Embed Size (px)
Citation preview

1452 Chem. Commun., 2013, 49, 1452--1454 This journal is c The Royal Society of Chemistry 2013
Cite this: Chem. Commun.,2013,49, 1452
Functional conversion of nickel-containingmetalloproteins via molecular design: from a truncatedacetyl-coenzyme A synthase to a nickel superoxidedismutase†
Yi Liu,a Qingli Wang,b Yaozhu Wei,a Ying-Wu Lin,c Wei Li,a Ji-Hu Su,d Zhen Wang,e
Yang Tian,e Zhong-Xian Huanga and Xiangshi Tan*a
Truncated acetyl-coenzyme A synthase (ACS) was successfully
converted into functional nickel superoxide dismutase (Ni-SOD)
by molecular design and the designed metalloproteins possess
new spectroscopic, structural, and electrochemical characteristics
required for catalyzing O2�� disproportionation, and exhibit
impressive Ni-SOD activity.
The diversity of functions for metalloproteins is mainly due tothe incorporated metal centers with various coordination envir-onments.1 Rational design of functional metalloproteins is apowerful strategy to reveal undiscovered structural and mecha-nistic features.1c The functional conversion of metalloproteinsby molecular design is a challenge to explain the structure–function relationship,1a and provides new insight into thefunctional evolution of metalloproteins. Significant advanceshave been made in functional design of new metalloproteinsthroughout the past few years,1c whereas most of the studieswere performed on iron-containing hemeproteins.2 We hereinreport the first investigation into a nickel-containing protein asthe engineering target, and successfully convert a truncatedacetyl-coenzyme A synthase (ACS)3 into Ni-SOD-like4 metallo-proteins by molecular design at the nickel site.
ACS and Ni-SOD have distinct properties and functions.Ni-SOD, which has been characterized in both reduced Ni2+
and oxidized Ni3+ states,4 catalyzes the disproportionation ofsuperoxide anions to dioxygen and hydrogen peroxide. In thereduced state, it displays a square-planar coordination
geometry, which resembles the [NiN2S2] coordination environ-ment found in the Nid site of ACS (Nid-ACS). Unlike Nid-ACS,the Ni ion in Ni-SOD involves in redox chemistry and switchesbetween Ni3+ and Ni2+ states during the catalytic cycle. WhenNi-SOD is in the oxidized state, the coordination geometry ofthe Ni3+ center is converted to square-pyramidal with His1moved to an axial position forming the fifth coordination bond.The axial Ni–N bond distance is relatively longer (2.3–2.6 Å)(Fig. 1B) than the regular one.4 Upon substitution of His1 withAsp or Ala, the catalytic activity of the Ni-SOD is completelylost,5 indicating the critical role of the axial His ligand.
ACS catalyzes the synthesis of acetyl-coenzyme A by conden-sing CO, coenzyme A, and a methyl group at the A-cluster,3
which consists of an [Fe4S4] cubane bridged to a [NipNid]subcomponent via the thiolate of Cys509 (Fig. S1, ESI†).3 Thedistal square-planar nickel (Nid) is ligated in a Cys-Gly-Cysmotif consisting of two deprotonated amides and two cysteinethiolates [NidN2S2]. The Nip is the catalytic metal for acetyl-CoAsynthesis, whereas the Nid is a redox-inactive metal andremains in the Ni2+ state throughout the catalytic cycle.3b
Fig. 1 Structures of (A) the truncated ACS protein ACS-a15 (PDB entry 3S2X) and(B) WT Ni-SOD (PDB entry 1T6U), and (C) the molecular model of the designed Ni-SOD-like metalloprotein F598H.
a Department of Chemistry & Institutes of Biomedical Sciences, Fudan University,
Shanghai 200433, China. E-mail: [email protected]; Fax: +86-21-65641740;
Tel: +86-21-55664475b College of Chemistry, Chemical Engineering and Materials Science,
Shandong Normal University, Jinan 250014, Chinac School of Chemistry and Chemical Engineering, University of South China,
Hengyang 421001, Chinad Department of Modern Physics, University of Science and Technology of China,
Hefei 230026, Chinae Department of Chemistry, Tongji University, Shanghai, China
† Electronic supplementary information (ESI) available: Experimental procedures.See DOI: 10.1039/c2cc38224e
Received 14th November 2012,Accepted 2nd January 2013
DOI: 10.1039/c2cc38224e
www.rsc.org/chemcomm
ChemComm
COMMUNICATION
Dow
nloa
ded
by F
udan
Uni
vers
ity o
n 02
Mar
ch 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2CC
3822
4E
View Article OnlineView Journal | View Issue
This journal is c The Royal Society of Chemistry 2013 Chem. Commun., 2013, 49, 1452--1454 1453
The truncated ACS (named ACS-a15, Fig. 1A),6 which containsthe C-terminal 136 residues of ACS, with the Nid-site butwithout the [Fe4S4] cluster, lost the ACS catalytic activity.
