructs, transienttransfection and reporter assay, and DNA pull-down assay.
An expanded Materials and Methods section is available in theonline-only Data Supplement.
ResultsAtox1 Expression Is Increased in Aortas of AngII–Infused MiceWe first examined the Atox1 expression in aortas from micewith Ang II–induced hypertension. Western (Figure 1A) andimmunohistochemical (Figure 1B) analysis showed that AngII infusion (0.7 mg/kg per day) for 7 days significantly
Immunohistochemical analysis of Atox-1
WT++Ang II
Atox-KO+Ang II
WT
Atox-KO
N.C.
Control Ang II infusion
Atox1
Fold
Incr
ease
0
1
2
3
4*
#1 #2 #3 #4 #1 #2 #3
Atox1 protein expression
Cont Ang II
BA
Figure 1. Effect of angiotensin II (Ang II) infusion on Antioxidant 1 (Atox1) expression in aortas of wild-type (WT) and Atox1�/� mice invivo by immunoblotting (A) and immunohistochemistry (B). Aortas of C57Bl/6 mice were harvested after a 7-day infusion of Ang II (0.7mg/kg per day) or vehicle SC with osmotic minipumps and subjected to Western analysis with antibodies specific to Atox1. Represen-tative blots were from 3 individual experiments. *P�0.001 vs WT mice. B, Immunohistochemical analysis of Atox1 expression in aortasfrom Ang II–treated C57Bl/6 (WT) and Atox1�/� (Atox1-KO) mice. Arrows in inset shows nuclear staining of Atox1. All of the imageswere taken at 5 different fields, and the cell images are representative of �3 different experiments.
2 Hypertension August 2012
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

increased Atox1 protein expression in the medial layer of theaorta. Importantly, some Atox1-positive cells were found inthe nucleus (Figure 1B).
Effect of Ang II Infusion on SOD3 Expression andActivity in Aortas of WT and Atox1�/� MiceAtox1 has been shown to function as a copper chaperone forsecretory copper-containing enzymes20,21 and a copper-dependent transcription factor in vitro.9,10,12 Thus, we exam-ined the SOD3 expression and activity in aortas of AngII–infused Atox1�/� mice. The body weights and variousorgan weights (heart, kidney, lung, and brain) were not
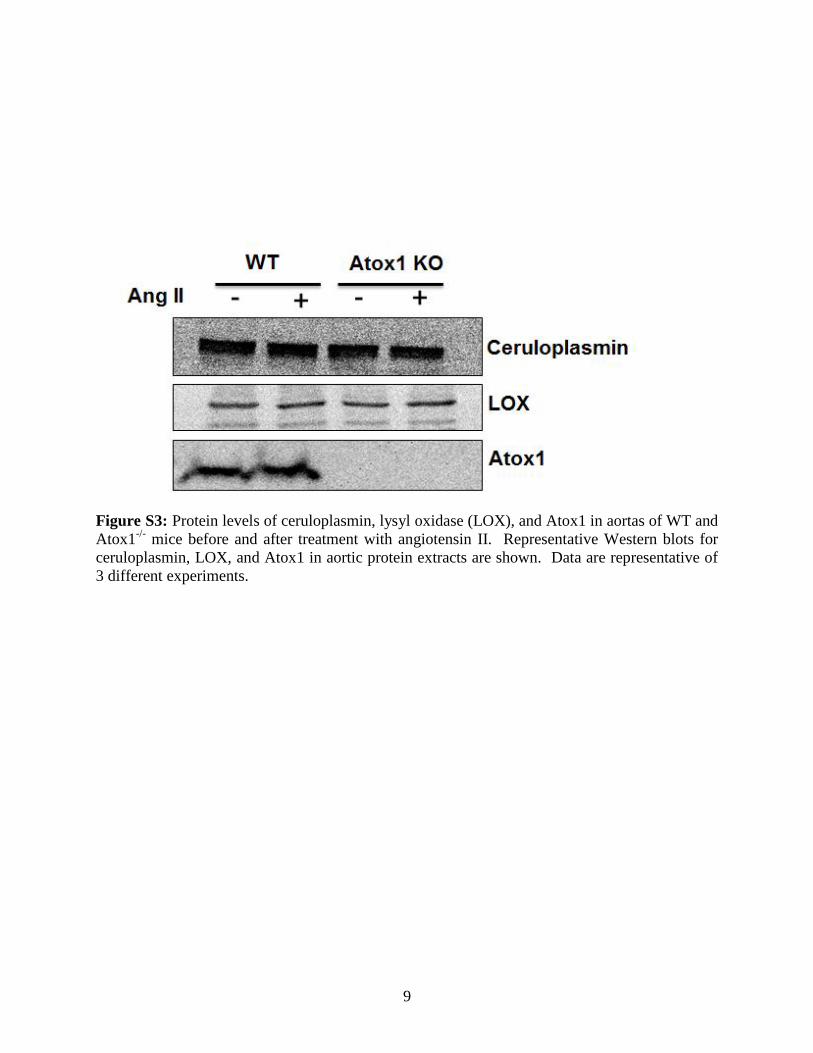
different between C57Bl/6 WT and Atox1�/� mice (FigureS1, available in the online-only Data Supplement). Ang IIinfusion for 7 days significantly increased mRNA and proteinlevels (Figure 2A and 2B), as well as activity of SOD3(Figure 2C), but not those of SOD1, in aortas from WT mice.These Ang II–induced effects were almost completely inhib-ited in Atox1�/� mice (Figure 2). At baseline, SOD3 proteinand mRNA levels, as well as activity, were decreased inAtox1�/� compared with WT aortas. In contrast, total SODactivity and protein expression of other major Cu enzymes(including ceruloplasmin or lysyl oxidase) in aortas weresimilar between WT and Atox1�/� mice at baseline and after
ActivitySOD3 SOD1
+ +
SOD
act
ivity
(U/m
g to
tal p
rote
in)
0
2
4
6
8
10
Ang II - -
WT Atox1 KO
+0
2
4
6
8
10
12
Ang II - + -
WT Atox1 KO
SOD
act
ivity
(U/m
g to
tal p
rote
in)
C
B
Ang II0
Fold
incr
ease
1
2
3
4
- + - +
Fold
incr
ease
0
1
2
3
4
Ang II - + - +
WT Atox1-KO WT Atox1-KOSOD3
ProteinSOD1
SOD3 SOD1mRNAA
0
0.4
0.8
1.2
1.6
SOD
1 m
RN
A/ g
apdh
(Fol
d ch
ange
s)0
0.4
0.8
1.2
1.6
2.0
SOD
3 m
RN
A/ g
apdh
(Fol
d ch
ange
s)
WT Atox1-KO WT Atox1-KO
Ang II - + - + Ang II - + - +
*
*#
#
*
NS
NS NS
NS
NS NS
NS
NS NS
#
Figure 2. Effect of angiotensin II (Ang II)infusion on mRNA expression (A), proteinlevel (B), and activity (C) of superoxide dis-mutase (SOD) 3 and SOD1 in aortas fromantioxidant 1 (Atox1)�/� mice. Mice wereinfused with Ang II as described in Figure 1.A and B, mRNA expression and protein lev-els of SOD1 and SOD3 in aortas of C57Bl/6and Atox1�/� mice were measured by real-time PCR and by Western analysis withantibodies specific to either SOD1 orSOD3. C, Activity of SOD3 and SOD1 inaortas were analyzed by the inhibition ofcytochrome C reduction by xanthine/xanthine oxidase. SOD3 activity wasdetermined after separation with ConA-Sepharose. The results are presented asmean�SE from 4 separate experiments.Representative blots are from 3 individualexperiments. *P�0.001, #P�0.01 vs eithervehicle-infused WT or Atox1�/� mice. NSindicates not significant.
Ozumi et al Atox1, SOD3, and Hypertension 3
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Ang II infusion (Figures S2, S3, and 2). These findingssuggest that Atox1 plays an important role in regulatingexpression and activity of SOD3 in vessels of Ang II–infusedmice.
Effect of Ang II on Blood Pressure and VascularO2
·� Production in WT and Atox1�/� MiceWe next examined the functional role of Atox1 in AngII–induced hypertension and O2
·� levels using Atox1�/� mice.The basal blood pressure and the initial hypertensive responseto Ang II by day 4 were similar in the Atox1�/� and WTmice. However, Ang II infusion for 7 days increased systolicblood pressure to a greater extent in Atox1�/� mice than inWT mice (Figure 3A). Furthermore, lucigenin chemilumines-cence analysis showed that Ang II–induced increase in O2
·�
production was significantly enhanced in Atox1�/� aortas ascompared with WT mice (Figure 3B), whereas basal O2
·�
production was modestly but not significantly increased inAtox1�/� aortas. Ang II–induced alteration of blood pressure
and vascular O2·� production in Atox1�/� mice and WT mice
was rescued by coinfusion of the SOD mimetic Tempol(Figure 3A and 3B). These results suggest that augmentationof Ang II–induced increase in blood pressure in Atox1�/�
mice is attributed to increased O2·� levels.
