Embed Size (px)
Citation preview

Strengthened glycolysis under hypoxia supportstumor symbiosis and hexosamine biosynthesisin pancreatic adenocarcinomaFabienne Guillaumonda,b,c,d, Julie Lecaa,b,c,d,1, Orianne Olivaresa,b,c,d,1, Marie-Noëlle Lavauta,b,c,d, Nicolas Vidale,Patrice Berthezènea,b,c,d, Nelson Javier Dusettia,b,c,d, Céline Lonclea,b,c,d, Ezequiel Calvof, Olivier Turrinib,Juan Lucio Iovannaa,b,c,d, Richard Tomasinia,b,c,d, and Sophie Vasseura,b,c,d,2
aCentre de Recherche en Cancérologie de Marseille (CRCM), Unité 1068, Institut National de la Santé et de la Recherche Médicale, F-13009 Marseille, France;bInstitut Paoli-Calmettes (IPC), F-13009 Marseille, France; cUnité Mixte de Recherche (UMR) 7258, Centre National de la Recherche Scientifique (CNRS), F-13009Marseille, France; dUniversité Aix-Marseille, F-13284 Marseille, France; eInstitut de Chimie Radicalaire, UMR 7273, CNRS, F-13397 Marseille, France; andfMolecular Endocrinology and Oncology Research Center, Research Center of Centre Hospitalier Universitaire de Laval, Quebec City, QC, Canada G1V 4G2
Edited by Tak W. Mak, The Campbell Family Institute for Breast Cancer Research, Ontario Cancer Institute at Princess Margaret Hospital, University HealthNetwork, Toronto, ON, Canada, and approved January 17, 2013 (received for review November 11, 2012)
Pancreatic ductal adenocarcinoma is one of the most intractableand fatal cancer. The decreased blood vessel density displayed bythis tumor not only favors its resistance to chemotherapy but alsoparticipates in its aggressiveness due to the consequent highdegree of hypoxia. It is indeed clear that hypoxia promotesselective pressure on malignant cells that must develop adaptivemetabolic responses to reach their energetic and biosyntheticdemands. Here, using a well-defined mouse model of pancreaticcancer, we report that hypoxic areas from pancreatic ductaladenocarcinoma are mainly composed of epithelial cells harboringepithelial-mesenchymal transition features and expressing glyco-lytic markers, two characteristics associated with tumor aggres-siveness. We also show that hypoxia increases the “glycolytic”switch of pancreatic cancer cells from oxydative phosphorylationto lactate production and we demonstrate that increased lactateefflux from hypoxic cancer cells favors the growth of normoxiccancer cells. In addition, we show that glutamine metabolizationby hypoxic pancreatic tumor cells is necessary for their survival.Metabolized glucose and glutamine converge toward a commonpathway, termed hexosamine biosynthetic pathway, which allowsO-linked N-acetylglucosamine modifications of proteins. Here, wereport that hypoxia increases transcription of hexosamine biosyn-thetic pathway genes as well as levels of O-glycosylated proteinsand thatO-linkedN-acetylglucosaminylationof proteins is a processrequired for hypoxic pancreatic cancer cell survival. Our resultsdemonstrate that hypoxia-driven metabolic adaptive processes,such as high glycolytic rate and hexosamine biosynthetic pathwayactivation, favor hypoxic and normoxic cancer cell survival and cor-relate with pancreatic ductal adenocarcinoma aggressiveness.
pancreas | malignancy | metabolism | glutamate
Ninety-five percent of patients with pancreatic cancer harbortumors classified as pancreatic ductal adenocarcinoma
(PDAC). Commonly described as a silent killer regarding its latediagnosis, PDAC is noted for its aggressiveness and its intrinsicresistance to standard chemotherapy. This specificity is probablydue to a low vascular density and a prominent nontumoral cellcompartment (stroma), which impact on intratumoral perfusion,therapeutic delivery, and patient outcome (1). Indeed, PDAC ischaracterized by numerous and severe hypoxic regions (2), afeature that has been proven to be correlated with tumor ag-gressiveness and poor prognosis compared with well-oxygenatedtumors (3). Moreover, combined with hypoxia, the subsequentnutrient-devoid environment provides physiological selectivepressure promoting expansion of the most aggressive malignantcells, particularly those acquiring mutations in genes encodingtumor suppressor protein p53 (TP53) and v-Ki-ras2 Kirsten ratsarcoma viral oncogene homolog protein (KRAS) (4, 5), two ofthe main mutations present in PDAC patients. Regarding such
statements, it appears relevant to deeply explore consequences ofhypoxia on PDAC carcinogenesis. In the past decade, it has beenclearly highlighted that under hypoxia cancer cells develop anefficient adaptive metabolic response to ensure their survival andproliferation. Indeed, hypoxic cancer cells activate glucose up-take and glycolysis to produce pyruvate, which is then convertedinto lactate instead of being oxidized via the tricarboxylic acid(TCA) cycle and oxidative phosphorylation (OXPHOS) (6). Thismetabolic shift is driven by the hypoxia-inducible factor–1 (HIF1)through transcriptional activation of glycolytic genes and inhibitionof those promotingOXPHOS (7).More recently, it has been shownthat hypoxic cancer cells also use glutamine as a carbon fuel sourcefor survival. After successive conversion into glutamate andα-ketoglutarate, glutamine promotes citrate synthesis and de novolipogenesis via the isocitrate dehydrogenase–1 and –2 (IDH1 and2)–dependent reductive carboxylation pathway (8, 9) and alsosupports cell proliferation through a glucose-independent TCAcycle pathway (10). Hypoxia within PDAC could then also favora switch to both glucose- and glutamine-dependent anaerobicmetabolic pathways, allowing adaptation and resistance of cancercells to this hostile tumor microenvironment and promotingtumor development.This metabolic switch has only been described under normoxic
conditions in pancreatic cancer cell lines. Indeed, normoxic pan-creatic cancer cells display high glycolytic activity and are able tometabolize glutamine (11). Furthermore, oncogenic Kras (KrasG12D)in these cells funnels glucose into either the pentose phosphatepathway (PPP) or the hexosamine biosynthetic pathway (HBP) (12).The latter uses glucose and glutamine to generate UDP–N-acetylglucosamine (UDP–GlcNAc), a donor substrate for gly-cosylation reactions. Except a recent study showing a glutamineuptake following hypoxic stress in vitro and an enhanced glycolysisin hypoxic regions of PDAC (13), little effort has been investedso far to precisely determine the metabolic changes involved inhypoxia resistance and PDAC progression.Here, we perform an exhaustive characterization of hypoxic
regions in mouse PDAC and precisely define the phenotypicand metabolic features of related tumoral cells, with particular
Author contributions: F.G., J.L., O.O., J.L.I., R.T., and S.V. designed research; F.G., J.L., O.O.,M.-N.L., N.V., N.J.D., C.L., E.C., O.T., R.T., and S.V. performed research; N.V. contributednew reagents/analytic tools; F.G., J.L., O.O., N.V., P.B., E.C., R.T., and S.V. analyzed data;and F.G., R.T., and S.V. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.1J.L. and O.O. contributed equally to this work.2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219555110/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1219555110 PNAS | March 5, 2013 | vol. 110 | no. 10 | 3919–3924
CELL
BIOLO
GY
attention on glucose- and glutamine-dependent hypoxic meta-bolic processes.