Although the recombinant ACS-a15 resembles the [NiN2S2]coordination environment of the reduced Ni-SOD, it does notexhibit any SOD activity. Upon addition of imidazole to serve asthe axial ligand of the Nid-ACS, ACS-a15 did not exhibit detectableSOD activity, which suggests that the imidazole in solution failedto be a functional axial ligand of the Nid-site. The flexibility of theaxial His1 imidazole in Ni-SOD is vitally important for stabilizingthe Ni3+ intermediate species4b,5 required for catalyzing O2
��
disproportionation. Therefore, we tried to introduce an axial Hisligand in the peptide chain of ACS-a15 by site-directed mutagen-esis. The crystal structure of ACS-a15 shows that Phe598 (accord-ing to the residue sequence number in ACS) is located aroundthe Nid-site within a distance of B4 Å to the Nid (Fig. 1A),6 andmolecular dynamics simulation shows that the replacement ofPhe598 by His could place the N atom of His at B2.73 Å from thenickel ion (Fig. 1C). This bond length obtained by energyminimization and equilibration is similar to that between theNd of His1 and the Ni ion in native Ni-SOD (2.67 Å, Fig. 1B).4b
Compared to ACS-a15, F598H exhibits a green color similar to thebis-amidate Ni2+ complex.7a The UV-Vis absorption spectrum ofF598H (Fig. S3, ESI†) displays two ligand field transitions at425 nm (B1495 M�1 cm�1) and 582 nm (B752 M�1 cm�1),respectively, which shift slightly to higher energy bands com-pared to those of ACS-a15 (Fig. S2, ESI†). The electronic absorp-tion at around B440 nm is due to a Ni(3dxy)/S(p)* - Ni(3dx2�y2/S/N(s))* transition,7 a characteristic of square-planar [NiN2S2]complexes. The differences in the absorption features can beexplained by destabilization of the Ni(3dx2�y2/S/N(s))* LUMO.7
Thus, the UV-Vis spectra indicated that the coordinationenvironment of the Ni ion in F598H is different from that ofNid in ACS-a15. In addition, the Ni2+ affinity constants (Kd) ofF598H (6.9 � 0.4 mM) and ACS-a15 (11.8 � 0.5 mM), which weredetermined by isothermal titration calorimetry (ITC) and spectro-scopic titration assays,8 indicated that F598H has a higher Ni2+-binding affinity than ACS-a15 (Fig. S4 and S5; Table S1, ESI†). Thesubstitution of Phe598 by His may form a possible five-coordinatedNi3+ species of F598H. As a result, the F598H exhibits a Ni-SOD-likeactivity of 2850 U mM�1 with controls of NiCl2 and the apo-F598H.This value is B16-fold lower than that of the native Ni-SOD fromS. coelicolor (45 292 U mM�1),9 but it is comparable to that of themetallopeptide with the complete natural ‘‘Ni-hook’’ structure(B2100 U mM�1) reported by Weston et al.9b The defective SODactivity of F598H might be due to the difference in the nickelcoordination environment, and/or the lack of the H-bond networkin the second sphere of the Ni site.