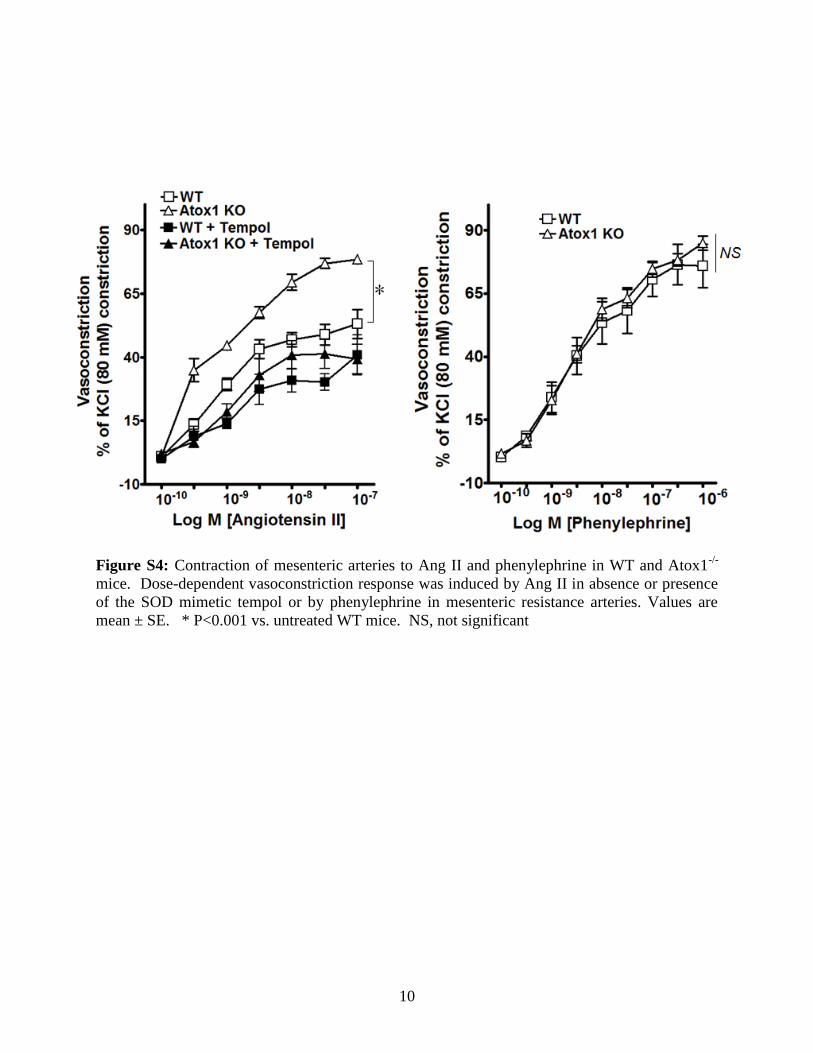
Effect of Ang II on Mesenteric Vascular Functionin WT Mice and Atox1�/� MiceTo determine the role of Atox1 in endothelium-dependentvasodilation, we used mesenteric arteries (200 �m in diam-eter) with the wire myograph approach. Under basal condi-tions, endothelium-dependent relaxations to acetylcholinewere impaired in Atox1�/� mice compared with those of WTmice (Figure 4A, left). Ang II caused a marked impairment ofacetylcholine-evoked relaxation to a greater extent in arteriesof Atox1�/� than WT mice (max relaxation, 44.8�1.72%versus 61.3�3.02%; P�0.01). Relaxations to endothelium-independent vasodilator sodium nitroprusside were similar inWT and Atox1�/� mice at baseline and were not altered by
(mmHg)Sy
stol
ic B
lood
pre
ssur
e
WTAtox1 KO
Atox1 KO +AngII
WT +AngII
Atox1 KO +AngII + TempolWT +AngII + Tempol
Days of Angiotensin II Infusionday 0 day 4 day 7
80
100
120
140
160#
A (× 105)
0
220
440
660
80
(CPM
/g d
ry ti
ssue
wei
ght)
Vasc
ular
Sup
erox
ide
prod
uctio
n
WT
Ang II
Atox1-KO#
Control Ang+Tempol
B
Figure 3. Effect of angiotensin II (Ang II) on blood pressure and vascular superoxide (O2·�) production in antioxidant 1 (Atox1)�/� mice.
A, Either Ang II (0.7 mg/kg per day) or Ang II and the superoxide dismutase (SOD) mimetic Tempol (50 mg/kg per day) or vehicles wereinfused, and blood pressure was measured before and 4 and 7 days after minipumps implantation (n�5 per group). #P�0.01 vs wild-type (WT) or Atox1�/� mice with Tempol treatment. B, O2
·� production in aorta from C57Bl/6 (WT) and Atox1�/� mice, as determinedwith lucigenin-enhanced chemiluminescence (5 �mol/L). #P�0.01 vs WT mice (n�6).
-10
15
40
65
90
WT + ANG IIWT + ANG II + TempolAtox1 KO + ANG IIAtox1 KO + ANG II + Tempol
*
#
10-9 10-8 10-7 10-6 10-5
Log M [ACh]
% R
elax
atio
n
-10
15
40
65
90
Wild Type
Atox-1 KOWT + ANG II
Atox-1 KO + ANG II
*
*
10-9 10-8 10-7 10-6 10-5
Log M [ACh]
% R
elax
atio
n
A
* *
###
0
20
40
60
80
100
120
WT
Atox-1 KOAtox1 KO + ANG II
WT + ANG II
10-9 10-8 10-7 10-6 10-5
Log M [SNP]
% R
elax
atio
n
B
NS
Figure 4. Effect of angiotensin II (Ang II) infusion on endothelium-dependent vascular relaxation in mesenteric arteries from antioxidant 1(Atox1)�/� (Atox-1 KO) and wild-type (WT) mice. A and B, First or second-order mesenteric resistance arteries were studied using a wiremyograph and precontracted with phenylephrine. Vasodilation was evoked by acetylcholine (Ach) in the absence or presence of the SODmimetic Tempol (A) and sodium nitroprusside (SNP; B). Data are expressed as mean�SE (n�6–8 per group). *P�0.001 vs remaining 3groups, #P�0.01 vs either untreated WT mice or Ang II–treated WT mice with Tempol treatment. NS indicates not significant.
4 Hypertension August 2012
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Ang II (Figure 4B). The impairment of acetylcholine-inducedvasorelaxation in Ang II–infused Atox1�/� mice and WTmice was rescued by coinfusion of the SOD mimetic Tempol(Figure 4A, right). In mice without Ang II infusion, directcontractile responses to Ang II were markedly increased inmesenteric arteries of Atox1�/� mice compared with WTmice, which was rescued by the SOD mimetic Tempol(Figure S4). By contrast, contractions induced by phenyleph-rine, which did not produce O2
·�, or KCl were not differentin WT and Atox1�/� mice. Thus, non-KCl normalized datafor Ang II–induced vasoconstriction in WT and Atox1�/�
mice was similar to that after normalization (data notshown). Thus, these results suggest that Atox1 is requiredfor full activity of SOD3, thus decreasing O2
·� level,thereby inhibiting Ang II–induced endothelial dysfunctionand vasoconstriction.
Ang II Promotes Translocation of Atox1 Into theNucleus in VSMCsTo examine the molecular mechanisms by which Atox1regulates SOD3 expression and activity in the vasculature, we
next examined the subcellular localization of Atox1. Immu-nofluorescence analysis in VSMCs shows that Ang II stim-ulation promoted translocation of Atox1 from the cytosol tothe nucleus in a time-dependent manner with peak at 30minutes (Figure 5A). Consistently, subcellular fractionationshowed that Atox1 protein level is increased in nuclearfraction, with a peak at 30 minutes after Ang II stimulation,and then decreased after 120 minutes (Figure 5B). Weconfirmed the purity of nuclear and cytosolic fractions bydetecting the markers, laminin B and tubulin, respectively.