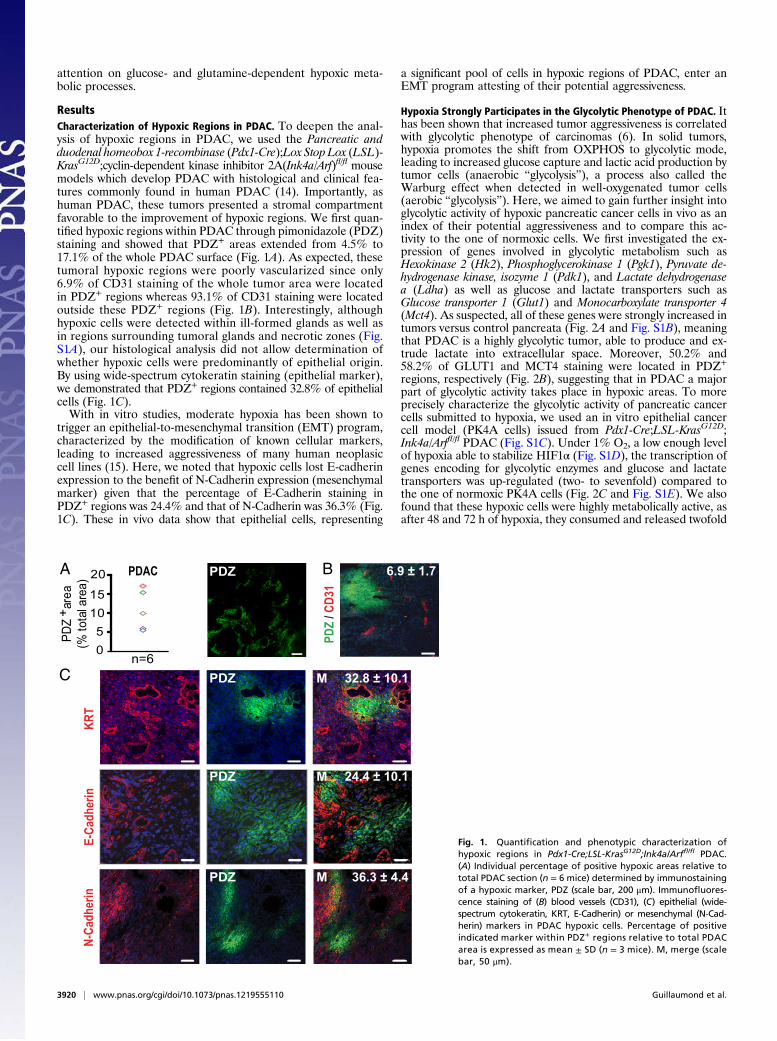
ResultsCharacterization of Hypoxic Regions in PDAC. To deepen the anal-ysis of hypoxic regions in PDAC, we used the Pancreatic andduodenal homeobox 1-recombinase (Pdx1-Cre);Lox Stop Lox (LSL)-KrasG12D;cyclin-dependent kinase inhibitor 2A(Ink4a/Arf)fl/fl mousemodels which develop PDAC with histological and clinical fea-tures commonly found in human PDAC (14). Importantly, ashuman PDAC, these tumors presented a stromal compartmentfavorable to the improvement of hypoxic regions. We first quan-tified hypoxic regions within PDAC through pimonidazole (PDZ)staining and showed that PDZ+ areas extended from 4.5% to17.1% of the whole PDAC surface (Fig. 1A). As expected, thesetumoral hypoxic regions were poorly vascularized since only6.9% of CD31 staining of the whole tumor area were locatedin PDZ+ regions whereas 93.1% of CD31 staining were locatedoutside these PDZ+ regions (Fig. 1B). Interestingly, althoughhypoxic cells were detected within ill-formed glands as well asin regions surrounding tumoral glands and necrotic zones (Fig.S1A), our histological analysis did not allow determination ofwhether hypoxic cells were predominantly of epithelial origin.By using wide-spectrum cytokeratin staining (epithelial marker),we demonstrated that PDZ+ regions contained 32.8% of epithelialcells (Fig. 1C).With in vitro studies, moderate hypoxia has been shown to
trigger an epithelial-to-mesenchymal transition (EMT) program,characterized by the modification of known cellular markers,leading to increased aggressiveness of many human neoplasiccell lines (15). Here, we noted that hypoxic cells lost E-cadherinexpression to the benefit of N-Cadherin expression (mesenchymalmarker) given that the percentage of E-Cadherin staining inPDZ+ regions was 24.4% and that of N-Cadherin was 36.3% (Fig.1C). These in vivo data show that epithelial cells, representing
a significant pool of cells in hypoxic regions of PDAC, enter anEMT program attesting of their potential aggressiveness.
