Embed Size (px)
Citation preview

www.elsevier.com/locate/ymcne
Mol. Cell. Neurosci. 35 (2007) 1–13Mutations in the SPG3A gene encoding the GTPase atlastininterfere with vesicle trafficking in the ER/Golgi interfaceand Golgi morphogenesis
M. Namekawa,a,1 M.-P. Muriel,a,b A. Janer,a,b M. Latouche,a,b A. Dauphin,c T. Debeir,a,b
E. Martin,a,b C. Duyckaerts,a,b A. Prigent,a,b C. Depienne,a,b,d A. Sittler,a,b
A. Brice,a,b,d,e,⁎ and M. Ruberg,a,b
aINSERM, U679, Paris F-75013, FrancebUniversité Pierre et Marie Curie-Paris6, Federative Institute for Neuroscience Research (IFR70), UMR-S679, Paris F-75012, FrancecAP-HP, Hôpital Pitié-Salpêtrière, Plateforme de l'imagerie cellulaire, Paris F-75013, FrancedAP-HP, Hôpital Pitié-Salpêtrière, Federation de Neurology, Paris F-75013, FranceeAP-HP, Hôpital Pitié-Salpêtrière, Department of Genetics and Cytogenetics, Paris F-75013, France
Received 10 August 2006; revised 22 January 2007; accepted 22 January 2007Available online 26 January 2007
Mutations in SPG3A causing autosomal dominant pure spasticparaplegia led to identification of atlastin, a new dynamin-like largeGTPase. Atlastin is localized in the endoplasmic reticulum, the Golgi,neurites and growth cones and has been implicated in neuriteoutgrowth. To investigate whether it exerts its activity in the earlysecretory system, we expressed normal and mutant atlastin in cellculture. Pathogenic mutations in the GTPase domain interfered withthe maturation of Golgi complexes by preventing the budding ofvesicles from the endoplasmic reticulum, whereas mutations in otherregions of the protein disrupted fission of endoplasmic reticulum-derived vesicles or their migration to their Golgi target. Atlastin,therefore, plays a role in vesicle trafficking in the ER/Golgi interface.Furthermore, atlastin partially co-localized with proteins of the p24/emp/gp25L family that regulate vesicle budding and trafficking in theearly secretory pathway, and co-immunoprecipitated p24, suggesting afunctional relationship that should be further explored.© 2007 Elsevier Inc. All rights reserved.
Keywords: Spastic paraplegia; Spastin; Endoplasmic reticulum; Spinalcord; Motor neurons; HEK293 cells; Cortical neurons; Brefeldin A
⁎ Corresponding author. INSERM, U679, Groupe Pitié-Salpêtrière, 47,Boulevard de l’Hôpital, Paris F-75013, France. Fax: +33 1 44 24 36 58.
E-mail address: [email protected] (A. Brice).1 Present address: Department of Neurology, Jichi Medical University,
Tochigi, Japan.Available online on ScienceDirect (www.sciencedirect.com).
1044-7431/$ - see front matter © 2007 Elsevier Inc. All rights reserved.doi:10.1016/j.mcn.2007.01.012
Introduction
Hereditary spastic paraplegias (HSPs) are a group of geneticallyheterogeneous neurological disorders, characterized by progressivespasticity and weakness of the lower limbs, that are caused bydegeneration, or failure to develop, of the corticospinal tract (Reid,2003). Over 30 loci for HSP have been found, but only 15 of thecausative genes have been identified. Mutations in the SPG3A geneon chromosome 14 were identified for the first time in HSP patientsin 2001 (Zhao et al., 2001) and cause the most frequent autosomaldominant form of pure HSP with very early onset and the secondmost frequent of all autosomal dominant forms of this disease (Dürret al., 2004; Namekawa et al., 2006). The gene encodes a 558 amino-acid protein, atlastin (Zhao et al., 2001), a new member of thedynamin superfamily of large GTPases (Praefcke and McMahon,2004), with an N-terminal GTPase domain that contains the fourcanonical GTP binding motifs (Praefcke and McMahon, 2004), amid-portion, two transmembrane domains and a short C-terminalregion. One of a subfamily of three homologous proteins, it wasrecently renamed atlastin-1 (Zhu et al., 2003).
Twenty different missense mutations and one insertion have beenreported in the SPG3A gene (Namekawa et al., 2006). Thepredominance of missense mutations suggests that pathogenesismay be due to either a toxic gain of function, or a dominant negativeeffect, but haploinsufficiency cannot be excluded. Consistent withthe selective dysfunction of the corticospinal pathway in patients,atlastin is highly expressed in motor neurons in the rat (Zhu et al.,2003) and has been localized in the Golgi, the endoplasmicreticulum (ER), neurites and growth cones (Zhu et al., 2003, 2006).
Atlastin was shown by shRNA knock-down to play a role inneurite outgrowth (Zhu et al., 2006), but it is not clear whether lossof atlastin affects neurite outgrowth locally in the neurite or growth

Fig. 1. Intracellular distribution of atlastin in human brain. (A) Left panel:polyclonal antibody 5409 against the N-terminal of human atlastin (CraigBlackstone, NIH, Bethesda, MD, USA) labels a single band corresponding toendogenous atlastin in extracts of cerebral cortex from two normal humansubjects (lanes 1 and 2), V5- (lane 3) and GFP-tagged (lane 4) human atlastinextracted from HEK293 cells, and endogenous atlastin in HEK293 cells(lanes 4 and 5). Lane 6 shows V5-tagged human atlastin labeled with an anti-V5 antibody. Right panel: preabsorption of the 5409 antibody with thepeptide used for immunization abolishes immunohistochemical staining;shown on a transverse section of spinal cord. (B) Immunohistochemicallabeling of humanmotor cortex and lumbar spinal cord (DAB staining) at low(a, c) and high (b, d) magnification. Note the punctate distribution of atlastinin Betz cells in layer Vof the motor cortex (b) and in lower motor neurons inthe spinal cord (d). Atlastin-positive axons in the lateral corticospinal tract cuttransversely (e). The punctate distribution can also be seen in an axon in thelumbar spinal cord cut longitudinally (f). Scale bars=10 μm (a, c, e), 5 μm(b, d, f).
2 M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
cone or affects a function further upstream at the level of the ER orGolgi. We have, therefore, analyzed the effects of pathogenicmutations on the localization or function of atlastin in the earlysecretory system. We provide evidence that atlastin is implicated invesicle trafficking in the ER/Golgi interface and maturation of theGolgi complex, which is compromised by disease-causingmutations. We have also found that atlastin is localized in thesame functional compartment as members of the p24 (emp/gp25L)family of transmembrane proteins, hetero-oligomeric proteins thatplay a role in vesicle transport between the ER and Golgi (Stamneset al., 1995; Dominguez et al., 1998) and co-immunoprecipitatesp24. Identification of these putative partners will help to elucidatethe biochemical function of the atlastin GTPase in vesicletrafficking.
Results
Distribution of endogenous atlastin in human motor cortex andspinal cord
We first verified that overexpressed atlastin had the samedistribution in our cell culture model, HEK293 cells, as in humanmotor neurons. Atlastin was detected on sections of human brain andspinal cord with the 5409 polyclonal antibody developed by Dr.Craig Blackstone (Zhu et al., 2003). The antibody labeled a singleband on Western blots of human brain extract corresponding toendogenous atlastin, as well as to V5- and GFP-tagged recombinantatlastin expressed in HEK293 cells (Fig. 1A). A low level ofendogenous atlastin was also detected in HEK293 cell extracts(Fig. 1A). Absorption of the antibody with a peptide identical to theone used to generate the antibody abolished labeling on humannervous system tissue (Fig. 1A), confirming the specificity of thelabeling. Fig. 1B shows low (a, c) and high (b, d) power images ofatlastin-like immunoreactivity in human cerebral cortex and spinalcord. Atlastin-like immunoreactivity was distributed in a reticularpattern throughout the cell bodies of corticospinal neurons in themotor cortex, illustrated by a Betz cell in layer V (Fig. 1Bb) and alower motor neuron in the anterior horn of the spinal cord (Fig. 1Bd).Although not all neurons or fibers in the spinal cord contained theprotein, atlastin-immunoreactive structures were relatively dense, asshown in the lateral corticospinal tract (Fig. 1Be). The distribution ofatlastin in the axons was punctate, as seen on longitudinal sections ofthe lumbar spinal cord (Fig. 1Bf). This distribution of endogenousatlastin in human upper and lower motor neurons was similar to whathas been reported in rat brain sections and cultured neurons (Zhuet al., 2003, 2006).