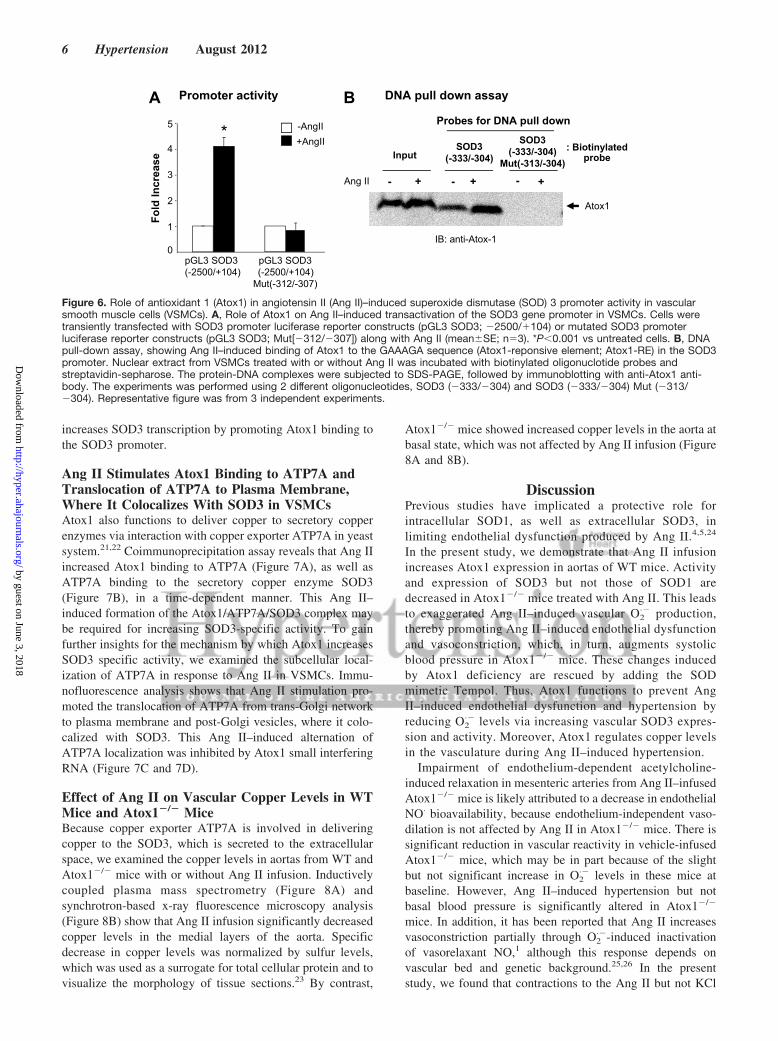
Ang II Stimulates Atox1 Binding to the SOD3Promoter and Its Promoter Activity in VSMCsTo determine whether Atox1 functions as a transcriptionfactor for SOD3 in Ang II–stimulated VSMCs, we performedluciferase reporter gene assays. Ang II stimulation increasedSOD3 promoter activity, which was blocked by the mutationof Atox1 responsive elements12 in the SOD3 promoter(Figure 6A). DNA pull-down assays show that Atox1 directlybound to the Atox1 responsive element, including the DNAsegment (Figure 6B). These results suggest that Ang II
Atox1
DAPI
Merge
Ang II
0min 15min 120min30min
(min)Nuclear
Laminin B1
Atox-1
0 30 60 120
Nuclear+AngII
Tubulin
0 30 60 120
Non-Nuclear
0
1
2
3
4
0 30 60 120
Fold
incr
ease
(min)
Fold
incr
ease
1200
0.4
0.8
1.2
0 30 60
Non-nuclear
(min)
A
B
Ang II treatment0 5 15 30 60 120 min
%
0
20
40
60
80
*
*
Rat
io o
f Nuc
lear
>Cyt
osol
ic A
tox1
Figure 5. Effect of angiotensin II (Ang II) on nuclear translocation of antioxidant 1 (Atox1) in vascular smooth muscle cells (VSMCs). A,VSMCs were stimulated with Ang II (100 nmol/L) for indicated time, and cells were stained with anti-Atox 1 antibody and the nuclearmarker, 4=,6-diamidino-2-phenylindole (DAPI). In each image, ratio of nuclear�cytosolic Atox1 (immunofluorescence of nuclear Atox1 ishigher than that of cytosolic Atox1) was calculated from 5 randomized views, and the cell images are representative of �3 differentexperiments. *P�0.001 vs no treated cells. White arrows indicate nuclear Atox1 predominant cells. B, Nuclear and cytoplasmic frac-tions were immunoblotted with anti-Atox 1, tubulin (cytoplasmic marker), and laminin B1 (nuclear marker) antibody. Right panel showsaveraged data for nuclear and nonnuclear expression of Atox1 protein, expressed as fold increased against control VSMCs (mean�SE,n�3). *P�0.001 vs untreated cells.
Ozumi et al Atox1, SOD3, and Hypertension 5
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
increases SOD3 transcription by promoting Atox1 binding tothe SOD3 promoter.
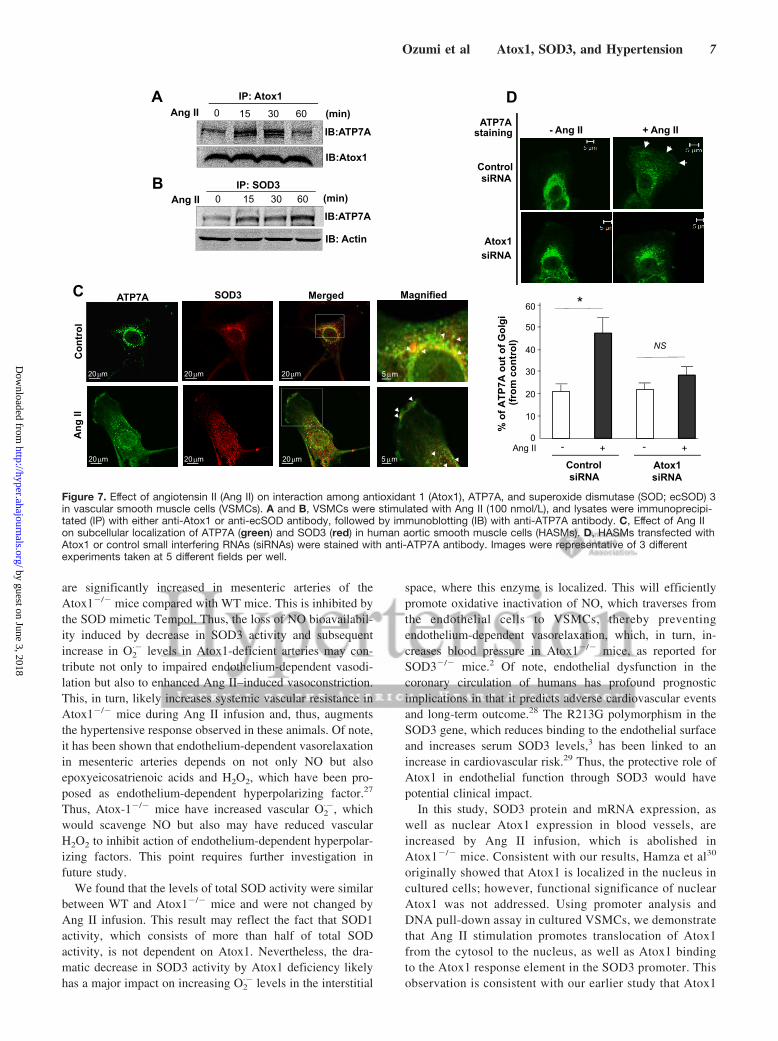
Ang II Stimulates Atox1 Binding to ATP7A andTranslocation of ATP7A to Plasma Membrane,Where It Colocalizes With SOD3 in VSMCsAtox1 also functions to deliver copper to secretory copperenzymes via interaction with copper exporter ATP7A in yeastsystem.21,22 Coimmunoprecipitation assay reveals that Ang IIincreased Atox1 binding to ATP7A (Figure 7A), as well asATP7A binding to the secretory copper enzyme SOD3(Figure 7B), in a time-dependent manner. This Ang II–induced formation of the Atox1/ATP7A/SOD3 complex maybe required for increasing SOD3-specific activity. To gainfurther insights for the mechanism by which Atox1 increasesSOD3 specific activity, we examined the subcellular local-ization of ATP7A in response to Ang II in VSMCs. Immu-nofluorescence analysis shows that Ang II stimulation pro-moted the translocation of ATP7A from trans-Golgi networkto plasma membrane and post-Golgi vesicles, where it colo-calized with SOD3. This Ang II–induced alternation ofATP7A localization was inhibited by Atox1 small interferingRNA (Figure 7C and 7D).
Effect of Ang II on Vascular Copper Levels in WTMice and Atox1�/� MiceBecause copper exporter ATP7A is involved in deliveringcopper to the SOD3, which is secreted to the extracellularspace, we examined the copper levels in aortas from WT andAtox1�/� mice with or without Ang II infusion. Inductivelycoupled plasma mass spectrometry (Figure 8A) andsynchrotron-based x-ray fluorescence microscopy analysis(Figure 8B) show that Ang II infusion significantly decreasedcopper levels in the medial layers of the aorta. Specificdecrease in copper levels was normalized by sulfur levels,which was used as a surrogate for total cellular protein and tovisualize the morphology of tissue sections.23 By contrast,
Atox1�/� mice showed increased copper levels in the aorta atbasal state, which was not affected by Ang II infusion (Figure8A and 8B).