Hypoxia Strongly Participates in the Glycolytic Phenotype of PDAC. Ithas been shown that increased tumor aggressiveness is correlatedwith glycolytic phenotype of carcinomas (6). In solid tumors,hypoxia promotes the shift from OXPHOS to glycolytic mode,leading to increased glucose capture and lactic acid production bytumor cells (anaerobic “glycolysis”), a process also called theWarburg effect when detected in well-oxygenated tumor cells(aerobic “glycolysis”). Here, we aimed to gain further insight intoglycolytic activity of hypoxic pancreatic cancer cells in vivo as anindex of their potential aggressiveness and to compare this ac-tivity to the one of normoxic cells. We first investigated the ex-pression of genes involved in glycolytic metabolism such asHexokinase 2 (Hk2), Phosphoglycerokinase 1 (Pgk1), Pyruvate de-hydrogenase kinase, isozyme 1 (Pdk1), and Lactate dehydrogenasea (Ldha) as well as glucose and lactate transporters such asGlucose transporter 1 (Glut1) and Monocarboxylate transporter 4(Mct4). As suspected, all of these genes were strongly increased intumors versus control pancreata (Fig. 2A and Fig. S1B), meaningthat PDAC is a highly glycolytic tumor, able to produce and ex-trude lactate into extracellular space. Moreover, 50.2% and58.2% of GLUT1 and MCT4 staining were located in PDZ+
regions, respectively (Fig. 2B), suggesting that in PDAC a majorpart of glycolytic activity takes place in hypoxic areas. To moreprecisely characterize the glycolytic activity of pancreatic cancercells submitted to hypoxia, we used an in vitro epithelial cancercell model (PK4A cells) issued from Pdx1-Cre;LSL-KrasG12D;Ink4a/Arffl/fl PDAC (Fig. S1C). Under 1% O2, a low enough levelof hypoxia able to stabilize HIF1α (Fig. S1D), the transcription ofgenes encoding for glycolytic enzymes and glucose and lactatetransporters was up-regulated (two- to sevenfold) compared tothe one of normoxic PK4A cells (Fig. 2C and Fig. S1E). We alsofound that these hypoxic cells were highly metabolically active, asafter 48 and 72 h of hypoxia, they consumed and released twofold
05
1015
20
n=6
PDZ
are
a(%
tota
l are
a)+
A B
C
PDZ
/ CD3
1
KRT
E-Ca
dher
inN-
Cadh
erin
24.4 ± 10.1PDZ M
M
PDZ M
PDZ
6.9 ± 1.7PDAC
36.3 ± 4.4
32.8 ± 10.1
PDZ
Fig. 1. Quantification and phenotypic characterization ofhypoxic regions in Pdx1-Cre;LSL-KrasG12D;Ink4a/Arffl/fl PDAC.(A) Individual percentage of positive hypoxic areas relative tototal PDAC section (n = 6 mice) determined by immunostainingof a hypoxic marker, PDZ (scale bar, 200 μm). Immunofluores-cence staining of (B) blood vessels (CD31), (C) epithelial (wide-spectrum cytokeratin, KRT, E-Cadherin) or mesenchymal (N-Cad-herin) markers in PDAC hypoxic cells. Percentage of positiveindicated marker within PDZ+ regions relative to total PDACarea is expressed as mean ± SD (n = 3 mice). M, merge (scalebar, 50 μm).
3920 | www.pnas.org/cgi/doi/10.1073/pnas.1219555110 Guillaumond et al.
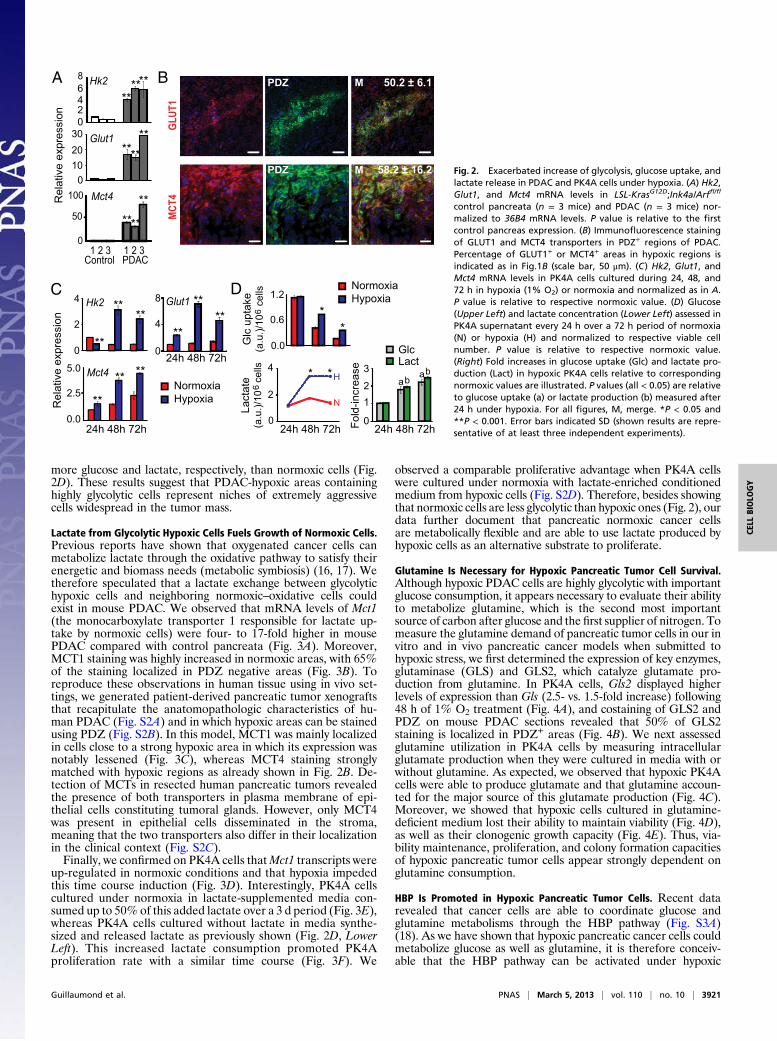
more glucose and lactate, respectively, than normoxic cells (Fig.2D). These results suggest that PDAC-hypoxic areas containinghighly glycolytic cells represent niches of extremely aggressivecells widespread in the tumor mass.
