Embed Size (px)
Citation preview

TRISOMIA XXI Estefania Vintimilla

SÍNDROME DE DOWN
Se trata de la primera cromosomopatía descubierta en ser humano en 1958,su clínica es conocida desde la descripción realizada en 1866 por J. LangdonDown.
Es el trastorno cromosómico más frecuente y la causa genéticaindependiente más común de retraso mental moderado. Su incidencia ennacidos vivos es de alrededor de 1/750.
La aparición de trisomía 21, así como de otras trisomías autosómicas,aumenta con la edad materna avanzada (≥35 años).

PATOGENIA
Regular
94%
Traslocación
4%
Mosaico
2%
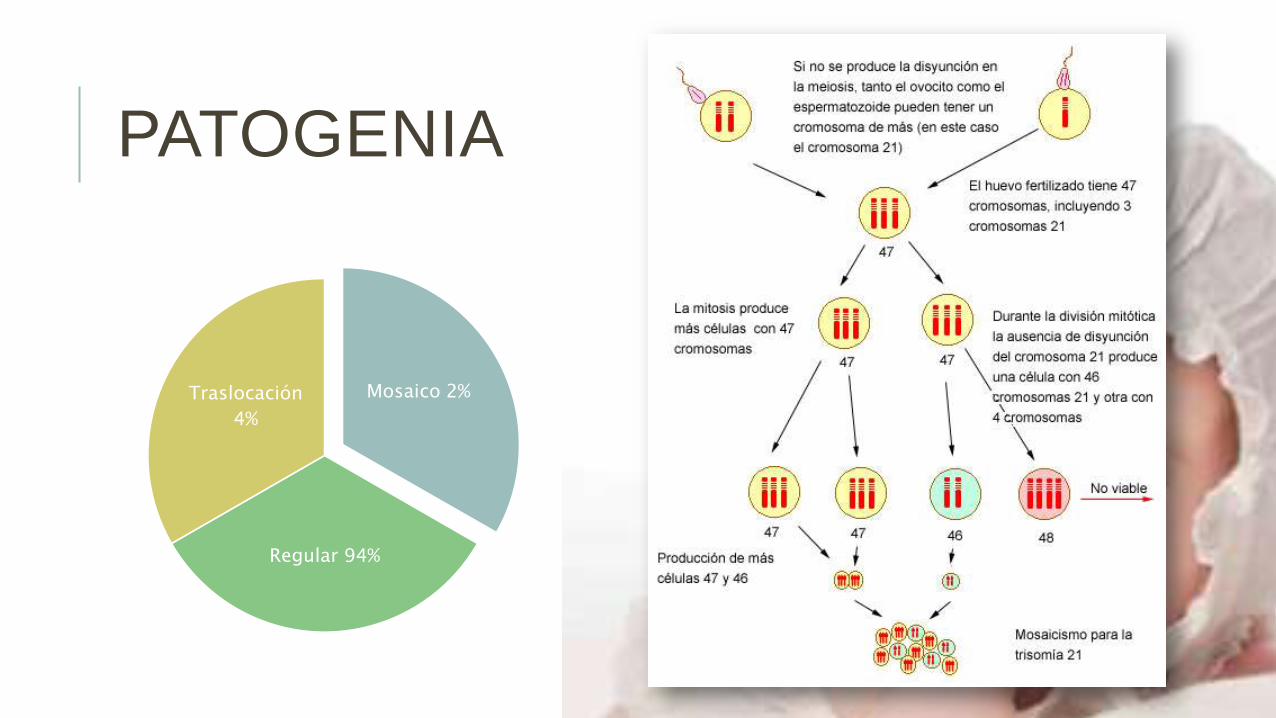
80% de casos se deben a nodisyunción materna (en meiosis I el80% y el meiosis II el 20%).20% de los casos lo son por nodisyunción paterna (en meiosis I el60% y en meiosis II el 40%).