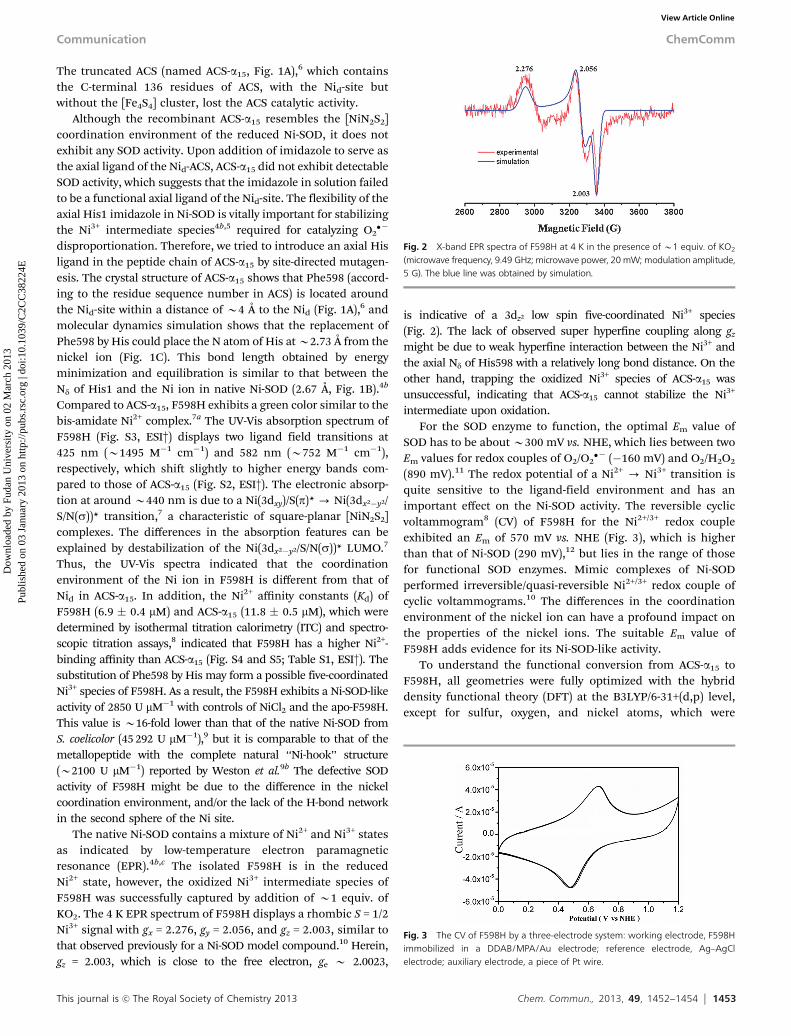
The native Ni-SOD contains a mixture of Ni2+ and Ni3+ statesas indicated by low-temperature electron paramagneticresonance (EPR).4b,c The isolated F598H is in the reducedNi2+ state, however, the oxidized Ni3+ intermediate species ofF598H was successfully captured by addition of B1 equiv. ofKO2. The 4 K EPR spectrum of F598H displays a rhombic S = 1/2Ni3+ signal with gx = 2.276, gy = 2.056, and gz = 2.003, similar tothat observed previously for a Ni-SOD model compound.10 Herein,gz = 2.003, which is close to the free electron, ge B 2.0023,
is indicative of a 3dz2 low spin five-coordinated Ni3+ species(Fig. 2). The lack of observed super hyperfine coupling along gz
might be due to weak hyperfine interaction between the Ni3+ andthe axial Nd of His598 with a relatively long bond distance. On theother hand, trapping the oxidized Ni3+ species of ACS-a15 wasunsuccessful, indicating that ACS-a15 cannot stabilize the Ni3+
intermediate upon oxidation.For the SOD enzyme to function, the optimal Em value of
SOD has to be about B300 mV vs. NHE, which lies between twoEm values for redox couples of O2/O2
�� (�160 mV) and O2/H2O2
(890 mV).11 The redox potential of a Ni2+ - Ni3+ transition isquite sensitive to the ligand-field environment and has animportant effect on the Ni-SOD activity. The reversible cyclicvoltammogram8 (CV) of F598H for the Ni2+/3+ redox coupleexhibited an Em of 570 mV vs. NHE (Fig. 3), which is higherthan that of Ni-SOD (290 mV),12 but lies in the range of thosefor functional SOD enzymes. Mimic complexes of Ni-SODperformed irreversible/quasi-reversible Ni2+/3+ redox couple ofcyclic voltammograms.10 The differences in the coordinationenvironment of the nickel ion can have a profound impact onthe properties of the nickel ions. The suitable Em value ofF598H adds evidence for its Ni-SOD-like activity.