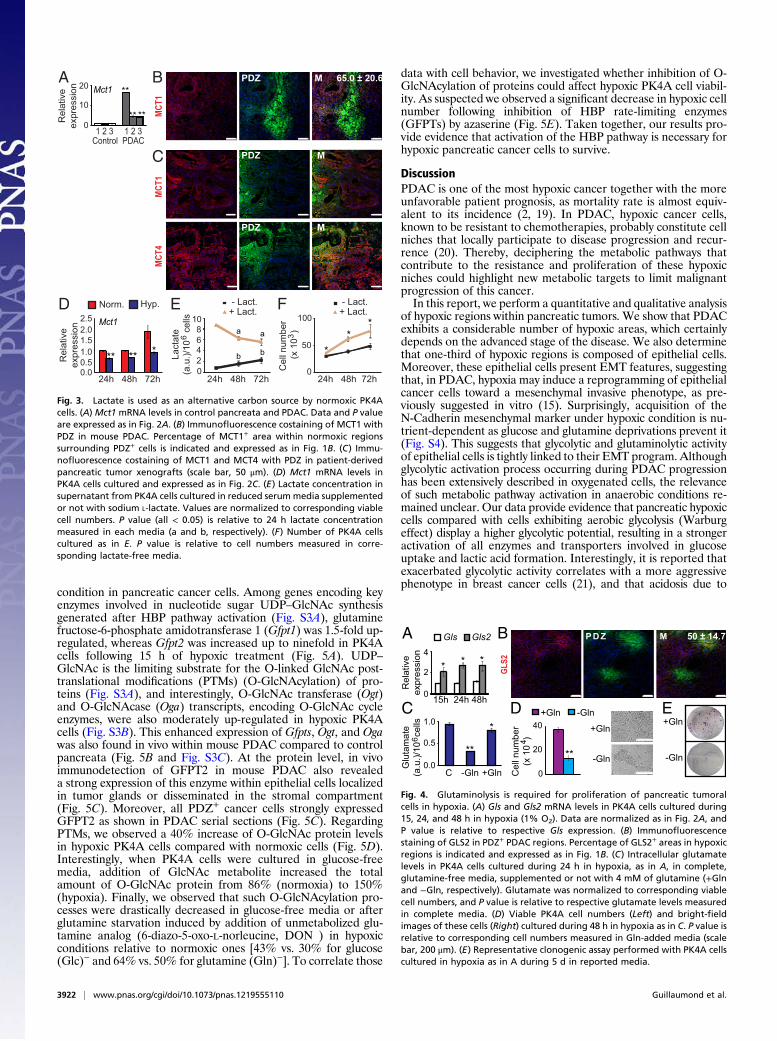
Lactate from Glycolytic Hypoxic Cells Fuels Growth of Normoxic Cells.Previous reports have shown that oxygenated cancer cells canmetabolize lactate through the oxidative pathway to satisfy theirenergetic and biomass needs (metabolic symbiosis) (16, 17). Wetherefore speculated that a lactate exchange between glycolytichypoxic cells and neighboring normoxic–oxidative cells couldexist in mouse PDAC. We observed that mRNA levels of Mct1(the monocarboxylate transporter 1 responsible for lactate up-take by normoxic cells) were four- to 17-fold higher in mousePDAC compared with control pancreata (Fig. 3A). Moreover,MCT1 staining was highly increased in normoxic areas, with 65%of the staining localized in PDZ negative areas (Fig. 3B). Toreproduce these observations in human tissue using in vivo set-tings, we generated patient-derived pancreatic tumor xenograftsthat recapitulate the anatomopathologic characteristics of hu-man PDAC (Fig. S2A) and in which hypoxic areas can be stainedusing PDZ (Fig. S2B). In this model, MCT1 was mainly localizedin cells close to a strong hypoxic area in which its expression wasnotably lessened (Fig. 3C), whereas MCT4 staining stronglymatched with hypoxic regions as already shown in Fig. 2B. De-tection of MCTs in resected human pancreatic tumors revealedthe presence of both transporters in plasma membrane of epi-thelial cells constituting tumoral glands. However, only MCT4was present in epithelial cells disseminated in the stroma,meaning that the two transporters also differ in their localizationin the clinical context (Fig. S2C).Finally, we confirmed on PK4A cells thatMct1 transcripts were
up-regulated in normoxic conditions and that hypoxia impededthis time course induction (Fig. 3D). Interestingly, PK4A cellscultured under normoxia in lactate-supplemented media con-sumed up to 50% of this added lactate over a 3 d period (Fig. 3E),whereas PK4A cells cultured without lactate in media synthe-sized and released lactate as previously shown (Fig. 2D, LowerLeft). This increased lactate consumption promoted PK4Aproliferation rate with a similar time course (Fig. 3F). We
observed a comparable proliferative advantage when PK4A cellswere cultured under normoxia with lactate-enriched conditionedmedium from hypoxic cells (Fig. S2D). Therefore, besides showingthat normoxic cells are less glycolytic than hypoxic ones (Fig. 2), ourdata further document that pancreatic normoxic cancer cellsare metabolically flexible and are able to use lactate produced byhypoxic cells as an alternative substrate to proliferate.
Glutamine Is Necessary for Hypoxic Pancreatic Tumor Cell Survival.Although hypoxic PDAC cells are highly glycolytic with importantglucose consumption, it appears necessary to evaluate their abilityto metabolize glutamine, which is the second most importantsource of carbon after glucose and the first supplier of nitrogen. Tomeasure the glutamine demand of pancreatic tumor cells in our invitro and in vivo pancreatic cancer models when submitted tohypoxic stress, we first determined the expression of key enzymes,glutaminase (GLS) and GLS2, which catalyze glutamate pro-duction from glutamine. In PK4A cells, Gls2 displayed higherlevels of expression than Gls (2.5- vs. 1.5-fold increase) following48 h of 1% O2 treatment (Fig. 4A), and costaining of GLS2 andPDZ on mouse PDAC sections revealed that 50% of GLS2staining is localized in PDZ+ areas (Fig. 4B). We next assessedglutamine utilization in PK4A cells by measuring intracellularglutamate production when they were cultured in media with orwithout glutamine. As expected, we observed that hypoxic PK4Acells were able to produce glutamate and that glutamine accoun-ted for the major source of this glutamate production (Fig. 4C).Moreover, we showed that hypoxic cells cultured in glutamine-deficient medium lost their ability to maintain viability (Fig. 4D),as well as their clonogenic growth capacity (Fig. 4E). Thus, via-bility maintenance, proliferation, and colony formation capacitiesof hypoxic pancreatic tumor cells appear strongly dependent onglutamine consumption.