Intracellular distribution of epitope-tagged atlastin
Full-length wild-type atlastin with either an N-terminal Xpresstag or a C-terminal V5 tag was expressed in HEK293 cells, and itsdistribution verified by immunofluorescence with antibodiesagainst the tags. As in human brain and in previously reportedcultures of primary neurons (Zhu et al., 2003, 2006), V5-taggedatlastin was distributed in a diffuse reticular pattern, interspersedwith small puncta, throughout the cell bodies and the long neurite-like processes of HEK293 cells; immunolabeling was particularlyintense in the perinuclear region and at the ends of the processes(Fig. 2A). The distributions of both V5- and Xpress-tagged atlastinwere similar (Fig. 2B), indicating that the position of the tag didnot affect the localization of the expressed proteins.

3M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
To identify the subcellular structure in which V5-tagged atlastinwas localized in HEK293 cells, we co-immunolabeled transfectedHEK293 cells with antibodies against the V5 tag and againstcalreticulin and calnexin, markers of the endoplasmic reticulum(ER), giantin, a marker of the cis- and medial Golgi. As observedby confocal microscopy, atlastin-V5 had the same cellulardistribution as the ER lumen marker calreticulin (Fig. 2C) andcalnexin (not shown), suggesting a localization within or in closeproximity to the ER. Some punctiform atlastin-positive structuresthat did not express the ER marker were interspersed among theatlastin/calreticulin-positive structures, suggesting the presence of acontiguous, probably vesicular, structure that contains atlastin butis independent of the ER. Atlastin was also partially co-localizedwith the Golgi marker giantin (Fig. 2C). The co-localization of
Fig. 2. Intracellular distribution of epitope-tagged atlastin in HEK293 cells.(A) Immunofluorescent labeled V5-tagged human atlastin in the cell bodyand processes of an HEK293 cell. Note the fine reticular distribution, thesmall puncta in the cell body and processes and the strong labeling in theperinuclear region and at the end of a cell process. (B) C-terminal taggedatlastin-V5 and N-terminal tagged atlastin-Xpress have similar distributionsin HEK293 cells. (C) Confocal microscopic images showing co-localizationof epitope-tagged atlastin with the ER marker calreticulin, and partially withthe cis-Golgi marker giantin. Scale bars=5 μm.
atlastin with ER markers and its partial co-localization with the cis-Golgi markers was similar to the localization of epitope-taggedatlastin in HeLa cells and in the motor neuron–neuroblastomahybrid NSC-34 cells (Sanderson et al., 2006).
GTPase domain mutations modify the morphology of the ER
We next examined the effects of disease-causing mutations onatlastin-V5 expression and subcellular distribution. The mutationsstudied were distributed throughout the protein and were introducedinto the wild-type atlastin-V5 cDNA sequence by in vitromutagenesis; they are represented schematically in Fig. 3A. Fivemutations affected the GTPase domain (F151S, A161P, T162P,R217Q, R239C), and one each the mid-portion (S398Y), the secondtransmembrane domain (R495W) and the C-terminal (S519N).
The levels of expression of wild-type and mutant atlastin proteinin HEK293 cell cultures, relative to the actin loading control, weresimilar as shown by Western blot, except for R217Q, which wasexpressed at lower levels (Fig. 3B). A cluster of three mutations,F151S, A161P and T162P, changed the distribution of atlastin fromthe reticular pattern, shown in Fig. 2, to aggregate-like structures(Fig. 3C), the size of which varied according to the mutation; thoseproduced by the F151S mutation were particularly large anddisruptive (Fig. 3C). These mutations are located right after theDxxG motif, one of four conserved motifs forming the GTP bindingsite in dynamin, Ras family and other small GTPases, that has beenshown to be critical for GTP binding or hydrolysis (Praefcke et al.,2004). The other two GTPase mutations, R217Q, which affects theRD motif also important for GTPase activity (Praefcke et al., 2004),and R239C, and the mutations affecting downstream domains ofatlastin had little or no effect on the distribution of atlastin-V5, whichlargely retained its reticular character (Fig. 3C).
To determine whether the change in the cellular distribution ofatlastin with the F151S, A161P and T162P mutations was due to achange in its subcellular localization or to a change in themorphology of the ER, we co-immunolabeled atlastin-V5 andcalreticulin. The aggregate-like structures formed by atlastin withthe three clustered GTPase mutations were co-labeled with theantibody against calreticulin (Fig. 3D), demonstrating that theywere not aggregates of overexpressed protein but that mutationswhich may alter the active site of the atlastin GTPase also modifiedthe morphology of the ER.
GTPase mutations interfere with the development of theGolgi apparatus
To determine the consequences of the change in ER morphologyin HEK293 cells expressing atlastin with the GTPase mutations,F151S, A161P and T162P, the ultrastructure of the ER and Golgicomplexes in these cells was examined by electron microscopy.Sister cultures were immunolabeled to verify the efficacy oftransfection (40–60%) and the expression of the V5-taggedproteins. Untransfected cells, cells expressing wild-type atlastinand cells expressing an unrelated protein, the neuron-specificpotassium-chloride co-transporter KCC-2 (gene SLC12A5, acces-sion no. NM_020708) that is addressed to the plasma membrane,were used to control for possible effects of both cellular location andexpression levels.
Representative images are shown in Fig. 4. It was not possibleto distinguish cells expressing wild-type atlastin or KCC-2 fromuntransfected cells (Fig. 4A). In all of these cells, the ER was

Fig. 3. A cluster of disease-causing mutations in the GTPase domain of atlastin change its intracellular distribution in HEK293 cells. (A) Schematic representationof the disease-causing atlastin mutations studied (in bold with arrows) with respect to the GTPase, mid-portion (mid), transmembrane (TM) and C-terminaldomains of the protein. The numbers of the amino acids bounding the domains are indicated. The expansion of the GTPase domain shows the four consensus GTPbinding motifs defining the subfamily of dynamin-like large GTPases to which atlastin belongs (Praefcke and McMahon, 2004). The first three motifs are sharedby all dynamin family and other GTPases, the RDmotif by atlastin andGBP1. (B)Western blot comparing the levels of expression of wild-type andmutant atlastinin HEK293 cells. Equal amounts of plasmid DNAwere transfected, and equal quantities of cell lysate (20 μg of protein) were loaded in each lane. Actin wasimmunolabeled as a loading control. (C) Compared to the reticular distribution of wild-type atlastin (Figs. 2A, B), V5-tagged atlastin with the GTPase domainmutations F151S, A161P and T162P forms large aggregate-like structures inHEK293.GTPasemutations R217Q andR239C and themid-portion, transmembraneand C-terminal domain mutations have little or no effect on the reticular distribution of atlastin. (D) Confocal microscopic images showing that the aggregate-likestructures formed by atlastin with the F151S, A161P and T162P mutations still contain the ER marker calreticulin, indicating that these mutations change themorphology of the ER. Scale bars=5 μm.
4 M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
separated from the Golgi complexes by vesicle-containing zonesand the Golgi complexes consisted of stacks of numerous thintubules surrounded by vesicles, some of which could be seenbudding from the tubules.
Expression of the GTPase mutants F151S, A161P and T162P,however, led to evident alterations of Golgi morphology (Fig. 4B).The effects of these mutations differed, however. The F151Smutation, which caused the most severe change in the immuno-
fluorescence labeling of atlastin and the ER marker calreticulin (seeFig. 3B), also caused themost severe ultrastructural alterations of theER and Golgi: segments of swollen ER coalesced around, andappeared to be incorporated into, a large inhomogeneousmembrane-bound lysosome-like structure containing vesicles and what appearsto be fragments of ER (Fig. 4B); the only identifiable Golgi complexin this cell consisted of a few swollen tubules with no apparentstructure (not shown). The contiguous A161P and T162P mutations

5M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
caused less severe changes that were the same in both cases:segments of rough ER were attached to a few Golgi tubules thatremained attached to each other and the vesicle-containing zone wasno longer visible (Fig. 4B), as if vesicle budding from the ER hadbeen prevented.