PATOGENIA
Traslocación
4%
Regular 94%
Mosaico 2%
PATOGENIA
Mosaico 2%
Regular 94%
Traslocación
4%

PATOGENIA
El cromosoma 21 contiene unos 46 millones de pares de bases de ADN, con 386 genes y pseudogenes.
La trisomía de los genes localizados en los brazos largos del cromosoma y, especialmente, los de la región21q22, la llamada región crítica, tienen una importante intervención en la génesis de las complejasalteraciones anatómicas y funcionales del síndrome.
CUADRO CLÍNICO
Fenotipo
Malformaciones
Dermatoglifos
Alteraciones
funcionales
FENOTIPO
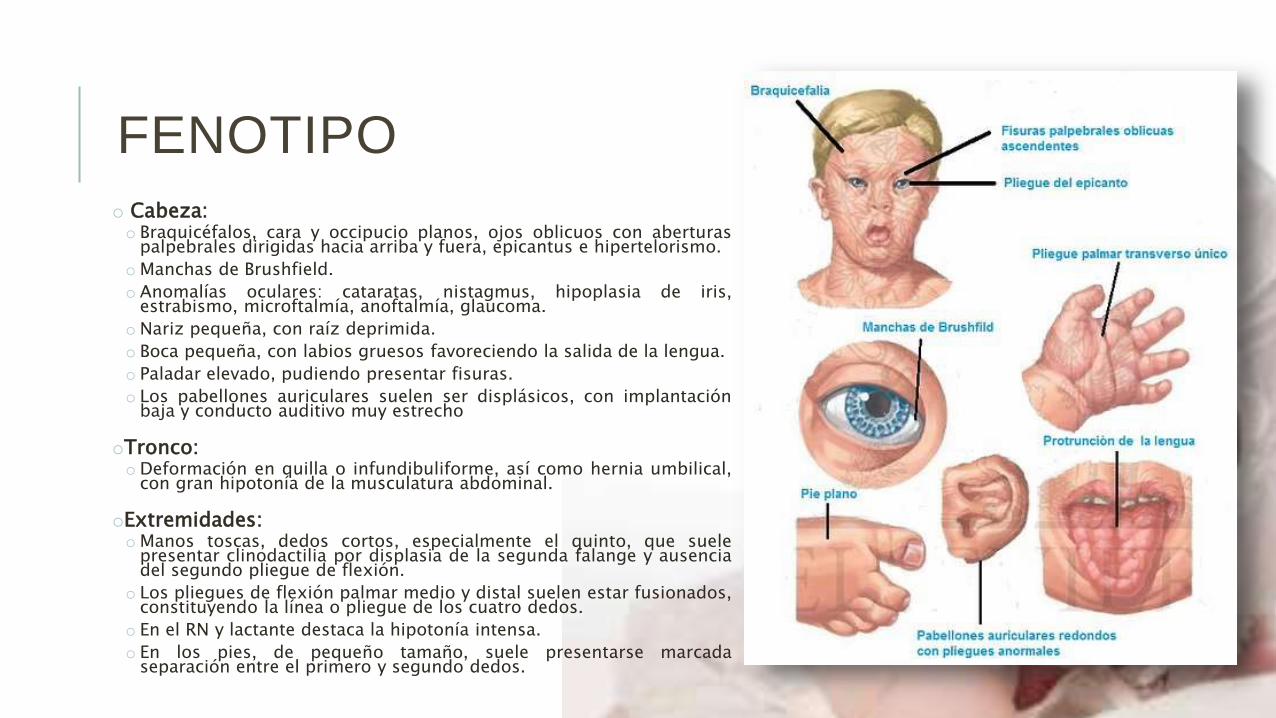
o Cabeza:o Braquicéfalos, cara y occipucio planos, ojos oblicuos con aberturas
palpebrales dirigidas hacia arriba y fuera, epicantus e hipertelorismo.
o Manchas de Brushfield.
o Anomalías oculares: cataratas, nistagmus, hipoplasia de iris,estrabismo, microftalmía, anoftalmía, glaucoma.
o Nariz pequeña, con raíz deprimida.