HBP Is Promoted in Hypoxic Pancreatic Tumor Cells. Recent datarevealed that cancer cells are able to coordinate glucose andglutamine metabolisms through the HBP pathway (Fig. S3A)(18). As we have shown that hypoxic pancreatic cancer cells couldmetabolize glucose as well as glutamine, it is therefore conceiv-able that the HBP pathway can be activated under hypoxic
100
50
0
Mct4
ba
ab2
3
1
0Fold
-incr
ease
Glc Lact
N
H
Glc
upt
ake
(a.u
.)/10
ce
lls6
1.2
0.0
0.6*
*
D
2.5
0.0
5.0
4
2
0
Mct4
Hk2
**
** ****
**
A 8
46
20
Hk2 B50.2 ± 6.1MPDZ
GLUT
158.2 ± 16.2PDZ M
MCT4
30
1020
0
Glut1
******
******
****
**
24h 48h 72h
8
4
0
Glut1
**
**
**
1 2 3 1 2 3Control PDAC
Rel
ativ
e ex
pres
sion
**4
2
0
Lact
ate
(a.u
.)/10
ce
lls6
24h 48h 72h
24h 48h 72h
NormoxiaHypoxia
C
Rel
ativ
e ex
pres
sion
**
NormoxiaHypoxia
24h 48h 72h
Fig. 2. Exacerbated increase of glycolysis, glucose uptake, andlactate release in PDAC and PK4A cells under hypoxia. (A) Hk2,Glut1, and Mct4 mRNA levels in LSL-KrasG12D;Ink4a/Arffl/fl
control pancreata (n = 3 mice) and PDAC (n = 3 mice) nor-malized to 36B4 mRNA levels. P value is relative to the firstcontrol pancreas expression. (B) Immunofluorescence stainingof GLUT1 and MCT4 transporters in PDZ+ regions of PDAC.Percentage of GLUT1+ or MCT4+ areas in hypoxic regions isindicated as in Fig.1B (scale bar, 50 μm). (C) Hk2, Glut1, andMct4 mRNA levels in PK4A cells cultured during 24, 48, and72 h in hypoxia (1% O2) or normoxia and normalized as in A.P value is relative to respective normoxic value. (D) Glucose(Upper Left) and lactate concentration (Lower Left) assessed inPK4A supernatant every 24 h over a 72 h period of normoxia(N) or hypoxia (H) and normalized to respective viable cellnumber. P value is relative to respective normoxic value.(Right) Fold increases in glucose uptake (Glc) and lactate pro-duction (Lact) in hypoxic PK4A cells relative to correspondingnormoxic values are illustrated. P values (all < 0.05) are relativeto glucose uptake (a) or lactate production (b) measured after24 h under hypoxia. For all figures, M, merge. *P < 0.05 and**P < 0.001. Error bars indicated SD (shown results are repre-sentative of at least three independent experiments).
Guillaumond et al. PNAS | March 5, 2013 | vol. 110 | no. 10 | 3921
CELL
BIOLO
GY
condition in pancreatic cancer cells. Among genes encoding keyenzymes involved in nucleotide sugar UDP–GlcNAc synthesisgenerated after HBP pathway activation (Fig. S3A), glutaminefructose-6-phosphate amidotransferase 1 (Gfpt1) was 1.5-fold up-regulated, whereas Gfpt2 was increased up to ninefold in PK4Acells following 15 h of hypoxic treatment (Fig. 5A). UDP–GlcNAc is the limiting substrate for the O-linked GlcNAc post-translational modifications (PTMs) (O-GlcNAcylation) of pro-teins (Fig. S3A), and interestingly, O-GlcNAc transferase (Ogt)and O-GlcNAcase (Oga) transcripts, encoding O-GlcNAc cycleenzymes, were also moderately up-regulated in hypoxic PK4Acells (Fig. S3B). This enhanced expression of Gfpts, Ogt, and Ogawas also found in vivo within mouse PDAC compared to controlpancreata (Fig. 5B and Fig. S3C). At the protein level, in vivoimmunodetection of GFPT2 in mouse PDAC also revealeda strong expression of this enzyme within epithelial cells localizedin tumor glands or disseminated in the stromal compartment(Fig. 5C). Moreover, all PDZ+ cancer cells strongly expressedGFPT2 as shown in PDAC serial sections (Fig. 5C). RegardingPTMs, we observed a 40% increase of O-GlcNAc protein levelsin hypoxic PK4A cells compared with normoxic cells (Fig. 5D).Interestingly, when PK4A cells were cultured in glucose-freemedia, addition of GlcNAc metabolite increased the totalamount of O-GlcNAc protein from 86% (normoxia) to 150%(hypoxia). Finally, we observed that such O-GlcNAcylation pro-cesses were drastically decreased in glucose-free media or afterglutamine starvation induced by addition of unmetabolized glu-tamine analog (6-diazo-5-oxo-L-norleucine, DON ) in hypoxicconditions relative to normoxic ones [43% vs. 30% for glucose(Glc)− and 64% vs. 50% for glutamine (Gln)−]. To correlate those
data with cell behavior, we investigated whether inhibition of O-GlcNAcylation of proteins could affect hypoxic PK4A cell viabil-ity. As suspected we observed a significant decrease in hypoxic cellnumber following inhibition of HBP rate-limiting enzymes(GFPTs) by azaserine (Fig. 5E). Taken together, our results pro-vide evidence that activation of the HBP pathway is necessary forhypoxic pancreatic cancer cells to survive.
DiscussionPDAC is one of the most hypoxic cancer together with the moreunfavorable patient prognosis, as mortality rate is almost equiv-alent to its incidence (2, 19). In PDAC, hypoxic cancer cells,known to be resistant to chemotherapies, probably constitute cellniches that locally participate to disease progression and recur-rence (20). Thereby, deciphering the metabolic pathways thatcontribute to the resistance and proliferation of these hypoxicniches could highlight new metabolic targets to limit malignantprogression of this cancer.In this report, we perform a quantitative and qualitative analysis
of hypoxic regions within pancreatic tumors. We show that PDACexhibits a considerable number of hypoxic areas, which certainlydepends on the advanced stage of the disease. We also determinethat one-third of hypoxic regions is composed of epithelial cells.Moreover, these epithelial cells present EMT features, suggestingthat, in PDAC, hypoxia may induce a reprogramming of epithelialcancer cells toward a mesenchymal invasive phenotype, as pre-viously suggested in vitro (15). Surprisingly, acquisition of theN-Cadherin mesenchymal marker under hypoxic condition is nu-trient-dependent as glucose and glutamine deprivations prevent it(Fig. S4). This suggests that glycolytic and glutaminolytic activityof epithelial cells is tightly linked to their EMT program. Althoughglycolytic activation process occurring during PDAC progressionhas been extensively described in oxygenated cells, the relevanceof such metabolic pathway activation in anaerobic conditions re-mained unclear. Our data provide evidence that pancreatic hypoxiccells compared with cells exhibiting aerobic glycolysis (Warburgeffect) display a higher glycolytic potential, resulting in a strongeractivation of all enzymes and transporters involved in glucoseuptake and lactic acid formation. Interestingly, it is reported thatexacerbated glycolytic activity correlates with a more aggressivephenotype in breast cancer cells (21), and that acidosis due to
A B
C
2.0
1.01.5
0.50.0
2.5Norm. Hyp.