The other mutations, illustrated by R239C, S398Y andR495W, also affected ER and Golgi morphology (Fig. 4C), ina manner that differed from the GTPase mutations. In these cases,there was an overabundance of vesicles that could be seenbudding from deformed ER. The vesicles did not appear,however, to migrate from their site of formation and, again,Golgi complexes did not develop (Fig. 4C).
Fig. 4. Disease-causing mutations in atlastin disrupt the morphology of the Golgi apatlastin with mutations in the GTPase (F151S, A161P, T162P) and other domainscells, cells expressing wild-type atlastin or the plasmamembrane potassium-chlorideor cells expressing wild-type atlastin and KCC-2. Transfected cells could not be disticlearly defined Golgi apparatus (G) with budding vesicles (bV). (B) Effects of the thrthat coalesced around and appeared to be incorporated into a membrane-bound lysocell (not shown). In cells expressing the contiguous A161P and T162P mutations, thER appeared fused to an underdeveloped Golgi complex. (C) In contrast to the GTPaER that appeared to be actively producing vesicles (arrows) that appeared to accumGolgi complexes.
Distribution and effects of wild-type and mutant atlastin on the ERand the Golgi analyzed by brefeldin A treatment
To further evaluate the localization of atlastin in the ER and theGolgi and the effects of the mutants on Golgi development, weexamined the effects of brefeldin A (BFA) treatment, whichredistributes Golgi proteins to the ER, on the distribution of atlastinand on markers of the ER and the cis-Golgi complex (Fig. 5). Inaddition to wild-type V5-labeled atlastin, two representativemutations were studied: T162P, a GTPase domain mutation thatstrongly affected the distribution of atlastin and the ER markercalreticulin (see Figs. 3C, D above) and the formation of the Golgi,
paratus. Ultrastructure of the ER/Golgi interface in HEK293 cells expressing(R293C, S398Y, R495W) compared to control cells (untransfected HEK293co-transporter KCC-2). (A) Golgi apparatus in cultures of untransfected cellsnguished from untransfected cells; all had a vesicle-containing zone (V) and aee GTPasemutations. The F151Smutation caused distension of ER segmentssome-like (L-l) structure; recognizable Golgi complexes did not form in thise vesicle-filled region between the ER and cis-Golgi no longer existed and these mutations, the mutations R239C, R398Yand R495Whad highly deformedulate at their site of production, but no identifiable, or highly disorganized,
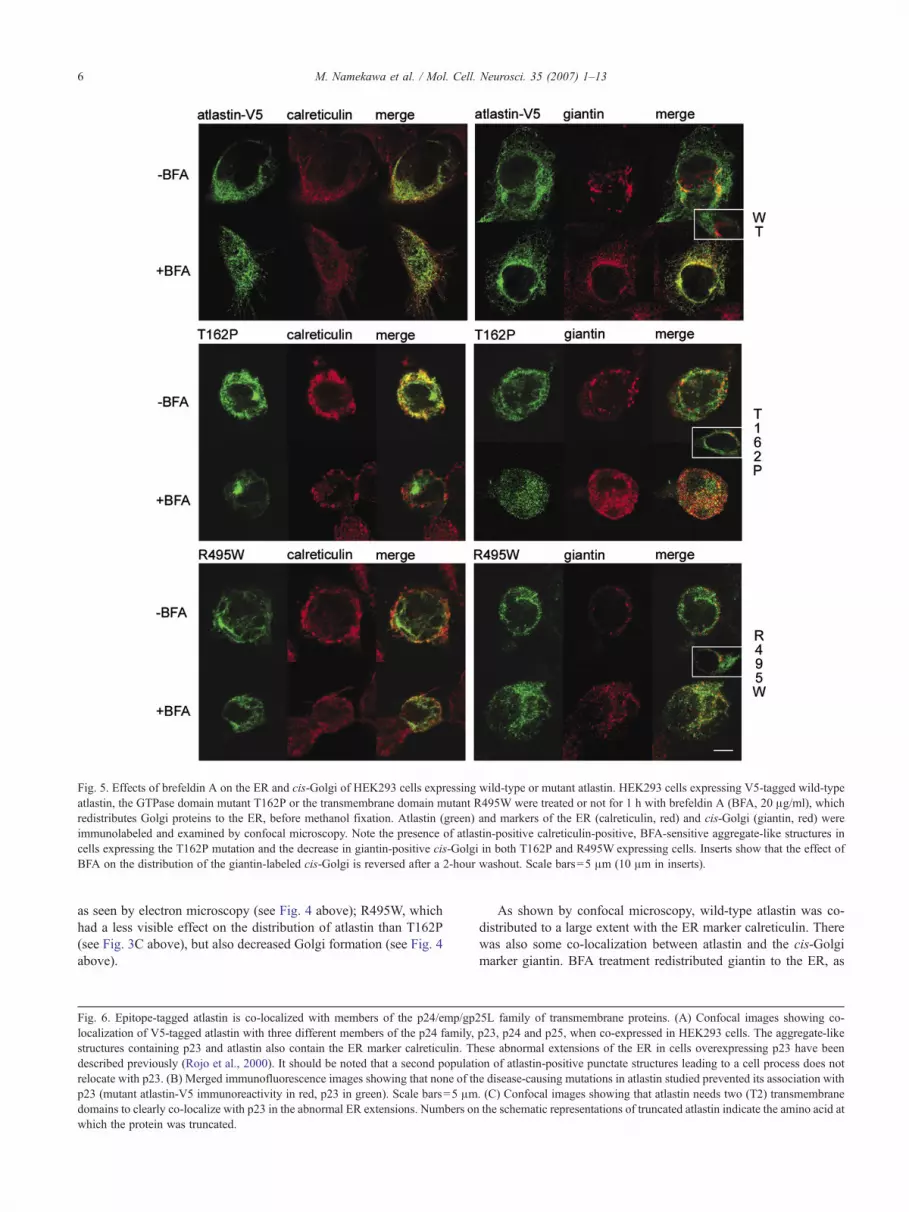
Fig. 5. Effects of brefeldin A on the ER and cis-Golgi of HEK293 cells expressing wild-type or mutant atlastin. HEK293 cells expressing V5-tagged wild-typeatlastin, the GTPase domain mutant T162P or the transmembrane domain mutant R495W were treated or not for 1 h with brefeldin A (BFA, 20 μg/ml), whichredistributes Golgi proteins to the ER, before methanol fixation. Atlastin (green) and markers of the ER (calreticulin, red) and cis-Golgi (giantin, red) wereimmunolabeled and examined by confocal microscopy. Note the presence of atlastin-positive calreticulin-positive, BFA-sensitive aggregate-like structures incells expressing the T162P mutation and the decrease in giantin-positive cis-Golgi in both T162P and R495W expressing cells. Inserts show that the effect ofBFA on the distribution of the giantin-labeled cis-Golgi is reversed after a 2-hour washout. Scale bars=5 μm (10 μm in inserts).
6 M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
as seen by electron microscopy (see Fig. 4 above); R495W, whichhad a less visible effect on the distribution of atlastin than T162P(see Fig. 3C above), but also decreased Golgi formation (see Fig. 4above).
Fig. 6. Epitope-tagged atlastin is co-localized with members of the p24/emp/gp2localization of V5-tagged atlastin with three different members of the p24 family,structures containing p23 and atlastin also contain the ER marker calreticulin. Thdescribed previously (Rojo et al., 2000). It should be noted that a second populatirelocate with p23. (B) Merged immunofluorescence images showing that none of thp23 (mutant atlastin-V5 immunoreactivity in red, p23 in green). Scale bars=5 μmdomains to clearly co-localize with p23 in the abnormal ER extensions. Numbers onwhich the protein was truncated.