To understand the functional conversion from ACS-a15 toF598H, all geometries were fully optimized with the hybriddensity functional theory (DFT) at the B3LYP/6-31+(d,p) level,except for sulfur, oxygen, and nickel atoms, which were
Fig. 2 X-band EPR spectra of F598H at 4 K in the presence of B1 equiv. of KO2
(microwave frequency, 9.49 GHz; microwave power, 20 mW; modulation amplitude,5 G). The blue line was obtained by simulation.
Fig. 3 The CV of F598H by a three-electrode system: working electrode, F598Himmobilized in a DDAB/MPA/Au electrode; reference electrode, Ag–AgClelectrode; auxiliary electrode, a piece of Pt wire.
Communication ChemComm
Dow
nloa
ded
by F
udan
Uni
vers
ity o
n 02
Mar
ch 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2CC
3822
4EView Article Online

1454 Chem. Commun., 2013, 49, 1452--1454 This journal is c The Royal Society of Chemistry 2013
optimized using the B3LYP/6-31+(2d,p) method.13,14 DFToptimized models of F598H and ACS-a15 are shown in Fig. S9(ESI†). The frontier molecular orbitals (FMO) of ACS-a15,F598H, and �OOH are depicted in Fig. 4 (in eV). The gapbetween HOMO F598H and LUMO �OOH is smaller than thecorresponding gap between HOMO ACS-a15 and LUMO �OOH.F598H has more flexibility than ACS-a15 in both oxidized andreduced states (Table S8, ESI†). These results indicate thatF598H can potentially catalyze O2
�� disproportionation easierthan ACS-a15. It also demonstrates the importance of the axialhistidine ligand in fine-tuning the reactive properties of theNi-SOD-like metalloproteins.
Moreover, the difference between F598H and native Ni-SODin the second sphere of the nickel active site should have aneffect on SOD activity. In native Ni-SOD, Tyr 9 (Fig. 1B) is nearthe vacant coordination site, opposite the His1 axial ligand inthe His-on structure and is involved in the H-bond networkwith two water molecules.4 The Tyr 9 residue is thought to playa key role in the catalysis by providing an H-bond to helppositioning the O2
�� substrate.12 Thus, to better replicate thesecondary coordination environment of the nickel ion, wedesigned two double-site mutants, S594Y/F598H and S594E/F598H (Fig. S7 and S8; Table S3, ESI†). Unfortunately, theattempts were unsuccessful for improving the SOD-activity.Unlike native Ni-SOD, either Tyr594 in S594Y/F598H orGlu594 in S594E/F598H, which is located at the same side asHis598 (Fig. S7, ESI†), likely prevents O2
�� from approachingthe Ni ion, due to the negative charge interaction.
In summary, the truncated metalloprotein ACS-a15 wassuccessfully converted into functional Ni-SOD-like enzyme byaltering the coordination environment of the Nid ion. Thedesigned metalloproteins develop new spectroscopic character-istics (UV-Vis and EPR), obtain redox properties required forcatalyzing the O2
�� disproportionation reaction, and exhibitimpressive Ni-SOD activity. Furthermore, molecular dynamicssimulation and DFT calculations provide evidential confirmationof the claim that F598H could catalyze the disproportionation ofsuperoxide radical anions. The axial histidine ligand is crucial forstabilizing the Ni3+-state and fine-tuning the reactive properties of
the Ni-SOD-like metalloproteins. The metal centers and theircoordination micro-environments, including coordinatedresidues and hydrogen bonds around the active site, determinethe functions of metalloproteins. This study presents a successfulmodel for the functional conversion of Ni-containing metallo-proteins by molecular design and provides new insight into thestructural and mechanistic features of Ni-SOD metalloproteins.
This work was supported partly by the National NaturalScience Foundation of China (No. 91013001, No. 20771029,No. 31270869 and No. 31070211), Shanghai Leading AcademicDiscipline Project (B108), the PhD Program of the EducationMinistry of China (20100071110011), the high magnetic fieldlaboratory of Chinese Academy of Science, and ShanghaiSynchrotron Radiation Facility (SSRF).
References1 (a) J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and
A. Volbeda, Nature, 2009, 460, 814–822; (b) K. J. Waldron,J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460,823–830; (c) Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature,2009, 460, 855–862; (d) R. Lill, Nature, 2009, 460, 831–838;(e) J. P. Collman, Y. Yang, A. Dey, R. A. Decreau, S. Ghosh, T. Ohtaand E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105,15660–15665.
