Embed Size (px)
Citation preview

UMEÅ UNIVERSITY MEDICAL DISSERTATION
New series No 1190 ISBN No 978-91-7264-582-0 ISSN 0346-6612
___________________________________________________________________
From the Department of Public Health and Clinical Medicine, Respiratory Medicine and Allergy
Umeå University, Sweden
Respiratory and Cardiovascular Responses to Diesel Exhaust Exposure
Håkan Törnqvist
Umeå 2008

Copyright © 2008 by Håkan Törnqvist ISSN 0346-6612, ISBN No 978-91-7264-582-0 Printed by Print och Media, 2008 Department of Public Health and Clinical Medicine, Respiratory Medicine and Allergy Umeå University SE-901 85 Umeå, Sweden

To my family, the dearest in my life!
To my wife Anette and our two lovely sons Joacim and Marcus,
with love!


5
TABLE OF CONTENTS
ABSTRACT ................................................................................................................ 7 SVENSK SAMMANFATTNING .................................................................................. 9 SELECTED ABBREVIATIONS ................................................................................. 11 ORIGINAL PAPERS ................................................................................................. 13 INTRODUCTION ...................................................................................................... 15
BACKGROUND .................................................................................................... 15 Particulate matter air pollution ........................................................................... 15 Epidemiological studies on health effects of PM pollution ................................. 17 Health effects in infants by PM pollution ............................................................ 18 Impaired lung growth and lung function in children ........................................... 19 Asthma .............................................................................................................. 19 COPD ................................................................................................................ 20 COPD and occupational exposure .................................................................... 20 Cardiovascular Disease ..................................................................................... 21
EXPERIMENTAL STUDIES OF PM POLLUTION ................................................ 22 Diesel exhaust ................................................................................................... 22 Controlled exposure to particulate matter air pollution ...................................... 22 In-vitro and animal studies ................................................................................. 23 Experimental studies in humans ........................................................................ 25
AIMS ......................................................................................................................... 29 SUBJECTS AND METHODS ................................................................................... 31
STUDY DESIGN ................................................................................................... 31 Subjects ............................................................................................................. 31 Subject Preparation ........................................................................................... 32 Chamber exposures .......................................................................................... 33 Lung function measurements ............................................................................ 35 Sputum induction and processing ..................................................................... 35 Sputum analyses ............................................................................................... 36 Analysis in peripheral blood in study II .............................................................. 36 Venous occlusion pletysmography studies III-V ................................................ 38 Venous occlusion plethysmography .................................................................. 39

6
Venous sampling and laboratory assays studies III-V ....................................... 41 Data analysis and statistics ............................................................................... 43
MAIN RESULTS ....................................................................................................... 45 STUDY I ................................................................................................................ 45 STUDY II ............................................................................................................... 45 STUDY III .............................................................................................................. 45 STUDY IV ............................................................................................................. 45 STUDY V .............................................................................................................. 46
DISCUSSION ........................................................................................................... 49 Studies I and II ...................................................................................................... 49
Diesel exhaust effects in COPD patients ........................................................... 49 Discussion of Results ........................................................................................ 49 Conclusions from diesel exhaust exposures in COPD ...................................... 53
Studies III-V .......................................................................................................... 53 Diesel exhaust effects in Cardiovascular patients ............................................. 53
FINAL COMMENTS ................................................................................................. 63 CONCLUSIONS ....................................................................................................... 67 ACKNOWLEDGEMENTS ......................................................................................... 68 REFERENCES ......................................................................................................... 71

7
ABSTRACT Background: Exposure to traffic-derived air pollution is associated to high incidence of respiratory and cardio-vascular morbidity and mortality. Diesel engines and fossil fuel contribute to a great amount to the ambient particulate matter pollution. Exposure to diesel exhaust in healthy volunteers is known to cause inflammatory and oxidative responses in the airways. In contrast, very little is known about the air pollution-related mechanisms behind the adverse cardiovascular effects and why patients with cardio-respiratory disease are more susceptible to the adverse effect of particulate matter air pollution. Methods: Volunteers were exposed to diesel exhaust at a particulate matter concentration of 300 μg/m3 and filtered air for one hour in random order. In studies I-II, patients with moderately severe, stable COPD were examined with lung function, induced sputum and peripheral blood samples. In studies III-V, vascular assessment was performed using venous occlusion plethysmography. Vascular responses to intra-arterially infused endothelial dependent and independent vasodilators were determined, together with endogenous fibrinolysis, systemic inflammation and long-term ECG registration. These vascular studies were carried out in healthy volunteers and patients with stable coronary heart disease. Results: In healthy subjects, diesel exhaust exposure induced an acute vasomotor dysfunction, which was partly sustained at 24 hours. Endogenous fibrinolysis reflected by tissue plasminogen activator (t-Pa) levels and activity were reduced at 6 hours post exposure both in healthy subjects and patients with stable PCI-treated coronary heart disease. During diesel exhaust exposure, ECG analyses demonstrated significant exercise-induced ST-T segment depression in patients with coronary heart disease. These findings occurred at a moderately increased heart rate of approximately 90 beats per minute during both diesel and air exposures. The investigated group of stable COPD patients did not demonstrate any further deterioration of lung function, induced sputum or systemic inflammatory parameters within the investigated time frame. Conclusion: Inhalation of diesel exhaust impaired two important and complementary aspects of vascular function in healthy subjects; regulation of vascular tone and endogenous fibrinolysis. In men with stable coronary heart disease, exposure to diesel exhaust induced signs of myocardial ischemia, along with impaired endogenous fibrinolytic capacity, despite full secondary preventive medication. These exposure studies support the epidemiological evidence of an association between particulate matter air pollution and adverse cardiovascular effects and demonstrate important underlying mechanisms.

8

9
SVENSK SAMMANFATTNING Partiklar i dieselavgaser är en betydande orsak till de negativa hälsoeffekter som ses av luftföroreningar i framför allt trafikmiljöer. Dieselavgaserna innehåller en mängd ytterst små partiklar på ca 1/10 000 mm i diameter. Dessa partiklar har kemiska ämnen bundna till ytan t ex kolväten och metaller. Detta har föreslagits ligga bakom partiklarnas förmåga att ge skadliga hälsoeffekter. Individer med lung- eller hjärtkärlsjukdom är särskilt sårbara och påverkas mycket negativt under perioder med höga halter av luftföroreningar. I denna avhandling har effekter av dieselavgaser studerats på friska samt patientgrupper med kroniskt obstruktiv lungsjukdom (KOL) respektive åderförkalkning i hjärtats kranskärl Målsättningen med studierna i avhandlingen har varit, att genom kontrollerade exponeringsstudier försöka klarlägga de mekanismer som skulle kunna förklara varför dieselavgaspartiklar i luftföroreningar ger upphov till ökad sjuklighet i både lung- och hjärtsjukdomar. Studierna har genomförts i en exponeringskammare, där samtliga forskningspersoner har exponerats för dieselavgaser med en partikelkoncentration på 300 µg/m3 respektive filtrerad luft under en timme. De två exponeringarna har skett i slumpvis ordning, och forskningspersonerna har således varit sina egna kontroller. I studie I undersöktes om exponering för dieselavgaser skulle kunna ge försämring av lungfunktionen och ökad luftvägsinflammation mätt i upphostningsprov hos patienter med måttligt svår men stabil KOL. Analyser av upphostningsproven kunde inte påvisa någon ökad luftvägsinflammation, och inte heller noterades någon försämring av lungfunktionen. I studie II undersöktes samma patienter med stabil KOL som i studie I. Frågeställningen var om dieselavgasexponering kunde ge ökad generell inflammation, påverkan på blodlevringsförmåga eller lungepitelskada mätt i blodet. Ingen generellt ökad blodlevringstendens kunde påvisas, och inte heller noterades några tecken på ökad inflammation i blodet. I studie III studerades om exponering för dieselavgaser kunde påverka kärlfunktionen, mätt som försämrad kärlvidgande och blodproppsupplösande förmåga, hos en grupp av friska individer 2 och 6 timmar efter exponering. I denna grupp av unga, friska individer försämrade exponering för dieselavgaser två viktiga och kompletterande blodkärlsfunktioner, nämligen regleringen av blodkärlens vidd och kroppens egen förmåga att lösa upp blodproppar. En försämrad vidgning av blodkärlen demonstrerades akut (2 timmar) efter dieselexponering, men även 6 timmar efter avslutad exponering. En försämrad blodproppsupplösande kapacitet (fibrinolys) hade också utvecklats efter 6 timmar, vilket tyder på en ökad risk för blodproppsbildning efter exponering för dieselavgaser.