o Boca pequeña, con labios gruesos favoreciendo la salida de la lengua.
o Paladar elevado, pudiendo presentar fisuras.
o Los pabellones auriculares suelen ser displásicos, con implantaciónbaja y conducto auditivo muy estrecho
oTronco:o Deformación en quilla o infundibuliforme, así como hernia umbilical,
con gran hipotonía de la musculatura abdominal.
oExtremidades:o Manos toscas, dedos cortos, especialmente el quinto, que suele
presentar clinodactilia por displasia de la segunda falange y ausenciadel segundo pliegue de flexión.
o Los pliegues de flexión palmar medio y distal suelen estar fusionados,constituyendo la línea o pliegue de los cuatro dedos.
o En el RN y lactante destaca la hipotonía intensa.
o En los pies, de pequeño tamaño, suele presentarse marcadaseparación entre el primero y segundo dedos.
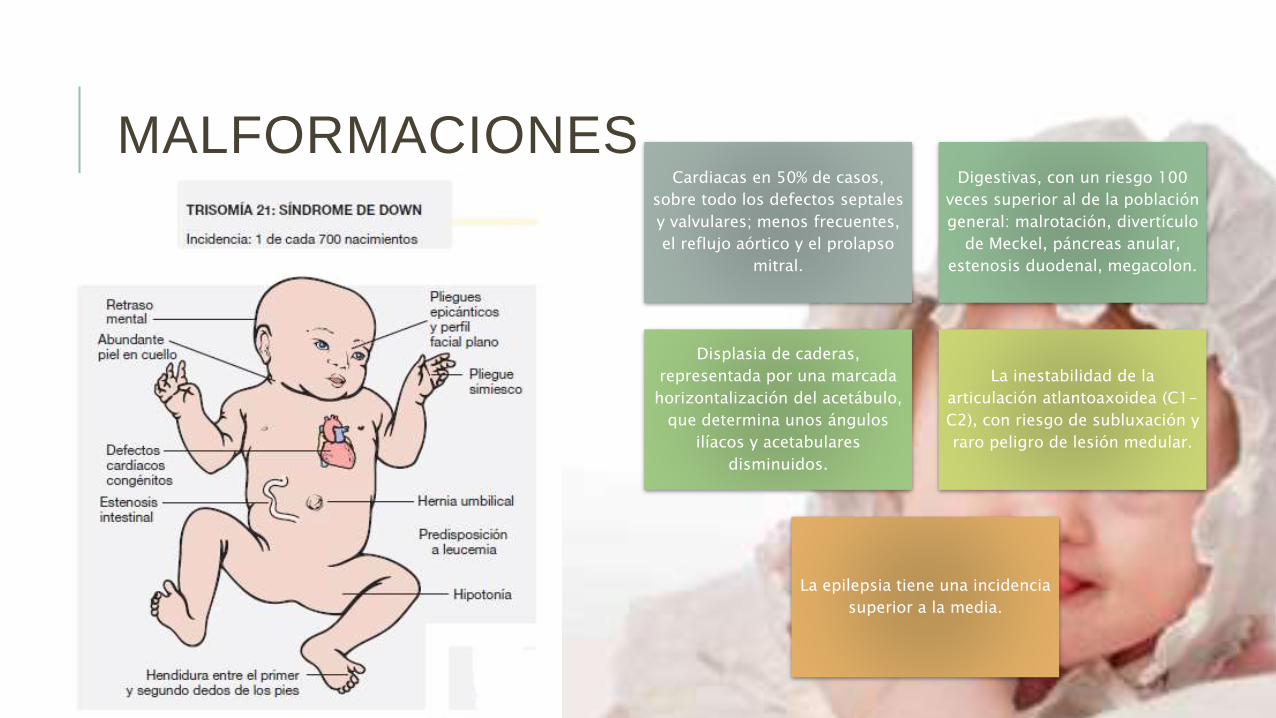
MALFORMACIONESCardiacas en 50% de casos,
sobre todo los defectos septales
y valvulares; menos frecuentes,
el reflujo aórtico y el prolapso
mitral.