2 (a) N. Yeung, Y. W. Lin, Y. G. Gao, X. Zhao, B. S. Russell, L. Lei,K. D. Miner, H. Robinson and Y. Lu, Nature, 2009, 462, 1079–1082;(b) S. I. Ozaki, M. P. Roach, T. Matsui and Y. Watanabe, Acc. Chem.Res., 2001, 34, 818–825.
3 (a) Y. Kung and C. L. Drennan, Curr. Opin. Chem. Biol., 2011,15, 276–283; (b) P. A. Lindahl, JBIC, J. Biol. Inorg. Chem., 2004,9, 516–524; (c) Y. Liu, X. F. Zhu, F. Wang, T. L. Ying, P. W. Li,Z.-X. Huang and X. S. Tan, Chem. Commun., 2011, 47,1291–1293.
4 (a) J. Wuerges, J.-W. Lee, Y.-I. Yim, H.-S. Yim, S.-O. Kang andK. D. Carugo, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8569–8574;(b) D. P. Barondeau, C. J. Kassmann, C. K. Bruns, J. A. Tainer andE. D. Getzoff, Biochemistry, 2004, 43, 8038–8047; (c) S. B. Choudhury,J. W. Lee, G. Davidson, Y. I. Yim, K. Bose, M. L. Sharma, S. O. Kangand D. E. Cabelli, Biochemistry, 1999, 38, 3744–3752.
5 P. A. Bryngelson, S. E. Arobo, J. L. Pinkham, D. E. Cabelli andM. J. Maroney, J. Am. Chem. Soc., 2004, 126, 460–461.
6 Y. Liu, F. Wang, P. W. Li and X. S. Tan, ChemBioChem, 2011, 12,1417–1421.
7 (a) J. Sheare and N. F. Zhao, Inorg. Chem., 2006, 45, 9637–9639;(b) A. T. Fiedler, P. A. Bryngelson, M. J. Maroney and T. C. Brunold,J. Am. Chem. Soc., 2005, 127, 5449–5462.
8 K. P. Neupane, K. Gearty, A. Francis and J. Shearer, J. Am. Chem. Soc.,2007, 129, 14605–14618.
9 (a) D. Tietze, M. Tischler, S. Voigt, D. Imhof, O. Ohlenschlager,M. Gorlach and G. Buntkowsky, Chem.–Eur. J., 2010, 16, 7572–7578;(b) M. Schmidt, S. Zahn, M. Carella, O. Ohlenschlager, M. Gorlach,E. Kothe and J. Weston, ChemBioChem, 2008, 9, 2135–2146.
10 M. Gennari, M. Orio, J. Pecaut, F. Neese, M.-C. Collomb andC. Duboc, Inorg. Chem., 2010, 49, 6399–6401.
11 J. A. Fee and J. S. Valentine, Chemistry of O2�, in Superoxide and
superoxide dismutases, ed. A. M. Michelsen, J. M. McCord andI. Fridovich, Academic Press, New York, 1977, pp. 25–28.
12 R. W. Herbst, A. Guce, P. A. Bryngelson, K. Higgins, K. C. Ryan,D. E. Cabelli, S. T. Garman and M. J. Maroney, Biochemistry, 2009,48, 3354–3369.
13 (a) K. Fukui, T. Yonezawa and H. J. Shingu, Chem. Phys., 1952,20, 722; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100.
14 Q. L. Wang, D.-Z. Chen, X. W. Liu and L. F. Zhang, Comput. Theor.Chem., 2011, 966, 357–363.
Fig. 4 The frontier molecular orbitals of ACS-a15, F598H and �OOH. Solid valuesare orbital energies (in eV).
ChemComm Communication
Dow
nloa
ded
by F
udan
Uni
vers
ity o
n 02
Mar
ch 2
013
Publ
ishe
d on
03
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
2CC
3822
4EView Article Online

















![Laser Raman Spectroscopic Studies on Hemeproteins in ... · Sir C. V. Raman. Raman and Krishnan [2] have worked jointly together and observed that some of the light scat- tered by](https://img.pdfslide.net/doc/110x75/5ed8b1db6714ca7f4768664d/laser-raman-spectroscopic-studies-on-hemeproteins-in-sir-c-v-raman-raman.jpg)