10
I studie IV klargjordes det sena förloppet av de dieselavgasutlösta blodkärlseffekterna hos friska försökspersoner. Även 24 timmar efter avgasexponering kvarstod en störning av blodkärlens vidgningsförmåga. Dessutom påvisades tecken till en systemisk inflammation mätt som ökning av inflammatoriska markörer i blod. Den tidigare visade nedsatta blodproppsupplösande kapaciteten (fibrinolys) hade vid denna tidpunkt normaliserats. I studie V undersöktes personer med kliniskt helt stabil kranskärlssjukdom. Frågeställningen var om dieselavgasexponering kunde påverka hjärtat, ge försämrad kärlrörlighet och minskad blodproppsupplösande förmåga i likhet med vad som visats på yngre och friska forskningspersoner. Hos denna grupp av kranskärlssjuka män noterades även en akut ökad hjärtmuskelbelastning vid fysisk ansträngning, visat som EKG förändringar, under exponering för dieselavgaser jämfört med luftexponering. Sammanfattningsvis, har studierna i avhandlingen bidragit till att visa på möjliga förklaringsmodeller till det överinsjuknande i hjärtkärlsjukdom, som beskrivits i relation till exponering för luftföroreningar. Sannolikt är en försämrad funktion hos blodkärlen av central betydelse när det gäller att förklara de negativa hälsoeffekter som relaterats till exponering för dieselavgaser, en av de viktigaste luftföroreningarna i trafikmiljö.

11
SELECTED ABBREVIATIONS ACE angiotensin converting enzyme ACH acetylcholine ARB angiotensin receptor blocker AP-1 activator protein-1 ATS american thoracic society BAL bronchoalveolar lavage Big-ET-1 big endothelin (ET)-1 BHR bronchial hyperresponsiveness BK bradykinin CAFE clean air for europe CAPs concentrated ambient particles CD4+ lymphocyte antigen for T-helper cells CD8+ lymphocyte antigen for T-suppressor cells CC16 clara cell protein CO, CO2 carbon monoxide, carbon dioxide COPD chronic obstructive pulmonary disease DE diesel exhaust DEPs diesel exhaust particles EGFR epithelial growth factor receptor ECG electro cardiografi EPR electron paramagnetic resonance ET-1 endothelin-1 FEV1 forced expiratory volume in one second FVC forced vital capacity FEF forced expiratory flow FBF fore arm blood flow GOLD global initiative obstructive lung disease HRV heart rate variability ICAM-1 intercellular adhesion molecule-1

12
IL- interleukin- ICD implantable cardiac defibrillators LDH lactate dehydrogenase MAPK mitogen activated protein-kinases mRNA messenger-Ribo Nucleid Acid MMEF maximal midexpiratory flow MPO myeloperoxidase NFĸB nuclear-factor kappa B NO2 NOx nitric oxide, nitrix oxides NO2 nitric dioxide O3 ozone O2-. superoxide radical OONO-. peroxynitrite PAI-1 plasminogen activator-1 PEF peak expiratory flow PM particulate matter PMN polymorph nuclear nucleus PAHs poly-aromatic hydrocarbons ROS reactive oxygen species SOD superoxide dismutase SNP sodium nitro-prusside TNF-α tumor necrosis alpha TEAC trolox equivalent antioxidant capacity t-PA tissue plasminogen activator VCAM-1 vascular cell adhesion molecule-1 VP verapamil WBC white blood cell count vWF von Willebrand factor

13
ORIGINAL PAPERS This thesis is based on the following papers, which will be referred to in the text by their Roman numerals.
I. Törnqvist H, Pourazar J, Ädelroth E, Sandström T, Blomberg A. Diesel exhaust exposure in subjects with chronic obstructive pulmonary disease. In manuscript
II. Blomberg A, Törnqvist H, Desmyter L, Deneys V, Hermans C. Exposure to diesel exhaust nanoparticles does not induce blood hypercoagulability in an at-risk population. J Thromb Haemost 2005;3(9):2103-5.
III. Mills NL*, Törnqvist H*, Robinson SD, Gonzalez M, Darnley K, MacNee W, Boon NA, Donaldson K, Blomberg A, Sandström T, Newby DE. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis: An explanation for the increased cardiovascular mortality associated with air pollution. Circulation 2005;112(25):3930-6.
IV. Törnqvist H*, Mills NL*, Gonzalez M, Robinson SD, Boon NA, MacNee W, Donaldson K, Newby DE, Sandström T, Blomberg A Prolonged endothelial dysfunction following diesel exhaust inhalation. Am J Respir Crit Care Med 2007;176:295-400.
V. Mills NL*, Törnqvist H*, Gonzalez M, Vink E, Robinson SD, Söderberg S, Boon NA, Donaldson K, Sandström T, Blomberg A, Newby DE. Ischemic and thrombotic effects of dilute diesel exhaust inhalation in men with coronary heart disease. N Engl J Med 2007;Sep 13;357(11):1075-82.
* Contributed equally to first authorship
The published papers have been reprinted with kind permission of the publishers.

14

15
INTRODUCTION
BACKGROUND During the 20th century, several air pollution disasters have been brought to attention, e.g. the historical events of the Meuse valley fog of 1930 in Belgium (1), the Donora disaster in Pennsylvania USA (2) and the London smog of 1952 (3, 4). Specific climatic and topographic conditions played important roles in the development of the disasters. The investigations of these incidents by the state and federal health officials resulted in the first meaningful federal and state laws to control air pollution and marked the beginning of modern efforts to assess and deal with the health threats from air pollution. Another early air pollution study demonstrating an association between the level of air pollution and mortality was published in 1954. The authors reported death from bronchitis to be associated with long-term exposure to sulphur dioxide in 35 English cities (5). The attention of the topic increased even further during the 1970`s, with studies showing a considerable impact on long-term air pollution exposure and consequences on mortality, morbidity as well as public economy (6). During that decade there were great efforts to reduce the total amount of ambient air pollution. The problems were considered “solved” by district heating, gas cleaning and cleaner fuels and many experts were doubtful regarding any remaining air pollution effect of importance (7). They were soon proven wrong.
During the two 1980:e`s and 90:e`s several epidemiological as well as experimental studies further strengthened the associations between gaseous and particulate matter pollution. The adverse health effects were seen as worsening of asthma and chronic obstructive pulmonary disease (COPD) as well as heart attacks and stroke. It also became more apparent that the pollutants were associated with an increase in mortality due to respiratory and cardiovascular causes (8, 9).
Short as well as long term exposure to traffic-related air pollution markedly increases the risk of disease and death.A topic of special interest is the findings that commuters with long travel distance or working in environments with heavy traffic, increase their exposures three times more than the general population (10).
The European Union has through the European framework program CAFE (Clean Air for Europe) estimated ambient particulate air pollution to be responsible for approximately 350,000 excess deaths in EU annually (11). In Sweden about 5,000 premature deaths occur each year because of particulate matter air pollution (12). A great proportion of these deaths of 3,500 people has been reported to be attributable to air pollution imported from long distances by winds. Local mainly traffic derived exhaust is according to the authors causing about 1,800 premature deaths among people in urban areas. Together with other European data including the large multinational investigation by Künzli et al, it is evident that a lot more people die from traffic-related air pollution than from traffic accidents (13).
Particulate matter air pollution Air pollutants consist of different potentially harmful contents, including combustion-derived nanoparticles, nitrogen dioxide, ozone, sulphur dioxide and volatile organic compounds. Both the WHO and United Nations have declared that the most significant global air pollution threat is posed by particulate matter (PM), of which a significant proportion is derived from

INTRODUCTION
16
combustion engines. The associations are strongest for fine particulate air pollutants (PM2.5) (14), of which the combustion-derived nanoparticulates from diesel exhaust are an important component (15,16).
PM air pollution can be found in different sizes from very large crustal particles to the very small ultrafine particles in the nanometer range. Particle size as well as the complex surface chemistry including, e.g. organic compounds and transition metals have been indicated to be of importance for health effects. Particles are of different size and origin and they are usually described in terms of aero dynamic size.
Figure 1. Since only fine particles can be inhaled deep into the lungs and national air quality standards have been based on the mass concentration of such ‘inhalable’ particles: typically defined as having an aerodynamic diameter centred below 2.5 (PM 2.5) or 10 (PM 10) µm. (Adapted from (17)).
Particles >10 μm
Particles with an aerodynamic diameter of more than 10 µm seldom reach the lung acini. They are filtered out during inspiration by wall impaction due to inertial forces at sites of turbulence in the nose, larynx or at branch points of conducting airways and are ultimately removed by ciliary transport.