Digestivas, con un riesgo 100
veces superior al de la población
general: malrotación, divertículo
de Meckel, páncreas anular,
estenosis duodenal, megacolon.
Displasia de caderas,
representada por una marcada
horizontalización del acetábulo,
que determina unos ángulos
ilíacos y acetabulares
disminuidos.
La inestabilidad de la
articulación atlantoaxoidea (C1-
C2), con riesgo de subluxación y
raro peligro de lesión medular.
La epilepsia tiene una incidencia
superior a la media.
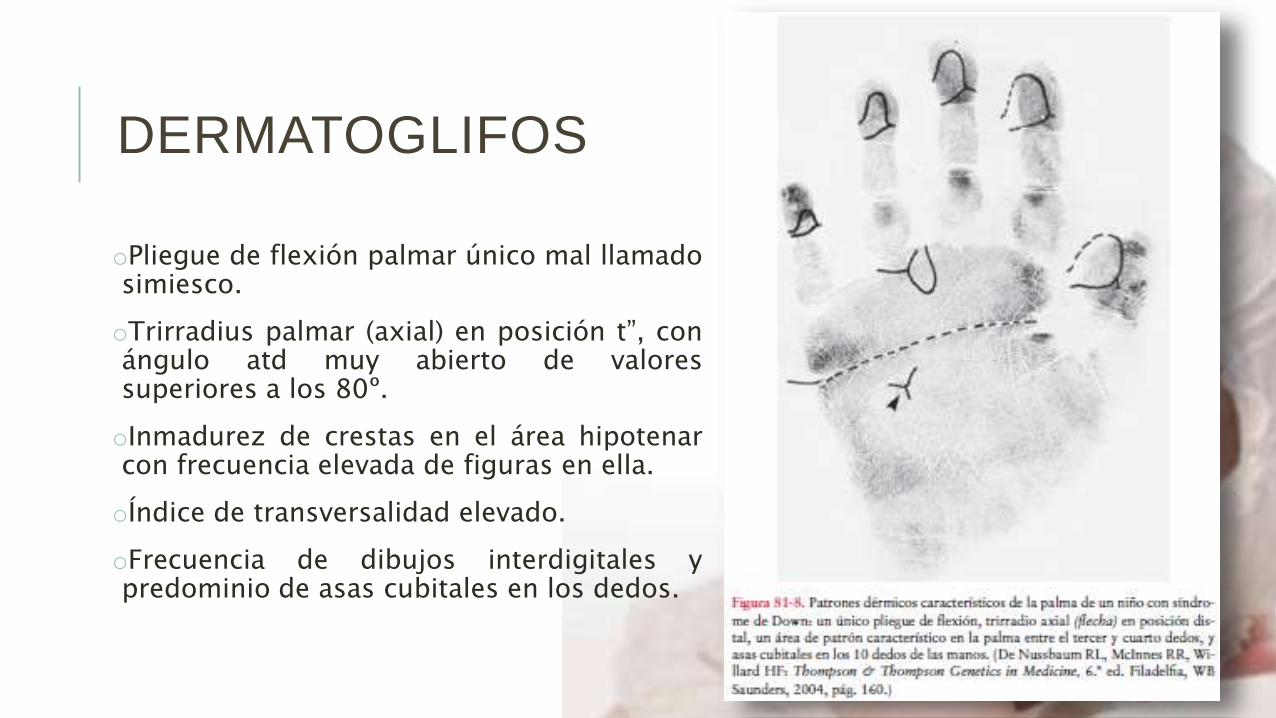
DERMATOGLIFOS
oPliegue de flexión palmar único mal llamadosimiesco.
oTrirradius palmar (axial) en posición t”, conángulo atd muy abierto de valoressuperiores a los 80º.
oInmadurez de crestas en el área hipotenarcon frecuencia elevada de figuras en ella.
oÍndice de transversalidad elevado.
oFrecuencia de dibujos interdigitales ypredominio de asas cubitales en los dedos.