24h 48h 72h
D FMct1
Rel
ativ
eex
pres
sion
** ** **
*50
0
100
Cel
l num
ber
(x 1
0 )3
24h 48h 72h
- Lact.+ Lact.▲
*
Lact
ate
(a.u
.)/10
ce
lls6 6
42
8
24h 48h 72h
0
10
20Mct1
1 2 3 1 2 3
Rel
ativ
eex
pres
sion
65.0 ± 20.6MPDZ
MCT1
** **
**
10
Control PDAC
0
bb
aa
- Lact.+ Lact.▲
MCT1
PDZ M
MPDZ
MCT4
E
Fig. 3. Lactate is used as an alternative carbon source by normoxic PK4Acells. (A) Mct1 mRNA levels in control pancreata and PDAC. Data and P valueare expressed as in Fig. 2A. (B) Immunofluorescence costaining of MCT1 withPDZ in mouse PDAC. Percentage of MCT1+ area within normoxic regionssurrounding PDZ+ cells is indicated and expressed as in Fig. 1B. (C) Immu-nofluorescence costaining of MCT1 and MCT4 with PDZ in patient-derivedpancreatic tumor xenografts (scale bar, 50 μm). (D) Mct1 mRNA levels inPK4A cells cultured and expressed as in Fig. 2C. (E) Lactate concentration insupernatant from PK4A cells cultured in reduced serummedia supplementedor not with sodium L-lactate. Values are normalized to corresponding viablecell numbers. P value (all < 0.05) is relative to 24 h lactate concentrationmeasured in each media (a and b, respectively). (F) Number of PK4A cellscultured as in E. P value is relative to cell numbers measured in corre-sponding lactate-free media.
A
E
-Gln
+Gln
C*
**0.0
1.0
Glu
tam
ate
(a.u
.)/10
cel
ls6
-GlnC
2
0
4
15h 24h 48h
B
Cel
l num
ber
(x 1
0 )4
0
40
20 **
-Gln+GlnD
GLS2
PD Z M 50 ± 14.7
+Gln
-Gln0.5
+Gln
* * *
Rel
ativ
eex
pres
sion
Gls Gls2
Fig. 4. Glutaminolysis is required for proliferation of pancreatic tumoralcells in hypoxia. (A) Gls and Gls2 mRNA levels in PK4A cells cultured during15, 24, and 48 h in hypoxia (1% O2). Data are normalized as in Fig. 2A, andP value is relative to respective Gls expression. (B) Immunofluorescencestaining of GLS2 in PDZ+ PDAC regions. Percentage of GLS2+ areas in hypoxicregions is indicated and expressed as in Fig. 1B. (C) Intracellular glutamatelevels in PK4A cells cultured during 24 h in hypoxia, as in A, in complete,glutamine-free media, supplemented or not with 4 mM of glutamine (+Glnand −Gln, respectively). Glutamate was normalized to corresponding viablecell numbers, and P value is relative to respective glutamate levels measuredin complete media. (D) Viable PK4A cell numbers (Left) and bright-fieldimages of these cells (Right) cultured during 48 h in hypoxia as in C. P value isrelative to corresponding cell numbers measured in Gln-added media (scalebar, 200 μm). (E) Representative clonogenic assay performed with PK4A cellscultured in hypoxia as in A during 5 d in reported media.
3922 | www.pnas.org/cgi/doi/10.1073/pnas.1219555110 Guillaumond et al.

increased lactic acid in tumor microenvironment is indirectly as-sociated to the extracellular matrix breakdown, a mechanism thatfavors invasiveness (6). Hence, in the context of PDAC, we suggestthat hypoxia, by promoting both glycolytic activity (and associatedlactate efflux) and invasive phenotype of pancreatic epithelial cells,is mainly responsible for the tumor aggressiveness.It is now well established that excess of lactate released by
highly glycolytic cells, besides promoting tumor invasion, canalso be used as a glucose-alternative carbon source by neigh-boring oxygenated cancer cells, as it has been recently describedin colon cancer cell lines (16). Here, we provide evidence thatsuch tumoral symbiosis between hypoxic and normoxic cancercells also exists in pancreatic cancer, as lactate present in culturemedia is taken up by oxygenated pancreatic cells that use it asa fuel source to increase their proliferation rate. These resultsare reinforced by the fact that, in mouse PDAC, expression ofMCT1, which is involved in the uptake of lactate, is switched onin normoxic cancer cells and reduced in hypoxic ones. The samepattern of expression of MCT1 is observed in patient-derivedxenografts, suggesting that such tumoral symbiosis may also existin human PDAC. All together, these data lead to the conclusionthat lactate constitutes a crucial energetic metabolite allowingfuel exchanges between hypoxic and normoxic compartments inthe pancreatic tumor. A recent study, showing that alteration ofMCT1 and MCT4 membrane trafficking by CD147 inhibitionreduces the proliferation rate of aerobic glycolytic pancreaticcancer cells (22), strengthens the fact that the export of lactate byhypoxic cells and its uptake by normoxic ones are essential forthe malignant phenotype of PDAC.
A role of oncogenic KRAS in reprogramming cancer cell me-tabolism through activation of glucose and glutamine metabolismand their derived pentose phosphate (PPP) and hexosaminepathways has been documented in various cancer cell lines in-cluding pancreatic ones (12, 23). Once taken up by cancer cells,glutamine is converted into glutamate partly by GLS enzymesthrough the glutaminolysis pathway. In pancreatic cancer cells,the isoform 2 (GLS2) is preferentially expressed in hypoxic areas,suggesting that glutamate production in hypoxic cells is essentiallydependent of GLS2 and not GLS. Furthermore, hypoxic pan-creatic cancer cells, in addition to their glutamine-dependent cellgrowth and their high glucose consumption, activate the HBPpathway mainly through the rate-limiting enzyme GFPT2, whichis the most activated GFPT enzyme under hypoxia. Finally, theincrease of O-GlcNAc protein levels under hypoxia comparedwith normoxia is clearly dependent on both glucose and glutamineavailability and it sustains tumor cell viability. O-GlcNAcylation isa PTM that stabilizes numerous factors involved in tumorigenicprocesses such as myc myelocytomatosis viral oncogene homologprotein (c-Myc), TP53, and β-catenin (24, 25). It also participatesin the phenotype of cancer cells by promoting aneuploidy and byactivating key oncogenic signaling pathways including insulin, fi-broblast growth factors, and transforming growth factor–β acti-vated pathways (26). Here, in the context of PDAC, we suggestthat GlcNAcylation allows cells to promptly react to microenvi-ronmental stress situations such as hypoxia through stabilization offactors yet to be determined and involved in resistance to hypoxiaand in its associated metabolic pathways. Very recently, Yi et al.