INTRODUCTION
17
Particles <10 μm. Thoracic particles
Particles with a mean aerodynamic diameter of less than 10 μm are defined as PM10. Includes particles from a range of different sources from crustal material to mechanically generated particles from e.g. road, tyre and break wear, but also combustion generated particles..
Coarse fraction (PM2.5-10)
The coarse fraction consists of particles with mean aerodynamic diameter of 2.5-10 μm. Road dust includes any particle component that may be found in the road environment no matter whether it has been generated from. The major fraction of wear particles and road dust are found in the coarse particle fraction. (18).
Fine fraction
The fine particle fraction often relates to particles with a mean aerodynamic diameter >0.1 µm and <2.5 µm. This particle fraction was invented to exclude the large particles of mineral content found in the coarse fraction. The fine fraction includes agglomerates of combustion particles while the primary particles usually are too small to be included.
Ultrafine fraction
Ultra fine particles have a diameter of less than <0.1 µm. As the particles are in the nanometer range, they are also called nanoparticles. Ultra fine particles are mainly combustion derived. In a recent review, toxicological studies indicate that these particles are especially reactive (19)
Epidemiological studies on health effects of PM pollution
Coarse vs. Fine PM
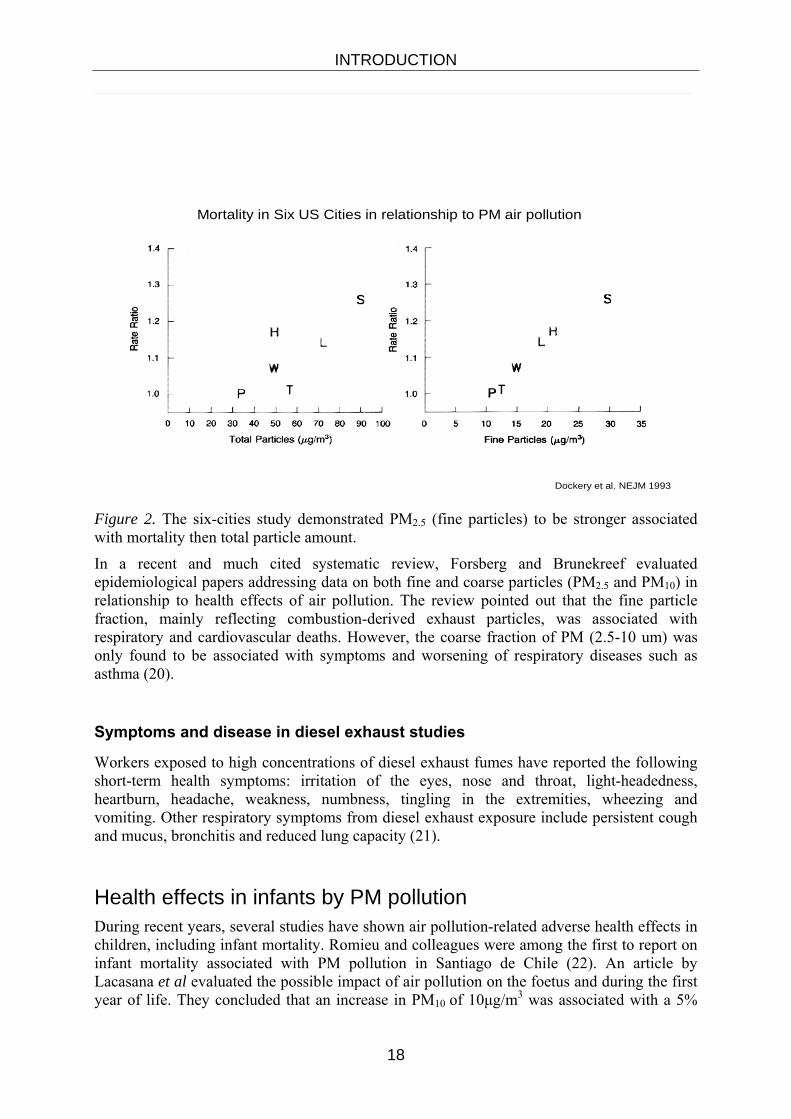
Epidemiological studies investigating effects of the variability of PM 10 have demonstrated a range of adverse respiratory and cardiovascular health effects including mortality. The important “Six-Cities study showed stronger relationship for the fine fraction, PM2.5, than PM 10 for mortality (8). See figure 2. This led to a widespread use of PM2.5 as an entity with some possibilities for source apportionment. PM2.5 largely excludes the coarse particles from crustal material, windblown dust and road wear. The fine fraction is usually associated with combustion and with traffic in urban areas.
INTRODUCTION
18
Dockery et al. NEJM 1993
Mortality in Six US Cities in relationship to PM air pollution
Figure 2. The six-cities study demonstrated PM2.5 (fine particles) to be stronger associated with mortality then total particle amount.
In a recent and much cited systematic review, Forsberg and Brunekreef evaluated epidemiological papers addressing data on both fine and coarse particles (PM2.5 and PM10) in relationship to health effects of air pollution. The review pointed out that the fine particle fraction, mainly reflecting combustion-derived exhaust particles, was associated with respiratory and cardiovascular deaths. However, the coarse fraction of PM (2.5-10 um) was only found to be associated with symptoms and worsening of respiratory diseases such as asthma (20).
Symptoms and disease in diesel exhaust studies
Workers exposed to high concentrations of diesel exhaust fumes have reported the following short-term health symptoms: irritation of the eyes, nose and throat, light-headedness, heartburn, headache, weakness, numbness, tingling in the extremities, wheezing and vomiting. Other respiratory symptoms from diesel exhaust exposure include persistent cough and mucus, bronchitis and reduced lung capacity (21).
Health effects in infants by PM pollution During recent years, several studies have shown air pollution-related adverse health effects in children, including infant mortality. Romieu and colleagues were among the first to report on infant mortality associated with PM pollution in Santiago de Chile (22). An article by Lacasana et al evaluated the possible impact of air pollution on the foetus and during the first year of life. They concluded that an increase in PM10 of 10μg/m3 was associated with a 5%

INTRODUCTION
19
increase in post neonatal mortality for all causes and a 22% increase in post neonatal mortality from respiratory diseases (23). An increase in infant mortality related to particulate matter (PM) air pollution has also been in children growing up in strongly polluted areas in California, USA (24).
Impaired lung growth and lung function in children Children are generally of special concern since their lungs are less mature and they may be more exposed to ambient air pollution than adults. Due to the shorter height they may be exposed to more primary exhaust. Children spend long periods of time outside and are physically active. (25).
Gauderman et al reported that increased exposure to PM10 is associated with impaired growth of lung function (26). During an 8 year follow up period in children from 10 to 18 years, the authors noticed that living within 500 m from a motor highway in southern California was associated to a significantly lower FEV1 and mid expiratory volumes as compared to other living more 1,500 m from the road, independently of regional air quality. In another study from Mexico city, Rojas-Martinez et al showed that long term exposure to O3, PM10 and NO2 in Mexico city was associated with a lung function decrease in FVC and FEV1 among school children (27) Gilliland
There is an increasing awareness that air pollution causes negative health effects in children and to lung function. Trenga et al demonstrated a decrease in maximal midexpiratory flow, (MMEF), in a Seattle cohort of untreated asthmatic children following increases of ambient air pollution without deteriorated PEF, FEV1 (28).
Kulkarni et al showed decreases in FEV1 in healthy and asthmatic children exposed to PM air pollution. The children underwent sputum induction, and sputum investigations found soot particles in macrophages that were inversely correlated with lung function (29).
Asthma Exposure to particulate matter air pollution induces asthma worsening in terms of an increase in asthma symptoms as well as need for medication, emergency room visits and hospitalisation (22, 30-32). The airway deposition of ultra fine particles at rest, which are supposed to be the most harmful particle fraction, has been suggested to be increased in asthmatics as compared to healthy subjects (33), indicating that this group might be especially vulnerable to the negative health effects of particulate matter air pollution exposure.
Long-term exposure to background air pollution is related to asthma-worsening in school children (34). Elderly individuals with both airway hyperresponsiveness and high total immunoglobulin E levels have been found especially susceptible to air pollution (35).
A recent and real life street-level exposure study in asthmatic subjects in London clearly demonstrated an acute small airway effect, which was more pronounced with increasing severity of the disease (36). Compared to a similar walk in Hyde Park, a two-hour walk at the side of the busy street (Oxford Street) induced a significant decrease in FEV1 up to 6.1% and FVC up to 5.4%. The changes were accompanied by increase in markers of inflammation. Walking along Oxford Street caused an increase in neutrophilic derived myeloperoxidase and a decrease in airway acidification as compared to a walk in Hyde Park. The results are remarkable, since the authors were demonstrating small, but significant differences in