ALTERACIONES FUNCIONALES

DIAGNÓSTICO
CLÍNICO:
oEl diagnóstico clínico no se basará en un solo síntoma, sino que alretraso psicomotor se debe añadir un mínimo de cuatro síntomasprincipales como: deficiencia mental, hipotonía, anomalías faciales yacromicria.
oEn algunos casos, con clínica atenuada y cariotipo normal, se hanobservado duplicaciones submicroscópicas, observables tras elempleo de citogenética molecular.
•síntomas principales o cardinales: hipotonía, hiperlaxitud
articular, posible ausencia o debilidad bilateral del reflejo de
Moro, cuello corto con piel abundante en la nuca, los rasgos
propios de cara y manos y la displasia de pelvis.
En el Rn
•Braquicefalia con occipucio plano, la oblicuidad palpebral con
epicantus, las anomalías dentarias, el retraso psíquico, la
lengua escrotal y el paladar ojival.
A partir de
los 2 años
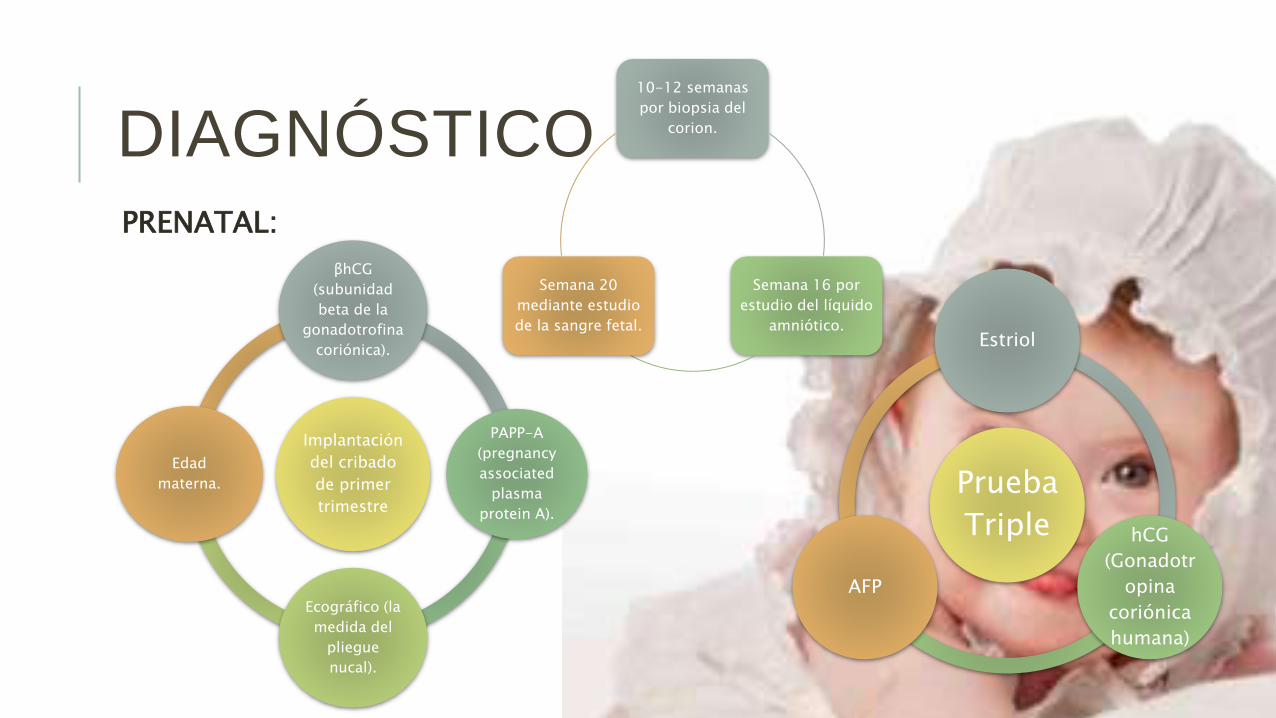
PRENATAL:
DIAGNÓSTICO10-12 semanas
por biopsia del
corion.