As shown by confocal microscopy, wild-type atlastin was co-distributed to a large extent with the ER marker calreticulin. Therewas also some co-localization between atlastin and the cis-Golgimarker giantin. BFA treatment redistributed giantin to the ER, as
5L family of transmembrane proteins. (A) Confocal images showing co-p23, p24 and p25, when co-expressed in HEK293 cells. The aggregate-likeese abnormal extensions of the ER in cells overexpressing p23 have beenon of atlastin-positive punctate structures leading to a cell process does note disease-causing mutations in atlastin studied prevented its association with. (C) Confocal images showing that atlastin needs two (T2) transmembranethe schematic representations of truncated atlastin indicate the amino acid at

7M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
expected, increasing its co-localization with atlastin. The effect ofBFAwas reversible. After a 2-hour washout, the giantin-labeled cis-Golgi resembled that of untransfected cells and cells expressingwild-type atlastin that had not been treated with BFA (Fig. 5, insert).
In cells expressing T162P atlastin, atlastin again strongly co-localized with calreticulin, but in aggregate-like structures, likethose seen above. Interestingly, BFA treatment dispersed bothatlastin and calreticulin in the cells and decreased their co-

8 M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
localization in the ER, suggesting that much of the association ofatlastin and calreticulin was in a BFA-sensitive Golgi compartmentthat may correspond to the aggregate-like structures observed.They may also correspond to the Golgi tubules attached toabnormal ER seen by electron microscopy. The amount of giantin-labeled atlastin-positive cis-Golgi was correspondingly decreasedin T162P-treated cells, and both atlastin and giantin were dispersedby BFA treatment in these cells, confirming the association ofT162P atlastin with a BFA-sensitive Golgi compartment. Asabove, the effect of BFA was reversible, but the effect of themutation was still observed. After a 2-hour washout, the giantin-labeled cis-Golgi resembled that of cells expressing T162P atlastinthat had not been treated with BFA (Fig. 5, insert).
The R495W mutation had a lesser effect on the morphology ofcalreticulin-labeled ER, but decreased its association with atlastin.It also greatly decreased the amount of giantin-labeled cis-Golgi.As for the T162P mutation, a 2-hour washout reversed the effect ofBFA on the giantin-labeled cis-Golgi, but did not change the effectof the R495W mutation (Fig. 5, insert).
These observations are consistent with the immunofluorescenceand ultrastructural observations above that showed a relativelynormal co-distribution of atlastin and calreticulin and the presenceof an actively budding ER, when the cells were examined byelectron microscopy. It may be hypothesized that the dissociationbetween R495W-atlastin and the calreticulin-positive ER in theBFA experiment might be responsible for the excessive vesiclebudding or the absence of fusion of the vesicles with Golgi targetmembranes observed by electron microscopy.
Atlastin co-localizes with members of the p24 family
The observations made by electron microscopy and theirconfirmation by calreticulin and giantin labeling in the BFAexperiments described above suggest that atlastin might play a rolein the transfer of membranous vesicles from the ER to the Golgi;mutations that affect critical sites in the GTPase domain appearedto prevent vesicle formation from the ER, whereas the othermutations allowed vesicle formation but prevented the vesiclesfrom reaching and integrating their putative Golgi target, perhapsby hindering interactions with essential partners in the complexesthat regulate vesicular transport in the early secretory system.Proteins of the p24 (emp24/gl25L) family are of interest in thisrespect, in particular p23, p24 and p25, that are reported to play arole in membrane budding (Stamnes et al., 1995) and trafficking inthe ER/Golgi interface and Golgi morphogenesis (Dominguezet al., 1998; Rojo et al., 1997, 2000; Emery et al., 2000, 2003).
To determine whether atlastin co-localizes with proteins of thep24 family, we co-transfected HEK293 cells with V5-taggedatlastin and myc-tagged p23 and p25 and HA-tagged p24. P25-myc, which has a canonical ER retention signal at its C-terminus,was distributed, as expected, in a reticular pattern, as was p24-HA(Fig. 5A), in accordance with previous reports (Stamnes et al.,1995; Blum et al., 1999). Overexpressed p23, however, formedaggregate-like structures (Fig. 6A). When co-expressed, atlastinco-localized with p24 and p25 (Fig. 6A). Interspersed p24- andp25-negative atlastin-positive structures were also observed, aswith the ER marker described above (Fig. 6A). When co-expressedwith p23, most atlastin was found in the aggregate-like structures(Fig. 6A). However, some atlastin in punctate structures leading tocell processes did not co-localize with p23 (Fig. 6A), confirmingthe previous report of the existence of more than one atlastin-
containing compartments, one of which is associated with theendoplasmic reticulum and Golgi, as shown here, another withneurite outgrowth (Zhu et al., 2006). The aggregate-like structuresin cells expressing both atlastin and p23 were also labeled by theanti-calreticulin antibody, suggesting that they might correspond tothe abnormal extensions of the ER formed under the effect of p23that were previously seen by immunofluorescence and electronmicroscopy (Rojo et al., 2000). Interestingly, these structuresresembled those formed by atlastin with critical GTPase mutations.None of the mutations, however, whether in the GTPase domain orelsewhere in the protein, prevented the co-localization of atlastinwith p23 in the aggregate-like ER structures (Fig. 6B). The effectsof the mutations cannot therefore be attributed to interference withan interaction between p23 and atlastin.
Does atlastin interact with p24 family proteins?
To determine whether atlastin co-localized with p23 becausethey are in the same membrane compartment or whether they alsointeract, we co-expressed p23 with either full-length atlastin (F) ortruncated forms of atlastin in which the C-terminal (T2) or one (T1)or both transmembrane domains (T0) were deleted (Fig. 6C). Onlyatlastin with transmembrane domains (T1 or T2) co-localized withp23 (Fig. 6C), indicating that atlastin needed to be inserted in theER membrane to localize with p23. This does not indicate,however, that the proteins necessarily interact directly.
To further investigate the relationship between atlastin and p24family proteins, we performed co-immunoprecipitation experi-ments with epitope-tagged atlastin co-expressed in HEK293 cellswith p23-myc, p24-HA and p25-myc. No evidence of aninteraction between atlastin and p23 or p25 was obtained by co-immunoprecipitation of either V5-tagged or Xpress-tagged atlastin(Fig. 7). Xpress-tagged atlastin, however, co-immunoprecipitatedp24 (Fig. 7), suggesting that these proteins might interact directlyor function in the same multiprotein complexes. High molecularweight complexes containing atlastin have been detected by FPLCgel-exclusion chromatography that may be larger than expected ifthey contained atlastin tetramers alone (Zhu et al., 2006).
Atlastin in primary cultures of cortical neurons
To determine whether the observations made in the HEK293cell line, which expresses low levels of endogenous atlastin, arerelevant to a protein implicated in a pathology affecting thecorticospinal tract, we sought to confirm the co-localization ofwild-type and mutant atlastin with calreticulin, as above, inprimary cultures of neurons from embryonic day 16 rats. Theefficiency of transfection was less than 1%, but both the wild-typeand the mutant DNA were reproducibly expressed.
Since atlastin is expressed in cortical neurons at higher levelsthan in HEK293 cells, we first confirmed that overexpressedatlastin was found in the same cellular compartment as theendogenous protein. The co-localization of these proteins wasvirtually complete (Fig. 8A), confirmed by a Pearson correlationcoefficient of 0.917, demonstrating that the distribution ofoverexpressed V5-tagged wild-type atlastin in transfected cells isnot an artefact of overexpression.