DiscussionPrevious studies have implicated a protective role forintracellular SOD1, as well as extracellular SOD3, inlimiting endothelial dysfunction produced by Ang II.4,5,24
In the present study, we demonstrate that Ang II infusionincreases Atox1 expression in aortas of WT mice. Activityand expression of SOD3 but not those of SOD1 aredecreased in Atox1�/� mice treated with Ang II. This leadsto exaggerated Ang II–induced vascular O2
·� production,thereby promoting Ang II–induced endothelial dysfunctionand vasoconstriction, which, in turn, augments systolicblood pressure in Atox1�/� mice. These changes inducedby Atox1 deficiency are rescued by adding the SODmimetic Tempol. Thus, Atox1 functions to prevent AngII–induced endothelial dysfunction and hypertension byreducing O2
·� levels via increasing vascular SOD3 expres-sion and activity. Moreover, Atox1 regulates copper levelsin the vasculature during Ang II–induced hypertension.
Impairment of endothelium-dependent acetylcholine-induced relaxation in mesenteric arteries from Ang II–infusedAtox1�/� mice is likely attributed to a decrease in endothelialNO· bioavailability, because endothelium-independent vaso-dilation is not affected by Ang II in Atox1�/� mice. There issignificant reduction in vascular reactivity in vehicle-infusedAtox1�/� mice, which may be in part because of the slightbut not significant increase in O2
·� levels in these mice atbaseline. However, Ang II–induced hypertension but notbasal blood pressure is significantly altered in Atox1�/�
mice. In addition, it has been reported that Ang II increasesvasoconstriction partially through O2
·�-induced inactivationof vasorelaxant NO,1 although this response depends onvascular bed and genetic background.25,26 In the presentstudy, we found that contractions to the Ang II but not KCl
Promoter activity BA DNA pull down assay
Input
Probes for DNA pull down
: Biotinylatedprobe
IB: anti-Atox-1
Ang II - + - + - +
Atox1
SOD3(-333/-304)
SOD3(-333/-304)
Mut(-313/-304)
0
1
2
3
4
5
Fold
Incr
ease
pGL3 SOD3(-2500/+104)
-AngII+AngII
pGL3 SOD3(-2500/+104)
Mut(-312/-307)
*
Figure 6. Role of antioxidant 1 (Atox1) in angiotensin II (Ang II)–induced superoxide dismutase (SOD) 3 promoter activity in vascularsmooth muscle cells (VSMCs). A, Role of Atox1 on Ang II–induced transactivation of the SOD3 gene promoter in VSMCs. Cells weretransiently transfected with SOD3 promoter luciferase reporter constructs (pGL3 SOD3; �2500/�104) or mutated SOD3 promoterluciferase reporter constructs (pGL3 SOD3; Mut[�312/�307]) along with Ang II (mean�SE; n�3). *P�0.001 vs untreated cells. B, DNApull-down assay, showing Ang II–induced binding of Atox1 to the GAAAGA sequence (Atox1-reponsive element; Atox1-RE) in the SOD3promoter. Nuclear extract from VSMCs treated with or without Ang II was incubated with biotinylated oligonuclotide probes andstreptavidin-sepharose. The protein-DNA complexes were subjected to SDS-PAGE, followed by immunoblotting with anti-Atox1 anti-body. The experiments was performed using 2 different oligonucleotides, SOD3 (�333/�304) and SOD3 (�333/�304) Mut (�313/�304). Representative figure was from 3 independent experiments.
6 Hypertension August 2012
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from
are significantly increased in mesenteric arteries of theAtox1�/� mice compared with WT mice. This is inhibited bythe SOD mimetic Tempol. Thus, the loss of NO bioavailabil-ity induced by decrease in SOD3 activity and subsequentincrease in O2
·� levels in Atox1-deficient arteries may con-tribute not only to impaired endothelium-dependent vasodi-lation but also to enhanced Ang II–induced vasoconstriction.This, in turn, likely increases systemic vascular resistance inAtox1�/� mice during Ang II infusion and, thus, augmentsthe hypertensive response observed in these animals. Of note,it has been shown that endothelium-dependent vasorelaxationin mesenteric arteries depends on not only NO but alsoepoxyeicosatrienoic acids and H2O2, which have been pro-posed as endothelium-dependent hyperpolarizing factor.27
Thus, Atox-1�/� mice have increased vascular O2·�, which
would scavenge NO but also may have reduced vascularH2O2 to inhibit action of endothelium-dependent hyperpolar-izing factors. This point requires further investigation infuture study.
We found that the levels of total SOD activity were similarbetween WT and Atox1�/� mice and were not changed byAng II infusion. This result may reflect the fact that SOD1activity, which consists of more than half of total SODactivity, is not dependent on Atox1. Nevertheless, the dra-matic decrease in SOD3 activity by Atox1 deficiency likelyhas a major impact on increasing O2
·� levels in the interstitial
space, where this enzyme is localized. This will efficientlypromote oxidative inactivation of NO, which traverses fromthe endothelial cells to VSMCs, thereby preventingendothelium-dependent vasorelaxation, which, in turn, in-creases blood pressure in Atox1�/� mice, as reported forSOD3�/� mice.2 Of note, endothelial dysfunction in thecoronary circulation of humans has profound prognosticimplications in that it predicts adverse cardiovascular eventsand long-term outcome.28 The R213G polymorphism in theSOD3 gene, which reduces binding to the endothelial surfaceand increases serum SOD3 levels,3 has been linked to anincrease in cardiovascular risk.29 Thus, the protective role ofAtox1 in endothelial function through SOD3 would havepotential clinical impact.
In this study, SOD3 protein and mRNA expression, aswell as nuclear Atox1 expression in blood vessels, areincreased by Ang II infusion, which is abolished inAtox1�/� mice. Consistent with our results, Hamza et al30
originally showed that Atox1 is localized in the nucleus incultured cells; however, functional significance of nuclearAtox1 was not addressed. Using promoter analysis andDNA pull-down assay in cultured VSMCs, we demonstratethat Ang II stimulation promotes translocation of Atox1from the cytosol to the nucleus, as well as Atox1 bindingto the Atox1 response element in the SOD3 promoter. Thisobservation is consistent with our earlier study that Atox1
A
IB:ATP7A
Ang II 0 15 30 60
IP: Atox1
IB:Atox1
(min)
B
IB:ATP7AAng II 0 15 30 60 (min)
IB: Actin
IP: SOD3
C
ATP7A staining
ControlsiRNA
Atox1siRNA
- Ang II + Ang II
II
DC
ontr
olA
ng II
ATP7A SOD3 Merged Magnified
5µm
20µm 5µm
20µm
20µm
20µm 20µm
20µmAng II - + - +
ControlsiRNA
Atox1siRNA
% o
f ATP
7A o
ut o
f Gol
gi
(from
con
trol
) NS
*
0
10
20
30
40
50
60
Figure 7. Effect of angiotensin II (Ang II) on interaction among antioxidant 1 (Atox1), ATP7A, and superoxide dismutase (SOD; ecSOD) 3in vascular smooth muscle cells (VSMCs). A and B, VSMCs were stimulated with Ang II (100 nmol/L), and lysates were immunoprecipi-tated (IP) with either anti-Atox1 or anti-ecSOD antibody, followed by immunoblotting (IB) with anti-ATP7A antibody. C, Effect of Ang IIon subcellular localization of ATP7A (green) and SOD3 (red) in human aortic smooth muscle cells (HASMs). D, HASMs transfected withAtox1 or control small interfering RNAs (siRNAs) were stained with anti-ATP7A antibody. Images were representative of 3 differentexperiments taken at 5 different fields per well.
Ozumi et al Atox1, SOD3, and Hypertension 7
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

serves as a transcription factor for copper-induced increasein SOD3 and cyclin D1 expression in fibroblasts.9,12 Thepresent study also found that expression of other copper-containing enzymes, including lysyl oxidase, ceruloplas-min, or SOD1, was not changed in Atox1�/� arteries ascompared with WT with or without Ang II infusion. Thisis consistent with the fact that SOD3 but not lysyl oxidase,ceruloplasmin, or SOD1 has Atox1-responsive elements inits promoters. Thus, the current study provides the firstevidence that “agonist-induced” transcription factor func-tion of Atox1 for SOD3 plays a potentially important rolein Ang II–induced upregulation of SOD3 expression inblood vessels, thereby modulating O2
·� levels and hyper-tensive responses induced by Ang II (Figure 9).