Semana 16 por
estudio del líquido
amniótico.
Semana 20
mediante estudio
de la sangre fetal.
Prueba
Triple
Estriol
hCG
(Gonadotr
opina
coriónica
humana)
AFP
Implantación
del cribado
de primer
trimestre
βhCG
(subunidad
beta de la
gonadotrofina
coriónica).
PAPP-A
(pregnancy
associated
plasma
protein A).
Ecográfico (la
medida del
pliegue
nucal).
Edad
materna.
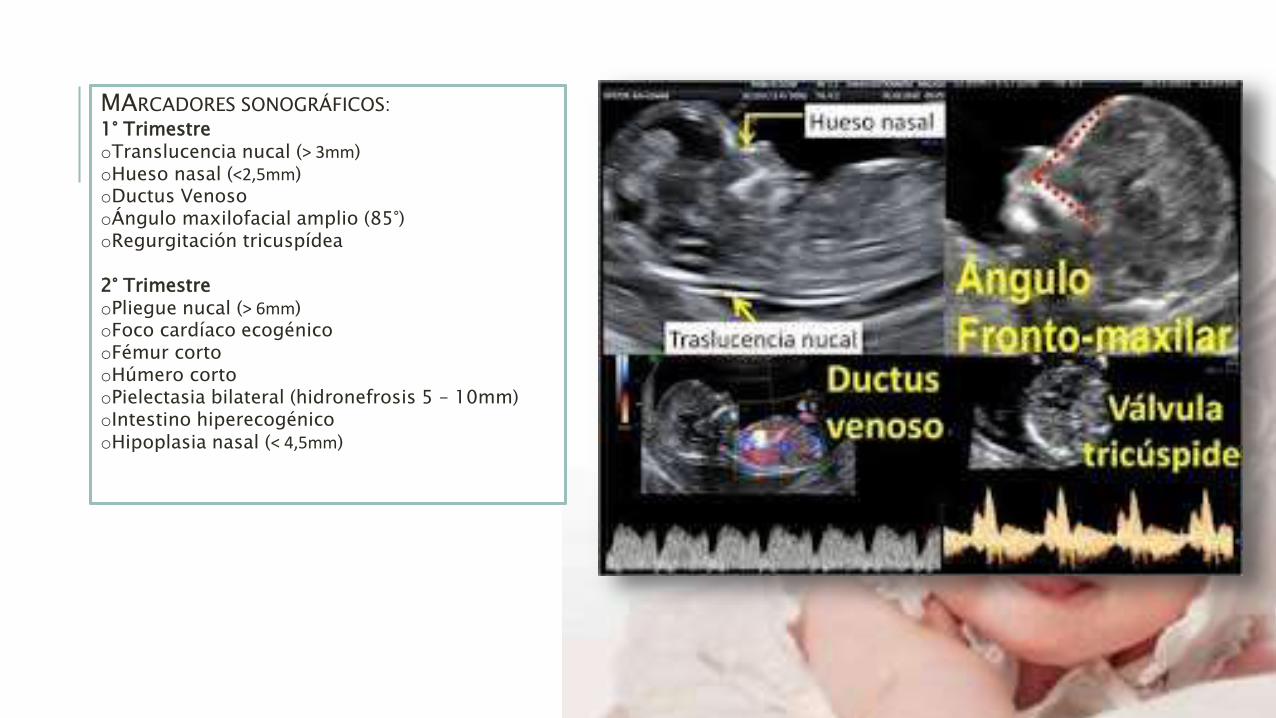
MARCADORES SONOGRÁFICOS:
1° TrimestreoTranslucencia nucal (˃ 3mm)oHueso nasal (˂2,5mm)oDuctus VenosooÁngulo maxilofacial amplio (85°)oRegurgitación tricuspídea
2° TrimestreoPliegue nucal (˃ 6mm)oFoco cardíaco ecogénicooFémur cortooHúmero cortooPielectasia bilateral (hidronefrosis 5 – 10mm)oIntestino hiperecogénicooHipoplasia nasal (˂ 4,5mm)

EVOLUCIÓN Y PRONÓSTICOoPatrón de crecimiento peculiar: La media es de 152 cm en el varón y de 145 en la mujer, pero con tendencia a mejorar.