As in HEK293 cells, overexpressed atlastin co-localized to alarge extent with the ER marker calreticulin, and its distribution,and that of calreticulin, was changed by certain mutants (Fig. 8B).The three GTPase mutations, F151S, A161P, T162P, caused the

Fig. 8. Wild-type and mutant atlastin in primary cultures of cortical neurons.Cultures of post-mitotic rat embryonic neurons expressing V5-tagged wild-type or mutant atlastin were co-immunolabeled with antibodies against the
Fig. 7. Co-immunoprecipitation of p24, but not p23 or p25, with atlastin.The proteins expressed are indicated beneath the wells. The input (extracts oftransfected HEK293 cells expressing p24-HA, p23-myc or p25-myc alone orwith Xpress or V5-tagged atlastin) or the antibodies used to immunopre-cipitate the tagged proteins (anti-HA or anti-myc for the p24 family proteins,anti Xpress or anti-V5 for atlastin) are indicated above the lanes. The blotswere immunolabeled with anti-HA for p24 and anti-myc for p23 and p25and show the presence of p24-HA, p23-myc and p25-myc in the positivecontrols (immunoprecipitated with anti-HA or anti-myc) and in the inputs.P24 was co-immunoprecipitated with atlastin by the anti-Xpress antibody,but not when expressed alone. Neither p23 nor p25 were co-immunopre-cipitated with atlastin-V5 or by the anti-V5 antibody when expressed alone.
9M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
formation of aberrant atlastin-positive aggregate-like structures,whereas the downstream mutations had little or no effect on thereticular distribution of atlastin and calreticulin, except for theC-terminal S519N mutation, which caused the formation of cord-like calreticulin-positive structures. The C-terminal of atlastin,which might be expected to participate in intermolecular interac-tions, would therefore be of particular interest for further studies onthe role of atlastin in the ER. In the neuronal cultures, as inHEK293 cells, overexpressed p23 induced the formation ofaggregate-like structures in which p23 and atlastin were highlyco-localized (Fig. 8C). Partial co-localization with calreticulin wasalso observed (Fig. 8C).
V5 tag (green) and the ER marker calreticulin (red) and were analyzed byconfocal microscopy. (A) V5-tagged wild-type atlastin (green) is strictly co-localized with endogenous atlastin (red) labeled with the 5409 anti-atlastinantibody (Craig Blackstone, NIH, Bethesda, MD). (B) As in HEK293 cells(see Fig. 3C), the GTPase mutations F151S, A161P and T162P caused theformation of calreticulin-positive, atlastin-positive aggregate-like structures,whereas the distribution of calreticulin is little or not affected by thedownstream mutations, except for the C-terminal N519S mutation thatinduced the formation of a calreticulin-positive cord-like network. (C)Overexpression of p23 in cortical neurons, as in HEK293 cells (see Fig. 6),caused the formation of atlastin-positive calreticulin-positive aggregate-likestructures. Scale bar=5 μm.
Discussion
We have shown that atlastin is distributed throughout the cellbodies and processes of human motor neurons, and in HEK293cells when overexpressed, in association with ER and Golgimarkers. A cluster of mutations at a critical site in the GTPasedomain of the protein altered the morphology of the ER, whichformed aggregate-like structures. Ultrastructural analysis showedthat all the mutations studied prevented maturation of the Golgi
complex, those in the GTPase domain by preventing vesiclebudding from the ER, the others by preventing new vesicles fromleaving their site of formation and contributing to Golgimorphogenesis. These observations were confirmed by experi-ments with BFA-treated cultures of HEK293 cells co-immunola-beled with antibodies against V5-tagged atlastin and markers of the

10 M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
ER and the cis-Golgi. Finally, atlastin partially co-localized withp24 and p25 in normal ER. Some atlastin also co-localized withp23 in the abnormal extensions formed when this protein wasoverexpressed, and which resembled those formed by atlastin withGTPase mutations. Co-immunoprecipitation of atlastin and p24suggests that these proteins may interact or be found in the samemultiprotein complex. Since these proteins are known to beinvolved in vesicle trafficking between the ER and the Golgi, theseinteractions need further study.
The cellular distribution of atlastin
Overexpressed atlastin was markedly associated with the ER inour HEK293 cells, as previously reported in HeLa and NSC-34cells (Sanderson et al., 2006). Although endogenous atlastin wasreported to be most prominently co-localized with Golgi markers(Zhu et al., 2003), electron microscopy (Zhu et al., 2006) andcellular fractionation of cortical neurons (Zhu et al., 2003, 2006)showed that it also was present in the ER, indicating that thelocalization of atlastin in the ER in our study was not an artefact ofoverexpression or specific to our cell culture model. Thedistribution of atlastin in human brain was also compatible with alocalization in the ER. We confirmed the dual localization of atlastinin the ER and the Golgi of HEK293 cells by BFA treatment ofcultures that were then labeled with markers of the ER and the cis-Golgi, calreticulin and giantin. We investigated the possibility,however, that epitope-tagged atlastin was retained in the ER for lackof a partner necessary for its transport to the cis-Golgi, as previouslydescribed for p23 (Rojo et al., 2000). Co-expression of p24 and p23was reported to be sufficient for both of these proteins to betransported to the Golgi (Emery et al., 2000), but co-expression ofcombinations of atlastin, p23 and p24 did not displace any of theseproteins from the ER under our experimental conditions (notshown). The fact that atlastin was found in both ER and Golgi, aswell as in punctiform, probably vesicular, structures in all the celltypes studied is compatible, however, with a role in vesicletrafficking between the ER and Golgi, and perhaps beyond, assuggested by its presence in the processes of HEK293 cells and inneurites of cortical neurons in the human brain and spinal cord andin cultures of embryonic rat cortex (this study; Zhu et al., 2006).
Mutations in the GTPase domain of atlastin cause morphologicalabnormalities in the ER and prevent maturation of the Golgicomplex
Expression of a cluster of disease-causing mutations betweenamino acid 151 and 162 in the GTPase domain of atlastin resultedin the formation of large aggregate-like structures that containedER markers. This suggested that these structures might have beenformed as a result of aborted vesiculation, a hypothesis that wasconfirmed by ultrastructural analysis. The atlastin GTPase domaincontains three consensus GTP binding motifs, G1 (P-loop), G2(switch 1) and G3 (switch 2), like homologous domains in othermembers of dynamin-like and other GTPases, and a G4 motif, RD,which distinguishes atlastin and guanylate binding protein 1(GBP1) from the other GTPases that have an NKxD motif at thissite. The F151S, A161P and T162P mutations follow the G3 DxxGmotif, which has been shown to be critical for Ras (Der et al.,1986) and dynamin (Marks et al., 2001) GTPase activity, althoughmutations at analogous sites in GBP1 did not affect GTP bindingor hydrolysis (Praefcke et al., 2004).
The results of the ultrastructural analysis were consistent withthe hypothesis that these GTPase domain mutations cause a deficitin vesicle budding from the ER that prevented maturation of Golgicomplexes. Furthermore, there was a correlation between theseverity of the effect of the mutations on the ER, observed byfluorescence immunocytochemistry, and their effect on thedevelopment of the Golgi, the F151S mutation having the strongesteffects on both. The contiguous A161P and T162P mutations hadless drastic effects on the ER than F151, but the consequences onGolgi development were as severe. The fact that all three mutationshad similar effects on Golgi structure supports the hypothesis thatthe GTPase activity of atlastin may be implicated in vesiclebudding from the ER.
Although the other mutations studied had minimal or no effectson the ER when observed by fluorescence microscopy, they alsoaffected Golgi development when observed at the ultrastructurallevel. However, instead of preventing vesicle formation, as seemedto be the case with the GTPase mutations, these mutationsappeared to prevent their severing, migration or fusion with Golgicomplexes, which again could not develop. The similarity betweenthe effects of the two types of mutations supports the hypothesisthat atlastin plays a role in regulating coated vesicle traffic in theER/Golgi interface, a conclusion supported by the experimentsshowing the association between a small proportion of cellularatlastin with a BFA-sensitive cis-Golgi structure. A previous studyhas shown that mutations in the GTPase domain of atlastin, orelsewhere in the protein, all decrease its GTPase activity, probablythrough a dominant-negative effect of mutant atlastin in mixedhomo-oligomers (Zhu et al., 2006), or by competition with normalatlastin for essential partners, a mechanism that might also explainour observations.
It remains to be determined what the atlastin GTPase does. It ishighly homologous to, and has the same predicted membranetopology as mitofusin (N-terminal GTPase and short C-terminalsequences in the cytoplasm) that is involved in the fusion ofmitochondrial outer membranes (Legros et al., 2002), but likedynamin, which severs clathrin-coated vesicles (Sweitzer andHinshaw, 1998; Stowell et al., 1999; Sever et al., 1999), it mightparticipate in vesicle fission, as suggested by the effects of some ofthe mutations in our ultrastructure study.