GlcNAc
Glc GlcNAc DON
+--
---
-+-
+-+
+--
---
-+-
+-+
Normoxia Hypoxia
64
98kDa
***
Gfpt1 Gfpt2
*
**
******** ****
Gfpt1 Gfpt2
40%
86% 150%
64%
43%
50%
30%
*
PDZ
GFPT
2
1.0 1.3 1.4 2.0 0.7 0.5 0.8 0.5
6
0
108
42
8h 15h 24h
Cel
l num
ber
(x 1
0 )4
0
20
10
40
30
CompleteAzaserine
A B
D
E
C
Hypoxia
1015
50
1.25
0.00
2.50
Rel
ativ
eex
pres
sion
Control PDAC 1 2 3 1 2 3
Rel
ativ
eex
pres
sion
Control PDAC 1 2 3 1 2 3
Fig. 5. Activation of the HBP in hypoxic pancreatic cancer cells. (A)Gfpt1 andGfpt2 mRNA levels in PK4A cells cultured during 8, 15, and 24 h in hypoxia(1% O2). Data are expressed as in Fig. 4A. P value is relative to respective 8 hexpression. (B) Gfpt1 and Gfpt2 mRNA level in PDAC and control pancreata.Data are expressed as in Fig. 2A. (C) GFPT2 and PDZ staining of serial mousePDAC sections. Hypoxic regions are delineated by red line, and GFPT2+ cells(►) in tumor glands or disseminated in stroma are indicated (scale bar,50 μm). (D) O-GlcNAc proteins expression in PK4A cells cultured during 24 hin normoxia or hypoxia in complete media supplemented or not with DON(30 μM) and in glucose-freemedia supplemented or not with GlcNAc (15mM).O-GlcNAc–β-actin protein ratios are expressed relative to complete mediaratio measured in normoxia and are indicated below immunoblots. (E)Number of viable PK4A cells cultured during 48 h in hypoxia in completemedia supplemented or not with azaserine (10 μM). P value is relative tocorresponding viable cell numbers determined in complete media.
GlutamineGlucose
Pyruvate
Lactate
Glycolysis Glutaminolysis
Hexosaminebiosynthe�c
pathway
GlcNAc-proteinmodifica�ons
Glutamate
Invasion(acidosis)
TCA cycle
Biomass
Cell growth
PDAC PROGRESSION
HYPOXIA
Normoxic cells
Cell growth
Hypoxic cells
Fig. 6. Schematic representation of glucose- and glutamine-derived meta-bolic pathways activated by hypoxia in pancreatic cancer cells. Hypoxiaenables PDAC progression through (i) the increased glucose flux and sub-sequent glycolysis and lactate production [lactate, released in extracellularspace, increases invasive potency of tumoral cells through acidification ofpericellular pH (acidosis) and favors growth of normoxic cells that use it asa fuel source]; (ii) the increased glutaminolysis, which provides cells withglutamate that once converted into a TCA intermediate metabolite, pro-motes biomass production; and (iii) the HBP that requires both major ex-tracellular carbon sources, glucose and glutamine, to enable O-GlcNACmodifications of protumoral proteins. Activation of these various metabolicpathways by hypoxia contributes to PDAC progression.
Guillaumond et al. PNAS | March 5, 2013 | vol. 110 | no. 10 | 3923
CELL
BIOLO
GY

showed that phosphofructokinase 1, a key enzyme of glycolysisflux, is O-GlcNAcylated and that this PTM responsible for itsinactivation redirects glucose flux through pathways critically in-volved in tumor growth as PPP (27). Therefore, the crucial role ofHBP activation to maintain cell viability under hypoxic conditionsmight involve regulation of mediators including glycolytic enzymes.Taken together, our results suggest that enhanced glucose
metabolism in hypoxic pancreatic cancer cells represents a keyregulating pathway, as its end-product, lactate, is required forgrowth of neighboring normoxic cells, and its by-products co-operate with glutamine to enable PTMs, all favoring PDACprogression (Fig. 6). These results shed light on crucial metabolicpathways/enzymes activated in PDAC hypoxic regions that areconsidered as adaptive landscapes for selection of aggressiveand invasive cells. Therefore, targeting of those pathways couldeliminate hypoxic niches and thus limit PDAC progression andassociated metastases.
Materials and MethodsCells and Reagents. Cell culture media and conditions are described in SIMaterials and Methods. Hypoxia was performed at 1% (vol/vol) O2 and 5%(vol/vol) CO2 in nitrogen atmosphere.
Mouse Strains and Tissue Collection. Pdx1-Cre;Ink4a/Arffl/fl;LSL-KrasG12D micewere obtained by crossing the following strains: Pdx1-Cre;Ink4a/Arffl/fl andLSL-KrasG12D mice kindly provided by Dr. D. Melton (Harvard Stem Cell In-stitute, Cambridge, MA), Dr. R. Depinho (Dana-Farber Cancer Institute,Boston) and Dr. T Jacks (David H. Koch Institute for Integrative Cancer Re-search, Cambridge, MA), respectively. PDAC-bearing 8–12-wk-old malemice were killed with their mating control littermates. Pieces of tumor orcontrol pancreata were fixed in 4% (wt/vol) formaldehyde or frozen incold isopentane for further immunostaining analysis or directly homog-enized in 4 M guanidinium isothiocyanate lysis buffer for efficient pancre-atic RNA extraction according to Chirgwin’s procedure (28). Immunodetectionof cellular hypoxia required an i.p. injection of PDZ hydrochloride (60 mg/kg;Hypoxyprobe) 4 h before sacrifice. All animal care and experimental proce-dures were performed in agreement with the Animal Ethics Committee ofMarseille.