INTRODUCTION
20
pulmonary function, between traffic derived PM exposure from a highly diesel vehicle loaded street and a control exposure from a city park, located not far from major thoroughfares in London. The results of this study indicate that short-term exposure to traffic-derived PM air pollution mainly from diesel engines is related to acute negative health effects.
COPD In western countries COPD is induced mainly by tobacco smoking and is characterised by a chronic obstructive flow limitation as well as airway inflammation. The symptoms of COPD are related to bronchitis, bronchiolitis and emphysema, secondary to a chronic airway inflammation. In COPD metaplasi occurs within the conducting airways. Ciliated cells are destroyed and transformed into mucus producing cells thereby reducing the mucociliary clearance capacity. The peripheral airways as well as the conducting airways and lung parenchyma are affected by the inflammation. Neutrophils secrete mediators such as neutrophil elastase, matrix metalloproteinases and myeloperoxidase which are able to destroy alveolar septas, leading to the development of emphysema.
It is suggested that the low grade inflammation in the lungs in COPD “spills over” to the systemic circulation, leading to a variety of disease manifestations. A systemic inflammatory response in COPD is shown as increased serum levels of CRP and TNF-α which further increase with increased disease severity (37, 38).
There are numerous studies demonstrating increased frequency of deteriorations, exacerbations, emergency room visits, hospitalizations and mortality in COPD patients in association with short-term exposure to variations in air pollution (39, 40).
The traffic derived air pollution is seen as a risk factor of developing chronic obstructive disorders in adults. Long-term exposure to traffic-related particulate matter air pollution has been demonstrated as a risk factor of COPD and decreased lung function and the risk was even further increased when living within 100 m from a busy road (41).
COPD and occupational exposure There is an association between occupational exposures to air pollution and obstructive airway diseases. However, the risk estimations have not yet been fully clarified. Even after adjustments for smoking history, some occupational exposures and groups of employees are over-represented as regards to suffering from COPD. Examples of such working environments are sites with heavy diesel trucks, buses and trains as well as docks, garages and tunnels. In a study performed in workers on diesel-engine trains, Hart et al demonstrated a clear association between diesel exhaust exposure and increased COPD mortality, after correlating for ordinary risk factors such as age, race and smoking (42). As previously mentioned, tunnel workers experience decreases in lung function related to environmental air pollution. In a series of studies, Ulvestad et al found that exposure to particles and gases from diesel exhaust, blasting, drilling and rock transport in tunnel work enhances the risk for an accelerated decline in FEV1, respiratory symptoms and COPD in tunnel workers compared with other heavy construction workers (43-45).

INTRODUCTION
21
Cardiovascular Disease The increased cardiovascular morbidity and mortality associated with PM pollution are well documented. Air pollution has been implicated in the pathogenesis of cardiovascular disease based on e.g. observations of increased cardiovascular disease with fatal myocardial infarctions in patients living close to major roads (46, 47). The need for increased awareness of the cardiovascular risks associated with exposure to air pollution was recently highlighted in a scientific statement issued by the American Heart Association (17, 48, 49). The associations are strongest for fine particulate air pollutants (PM2.5) (14), of which the combustion-derived nanoparticulates from diesel exhaust are an important component (15) (16).
Short-term increases in fine particulate matter air pollution exacerbate cardio- vascular disease leading to hospitalization for conditions including acute myocardial infarction (50). Short-term exposure to PM air pollution is associated with an increased risk of early (2 h) and delayed (24 h) presentation with acute myocardial infarction, (51, 52) or re-hospitalisation for myocardial ischemia in patients with prior myocardial infarction (17, 53-55).
Long-term exposure to fine particulate matter air pollution increases the risk of cardiopulmonary mortality (8, 9). Recently Miller et al reported founding’s of negative health effects by long-term exposure to traffic-related air pollution. There was an important negative impact on mortality in women following exposure to fine particulate air pollution and the risk of death from cardiovascular disease increased by 76% (56). Moreover, a panel study in Los Angeles provided the first support to a link between chronic PM exposure and atherosclerosis in humans, a well known risk factor even for the development of stroke (57). A 10-μg/m3 increase in fine PM was associated with an increase in carotid intima-media thickness (an ultrasonic measurement) of atheroma suggesting that long-term ambient PM exposure may affect the progress of atherosclerosis in humans. Taken together, these data suggest that exposure to ambient air pollution can induce systemic effects, which can add to the progression of atherosclerosis.
There are no consistent associations, but some literature points out a possible association between arrhythmias and increased morbidity and mortality in cardiovascular disease. Most studies have been focused on heart rate variability (HRV), due to its impact on cardiovascular morbidity and mortality in both healthy individuals (58) and survivors of myocardial infarction (59). Liao and colleagues hypothesized that an effect of PM exposure on the autonomic control of heart rate and rhythm would explain the association between PM and adverse cardiovascular outcomes. They reported an association between fine particulate air pollution (PM2.5) and heart rate variability in a panel of elderly subjects (60). Another study found an association with an increase in the number of defibrillator interventions among patients with implantable cardiac defibrillators, ICDs. When analysis was restricted only to patients requiring frequent ICD interventions stronger associations were seen (61). Dockey et al demonstrated associations of ventricular tachyarrhythmia with fine particle mass, carbon monoxide, nitrogen dioxide, and black carbon suggest a link with motor vehicle pollutants. The associations with sulfate suggest a link with stationary fossil fuel combustion sources (62).
Further, in a recently published panel study the authors concluded that there was a significant increase in systolic blood pressure following an increase in total ambient air pollution and strengthened by co-exposure of traffic derived air pollution (63).

INTRODUCTION
22
EXPERIMENTAL STUDIES OF PM POLLUTION Despite well-designed epidemiological studies, it has not yet been possible to prove a causal relationship between road traffic and cardiovascular morbidity and mortality. There are a number of confounding factors to take into consideration. However, systems have been designed to deliver controlled amounts of air pollution and allow a mechanistic approach to determine the effects of inhaled PM.
Diesel exhaust Diesel engines are used in heavy vehicles and have increased in popularity in cars in Europe. Although diesel engines are more efficient and therefore more economic than conventional spark ignition gasoline engines, they may emit 8-100 more particulates than petrol engines (64) and is therefore a major contributor to atmospheric PM pollution in most places around the world. As a consequence, numbers of people are exposed directly in traffic or to DE components imported by winds from a long distance.
Diesel exhaust is a mixture of particulates (soot) and various gaseous, out of which the most prevalent are sulphur oxides, nitrogen oxides, carbon dioxide, carbon monoxide and various complex compounds such as aldehydes. Most of the particulate matter generated from diesel exhaust (50-80%) are extremely small with diameters of 0.02 µm to 0.5 µm and can reach the terminal alveoli and be deposited in the lungs. Agglomeration of the particles may occur depending on engine and exhaust conditions, but also to environmental factors (photochemical processes and humidity). Some factors influence the contribution of diesel emission to total airborne particulate matter, i.e. the percentage of diesel vehicles in the total volume of traffic, the type, age and maintenance of individual engines, fuel quality (e.g. sulphur content and aromaticity), emission control techniques and distance from roads (65) World Health Organisation. Diesel fuel and exhaust emissions. Environment Health Criteria No 171, 1996, Geneva: WHO.
Controlled exposure to particulate matter air pollution Controlled exposures to air pollutants can help to address the confounding factors by providing a defined exposure in a regulated environment and thus facilitate investigations of validated biomarkers and surrogate measures known to be involved in respiratory cardiovascular health. Studies utilising diesel exhaust are crucial in determining the health effects of exposure to the combustion-derived component of air pollution (66, 67).
The major advantage of chamber exposures is that they allow controlled and well-characterized exposure concentrations of both the gaseous and particulate fractions of diesel exhaust as well as a controlled exposure dose of the pollutant. Exposure to 300 μg/m3 of PM pollution for one hour increases a person’s average exposure during a 24-hour period by only 12 μg/m3. Changes of this magnitude may occur on a daily basis in even the least polluted of cities and are associated with increases in cardio-respiratory mortality (14). Health effects by diesel engine exhaust have been investigated in a series of Swedish studies, in which human subjects have been exposed in an experimental chamber. The exposure chamber system has been validated and allows for controlled and steady exposure concentrations of diluted diesel