Does atlastin interact with p24 family proteins?
We have shown that atlastin resides in the same membranecompartment and may also interact with the p24 family proteinsalready reported to play a role in the morphogenesis of the Golgicomplex (Rojo et al., 2000; Emery et al., 2003). The observationthat overexpressed p23 forms abnormal ER extensions, proposedto be due to the absence of an essential partner in sufficientconcentrations (Rojo et al., 2000), as does atlastin with criticalmutations in the GTPase domain, as shown in this study, supportsthe hypothesis that they may work together to perform thisfunction. As in our study, it was reported to be difficult to identifythe Golgi apparatus in cells overexpressing p23 (Rojo et al., 2000).
It is tempting to speculate that the p24 proteins, small type Itransmembrane proteins with similar structures but devoid ofenzymatic activity, form the scaffold to which atlastin is attachedvia their transmembrane and/or cytosolic C-terminal domains.Atlastin and p23 do not seem to interact directly. Only a fraction ofatlastin in the ER was associated with p23 in the abnormal ERcompartment formed when p23 was overexpressed, and only when

11M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
it was properly inserted in the ER membrane. Furthermore, if adirect association between atlastin and p23 was essential foratlastin function, at least one of the mutants studied might havebeen expected to interfere with this interaction. This was not thecase. Finally, we were unable to co-immunoprecipitate atlastin andp23. We were able, however, to co-immunoprecipitate atlastin andp24. This is intriguing in the light of reports that the cytosolic tailof p24 inhibits GTP hydrolysis by the GTP binding protein ADP-ribosylation factor 1 (ARF1) (Goldberg, 2000). Does p24 alsoregulate GTP hydrolysis by atlastin?
Atlastin, spastin and the p24 family
Atlastin has been reported to interact with the N-terminal of thecytoplasmic form of spastin (Sanderson et al., 2006; Evans et al.,2006), a microtubule severing protein responsible for the autosomaldominant HSP SPG4. The interaction appears to involve the C-terminal of atlastin (Evans et al., 2006), although evidence in favorof an interaction with the N-terminal has also been presented(Sanderson et al., 2006). The N-terminal of spastin is also reported tointeract with the microtubule cytoskeleton (Errico et al., 2002).Atlastin may therefore affect vesicle trafficking by regulatingmicrotubule dynamics.
Since the atlastin c1520insA mutation that interferes with theinteraction between atlastin and spastin (Evans et al., 2006) ispathogenic, disruption of spastin function may be responsible forHSP in patients with this mutation. This may possibly be due tomislocalization of spastin since atlastin reportedly has no effect onspastin’s enzymatic activity (Evans et al., 2006). The pathogenicspastin K388R mutation, which abrogates spastin’s ATPaseactivity, changed the localization of both atlastin and spastin,which were found in abnormal filamentous structures containingmicrotubules but also ER markers (Sanderson et al., 2006),suggesting that spastin, in conjunction with atlastin, plays a role inthe proper functioning of the ER. In light of our ultrastructuralobservations on HEK293 cells expressing mutant atlastin, it wouldbe interesting to see whether spastin mutations also affectmorphogenesis of the Golgi complex. This is not unlikely sincevesicles in the ER/Golgi interface migrate on microtubules(Mizuno and Singer, 1994; Lippincott-Schwartz et al., 1995).Atlastin-containing vesicles might be recruited to microtubules viaspastin, as previously suggested (Evans et al., 2006). Conversely,atlastin might recruit spastin or regulate its localization; atlastin isreported to recruit spastin to the ER when both are overexpressed(Evans et al., 2006). They also were reported to relocate to punctatestructures, vesicles or fragments of the Golgi that may result frommicrotubule severing by spastin (Evans et al., 2006).
Since spastin has been reported to interact with p25 (Reid et al.,2005), we hypothesize that the interaction between atlastin andspastin also links atlastin indirectly to p25, perhaps facilitating theinteraction with p24 under certain conditions. It is known thatvariable interactions among the p24 family proteins define the waythey circulate in the ER/Golgi interface (Jenne et al., 2002). Underwhat conditions atlastin, and perhaps spastin, interact with theseproteins, in the early secretory pathway, with what other partnersand to what end, remains to be determined.
Conclusion
This study has shown that all of the atlastin mutations studied,whatever their location in the protein, affected trafficking in the
ER/Golgi interface with the same result and impaired Golgimorphogenesis, suggesting that this may underlie the dying back ordysfunction of motor neuron axons in patients with SPG3A.Although atlastin has already been proposed to play a role invesicle trafficking, like spastin, spartin, and the kinesin subunitKIF5A that also cause autosomal dominant HSP when mutated(Reid, 2003), the effects of the mutations studied here haveprovided the first evidence that atlastin plays a role in vesiclebudding and membrane transport in the ER/Golgi interface.Atlastin now appears to have a place among the cascades of largeand small GTPases that regulate vesicle trafficking in the secretorypathway (Lee et al., 2004). Exactly what the atlastin GTPase doesstill remains to be elucidated. However, our evidence that somep24 family proteins are possible partners of atlastin provides auseful starting point for future studies.
Experimental methods
Expression vectors
Full-length atlastin (GenBank/EBI accession number NM_015915)was reverse transcribed and amplified by polymerase chain reaction (RT-PCR), according to standard methods, from mRNA extracted from asample of deep-frozen normal human cerebral cortex (Brain Bank,INSERM U679, Paris, France) and was subcloned downstream of anN-terminal Xpress tag or upstream of C-terminal V5 tag in either thepcDNA3.1/V5-HisTOPO or pcDNA4/HISMaxTOPO vectors (Invitrogen,San Diego, CA). GFP-tagged atlastin was generated by subcloning in thepEGFP-C1 vector (Clontech, Ozyme, Saint Quentin Yvelines, France).Truncated atlastin sequences were generated by PCR from the full-lengthatlastin cDNA and were subcloned into the pcDNA4/HISMaxTOPOvectors. Eight disease-causing point mutations (F151S, A161P, T162P,R217Q, R239C, S398Y, R495W, S519N) were introduced into V5-taggedatlastin by in vitro mutagenesis with the QuikChange site-directedmutagenesis kit (Stratagene, La Jolla, CA), according to the manufac-turer’s instructions. All constructs were verified by sequencing. Primersequences and PCR conditions are available on request. Vectors for theexpression of tagged p24 family proteins (p23-myc, p24-HA and p25-myc) were kindly offered by Dr. Manuel Rojo (INSERM U582, HôpitalPitié-Salpêtrière, Paris, France).
Antibodies
Overexpressed atlastin was detected with mouse monoclonal anti-V5and anti-Xpress antibodies (Invitrogen), or a polyclonal antibody (No.5409) against the N-terminal of atlastin, which was a kind gift from Dr.Craig Blackstone (NIH, Bethesda, MD, USA). All three antibodies wereaffinity purified. Myc-tagged p23 and p25 were detected by a mousemonoclonal (9E10, 1/600) or rabbit polyclonal (A-14, 1/600) anti-mycantibodies (Santa Cruz Biotechnology, Santa Cruz, CA), and p24-HA wasdetected with a mouse monoclonal anti-HA.11 antibody (1/1000)(Covance, Berkeley, CA).
Other primary antibodies were: rabbit anti-calnexin (1/200) and mouseanti-calreticulin (1/500) (Stressgen, Victoria, BC, Canada), rabbit anti-calreticulin (1/400), rabbit anti-giantin (1/1000, Abcam, Cambridge, UK),anti-actin (1/2000, Sigma-Aldrich, Lyon, France,). Secondary antibodiesconjugated to the fluorochromes FITC and Cy3 were from JacksonImmunoresearch Laboratories (West Grove, PA) or Sigma-Aldrich; Alexa488 conjugated antibodies were from Invitrogen.