Copper chaperone function of Atox1 to deliver copper toSOD3 in Ang II–treated VSMCs is also demonstrated inthis study. We found that Ang II promotes Atox1 bindingto the copper exporter ATP7A, which obtains copper fromAtox1. It also stimulates ATP7A translocation from trans-Golgi network to plasma membrane, where it colocalizeswith SOD3. Consistent with our results, Atox1 is shown todeliver copper to secretory copper enzymes via interaction
with ATP7A in yeast.21,22 Furthermore, ATP7A is reportedto deliver copper to the secretory copper enzymes in thepost-Golgi vesicles rather than in trans-Golgi network,where copper loading normally takes place.31,32 We re-ported previously that Ang II infusion–induced increase inblood pressure and vascular O2
·� production are augmentedin ATP7A mutant mice because of a decrease in SOD3activity.33 In line with this, the present study provides theadditional new evidence that Ang II promotes copperchaperone function of Atox1 by facilitating formation ofthe Atox1/ATP7A/SOD3 complex, thereby increasing spe-cific SOD3 activity (Figure 9).
The association of copper metabolism with hypertensionhas been implicated.13 However, there is no informationregarding vascular copper levels in hypertension or in-volvement of Atox1 in this response. The present studyshows that Ang II treatment significantly decreases vascu-lar copper levels, as assessed by inductively coupledplasma mass spectrometry and synchrotron-based x-rayfluorescence microscopy analysis, which is inhibited inAtox1�/� mice. This might in part reflect the Ang II–stimulated secretion of copper-loaded SOD3 to the circu-
0- + - +
WT Atox1 KO
Ang II
*
10
20
30
Aor
tic C
u ( µ
g/g
dry
wt.)
ICP-MS analysisA
#
Synchrotron X-ray Fluorescence (SXRF) AnalysisB
Ang II - + - +
WT Atox1 KO
0.002
0.003
0.004
0.005
0.006
0.007
Med
ial C
u/S
ratio
0.001
*Ang II
Cu S
Cu S
Cont
Atox1-KO
WT
Cu S
Cu S
min max
0.004-0.060 0.05-3.80 0.004-0.060 0.05-3.80 (µg/cm 2)
Figure 8. Effect of angiotensin II (Ang II)–induced hypertension on abundance and spatial distribution of copper in the vascular tis-sue from antioxidant 1 (Atox1) 1�/� and wild-type (WT) mice. A and B, Aortic copper content in Atox1�/� and WT mice infusedwith Ang II or vehicle for 7 days, as described in Figure 1, was measured by inductively coupled plasma mass spectrometry (ICP-MS) or synchrotron-based x-ray fluorescence (SXRF). SXRF scans (1–2 seconds per pixel) were performed in paraffin-embeddedtissue (left). The maximum and minimum threshold values in microgram per square centimeter are given above each frame. Mapof copper shows areas of the lowest to the highest content scaled to a rainbow color (bottom). Total sulfur is used as a surro-gate for total cellular protein and to visualize the morphology of tissue sections. Data are quantified using 3 samples for eachgroup, 2 to 3 scans per sample (right).
8 Hypertension August 2012
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

lation via the Atox1-ATP7A pathway, despite the fact thatthe majority of SOD3 will bind to the extracellular matrix.In addition, Ang II–induced decrease in vascular copperlevels may be also because of secretion of secretorycopper-containing proteins or excess copper to protectcells from its toxicity. This is consistent with a previousstudy that showed that levels of copper in some tissues,such as liver and kidney, are significantly lower inhypertensive rodents.14 The physiological consequence ofcopper export via ATP7A is placental copper transport tothe developing fetus during pregnancy or providing copperas part of a neuronal protective mechanism.34 Thus, thepresent study provides the first evidence that vascularcopper levels are altered during Ang II–induced hyperten-sion, which is at least in part regulated by Atox1. Detailedanalysis of molecular mechanism and functional signifi-cance of altered copper levels in hypertension requiresfuture investigation.
Previous studies have shown that SOD3�/� mice havenormal blood pressure at baseline, but Ang II–inducedincrease in blood pressure is augmented in these mice in atime-dependent manner with a peak at day 7, which remainedelevated at least until day 14.5 Consistent with this, thepresent study found that Atox1�/� mice showed similartime course of exaggerated hypertensive response to AngII, as reported for SOD3�/� mice. It has been reported thatdeletion of SOD3 in the circumventricular organ in thebrain increases blood pressure in the basal state and after
Ang II infusion, in part by modulating sympathetic out-flow.35 Intriguingly, Atox1 expression is observed inseveral regions of brain, including the choroid plexus,which belongs to the circumventricular organ, whereATP7A is also expressed.36 Thus, it is conceivable thatAtox1 may be involved in the regulation of blood pressure,either by regulation of SOD3 in the circumventricularorgan or by other secretory copper enzymes in the brain.Atox1 is also expressed in the kidney, including glomeruliin both the juxtramedullary and cortical nephrons andmedulla associated with the loops of Henle,37 whichregulates oxidative stress-dependent hypertension.38 Inparticular, O2
·� production in the brain is required for thegenesis of hypertension.35,39 In the current study, we useda noninvasive tail-cuff method to measure blood pressure.However, it does not give the information, such as 24-hourblood pressure and blood pressure variability. Thus,Atox1-SOD3–mediated regulation of Ang II–induced hy-pertension should be further confirmed using a telemetrysystem. Furthermore, investigating the role of Atox1 inother oxidative stress– dependent pathophysiologies, suchas diabetes mellitus, obesity, and atherosclerosis, in whichblood pressure is not consistently increased,38 is thesubject of future studies.
PerspectivesThe present study provides compelling evidence that Atox1plays an important role in regulating vascular function andhypertension induced by Ang II in vivo by decreasing
Ang II
O2•-
NO•
HypertensionEndothelial Dysfunction
Vasoconstriction
(active)Atox1
TGN
ecSODprotein
Cu+
(active)
Cu+
ecSODprotein
Cu+
Nuclear ecSODmRNA
Atox RE
transcription ecSOD geneCu+
ecSODprotein
Cu+
ATP7A
ATP7A
Cu+
CTR1 Cu+
Vascular Smooth Muscle
?
Ang II
AT1aR
Figure 9. Proposed model for protective role of antioxidant 1 (Atox1) in angiotensin II (Ang II)–induced hypertension through regulatingsuperoxide dismutase (SOD) 3 expression and activity. Atox1 plays an important role in Ang II–induced transcription and activity ofSOD3 by increasing Atox1 binding to the Atox1 response element in the SOD3 promoter, as well as to copper transporter ATP7A pro-tein, required for copper delivery to SOD3 for enhancing its specific activity. As its consequence, Atox1 contributes to the decrease inextracellular superoxide (O2
·�) levels in the vessel wall, thereby increasing available NO to preserve endothelial function and reduceblood pressure. TGN indicates trans-Golgi network.
Ozumi et al Atox1, SOD3, and Hypertension 9
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

extracellular O2·� and increasing bioavailability of NO
through copper chaperone and transcription factor functionfor SOD3 in blood vessels (Figure 9). Atox1 is also involvedin regulating copper homeostasis during Ang II–inducedhypertension. Our findings provide novel insight into Atox1as a potential therapeutic target for the treatment of hyper-tension and various other oxidative stress-dependent cardio-vascular diseases.
AcknowledgmentsUse of the Advanced Photon Source at Argonne National Laboratorywas supported by the US Department of Energy, Office of Science,Office of Basic Energy Sciences, under contract No. DE-AC02-06CH11357.
Sources of FundingThis research was supported by National Institutes of Health grantR01 HL070187 (to T.F.), a Veterans Affairs MERIT grant (toT.F.), and grants R01 HL077524 and HL077524-S1 (to M.U.-F.);American Heart Association postdoctoral fellowship11POST5740006 (to V.S.), and Ruth L. Kirschstein-NationalService Research Award T32 training grant (to G.-F.C.).
DisclosuresNone.
References1. Garrido AM, Griendling KK. NADPH oxidases and angiotensin II
receptor signaling. Mol Cell Endocrinol. 2009;302:148–158.2. Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling,
vascular function and diseases. Antioxid Redox Signal. 2011;15:1583–1606.
3. Chu Y, Alwahdani A, Iida S, Lund DD, Faraci FM, Heistad DD. Vasculareffects of the human extracellular superoxide dismutase R213G variant.Circulation. 2005;112:1047–1053.
4. Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP.Extracellular superoxide dismutase is a major determinant of nitric oxidebioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice.Circ Res. 2003;93:622–629.
5. Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S,Fukai T, Harrison DG. Role of extracellular superoxide dismutase inhypertension. Hypertension. 2006;48:473–481.
6. Fukai T, Siegfried MR, Ushio-Fukai M, Griendling KK, Harrison DG.Modulation of extracellular superoxide dismutase expression by angio-tensin II and hypertension. Circ Res. 1999;85:23–28.
7. Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Unde-tectable intracellular free copper: the requirement of a copper chaperonefor superoxide dismutase. Science. 1999;284:805–808.
8. Culotta VC, Yang M, O’Halloran TV. Activation of superoxide dis-mutases: putting the metal to the pedal. Biochim Biophys Acta. 2006;1763:747–758.