INTRODUCTION
23
exhaust (66, 68, 69). The exposure model is therefore relevant both when it comes to the composition and the magnitude of exposure for the assessment of short-term health effects in humans. As diesel exhaust is a complex mixture of gases and particles, we cannot, based on our findings, exclude a non-particulate cause of the adverse respiratory effects. The DE exposure model is not designed to allow evaluation of the individual gaseous and particulates, but ongoing studies with modern particle traps will eventually lead us further in this matter.
Another possibility to experimentally address the adverse effects or air pollution is to perform exposures to ambient particulates collected using a particle concentrator, which is able to enrich the mass of ambient fine particles in real time with little modification. They produce "real world" particles and they allow exposure at related masses. Limitations include variability in both particle mass and composition and some uncertainty about the best statistical approach to analyze the data (70).
In-vitro and animal studies In vitro and animal studies are important to examine effects following air pollutant challenges that could not be performed in humans. Result from in vitro and animal studies may be extrapolated into the human situation and thus play an important role when designing controlled human exposure studies.
The nanoparticle component in inhaled PM may potentially influence the cardiovascular system either through indirect effects mediated by lung inflammation or through the direct action of particles translocated into the circulation. A classic and potentially possible cause of the associations between air pollution and the adverse cardiovascular effects is an air pollution-induced systemic inflammatory response following oxidative stress and airway inflammation (71). It implies that the air pollution-produced increases in airway inflammatory cells and cytokines exert their effects in the systemic circulation in the same manner as demonstrated in the lungs.
Translocation of inhaled nanoparticles across the alveolar–blood barrier has been demonstrated in animal studies with nanoparticles delivered by both inhalation as well as by pulmonary and nasal instillation (72-76). However, the data on nanoparticle translocation in humans is so far not consistent. Nemmar Hoet 2002, (77). Wiebert et al could not give an account of 1 % of their labelled carbon particles in a similar study human study (78). It cannot be excluded that this small particle fraction may have been translocated and possibly enough to initiate adverse health effects. In animals it has furthermore been suggested that injured arteries can take up blood borne nanoparticles (79). A possible uptake of nanoparticulates into the vessel wall could play a role in the pathogenesis of atherosclerosis and initiate an inflammatory response, which is believed to increase and destabilize atherosclerotic plaques causing the detrimental outcomes, as suggested in APO-E knockout mice (80). Moreover, another animal study has demonstrated labelled nanoparticles to penetrate into the brain via nasal inhalation and suggested translocation via nervus olfactorius (81). Interestingly, Crutz et al showed that DE exposure in humans induced a significant deterioration in frontal cortex EEG pattern (82). However, the clinical significance of this finding is not yet possible to interpret.

INTRODUCTION
24
In vitro studies
In a recent study Fiorito et al demonstrated that a reduced particle emission does not automatically give a reduced human cytotoxicity (83). The authors have compared diesel particle effects from low emission engines with diesel particles from older engines on monocytes derived from peripheral human blood. They found that monocytes exposed to low emission engine particles demonstrated an increased rate of necrosis, degeneration and apoptosis. Through the changed surface structure of the smaller particles, these were more hydrophilic, thereby increasing the opportunity to interact with different bio molecules. In contrast, particles generated from older diesel engines were more inert and with a hydrophobic character. The smaller particles were more aggressive and aggregated to a larger extent causing increases in pro-inflammatory mediators such as IL-1, IL-6 and IL-10. This indicate that the smaller amounts of diesel exhaust particles generated in low emission engines filled with modern diesel fuel do not automatically reduce their well known negative health effects. Further investigations are important to understand these effects in humans.
Animal responses
Lung effects Exposure studies have established the oxidative and pro-inflammatory nature of combustion derived particulate matter and implied a role for oxidative stress in determining the toxicity of ambient air pollution such as diesel exhaust particulates (84-86). More recent studies have also investigated airway inflammation and bronchial hyperresponsiveness, which are features more in line with asthma, as well as suggested that diesel exhaust particles are able to enhance allergic responses (21, 87, 88).
Cardiovascular and systemic effects
Atherothrombotic development Preclinical models of exposure to particulate matter air pollution demonstrate accelerated atherosclerotic plaque development and increased in vitro (89) and in vivo (90) platelet aggregation. It is plausible that repeated exposure to PM might induce the vascular inflammation of atherosclerosis and promote plaque expansion or rupture. The first direct evidence to support this hypothesis was provided by Suwa and co-workers (91), who demonstrated that instillation of high doses of PM10 resulted in plaque progression and destabilization in an animal model of atherosclerosis. In the same studies, PM10 exposure accelerated monocyte release from the bone marrow (92). The amount of particulates phagocytosed by alveolar macrophages correlated with both the bone-marrow response and plaque volume (92), suggesting a role for pulmonary inflammation and systemic mediators in determining the pro-atherogenic effects of PM. In apo-lipoprotein E knockout mice, inhalation of ultra fine PM for 6 months increased atherosclerotic plaque volume, altered plaque composition and upregulated vascular inflammation (80, 93). These changes in plaque morphology were accompanied by abnormal vascular function characterized by exaggerated vasoconstriction and impaired endothelial-dependent vasodilatation (93). Further, increased thrombotic events in femoral veins have been demonstrated in hamsters 24 hours after intra-tracheal instillation of diesel exhaust particles (94).

INTRODUCTION
25
Arrhythmia A possible mechanism behind the observed negative cardiovascular outcomes following PM air pollution exposure is suggested to be a negative impact on the autonomic regulation. In rats, intra tracheal instillation of diesel exhaust particles has been shown to increase the risk for arrhythmias following induced coronary ischemia-reperfusion when investigated 24 and 48 hours after DEP-instillation. Serious ventricular arrhythmias also occurred (95). The genetic variant in lipoprotein metabolism, the ApoE-/- mouse, has been demonstrated to be especially sensitive for the development of cardiovascular disease. This model has been used to demonstrate ECG abnormalities measured by implantable radio telemetry devices during exposure to whole diesel exhaust emissions (96).
Experimental studies in humans
CAPs exposures
Exposure of healthy volunteers to concentrated ambient particles (CAPs) has been associated with decreases in both white blood cell (WBC) count and lactate dehydrogenase (LDH) as well as increased blood concentrations of fibrinogen (97). In a controlled human exposure of elderly subjects, CAPS induced changes in heart rate variability that was not seen in young subjects (98). Airway inflammatory responses have been demonstrated in bronchoscopy studies following inhalation of CAPs (Ghio et al., 2000; Holgate et al., 2003).
Pietropaoli and Frampton et al also demonstrated lung function decrements when exposing healthy subjects to inhalation of laboratory generated ultra fine particles with a mass concentration 50 µg/m3 (99). Furthermore, they demonstrated a reduction in mid expiratory flow rates and a reduction in carbon monoxide diffusing capacity without any signs of airway inflammation as reflected in induced sputum 21 hours after exposure.
Short-term inhalation of fine particulate air pollution via CAPs and ozone at concentrations that occur in urban environments has been found to cause conduit artery vasoconstriction (100). High resolution ultrasonography was used to measure alterations in brachial artery diameter, endothelial-dependent flow mediated vasodilatation (FMD) and endothelial-independent nitroglycerin-mediated dilatation (NMD). The authors claim that alterations in arterial tone and reactivity to PM2.5 and ozone play an important role in the research for biologic mechanisms linking air pollution to acute and, eventually, chronic cardiovascular events. Future studies in this field are needed to verify the findings in the coronary circulation and in subjects with coronary heart disease.
Road tunnel exposure
Exposure of healthy subjects to traffic-related air pollution in a road tunnel set-up in Stockholm, Sweden resulted in an airway inflammatory response with cell migration as reflected in BAL, together with signs of an initiated signal transduction in the bronchial epithelium (101). Further, an increase in allergen responsiveness has been demonstrated in asthmatics after a similar exposure (102).

INTRODUCTION
26
Carbon particle exposure
In a study in heart patients, neither exposure to carbon particles nor to sulphur dioxide was associated with decrements in heart rate variability (HRV) or markers of lung inflammation and systemic coagulation, (103). The authors performed a randomised double blind controlled study in healthy volunteers and patients with stable angina and exposed them to carbon particles (50 µg/m3) and SO2 (200 parts per billion (ppb). No significant changes in circulating markers of inflammation (white blood cell count and us-CRP) or coagulation (fibrinogen and D-dimer) were found at 4 and 24 hours after exposures in either group. Short-term exposure to pure carbon particles in healthy volunteers did not cause adverse effects on HRV or a systemic inflammatory response.
Diesel exhaust-induced effects on the respiratory system
Lung function effects Following inhalation, diesel engine exhaust causes a broncoconstrictive response reflected as increased airway resistance in both healthy and asthmatic subjects (104). Stenfors et al noticed a similar increase in airway resistance in both asthmatic and healthy following a relative low dose of diesel exhaust air pollution employing a PM concentration of 108 ug/m3.
Airway inflammatory responses Time course investigations studying effects of DE at PM10 concentrations of 100 and 300 µg/m3 suggest a highly established airway inflammation in human airways six hours after the higher exposure concentration of diesel engine exhaust particles. In healthy volunteers, significant increases in neutrophils and platelets were seen in peripheral blood following diesel exhaust exposure (69). Exposure to lower concentrations, 100 µg/m3, resulted in a slower onset of inflammation with the extent and amplitude at 18 hours corresponding to that at six hours after the higher (300 µg/m3) concentration. (105-107).
It has been indicated that the oxidative potential by diesel exhaust may trigger the airway inflammatory events. This is mediated by oxidative stress-sensitive transcription factors, such as NFκB and AP-1 and mitogen activated protein-kinases (MAPK) (108) and involves activation through tyrosine phosphorylation of the epithelial growth factor receptor (EGFR) (Pourazar et al Particle Fiber Toxicology, in press). This signalling pathway controls a range of neutrophil chemoattractants such as IL-8 and Gro-α, resulting in a neutrophilic infiltration in the airway mucosa and airway lumen. Neutrophils secrete a secondary oxidative enzyme, myeloperoxidase (MPO), which may add to tissue damage following DE exposure. Other inflammatory cells involved in the DE induced response are CD4+ and CD8+ T-cells and mast cells. The airway inflammatory response to diesel exhaust is compartmentalised, related to differing antioxidant responses in the conducting airway and alveolar regions, with protective antioxidant responses predominating at low doses and inflammation and injury only occurring at higher concentrations (107).
Compared to healthy subjects, there were differences in the inflammatory responses within the airways of asthmatic subjects following a DE challenge. Stenfors et al demonstrated airway neutrophilia and lymphocytosis in healthy subjects, together with increased inflammatory mediators IL-6 and IL-8 in BAL as well as increased mRNA-IL-8 in mucosal biopsies. Asthmatic individuals have been investigated and certain differences have been displayed in terms of bronchial epithelial cytokine response as compared to the inflammatory