Immunohistochemistry on human brain
Immunohistochemistry was performed on samples of human motorcortex and spinal cord from normal adults obtained from the NeuropathologyDepartment of the Salpêtrière Hospital, Paris. The samples, fixed in

12 M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
formaldehyde and embedded in paraffin, were cut in 7 μm sections on amicrotome, dewaxed in graded alcohols, blocked with 0.2% Tween-20/BSA2% in Tris buffer for 1 h at room temperature and incubated overnight at 4 °Cwith rabbit anti-atlastin (no. 5409) at a concentration of 5 μg/ml. Afterwashing, the sections were incubated with biotinylated anti-rabbit immu-noglobulins for 30 min at room temperature and the immunoreactivityrevealed with the streptavidin peroxidase complex (Vectastain, Abcys, Paris,France), as indicated by the manufacturer, with diaminobenzidine (DAB,Biogenex, San Ramon, CA) as the chromogen. After dehydration, thesections were mounted with EUKITT medium (Electron MicroscopySciences, Hatfield, PA). The specificity of the immunolabeling wasdetermined by: (1) Western blot on extracts of two deep-frozen samples ofhuman cerebral cortex, obtained from the Neuropathology Department of theSalpêtrière Hospital, (2) omitting the primary antibody and (3) absorbing theprimary antibody with the N-terminal atlastin peptide against which theantibodies were raised, in 2000/1 molar ratio (Zhu et al., 2003). DAB-stainedsections were observed and images acquired with Leitz Laborlux-Smicroscope equipped with a DXM1200 video camera and ACT-I software(Nikon, Champigny sur Marne, France).
Cell culture and transfection conditions
HEK293 cells, which express atlastin endogenously and emit longneurite-like processes, were used for most of the experiments in the presentstudy. They were plated on 24 well plates (Costar 3546, Corning, Corning,NY) or on collagen-coated coverslips, at a density of 60,000 cells per well, at37 °C in an atmosphere of 5% CO2 in 500 μl MEM, supplemented with 10%of fetal bovine serum, 100 U/ml of penicillin and 100 μg/ml streptomycin(GIBCO/Invitrogen). For experiments involving extraction of recombinantatlastin, cells were plated in 9 cm dishes at a density of 2–3×106 cells perdish.
Cells were transfected after 24 h in vitro, using DMRIE-C Reagent(Invitrogen), according to the manufacturer; transfection efficiency was 40–60%. To determinewhether levels of expression of atlastin and the p24 familyproteins had non-specific effects on the localization of the recombinantproteins, the amount of plasmid transfected per cell in single and multipletransfections was titrated down over two orders of magnitude, and thedistribution of the proteins in the cells was compared until a stable dis-tribution was achieved. Treatment with BFA (20 μg/ml for 1 h at 37 °C) wasperformed 24 h after transfection. To confirm that the effect of BFA wasreversible, the cells were rinsed once with BFA-free medium then incubatedin fresh medium for 2 h at 37 °C.
Post-mitotic neurons from the cortex of embryonic day 16 Wistar ratswere prepared as previously described (Michel and Agid, 1996). The ratswere treated in accordance with the Guide for the Care and Use ofLaboratory Animals (National Research Council 1996), European DirectiveNo. 86/609 and the guidelines of the local institutional animal care and usecommittee. Approximately 106 dissociated cells were plated on polyethyle-neimine-coated coverslips in 24 well plates in glutamate-free Neurobasalmedium containing 2% B27 (Invitrogen), 2 mM L-glutamine, 103 U/mlpenicillin and 103 μg/ml streptomycin. After 12 h in culture, glial cellproliferation was stopped with 2 μM cytosine arabinoside, and MK801(2 μM) was added to prevent excitotoxicity. After 96 h, the cells weretransfected with 1 μg of the expression vectors in Lipofectamine 2000(Invitrogen), according to the manufacturer’s instructions. Transfectionefficiency was less than 1%.
Immunofluorescence microscopy on cell cultures
After 24–48 h in culture, cells were fixed in 4% paraformaldehyde (PFA)for 30 min at room temperature then permeabilized and blocked for 1 h in0.1% Triton X-100/PBS containing 10% of normal goat or horse serumaccording to the antibody. For the BFA experiments, to better visualize theER and Golgi, the cells were fixed with methanol (−20 °C, 20 min), beforeblocking. Cells were then incubated overnight, or up to 72 h, with the primaryantibodies at 4 °C. After washing, the cells were incubated with appropriatesecondary antibodies (1/2000) in 2% serum for 45 min, washed again three
times and maintained at 4 °C. Coverslips were mounted with Fluoromount-GT (Electron Microscopy Science, Hatfield, PA). Fluorescent images ofmounted sections were acquired with an Axioplan 2 microscope (Carl ZeissS.A.S., Le Pecq, France) equipped with FluoUp image analysis systemsoftware (Explora Nova, La Rochelle, France). Confocal images wereacquired through a 63× objective on a Leica SP2AOBSmicroscope (Wetzlar,Germany), controlled by dedicated Leica software. Brightness and contrastwere adjusted with Photoshop or ImageJ software.
Transmission electron microscopy
HEK293 cells expressing epitope-labeled wild-type or mutant atlastinand p24 family proteins were fixed in the culture wells for 2 h at roomtemperature with 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4,then postfixed in 1% osmium tetroxide for 30 min at 4 °C and embedded inEpon. Ultrathin sections (50 nm) were cut and stained with uranyl acetate andlead citrate for examination under a JEOL 1200 EX electron microscope(Croissy-sur-Seine, France) at 80 kV.
Western blot
Transfected cultures grown in 9 cm plates (2×106 cells) were lysed in200 μl of buffer containing 50 mM Tris pH 8.0, 150 mMNaCl, 1 mM EDTAand 0.5% NP40, supplemented with a cocktail of protease inhibitors(Pefabloc, Roche, Fontenay, France), and the protein concentration wasdetermined by the Bradford method with a commercially available agent(Bio-Rad, Marne-la-Coquette, France). Equal quantities of extract (20 μg)were electrophoresed on 4–12% mini-gels (NuPage, Invitogen) and blottedonto Protran nitrocellulose membranes (Perkin-Elmer, Boston, MA). Afterblockingwith 3% driedmilk in PBS/0.01%Tween, the blot was incubated for2 h at room temperature with the primary antibody, washed 3 times in PBSthen incubated for 45 min with the appropriate secondary antibodyconjugated with horseradish peroxidase. Immunolabeling was revealed witha chemical luminescent substrate (Pierce, Perbio Science, Brebières, France)and exposure to Biomax Light film (Kodak).
Co-immunoprecipitation
HEK293 cells (2–3×106/9 cm plate) were transfected, as describedabove, with vectors expressing epitope-tagged atlastin and the p24 familyproteins either alone or in combination. Cells were harvested 42–45 h posttransfection and lysed on ice in 150 mM salt buffer (50 mM Tris pH 8.0,150 mM NaCl, 1 mM EDTA and 0.5% NP40) supplemented with a cocktailof protease inhibitors (Pefabloc, Roche, Fontenay, France). The total extractwas centrifuged at 10,000×g for 10 min at 4 °C and the supernatant was pre-cleared with sepharose A beads for 1 h at 4 °C. Pre-cleared supernatant andprotein G or A sepharose beads (Amersham Biosciences) coupled with theappropriate antibodies were incubated 2 h at 4 °C on a rotating wheel forimmunoprecipitation. Finally, the beads were washed 5 times with the lysisand the bound proteins were denatured by heating for 5 min at 95 °C in 30 μlLaemmli buffer, before analysis by Western blot and revelation by chemicalluminescence, as described above.
Acknowledgments
We gratefully acknowledge Drs. Manuel Rojo (INSERM U582,Hôpital Pitié-Salpêtrière, Paris, France), Craig Blackstone (NIH,Bethesda, MD, USA) and Francis A. Barr (Max-Planck Institute,Martinsreid, Germany), who generously provided us with materials,and the Plate-forme d’Imagerie Cellulaire Pitié-Salpêtrière, Paris,France, for their expert assistance with the confocal microscopy.This study was supported by grants from the French NationalInstitute for Health and Medical Research, the VERUM foundation(A.B.), Lilly (Japan, M.N.), the French ForeignMinistry (M.N.) andla Fondation Recherche Médicale (M.N.).