9. Itoh S, Ozumi K, Kim HW, Nakagawa O, McKinney RD, Folz RJ, ZelkoIN, Ushio-Fukai M, Fukai T. Novel mechanism for regulation of extra-cellular SOD transcription and activity by copper: role of antioxidant-1.Free Radic Biol Med. 2009;46:95–104.
10. Jeney V, Itoh S, Wendt M, Gradek Q, Ushio-Fukai M, Harrison DG,Fukai T. Role of antioxidant-1 in extracellular superoxide dismutasefunction and expression. Circ Res. 2005;96:723–729.
11. Qin Z, Itoh S, Jeney V, Ushio-Fukai M, Fukai T. Essential role forthe Menkes ATPase in activation of extracellular superoxide dis-mutase: implication for vascular oxidative stress. FASEB J. 2006;20:334–336.
12. Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, AkramK, McKinney RD, Ushio-Fukai M, Fukai T. Novel role of antioxidant-1(Atox1) as a copper-dependent transcription factor involved in cell pro-liferation. J Biol Chem. 2008;283:9157–9167.
13. Klevay LM. Cardiovascular disease from copper deficiency: a history.J Nutr. 2000;130:489S–492S.
14. Clegg MS, Ferrell F, Keen CL. Hypertension-induced alterations incopper and zinc metabolism in Dahl rats. Hypertension. 1987;9:624–628.
15. Garrow TA, Clegg MS, Metzler G, Keen CL. Influence of hypertensionand dietary copper on indexes of copper status in rats. Hypertension.1991;17:793–797.
16. Ahmed T, Sackner MA. Increased serum copper in primary pulmonaryhypertension: a possible pathogenic link? Respiration. 1985;47:243–246.
17. Olatunbosun DA, Bolodeoku JO, Cole TO, Adadevoh BK. Relationshipof serum copper and zinc to human hypertension in Nigerians. Bull WorldHealth Organ. 1976;53:134–135.
18. Medeiros DM, Lin KM, Liu C, Thorne BM. Pre-gestation dietary copperrestriction and blood pressure in the Long-Evans rat. Nutr Rep Int.1984;30:559–564.
19. Klevay LM. Hypertension in rats due to copper deficiency. Nutr Rep Int.1987;35:999–1005.
20. Hamza I, Faisst A, Prohaska J, Chen J, Gruss P, Gitlin JD. The metal-lochaperone Atox1 plays a critical role in perinatal copper homeostasis.Proc Natl Acad Sci U S A. 2001;98:6848–6852.
21. Hung IH, Casareno RL, Labesse G, Mathews FS, Gitlin JD. HAH1 is acopper-binding protein with distinct amino acid residues mediatingcopper homeostasis and antioxidant defense. J Biol Chem. 1998;273:1749–1754.
22. Pufahl RA, Singer CP, Peariso KL, Lin SJ, Schmidt PJ, Fahrni CJ,Culotta VC, Penner-Hahn JE, O’Halloran TV. Metal ion chaperonefunction of the soluble Cu(I) receptor Atx1. Science. 1997;278:853–856.
23. Malinouski M, Kehr S, Finney L, Vogt S, Carlson BA, Seravalli J, Jin R,Handy DE, Park TJ, Loscalzo J, Hatfield DL, Gladyshev VN. High-resolution imaging of selenium in kidneys: a localized selenium poolassociated with glutathione peroxidase 3. Antioxid Redox Signal. 2012;16:185–192.
24. Didion SP, Kinzenbaw DA, Faraci FM. Critical role for CuZn-superoxidedismutase in preventing angiotensin II-induced endothelial dysfunction.Hypertension. 2005;46:1147–1153.
25. Su J, Palen DI, Boulares H, Matrougui K. Role of ACE/AT2R complexin the control of mesenteric resistance artery contraction induced byACE/AT1R complex activation in response to Ang I. Mol Cell Biochem.2008;311:1–7.
26. Russell A, Watts S. Vascular reactivity of isolated thoracic aorta of theC57BL/6J mouse. J Pharmacol Exp Ther. 2000;294:598–604.
27. Feletou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond). 2009;117:139–155.
28. Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronaryvasodilator dysfunction on adverse long-term outcome of coronary heartdisease. Circulation. 2000;101:1899–1906.
29. Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R,Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxi-dative protection and increased ischemic heart disease risk: theCopenhagen City Heart Study. Circulation. 2004;109:59–65.
30. Hamza I, Schaefer M, Klomp LW, Gitlin JD. Interaction of the copperchaperone HAH1 with the Wilson disease protein is essential for copperhomeostasis. Proc Natl Acad Sci U S A. 1999;96:13363–13368.
31. Setty SR, Tenza D, Sviderskaya EV, Bennett DC, Raposo G, Marks MS.Cell-specific ATP7A transport sustains copper-dependent tyrosinaseactivity in melanosomes. Nature. 2008;454:1142–1146.
32. Ashino T, Sudhahar V, Urao N, Oshikawa J, Chen GF, Wang H, Huo Y,Finney L, Vogt S, McKinney RD, Maryon EB, Kaplan JH, Ushio-FukaiM, Fukai T. Unexpected role of the copper transporter ATP7A in PDGF-induced vascular smooth muscle cell migration. Circ Res. 2010;107:787–799.
33. Qin Z, Gongora MC, Ozumi K, Itoh S, Akram K, Ushio-Fukai M,Harrison DG, Fukai T. Role of Menkes ATPase in angiotensin II-inducedhypertension: a key modulator for extracellular superoxide dismutasefunction. Hypertension. 2008;52:945–951.
34. La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7Aand ATP7B: role in copper homeostasis. Arch Biochem Biophys. 2007;463:149–167.
35. Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C,Gordon FJ, Harrison DG. Induction of hypertension and peripheralinflammation by reduction of extracellular superoxide dismutase in thecentral nervous system. Hypertension. 2010;55:277–283.
10 Hypertension August 2012
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

36. Nishihara E, Furuyama T, Yamashita S, Mori N. Expression of coppertrafficking genes in the mouse brain. Neuroreport. 1998;9:3259–3263.
37. Moore SD, Helmle KE, Prat LM, Cox DW. Tissue localization of thecopper chaperone ATOX1 and its potential role in disease. MammGenome. 2002;13:563–568.
38. Harrison DG, Gongora MC. Oxidative stress and hypertension. Med ClinNorth Am. 2009;93:621–635.
39. Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role ofvascular extracellular superoxide dismutase in hypertension. Hypertension.2011;58:232–239.
Novelty and Significance
What Is New?● Copper transport protein Atox1 plays an important role in preventing
endothelial dysfunction, augmented vasoconstriction, and hypertensioninduced by Ang II in vivo.
● Vascular Atox1 modulates NO-mediated vascular function via copperchaperone and transcription factor for SOD3, which scavenges extra-cellular O2
·�.● Vascular Atox1 regulates copper homeostasis in Ang II–induced
hypertension.
What Is Relevant?● Atox1 regulates blood pressure and vascular function in Ang II–induced
hypertension by regulating vascular SOD3 expression and activity.
Summary
Our findings provide novel insight into Atox1 as a potentialtherapeutic target for various oxidative stress-dependent cardio-vascular diseases, such as hypertension.
Ozumi et al Atox1, SOD3, and Hypertension 11
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Finney, Stefan Vogt, Ronald D. McKinney, Masuko Ushio-Fukai and Tohru FukaiKiyoshi Ozumi, Varadarajan Sudhahar, Ha Won Kim, Gin-Fu Chen, Takashi Kohno, Lydia
A Key Regulator of Extracellular Superoxide DismutaseInduced Hypertension:−Role of Copper Transport Protein Antioxidant 1 in Angiotensin II
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 2012 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension published online July 2, 2012;Hypertension.
http://hyper.ahajournals.org/content/early/2012/06/29/HYPERTENSIONAHA.111.189571World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org/content/suppl/2012/07/02/HYPERTENSIONAHA.111.189571.DC1Data Supplement (unedited) at:
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 3, 2018http://hyper.ahajournals.org/
Dow
nloaded from

1
ONLINE SUPPLEMENT
Role of copper transport protein Antioxidant-1 in Angiotensin II-induced
hypertension: A key regulator of Extracellular SOD
#Kiyoshi Ozumi
1, # Varadarajan Sudhahar
1, Ha Won Kim
1, Gin-Fu Chen
1, Takashi Kohno
1,
Lydia Finney3,4
, Stefan Vogt4, Ronald D. McKinney
1,2, Masuko Ushio-Fukai
2, Tohru Fukai
1
From 1Departments of Medicine (Section of Cardiology) and Pharmacology, Center for
Cardiovascular Research, Center for Lung and Vascular Biology, University of Illinois at
Chicago, Chicago, IL60612, 2Department of Pharmacology, Center for Lung and Vascular Biology, Center for Cardiovascular
Research, University of Illinois at Chicago, Chicago, IL60612 3Biosciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, IL
60439, USA 4X-ray Science Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, IL
60439, USA
# These authors contributed equally to the manuscript.
Short running title: Atox1, SOD3, and Hypertension
Address correspondence to: Tohru Fukai, M.D., Ph.D.