INTRODUCTION
27
response in non-asthmatic healthy individuals. A four-fold increase in IL-10 was demonstrated in the bronchial epithelium of asthmatic subjects as compared with the reduction by half in healthy volunteers (106). No enhancement of the pre-existing asthmatic inflammation involving increased numbers of eosinophils and mast cells was seen. The demonstrated deterioration in BHR in asthmatics 24 hours after DE exposure is of special interest. Despite regular treatment with inhaled corticosteroids (average 1200 µg per day), the asthmatics showed an increase in BHR, a cardinal feature of asthma (109). The mean metacholine PC20 of the subjects changed from 3.4 mg to 1.7 mg representing a clinically potentially significant deterioration of the hyperresponsiveness. However, this may well explain why asthmatic subjects can experience exacerbations after exposure to traffic-related air pollution. Further studies are needed to clarify the local inflammatory events and functional changes in the airway smooth muscle that are responsible for these responses in asthmatic subjects.
Up to now no experimental exposure studies had evaluated the air pollution-induced airway inflammation and cardiovascular and systemic responses in patients with COPD.
Systemic effects of DE
Human studies There are limited studies of systemic effects of DE. In diesel exhaust exposure chamber studies similar to those in the present thesis, Kaufman et al investigated whether there are systemic effects following the DE challenges on inflammatory (CRP and fibrinogen) and hypercoagulability (D-dimer, vWF, PAI-1 and platelets) out comes (110), as well as affected autonomic function measured as HRV (111), without any significant changes. They used PM concentrations of DE of 100- 200 µg/m3 and followed the volunteers up to 22 hours after the exposures. However, in a small pilot study, DE exposure inhalation increased the ribo nuclei amino acid (RNA) to mononuclear white cells in peripheral human blood 2 hours after exposure, indicating an activated global gene expression of inflammation (112)

28
.

29
AIMS The overall aim of this thesis was:
to evaluate airway and cardiovascular effects of the common particulate matter air pollutant diesel exhaust in healthy subjects as well as in at-risk patients with chronic obstructive pulmonary disease (COPD) and stable coronary vascular disease.
The specific aims were: to determine whether exposure to diesel exhaust would cause lung
function deterioration and airway inflammatory responses as determined in induced sputum in patients with moderate COPD,
to evaluate whether diesel exhaust exposure would induce systemic inflammation, activation of blood coagulation, endothelial dysfunction or lung epithelial injury measurable in peripheral blood in individuals with stable COPD,
to investigate whether exposure to diesel exhaust would impair vascular function in terms of the regulation of vascular tone and endogenous fibrinolysis in healthy subjects,
to determine the time kinetics of the diesel exhaust-induced impairment of vascular function and systemic inflammation after DE exposure,
to clarify whether patients with clinically stable coronary heart disease would develop myocardial, vascular and fibrinolytic dysfunction following DE exposure.

30

31
SUBJECTS AND METHODS
STUDY DESIGN In order to understand the negative health effects in humans following exposure to diesel exhaust derived particulate matter air pollution, we conducted a series of studies in young and healthy subjects as well as in patients with COPD and stable coronary artery disease. To achieve a comprehensive picture of the airways and cardiovascular system following DE exposures, both non-invasive and invasive techniques were used. All healthy subjects were non-smokers, whereas COPD patients had a smoking history, but had stopped smoking more than one year previous to the start of the study. All but one of the coronary compromised volunteers had a smoking history but no one was a current smoker.
All five studies were performed using a double blinded cross over design with each subject serving as his/her own control. In order to reduce any possible carry over effect from one exposure to the other, they were conducted at least two weeks apart and in randomised sequence, with half the study population first being exposed to air and the other half first being exposed to diesel exhaust. The researchers and volunteers were blinded as to exposure details but the engineering staff supervising the exposure chamber was aware of exposure details.
In study I and II, volunteers alternated exercise on a bicycle ergo meter (VE =10-15 L/min/m2) and rest at twenty-minute intervals. In studies III and IV volunteers performed moderate exercise (minute ventilation, VE =25 L/min/m2) on a bicycle ergometer, alternated with rest at 15-minute intervals. In study V, the ergometer workload for each subject was titrated to achieve a minute ventilation of 15 L/min/m2 to ensure a similar exposure.
Subjects
Study I and II
Fifteen patients were recruited, out of which 12 (7 males and 5 females) completed the full protocol in study I. In study II all 15 participants completed the study. Mean age was 67 (range 56-72) and their smoking history was 32±5.1 pack years (mean ± SEM). Inclusion criteria were mild COPD (FEV1/FVC<70%, 50%<FEV1<80% predicted) according to GOLD (113), no steroid treatment, age 50-75 years, less than 12 % significant reversibility after bronchodilatation of 1.0 mg of nebulised Terbutalin, history of smoking (>15-20 pack-years) and smoking cessation >3 months ago. Exclusion criteria were; ischemic coronary heart disease or arrhythmia, diabetes mellitus, steroid treatment (oral or inhaled) within 8 weeks prior to or during study, history of asthma and/or atophy, and antioxidant as well as aspirin supplementation <2 weeks before exposure. All participants were free from airway infections or any exacerbation within six weeks prior to or during study. The only medication allowed was short-acting bronchodilators.

SUBJECTS AND METHODS
32
Study III and IV
In total, thirty healthy, male non smokers between 20 and 38 years old participated in these studies. Based on previous exposure (65) and systemic inflammatory (114) studies, vascular assessments were performed in 15 subjects at 2 and 6 hours after diesel or air exposure in study III. In light of the findings from study III, we subsequently determined vascular function in another 15 subjects at 24 hours after exposure to diesel exhaust or air (study IV). Subjects excluded were those taking regular medication and with clinical evidence of atherosclerotic vascular disease, arrhythmias, diabetes mellitus, hypertension, renal or hepatic failure, asthma, significant occupational exposure to air pollution or an undercurrent illness likely to be associated with inflammation were excluded from the study. Subjects had normal lung function and reported no symptoms of respiratory tract infection for at least 6 weeks before or during the study.
Study V
Twenty patients with stable coronary artery disease participated in this study. Mean age 60 ± 1 year (SEM). All patients had proven coronary heart disease with a previous myocardial infarction (> 6 months previously) treated by primary angioplasty and stenting, and were receiving standard secondary preventative therapy, i.e. aspirin, statins, beta-blockers and Angiotensin converting enzyme, (ACE) inhibitors / Angiotensin receptor blocker, (ARB). Patients with angina pectoris (Canadian Cardiovascular Society grade ≥2), a history of arrhythmia, diabetes mellitus, uncontrolled hypertension, renal or hepatic failure or those with unstable coronary disease (acute coronary syndrome or unstable symptoms within 3 months) were excluded. All volunteers were invited to a pre-study screening visit for exercise stress testing and patients unable to achieve stage 2 of the Bruce protocol, had marked ECG changes (left bundle branch block, early ST depression >2mm) or developed hypotension were excluded. Current smokers and those with asthma, significant occupational exposure to air pollution or an inter-current illness were also excluded from the study.
Subject Preparation In studies III-V subjects were requested to abstain from alcohol for 24 hours and food, caffeine-containing drinks and tobacco for at least 4 hours before each study. In study V, all medication was continued throughout the study period, with the exception of Angiotensin Converting Enzyme Inhibitors, which were withdrawn 7 days prior to each vascular study, as it augments bradykinin induced endothelial tissue plasminogen activator (t-PA) release (115)
Study I
In study I, it was hypothesized that patients with a pre-existing COPD-associated airway inflammation would experience an enhanced diesel exhaust-induced airway inflammatory response and whether diesel exhaust exposure would affect lung function. Static and dynamic spirometry including diffusion capacity was performed before and immediately after each exposure. Sputum induction with hypertonic saline was performed 6 h after each exposure.