oAutonomía personal en el 70% de los casos.
oInvolución psíquica tardía a partir de los 35 años, con un cuadro de regresión y sintomatología parecida a la enfermedad deAlzheimer con signos de envejecimiento precoz.
oProcesos respiratorios de repetición con importante participación de vías altas, adeno-amigdalitis y otitis que pueden favorecerla hipoacusia y las apneas del sueno
oEn el 5-10% de niños , alteraciones paroxísticas precoces (epilepsia rebelde).
oTrastornos de refracción ocular, como miopía o hipermetropía. Igualmente, los fenómenos de envejecimiento precoz: catarata,disminución de la elasticidad cutánea, penetración precoz de válvulas pulmonar y aortica y anomalías de lipoproteínas séricascon ateroesclerosis temprana
oRiesgo de hipotiroidismo hacia los 13 anos (17% de los pacientes), siendo el substrato habitual una tiroiditis de Hashimoto (sedeben vigilar cada 2 anos TSH y T4).
oLos trastornos inmunológicos (celiaquía, diabetes insulinodependiente, artritis, entre otros) pueden agravar el pronostico.
oTambién la osteoporosis puede interferir en la evolución.
oLa mayor incidencia de leucemia con una frecuencia 20 veces superior que la población general.
oVida media: 45 años; en el hombre hay hipogonadismo e infertilidad; mientras que la mujer es fértil.

TRATAMIENTO
oEstimuloterapia precoz
oPsicoterapia con educación especializada
oLa corrección de malformaciones (cardiopatía congénita, estrabismo, alteraciones del esqueleto)
oProfilaxis de sus posibles alteraciones evolutivas.
oMedidas preventivas de las patologías más habituales en estos niños: hipoacusia, cataratas, apneaobstructiva del sueño, patología digestiva, vigilancia de la articulación atlantoaxial, hipotiroidismo,inmunodeficiencias, y vigilar la sangre periférica a la búsqueda de posibles síndromesmielodisplásicos.
oLa medicación psicotropa (piriglutina, piracetam, cetoglutarato) puede ser de alguna ayuda en loscasos con epilepsia (5-10%).
oSuplementos (pre y postnatales) de folatos, vitaminas (B6, C, E), selenio y cinc, que podrían prevenir oretardar el comienzo de la demencia.
oTerapia génica.

RECURRENCIA
oHabiendo tenido una gestación previa con trisomía 21, el riesgo de recurrenciadependiente de la edad materna:o ˂30 años, el riesgo de recurrencia es el propio de la edad x 8
o ˃30 el riesgo se multiplica solo por dos.
oCuando los padres son portadores de una translocación equilibrada, el riesgo teóricode gametos desequilibrados es del 50%.
oCuando es la madre la portadora, el riesgo de descendencia viva con síndrome deDown es del 10%.
oCuando lo es el padre, el riesgo es solo del 1%.
oUna madre afecta del síndrome tendrá un riesgo teórico el 50% de hijos trisómicos yel 50% de sanos.

CONCLUSIÓN
El síndrome de Down no tienecura. Sin embargo, si, desde losprimeros momentos de vida, estosniños reciben una atenciónadecuada, que abarque todos losaspectos relacionados con eldesarrollo de las capacidades decada uno de ellos (aspectoscognitivos, psicomotrices,afectivos, educativos, sociales,etc), se lograran grandes mejorasen su bienestar, calidad de vida yen sus posibilidades dedesenvolverse en la vida conautonomía; ya que el desarrollocerebral no depende únicamentede factores genéticos, sino quetambién influyen los estímulosambientales.





