Depts. of Medicine and Pharmacology
University of Illinois at Chicago
835 S. Wolcott, M/C868, E403MSB
Chicago, IL 60612
Phone: 312-996-7631
Fax: 312-996-1225
Email: [email protected]

2
Supplemental Materials and Methods
Animals: Atox1-/-
mice (backcrossed eight times to C57Bl/6) were obtained from Mutant Mouse
Regional Resource Centers. Atox1-/-
mice were originally reported to have phenotypes which
show perinatal death (45% of pups) or survive more than one month.1 Our laboratory further
backcrossed to C57Bl/6 mice more than ten times, and thus used C57Bl/6 mice as control.
“Survivor” mice were intercrossed with more than 10 times and Atox1-/-
mice used in the present
study survived more than six months (90%). For the experiments, Atox1-/-
mice were weaned at
4 weeks of age and maintained on regular chow for 2 to 3 months. Diet and water were provided
ad libitum. All studies were approved by the Animal Care and Use Committee of the University
of Illinois-Chicago.
Mice were studied 3 month of age. Angiotensin II (Ang II) was delivered at a rate of 0.7 mg/ kg/
day for 7 days using osmotic minipumps (Alzet model 2002; Alza Corp) as previously
described.2,3
At some experiments, 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (Tempol)
dissolved in saline was administered in separate osmotic minipumps at a rate of 50 mg/kg/day.
Sham-operated animals underwent an identical surgical procedure, except that no pump was
implanted. Blood pressure was measured before and during infusion of Ang II (on days 7) using
the tail-cuff method (BP2000 Visitech System Inc.) as previously described.2,3
Immunohistochemical and Western blot analysis: Western analysis and immunohistochemical
analysis were performed, as previously described.4,5
The primary antibodies used includes a
rabbit polyclonal antibody against murine SOD3,
4 a sheep antibody against human SOD1, and
rabbit polyclonal antisera to Atox1. 6
Quantitative Real Time-polymerase chain reaction: Total RNA was isolated from aorta of
control and Atox1 KO mice using TRI reagent (Molecular Research Center, Inc) according to the
manufacturer’s instructions. 2 μg of RNA were used for synthesize first stranded cDNA with a
High-Capacity cDNA Reverse Transcription Kits. The PCR was performed according to the
manufacturer’s protocol using ABI PRISM® 7000 Sequence Detection System 26 (Applied
Biosystems, CA) and QuantiFast SYBR Green PCR Kit (Qiagen, CA). Amplification conditions
was performed with a 5 min preincubation at 95°C followed by 40 cycles of 10 s at 95°C and 30
s at 60°C. PCR products were subjected to melting curve analysis, using the ABI PRISM® 7000
Sequence Detection System, to exclude amplification of unspecific products, and separated on
agarose gel and stained with ethidium bromide. All real-time PCR primers were purchased from
predesigned primers of QuantiTect primer assays (Qiagen). Results were normalized by
glyceraldehyde-3-phosphate dehydrogenase expression levels.
Superoxide dismutase activity assays: Tissues were homogenized in 10 vol of 50 mmol/L
potassium phosphate (pH 7.4) containing 0.3 mol/L KBr and a cocktail of protease inhibitors
(Roche Applied Science, Indianapolis, Ind). SOD activity was measured in 50 mmol/L
phosphate buffer by inhibition of the reduction of cytochrome C (50 μmol/L) by superoxide
generated by xanthine (0.1 mmol/L) and xanthine oxidase (0.01 U/mL) at pH 7.4 as described
previously7. Cyanide (3 mmol/L) was used to distinguish between the cyanide-sensitive
isozymes Cu/Zn SOD and ecSOD and the cyanide-resistant Mn SOD. To isolate SOD3 from
vessels of MNKmut
mice and their control littermates, Con A-Sepharose chromatography
(Pharmacia Biotech, Piscataway, NJ) was used. Unlike Cu/Zn SOD and Mn SOD, the

3
glycoprotein in ecSOD binds to the lectin concanavalin A. The isolation procedure was
performed as previously described.8 Briefly, samples were applied to a Con A-Sepharose
column equilibrated with 50 mmol/L potassium phosphate buffer (pH 7.4) in 120 mmol/L NaCl.
After collecting the eluting fluid, which contains Cu/ZnSOD and MnSOD, ecSOD fraction was
eluted with 150 mmol/L α methyl mannoside in 50 mmol/L potassium phosphate buffer (pH
7.4).
Cell culture and nuclear fractionation: vascular smooth muscle cells (VSMCs) were isolated
from male Sprague-Dawley rat thoracic aortas by enzymatic digestion. Cells were grown in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine serum and used
at between passage 7 and 15. For some experiments, human aortic smooth muscle cells (HASM,
Invitrogen, Grand Island, NY) were studied. HASM were cultured in Smooth Muscle Basal
Medium (Invitrogen, Grand Island, NY). Experiments were performed with 0.05% serum and no
additives at passages 4 to 8. Before stimulation with Ang II, cells were starved for 24 hours with
serum free media. Nuclear/cytoplasmic fractionation of RASM was performed using an NE-
PER Nuclear and Cytoplasmic Extraction Reagents Kit (Pierce) according to manufacturer’s
protocol.
Immunofluorescence in VSMC: VSMCs on glass coverslips were rinsed quickly in ice-cold
PBS, fixed in freshly prepared 4% paraformaldehyde in PBS for 10 min at room temperature,
permeabilized in 0.05% Triton X-100 in PBS for 5 min, and rinsed sequentially in PBS, 50
mmol/L NH4Cl and PBS for 10 min each. The cells were incubated with a blocking buffer (3%
bovine serum albumin (BSA) in PBS), and subsequently incubated with Atox1 antibody for 18 h
at 4°C, rinsed in PBS/BSA, and then incubated in Alexa Fluor 546-conjugated goat anti-rabbit
IgG for 1 h at room temperature. Cells on cover slips were mounted onto glass slides in
Vectashield (Vector Laboratories) and were examined by confocal microscopy with Zeiss 510
LSCM system, using argon and green helium/neon laser excitation lines of 488 and
543 nm with
emission filters of BP 500-550 and LP 560.
Plasmid constructs: The promoter-reporter constructs consist of 5’ regions of the SOD3
promoter were inserted into the luciferase reporter vector pGL3-Basic (pGL3-SOD3 -2500/104)
as previously described 9. Site mutagenesis (GAAAGA to TCCCTA) of the SOD3 -2500/104
construct (-312 to -307 region) was done using Quick-change II XL (Agilent Technologies), as
previously described10
.
Transient transfection and Reporter Assay: The pGL3-SOD3 constructs were transfected into
VSMCs along with pRL-CMV (Promega), using Polyfect reagent (Qiagen), according to the
manufacturer’s protocol. Forty-eight hours after transfection, luciferase activity was measured
using a luminometer. Transfection efficiency was normalized on the basis of Renilla luciferase
activity.
DNA pull-down assay: Nuclear extracts from VSMC were prepared as mentioned above.
Synthetic complementary oligonucleotides were 3’-biotinylated using biotin 3’-end DNA
labeling kit (Pierce) according to the manufacturer’s instructions. After labeling, complementary
strands were mixed together in equimolar ratio and allowed to anneal for 1 h at 37°C to form the
double-stranded probe. The sequences of the oligonucleotides used were 5’-
TAATGAAAATGAAAGAAGATGCAGTCGCTG-3’ (-545/-516) and 5’-

4
TAATGAAAATTCCCTCAGATGCAGTCGC TG-3’ (probe3 -545/-516 with mutation of -535/-
530) were used. The DNA pull down assay was performed as previously described with minor
modifications 11
. Briefly, nuclear extract was incubated with each biotinylated double-stranded
DNA probes (200 pmol) and 20 ul of neutravidin coated agarose beads (Pierce) in the presence
of 50 ng/ul poly (dI-dC), 0.05% NP-40, 5 mM MgCl2, 10 mM EDTA, and 2.5% glycerol in 1X
binding buffer (LightShiftTM chemiluminescent EMSA kit, Pierce) for 6 h at 4 C. The protein-
DNA complexes were subjected to SDS-PAGE, followed by immunoblotting with anti-Atox1
antibody. Bound proteins were visualized by ECL (Pharmacia).