SUBJECTS AND METHODS
33
Study II
The aim of study II was to investigate whether an acute exposure to diesel exhaust nanoparticles would result in extra-pulmonary adverse effects including activation of blood coagulation, systemic inflammation, endothelial damage or alteration in pulmonary integrity, as reflected in peripheral blood. Therefore, blood samples were collected before exposures and 6 and 24 hours after the exposures.
Study III
Using the carefully characterized exposure system, we sought to assess the effect of diluted diesel exhaust inhalation on endothelial vasomotor and fibrinolytic functions in humans. Venous occlusion phletysmography was performed two and six hours after exposures to diesel exhaust or filtered air. Bilateral forearm blood flow, systemic inflammatory markers and endogenous fibrinolysis were measured before and during unilateral infusion of vasodilators. All subjects remained indoor between the exposure and vascular assessment.
Study IV
The aim of this study was to investigate whether there is systemic inflammation and sustained vascular dysfunction in healthy volunteers 24 hours after exposure to diesel exhaust. A venous occlusion phletysmography study was performed 24 hours after the exposures. All subjects remained indoors between the exposure and vascular assessment.
Study V
This study aimed to assess the effect of dilute diesel exhaust inhalation on myocardial, vascular and fibrinolytic function in an “at risk” population of patients with stable coronary heart disease. The study was identical to the six hours protocol in study III, apart from the volunteers and the workload used during exposures. All volunteers were fitted with 12-lead Holter electrocardiography monitors (Reynolds Medical Life card 12, Delmar Reynolds, UK). All subjects remained indoors between the exposure and vascular assessment.
Chamber exposures Diesel exhaust was generated from an idling Volvo diesel engine (Volvo TD45, 4.5L, 4 cylinders, 680 rpm) using Swedish Low Sulphur Gasoil E10 (Preem, Göteborg, Sweden), as described previously (65, 66, 116, 117). Over 90% of the exhaust was shunted away, and the remaining part diluted with ambient filtered air heated to 20º C (humidity ~50%) before being fed into a whole body exposure chamber (3 x 3 x 2.4 m) at a steady-state concentration. Throughout the exposures air from the breathing zone of the volunteers were collected and continuously monitored for the concentrations of nitric oxide NO, nitrogen dioxide, NO2 oxides of nitrogen, NOX and total gaseous hydrocarbons (indirect measured as propane). A Miran 1-A, an infrared-instrument (Foxboro Co, East Bridgewater, MA, USA), was used for analysis of CO. Oxides of nitrogen (NOx, NO, NO2) were analyzed with a chemiluminiscence instrument, (ECO-Physics CLD 700, Boo Instruments, Stockholm, Sweden). HC were analyzed with a FID-instrument, model 3-300 (J:U:M Engineering GmbH, Munich, Germany)

SUBJECTS AND METHODS
34
was determined by weighing particles collected on filters (PALL Life Sciences Teflo 47mm 2.0µm 25/Box 50/PK P/N R2PJ047 QTY 1 PK) particle mass (µg/m3). The chamber air was continuously changed every 2-3 minutes. The DE entering the chamber in Studies I-V was standardized to give a particle concentration of 300 µg/m3. See figure 3
Figure 3. The engine and shunt set-up for human exposure studies to dilute diesel exhaust. The exhaust is lead into the exposure chamber in the adjacent room, as depicted overleaf.

SUBJECTS AND METHODS
35
Figure 4. During exposures, the volunteers perform intermittent mild exercise in the exposure chamber.
Lung function measurements Dynamic spirometry, including forced expiratory volume in one second (FEV1), vital capacity (VC) and forced vital capacity (FVC) was performed at inclusion in all studies and during sputum induction in study I. (Vitalograph-COMPACT; Vitalograph Ltd., Buckingham, UK). In study I, pre and post-exposure lung function (FVC, FEV1 and DLCO) were measured using a computerized whole body pletysmograph (system 2800, Sensor Medics Corp., Ca, USA). The best of a minimum of three adequate performed measurements was used.
Sputum induction and processing Sputum induction was performed according to Pin et al (118). All subjects were pre-treated with an inhaled beta-2-agonist (1,0 mg of Terbutaline) Hypertonic saline was nebulised using an ultrasonic nebulizer, with an output of 1.5 mL/min. Inhalation was performed at intervals of 7 min with 3%, 4%, 5% of saline. Following each inhalation interval, subjects were advised to rinse their mouth with water and blow their nose before trying to cough sputum into a sterile plastic container. The obtained samples were kept on ice prior to processing.
The processing of the induced sputum were carried out using the method based on Pizzichini et al (119) and used in previous DE-exposure studies (109, 120). In short, portions appearing more viscid and dense were selected from the expectorated samples and transferred to a 10-mL siliconised tube. After adding of 0.1 % diothiotreitol (DTT) at a volume equal to four times the selected sputum weight, the sputum was rocked for 15 min to dissolve the mucus and disperse the cells. Phosphate buffered saline (PBS) was then added at a volume equal to

SUBJECTS AND METHODS
36
that of DTT and the rocking continued for 5 min. The mixture was filtered through a 48µm mesh nylon filter into another 10 mL tube and centrifuged at 300 x G for 10 min at 4° C.
The supernatant was separated from the cell pellet, re-centrifuged at 1,000 x G for further 10 min to remove debris, aspirated and stored in Eppendorf tubes at -70 °C for later analyses. The cell pellet was re-suspended in 1,000 µL PBS and total cell count and cell viability were determined in a haemocytometer using tryptan blue. Cell suspension was adjusted to 0,5 x 106 cells/mL and 50 µL were placed in each cup of a Shandon 3 centrifuge (Shandon Southern Instruments Inc., Sewickley, PA USA). Cytospins were made on pre-wet slides, prepared at 400 rpm for 5 min and stained with May-Grunewald-Giemsa.
Sputum analyses
Cell counts
Samples were considered adequate for analysis if the squamous cell contamination was < 20% and the viability >50%. Total cell count was calculated by dividing the total number of cells by the volume of processed sputum (1 mg=1 µL). At least 400 non-squamous cells were counted and the differential cell counts were expressed as a percentage of the total non-squamous cell count. Counting 400 additional cells and expressing this as a percentage of the total number of cells, obtained the proportion of squamous cells.
Soluble components
Interleukins (IL-8, IL-10) and myeloperoxidase (MPO) were determined in the supernatant using an immunosorbent assay kits (IL-8, IL-10) (R&D Systems Inc., Abingdon, UK) and a sensitive commercial radioimmunoassay kit (MPO) (Pharmacia & Upjohn, AB, Uppsala, Sweden).
Analysis in peripheral blood in study II
Sample Preparation
Venous blood samples were drawn at three time-points: before, 6 hours and 24 hours after the end of exposure. The samples were placed in melting ice and centrifuged at 4°C 2,000-2,500 g for 12 minutes within 1 hour. Most of the supernatant was transferred into a propylene plastic tube and centrifuged a second time in the same conditions to obtain platelet poor plasma (PPP). The supernatant plasma was transferred and stored in cryotubes in small aliquots of 0.5 mL. Aliquots were stored at minus 80°C to ensure rapid cooling and freezing until assays. Six aliquots were available for each subject: three of them corresponded to the three time-points relative to air exposure and the three other corresponded to the DE exposure.

SUBJECTS AND METHODS
37
Endothelial function-von Willebrand factor activity assay
The von Willebrand factor activity was determined by the assay Collagen Binding Assay (CBA). CBA is an enzyme immunoassay used for the qualitative determination of vWF function in human plasma. It quantifies the binding of vWF to collagen type III coated onto micro-titre wells. Collagen binding capacity of vWF is correlated with the higher molecular weight (HMW) forms of vWF, believed to be functionally more important in haemostasis than lower molecular forms (LMW). Therefore CBA may correlate more closely with vWF function and bleeding problems than regular ELISAs for vWF antigen determination which measure total (LMW+HMW) vWF.
During the first incubation step the vWF multimers present in the sample bind to the collagen which is attached to the surface of the micro-titre plate. A washing removes the unbound plasma protein. In a second reaction peroxidase conjugated anti-human vWF antibodies bind to vWF multimers. Excess antibodies are washed off and the substrate of the enzyme is added. The enzymatic reaction between hydrogen peroxide and the substrate is terminated by the addition of diluted sulphuric acid. The resulting colour intensity which is proportional to high molecular weight vWF multimers present in the sample, is determined photometrically (wavelength of reading: 450 and 690 nm). Calibrated standards are used to quantify the activity of the high molecular weight vWF multimers. The reagents used for CBA was supplied in a pack manufactured by Gradipore Ltd, Australia. The photometer used was the Multiskan EX from ThermoLabsystems.
Coagulopathy-Human Pro-thrombin Fragment 1+2 (F1+2) assays
Enzygnost® F1+2 micro is an enzyme immunoassay for the quantitative determination of human pro-thrombin fragment 1+2 as an aid in diagnosis, monitoring and evaluation of blood coagulation disorders involving changes in coagulation system activity. This assay is based on the sandwich principle. During the first incubation phase F1+2 antigens in the sample bind to the anti-F1+2 antibodies fixed to the surface of the micro-titre plate. Afterwards the plate is rinsed out. Peroxidase-conjugated antibodies are added and, in the second incubation step, bind to the free F1+2 determinants. The excess enzyme-conjugated antibodies are then washed and the substrate of the enzyme is subsequently added. The enzymatic reaction between hydrogen peroxide and chromogen is terminated by the addition of diluted sulphuric acid. The resultant colour intensity is proportional to the concentration of F1+2 and is determined photometrically (wavelength of reading: 492 nm). Calibration standards are used to quantify the pro-thrombin fragment 1+2 level. The reagents used for the quantification of the fragments 1+2 were supplied in a pack manufactured by Dade® Behring, Marburg, Germany. The photometer used was the Multiskan EX from ThermoLabsystems.
Coagulopathy-D-dimer assay
D-Dimer PLUS is a latex-enhanced turbidimetric test for the quantitative determination of cross-linked fibrin degradation products (D-dimer) in human plasma.
A mouse monoclonal antibody directed against the cross-linkage region of D-dimer is covalently linked to polystyrene particles that are agglutinated when mixed with samples containing D-dimer. The cross-linkage region has a stereosymmetrical structure i.e. the epitope for the monoclonal antibody occurs twice. Consequently, one antibody is sufficient to trigger an agglutination reaction which is then detected turbidimetrically. The reagents used