13M. Namekawa et al. / Mol. Cell. Neurosci. 35 (2007) 1–13
References
Blum, R., Pfeiffer, F., Feick, P., Nastainczyk, W., Kohler, B., Schäfer, K.H.,Schulz, I., 1999. Intracellular localization and in vivo trafficking ofp24A and p23. J. Cell Sci. 112, 537–548.
Der, C.J., Finkel, T., Cooper, G.M., 1986. Biological and biochemicalproperties of human rasH genes mutated at codon 61. Cell 44, 167–176.
Dominguez, M., Dejgaard, K., Fullekrug, J., Dahan, S., Fazel, A., Paccaud,J.P., Thomas, D.Y., Bergeron, J.J.M., Nilsson, T., 1998. gp25L/emp24/p24 protein family members of the cis-Golgi network bind both COP Iand II coatomer. J. Cell Biol. 140, 751–765.
Dürr, A., Camuzat, A., Colin, E., Tallaksen, C., Hannequin, D., Coutinho, P.,Fontaine, B., Rossi, A., Gil, R., Rousselle, C., Ruberg, M., Stevanin, G.,Brice, A., 2004. Atlastin1 mutations are frequent in young-onsetautosomal dominant spastic paraplegia. Arch. Neurol. 61, 1867–1872.
Emery, G., Rojo, M., Gruenberg, J., 2000. Coupled transport of p24 familymembers. J. Cell Sci. 113, 2507–2516.
Emery, G., Parton, R.G., Rojo, M., Gruenberg, J., 2003. The trans-membrane protein p25 forms highly specialized domains that regulatemembrane composition and dynamics. J. Cell Sci. 116, 4821–4832.
Errico, A., Ballabio, A., Rugarli, E.I., 2002. Spastin, the protein mutated inautosomal dominant hereditary spastic paraplegia, is involved inmicrotubule dynamics. Hum. Mol. Genet. 11, 153–163.
Evans, K., Keller, C., Pavur, K., Glasgow, K., Conn, B., Lauring, B., 2006.Interaction of two hereditary spastic paraplegia gene products, spastinand atlastin, suggests a common pathway for axonal maintenance. Proc.Natl. Acad. Sci. U. S. A. 103, 10666–10671.
Goldberg, J., 2000. Decoding of sorting signals by coatomer through aGTPase switch in the COPI coat complex. Cell 100, 671–679.
Jenne, N., Frey, K., Brügger, B., Wieland, F.T., 2002. Oligomeric state andstoichiometry of p24 proteins in the early secretory pathway. J. Biol. C277, 46504–46511.
Lee, M.C.S., Miller, E.A., Goldberg, J., Orci, L., Schekman, R., 2004.Bi-directional protein transport between the ER and Golgi. Annu.Rev. Cell Dev. Biol. 20, 87–123.
Legros, F., Lombes, A., Frachon, P., Rojo, M., 2002. Mitochondrial fusion inhuman cells is efficient, requires the inner membrane potential, and ismediated by mitofusins. Mol. Biol. Cell 13, 4343–4354.
Lippincott-Schwartz, J., Cole, N.B., Marotta, A., Conrad, P.A., Bloom, G.S.,1995. Kinesin is the motor for microtubule-mediated Golgi-to-ERmembrane traffic. J. Cell Biol. 128, 293–306.
Marks, B., Stowell, M.H.B., Vallis, Y., Mills, I.G., Gibson, A., Hopkins,C.R., McMahon, H.T., 2001. GTPase activity of dynamin and resultingconformation change are essential for endocytosis. Nature 410, 231–235.
Michel, P.P., Agid, Y., 1996. Chronic activation of the cyclic AMP signalingpathway promotes development and long-term survival of mesence-phalic dopaminergic neurons. J. Neurochem. 67, 1633–1642.
Mizuno, M., Singer, S.J., 1994. A possible role for stable microtubules inintracellular transport from the endoplasmic reticulum to the Golgiapparatus. J. Cell Sci. 107, 1321–1331.
Namekawa, M., Ribai, P., Nelson, I., Forlani, S., Fellmann, F., Goizet, C.,Depienne, C., Stevanin, G., Ruberg, M., Dürr, A., Brice, A., 2006.SPG3A is the most frequent cause of hereditary spastic paraplegia withonset before age 10 years. Neurology 66, 112–114.
Praefcke, G.J.K., McMahon, H.T., 2004. The dynamin superfamily:universal membrane tubulation and fission molecules? Nat. Rev., Mol.Cell Biol. 5, 133–147.
Praefcke, G.J.K., Kloep, S., Benscheid, U., Lilie, H., Prakash, B., Herrmann,C., 2004. Identification of residues in the human guanylate-bindingprotein 1 critical for nucleotide binding and cooperative GTP hydrolysis.J. Mol. Biol. 344, 257–269.
Reid, E., 2003. Science in motion: common molecular pathological themesemerge in the hereditary spastic paraplegias. J. Med. Genet. 40, 81–86.
Reid, E., Connell, J., Edwards, T.L., Duley, S., Brown, S.E., Sanderson,C.M., 2005. The hereditary spastic paraplegia protein spastin interactswith the ESCRT-III complex-associated endosomal protein CHMP1B.Hum. Mol. Genet. 14, 19–38.
Rojo, M., Pepperkok, R., Emery, G., Kellner, R., Stang, E., Parton, R.G.,Gruenberg, J., 1997. Involvement of the transmembrane protein p23 inbiosynthetic protein transport. J. Cell Biol. 139, 1119–1135.
Rojo, M., Emery, G., Marjomäki, V., McDowall, A.W., Parton, R.G.,Gruenberg, J., 2000. The transmembrane protein p23 contributes to theorganization of the Golgi apparatus. J. Cell Sci. 113, 1043–1057.
Sanderson, C.M., Connell, J.W., Edwards, T.L., Bright, N.A., Duley, S.,Thompson, A., Luzio, J.P., Reid, E., 2006. Spastin and atlastin, twoproteins mutated in autosomal hereditary spastic paraplegia, are bindingpartners. Hum. Mol. Genet. 15, 307–318.
Sever, S., Muhlberg, A.B., Schmid, S.L., 1999. Impairment of dynamin’sGAP domain stimulates receptor-mediated endocytosis. Nature 398,481–486.
Stamnes, M.A., Craighead, M.W., Hoe, M.H., Lampen, N., Geromanos, S.,Tempst, P., Rothman, J.E., 1995. An integral membrane component ofcoatomer-coated transport vesicles defines a family of proteins involvedin budding. Proc. Natl. Acad. Sci. U. S. A. 92, 8011–8015.
Stowell, M.H., Marks, B., Wigge, P., McMahon, H.T., 1999. Nucleotide-dependent conformational changes in dynamin: evidence for amechanochemical molecular spring. Nat. Cell Biol. 1, 27–32.
Sweitzer, S.M., Hinshaw, J.E., 1998. Dynamin undergoes a GTP-dependentconformational change causing vesiculation. Cell 93, 1021–1029.
Zhao, X., Alvarado, D., Rainier, S., Lemons, R., Hedera, P., Weber, C.H.,Tukel, T., Apak, M., Heiman-Patterson, T., Ming, L., Bui, M., Fink, J.K.,2001. Mutations in a newly identified GTPase gene cause autosomaldominant hereditary spastic paraplegia. Nat. Genet. 29, 326–331.
Zhu, P.P., Patterson, A., Lavoie, B., Stadler, J., Shoeb, M., Patel, R.,Blackstone, C., 2003. Cellular localization, oligomerization, andmembrane association of the hereditary spastic paraplegia 3A(SPG3A) protein atlastin. J. Biol. Chem. 278, 49063–49071.
Zhu, P.P., Soderblom, C., Tao-Cheng, J.-H., Stadler, J., Blackstone, C., 2006.SPG3A protein atlastin-1 is enriched in growth cones and promotes axonelongation during neuronal development.Hum.Mol.Genet. 15, 1343–1353.





