Xenografts. Patient-derived pancreatic tumor pieces (1 mm3) were embeddedin Matrigel before to be s.c. implanted into flank of adult male Swiss nudemice (Charles River Laboratories) under isoflurane anesthesia (induction, 4%(vol/vol); maintenance, 1.5% (vol/vol)). Tumors were measured weekly with acaliper until tumor volume reached 1 mm3. At 4 h after intratumoral in-jection of PDZ hydrochloride, pieces of tumor were removed, fixed in 4%(wt/vol) formaldehyde, or frozen in cold isopentane for further analysis.
Immunohistochemistry, Immunofluorescence, Western Blots, and PrimaryAntibodies. Protocols and antibodies used for immuno-detection of proteinsin tissue sections or in cell protein lysates are described in SI Materialsand Methods.
Image Acquisition and Analysis. Quantification of tissue sections stained forPDZ as well as various markers was determined as in SI Materials andMethodsusing ImageJ software (National Institutes of Health).
Real-Time PCR. RT-PCR was performed as in SI Materials and Methods. Pri-mers used are shown in Table S1.
Metabolites Assays. Measurements of glucose, lactate, and glutamate wereperformed by either Yelen (Marseille, France) or with a Biovision kit, asdescribed in SI Materials and Methods.
Statistical Analysis. Comparisons between experimental groups were per-formed either by Student t test or by ANOVA or MANOVA, with subsequentpost hoc test using SAS statistical software. P < 0.05 was considered statis-tically significant.
ACKNOWLEDGMENTS. We thank the cell culture platform and the micecolony facility (Cell stress, Unité 1068) for technical assistance as well as Jean-Pierre Cavaille for glucose and lactate measurements. Samples of humanorigin were obtained from the IPC/CRCM Tumour Bank (authorization no.AC-2007-33), granted by the French Ministry of Research. The project wasapproved by the IPC Institutional Review Board. This work was supported inpart by grants from the National Institut of Cancer, Cancéropôle Provence-Alpes-Côte d’Azur, and the Ligue contre le Cancer. F.G. was supported byFondation Santé, Sport et Développement Durable and Fondation de Francefellowships.
1. Neesse A, et al. (2011) Stromal biology and therapy in pancreatic cancer. Gut 60(6):861–868.
2. Koong AC, et al. (2000) Pancreatic tumors show high levels of hypoxia. Int J RadiatOncol Biol Phys 48(4):919–922.
3. Bristow RG, Hill RP (2008) Hypoxia and metabolism. Hypoxia, DNA repair and geneticinstability. Nat Rev Cancer 8(3):180–192.
4. Graeber TG, et al. (1996) Hypoxia-mediated selection of cells with diminished apo-ptotic potential in solid tumours. Nature 379(6560):88–91.
5. Yun J, et al. (2009) Glucose deprivation contributes to the development of KRASpathway mutations in tumor cells. Science 325(5947):1555–1559.
6. Chiche J, Brahimi-Horn MC, Pouysségur J (2010) Tumour hypoxia induces a metabolicshift causing acidosis: A common feature in cancer. J Cell Mol Med 14(4):771–794.
7. Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3(10):721–732.8. Wise DR, et al. (2011) Hypoxia promotes isocitrate dehydrogenase-dependent car-
boxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc NatlAcad Sci USA 108(49):19611–19616.
9. Metallo CM, et al. (2012) Reductive glutamine metabolism by IDH1 mediates lipo-genesis under hypoxia. Nature 481(7381):380–384.
10. Le A, et al. (2012) Glucose-independent glutamine metabolism via TCA cycling forproliferation and survival in B cells. Cell Metab 15(1):110–121.
11. Zhou W, et al. (2012) Proteomic analysis reveals Warburg effect and anomalousmetabolism of glutamine in pancreatic cancer cells. J Proteome Res 11(2):554–563.
12. Ying H, et al. (2012) Oncogenic Kras maintains pancreatic tumors through regulationof anabolic glucose metabolism. Cell 149(3):656–670.
13. Chaika NV, et al. (2012) MUC1 mucin stabilizes and activates hypoxia-inducible factor1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A 109(34):13787–13792.
14. Aguirre AJ, et al. (2003) Activated Kras and Ink4a/Arf deficiency cooperate to producemetastatic pancreatic ductal adenocarcinoma. Genes Dev 17(24):3112–3126.
15. Cannito S, et al. (2008) Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 29(12):2267–2278.
16. Sonveaux P, et al. (2008) Targeting lactate-fueled respiration selectively kills hypoxictumor cells in mice. J Clin Invest 118(12):3930–3942.
17. Kennedy KM, Dewhirst MW (2010) Tumor metabolism of lactate: The influence andtherapeutic potential for MCT and CD147 regulation. Future Oncol 6(1):127–148.
18. Metallo CM, Vander Heiden MG (2010) Metabolism strikes back: Metabolic fluxregulates cell signaling. Genes Dev 24(24):2717–2722.
19. Simard EP, Ward EM, Siegel R, Jemal A (2012) Cancers with increasing incidencetrends in the United States: 1999 through 2008. CA Cancer J Clin 62(2):118–128.
20. Schwartz DL, et al. (2010) Radiosensitization and stromal imaging response correlatesfor the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreaticcancer. Mol Cancer Ther 9(7):2057–2067.
21. Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat RevCancer 4(11):891–899.
22. Schneiderhan W, et al. (2009) CD147 silencing inhibits lactate transport and reducesmalignant potential of pancreatic cancer cells in in vivo and in vitro models. Gut58(10):1391–1398.
23. Gaglio D, et al. (2011) Oncogenic K-Ras decouples glucose and glutamine metabolismto support cancer cell growth. Mol Syst Biol 7:523.
24. Olivier-Van Stichelen S, et al. (2012) The hexosamine biosynthetic pathway andO-GlcNAcylation drive the expression of β-catenin and cell proliferation. Am J PhysiolEndocrinol Metab 302(4):E417–E424.
25. Slawson C, Copeland RJ, Hart GW (2010) O-GlcNAc signaling: A metabolic link be-tween diabetes and cancer? Trends Biochem Sci 35(10):547–555.
26. Hanover JA, Krause MW, Love DC (2012) Bittersweet memories: Linking metabolismto epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol 13(5):312–321.
27. Yi W, et al. (2012) Phosphofructokinase 1 glycosylation regulates cell growth andmetabolism. Science 337(6097):975–980.
28. Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ (1979) Isolation of biologicallyactive ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18(24):5294–5299.
3924 | www.pnas.org/cgi/doi/10.1073/pnas.1219555110 Guillaumond et al.





