Measurements of vascular superoxide production: Animals were euthanized by CO2
inhalation. vascular O2·-
production was determined using lucigenin-enhanced
chemiluminescence as described before12
. Low concentrations of lucigenin (5 µmol/L) was
used to minimize redox cycling, in which the lucigenin radical can react with oxygen to generate
O2-
13-15
. In addition, SOD mimetic tempol was used to prove that the signal is derived from
O2-
.
Vascular reactivity studies: Isometric tension of mesenteric resistance arteries were measured
using wire myograph (Model 610M, Danish Myo Technology, Denmark) 2. Briefly, the first or
second order branches of resistance arteries were isolated from mice mesenteric bed, cut into ~2
mm segment and stored in cold Krebs Physiological Salt Solution (PSS) (119.0 mM NaCl, 25.0
mM NaHCO3, 4.6 mM KCl, 1.2 mM MgSO4, 1.8 mM CaCl2, 11.0 mM glucose) at pH 7.4. The
vessel were mounted in between two hook using tungsten wire (25 µm in diameter) in organ
chamber which containing Krebs PSS bubbled with gas mixture containing 5% CO2 and 95% O2
at 37°C. Basal tension was set on arteries stretched to L100, where L100 is defined as the
circumference of the relaxed artery exposed to a transmural pressure of 100 mmHg and
equilibrated for 1 hr. After equilibration, the arteries were exposed to high concentration of KCl
(80 mM) and 10 μM noradrenalin for 2-3 min until reproducible maximal contractions occur.
The α-adrenergic receptor agonist, phenylephrine was added to increase basal tension to 60% to
80% of maximal KCl contraction. Cumulative concentration (0.01-10 μM) of acetylcholine
(ACh) were added to the bathing solution every 5 min. The vessels were rinsed and pretreated
with tempol, an SOD mimetic (1 mM) and the concentration response was repeated to determine
the contribution of O2-
in ACh induced vasodilation. At the end of the each experiment,
cumulative concentration of sodium nitropruside (0.01-10 μM) was added to the bath to
demonstrate the intact smooth muscle function. Results are expressed as percent relaxation of the
phenylephrine-treated arteries, with 100% relaxation representing basal tension. For
vasoconstriction study, dose-dependent vasoconstriction response curves Ang II (0.1 to 100 nM)
or Phenylephrine (0.0001-1 μM) were each studied in mesenteric resistance arteries. Results are
expressed as percent constriction of the phenylephrine-treated rings, with 100% relaxation
representing basal tension.
Copper Measurements by Inductively coupled plasma mass spectrometry (ICP-MS): Samples were diluted to 2.0 ml with deionized water containing 5 % v/v nitric acid. The copper
contents were analyzed by ICP-MS, using a PlasmaQuad3, as previously described 5. Copper
concentrations were calculated from calibration curves, and values for water
blank were
subtracted.

5
Synchrotron X-ray Fluorescence Microscopy: Sections (5 m thick) of formalin-fixed
paraffin-embedded vascular tissue were used. For X-ray imaging, the sections were mounted
intact on silicon nitride windows (area, 2 × 2 mm; thickness, 200 nm) manufactured by Silson
(Blisworth, U.K.) and attached by brief heating to 55°C, as previously described16
. Specimens
were imaged with the scanning X-ray fluorescence microprobe at beamline 2-ID-E of the
Advanced Photon Source (Argonne, IL). Undulator-generated x-rays of 10-keV incident energy
were monochromatized with a single bounce Si <111> monochromator and focused to a
measured spot size of 0.3 x 0.5 m using Fresnel zone plate optics (X-radia, Concord, CA).
Sections were raster-scanned in steps of 4.0 m, and fluorescence spectra were collected for 1- to
2-sec dwell times by using a single-element silicon drift detector (Vortex-EX, SII
Nanotechnology, CA). Quantitation and image-processing of the X-ray fluorescence (XRF) data
sets was performed with MAPS software 17
. Quantitation of elemental content was achieved by
fitting XRF spectra at each pixel, and comparing against a calibration curve derived from
measurements of thin-film standards NBS-1832 and NBS-1833 (National Bureau of Standards,
Gaithersburg, MD).
Statistical analysis: Statistical analysis was performed using the one-way analysis of variance
(ANOVA), with the Scheffe test for post hoc comparisons when significance was determined by
ANOVA or Student’s t-test at the analysis indicated in the figures. The accepted level of
significance was set at p < 0.05.

6
References
1. Hamza I, Faisst A, Prohaska J, Chen J, Gruss P, Gitlin JD. The metallochaperone Atox1
plays a critical role in perinatal copper homeostasis. Proc Natl Acad Sci U S A.
2001;98:6848-6852.
2. Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T,
Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension.
2006;48:473-481.
3. Qin Z, Gongora MC, Ozumi K, Itoh S, Akram K, Ushio-Fukai M, Harrison DG, Fukai T.
Role of Menkes ATPase in angiotensin II-induced hypertension: a key modulator for
extracellular superoxide dismutase function. Hypertension. 2008;52:945-951.
4. Fukai T, Galis ZS, Meng XP, Parthasarathy S, Harrison DG. Vascular expression of
extracellular superoxide dismutase in atherosclerosis. J Clin Invest. 1998;101:2101-2111.
5. Jeney V, Itoh S, Wendt M, Gradek Q, Ushio-Fukai M, Harrison DG, Fukai T. Role of
antioxidant-1 in extracellular superoxide dismutase function and expression. Circ Res.
2005;96:723-729.
6. Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, Akram K, McKinney RD,
Ushio-Fukai M, Fukai T. Novel role of antioxidant-1 (Atox1) as a copper-dependent
transcription factor involved in cell proliferation. J Biol Chem. 2008;283:9157-9167.
7. Crapo JD, McCord JM, Fridovich I. Preparation and Assay of Superoxide Dismutases.
Methods Enzymol. 1978;53:382-393.
8. Marklund SL. Extracellular superoxide dismutase in human tissues and human cell lines.
J Clin Invest. 1984;74:1398-1403.
9. Zelko IN, Folz RJ. Myeloid zinc finger (MZF)-like, Kruppel-like and Ets families of
transcription factors determine the cell-specific expression of mouse extracellular
superoxide dismutase. Biochem J. 2003;369:375-386.
10. Itoh S, Ozumi K, Kim HW, Nakagawa O, McKinney RD, Folz RJ, Zelko IN, Ushio-
Fukai M, Fukai T. Novel mechanism for regulation of extracellular SOD transcription
and activity by copper: role of antioxidant-1. Free Radic Biol Med. 2009;46:95-104.
11. Fukada T, Tonks NK. Identification of YB-1 as a regulator of PTP1B expression:
implications for regulation of insulin and cytokine signaling. EMBO J. 2003;22:479-493.
12. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE,
Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell
nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201-1209.
13. Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide,
superoxide, hydrogen peroxide, and peroxynitrite. Circ Res. 2001;89:224-236.
14. Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in
cardiovascular studies. Hypertension. 2007;49:717-727.
15. Li Y, Zhu H, Kuppusamy P, Roubaud V, Zweier JL, Trush MA. Validation of lucigenin
(bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion
radical production by enzymatic and cellular systems. J Biol Chem. 1998;273:2015-2023.
16. Finney L, Mandava S, Ursos L, Zhang W, Rodi D, Vogt S, Legnini D, Maser J, Ikpatt F,
Olopade OI, Glesne D. X-ray fluorescence microscopy reveals large-scale relocalization
and extracellular translocation of cellular copper during angiogenesis. Proc Natl Acad Sci
U S A. 2007;104:2247-2252.
17. Vogt S. MAPS: A set of software tools for analysis and visualization of 3D X-ray
fluorescence data sets. J Phys IV. 2003;104:635-638.

7
Figure S1: Body weight, the weight of various organs (heart, kidney, Lung, and brain) in
Atox1-/-
and WT mice. The results are presented as mean ± SE from three separate experiments.
NS, not significant

8
Figure S2: Total SOD activity in aortas of WT (C57Bl/6, white bars) and Atox1-/-
(black bars)
mice. Mice were treated as in Figure 1. SOD activity assay was performed as in Figure 2 and
the results are presented as mean ± SE from three separate experiments. NS, not significant vs
either untreated WT or untreated Atox1-/-
mice.
9
Figure S3: Protein levels of ceruloplasmin, lysyl oxidase (LOX), and Atox1 in aortas of WT and
Atox1-/-
mice before and after treatment with angiotensin II. Representative Western blots for
ceruloplasmin, LOX, and Atox1 in aortic protein extracts are shown. Data are representative of
3 different experiments.
10
Figure S4: Contraction of mesenteric arteries to Ang II and phenylephrine in WT and Atox1-/-
mice. Dose-dependent vasoconstriction response was induced by Ang II in absence or presence
of the SOD mimetic tempol or by phenylephrine in mesenteric resistance arteries. Values are
mean ± SE. * P<0.001 vs. untreated WT mice. NS, not significant





