SUBJECTS AND METHODS
38
for the quantification of the D-dimer were supplied in a pack manufactured by Dade® Behring, Marburg, Germany. The instrument used to determine its concentration was the Sysmex® CA- 1500 System, Dade Berhing,
Inflammatory C-reactive protein (CRP) assay
The concentration of CRP was determined by a high-sensitive immunoassay relying on the agglutination of polystyrene particles. Polystyrene particles coated with monoclonal antibodies to CRP are agglutinated when mixed with samples containing CRP. The intensity of the scattered light in the nephelometer depends on the CRP content of the sample and therefore the concentration can be determined versus dilutions of a standard of a known concentration. The reagents used came from a package made by Dade Behring, Marburg, Germany and the apparatus was the Behring Nephelometer II also supplied by this firm (121).
Inflammatory-Fibrinogen assay
The thrombin clotting time (TCT) of diluted plasma is inversely proportional to the fibrinogen concentration of the plasma. The enzyme thrombin converts the soluble plasma protein fibrinogen into its insoluble polymer fibrin. The clotting time of diluted plasma obtained after addition of thrombin is compared with that of a standardised fibrinogen preparation and fibrinogen concentration can be determined. Fibrinogen was measured by a modified Clauss method. The quantification of fibrinogen level required one reagent a bovine thrombin provided by Dade Berhing, Marburg, Germany. The coagulation analyzer used to determine the concentration in fibrinogen was the Sysmex® CA- 1500 System, Dade Behring, Germany (122).
Clara cell protein (CC16) assay
The concentrations of CC16 were determined by a sensitive immunoassay relying on the agglutination of latex particles. This method is based on the agglutination, by protein, of calibrated latex particles coated with a specific polyclonal antibody. The agglutination can then be quantified by nephelometry at a wavelength of 360 nm (123).
Venous occlusion pletysmography studies III-V Measuring the reaction to vasoactive substances released by, or those that interact with, the vascular endothelium in the forearm is a useful measure of endothelial function. Local intra-arterial drug infusion permits the direct assessment of vascular responses without invoking concomitant effects on other organs. In this way the vessels are studied in their physiological environment under the influence of neuronal, circulating and local mediators (124). Although measurement of coronary vascular response is of greatest clinical relevance, invasive coronary studies can only really be performed in patients undergoing angiography. The close correlation between coronary and peripheral endothelium-dependent responses (125) suggests that endothelial dysfunction may be a systemic state or that circulating factors have parallel effects in both coronary and peripheral arteries (126).
SUBJECTS AND METHODS
39
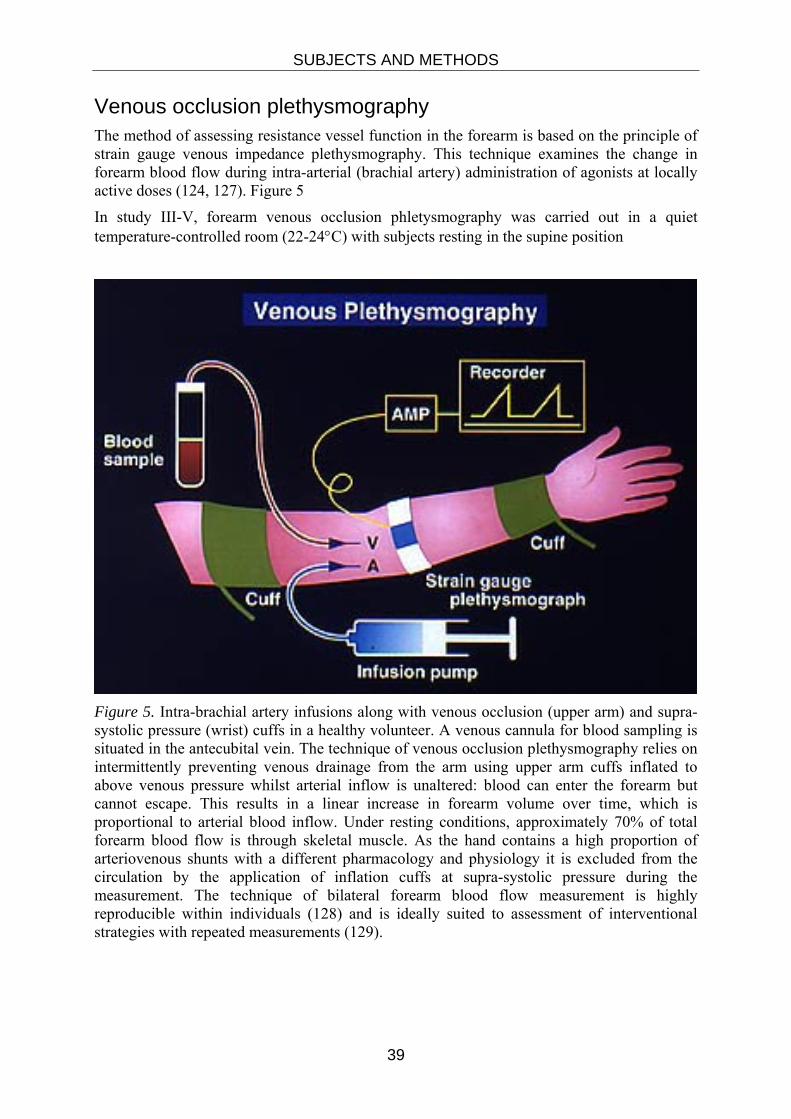
Venous occlusion plethysmography The method of assessing resistance vessel function in the forearm is based on the principle of strain gauge venous impedance plethysmography. This technique examines the change in forearm blood flow during intra-arterial (brachial artery) administration of agonists at locally active doses (124, 127). Figure 5
In study III-V, forearm venous occlusion phletysmography was carried out in a quiet temperature-controlled room (22-24°C) with subjects resting in the supine position
Figure 5. Intra-brachial artery infusions along with venous occlusion (upper arm) and supra-systolic pressure (wrist) cuffs in a healthy volunteer. A venous cannula for blood sampling is situated in the antecubital vein. The technique of venous occlusion plethysmography relies on intermittently preventing venous drainage from the arm using upper arm cuffs inflated to above venous pressure whilst arterial inflow is unaltered: blood can enter the forearm but cannot escape. This results in a linear increase in forearm volume over time, which is proportional to arterial blood inflow. Under resting conditions, approximately 70% of total forearm blood flow is through skeletal muscle. As the hand contains a high proportion of arteriovenous shunts with a different pharmacology and physiology it is excluded from the circulation by the application of inflation cuffs at supra-systolic pressure during the measurement. The technique of bilateral forearm blood flow measurement is highly reproducible within individuals (128) and is ideally suited to assessment of interventional strategies with repeated measurements (129).

SUBJECTS AND METHODS
40
Brachial Artery Cannulation
All subjects underwent brachial artery cannulation with a 27-standard wire gauge steel needle under controlled conditions.
Figure 6. Picture showing the thin arterial needle cannulated in arteria brachialis and a venous cannula.
After a 30-minute baseline saline infusion, acetylcholine at 5, 10, and 20 µg/min (endothelium-dependent vasodilator that does not release tissue plasminogen activator [t-PA]; Merck Biosciences); bradykinin at 100, 300, and 1000 pmol/min (endothelium-dependent vasodilator that releases t-PA; Merck Biosciences); and sodium nitroprusside at 2, 4, and 8 µg/min (endothelium-independent vasodilator that does not release t-PA; David Bull Laboratories) were infused for 6 minutes at each dose. The three vasodilators were separated by 20-minute saline infusions and given in a randomized order.
In the second cohort, (study III) with the early (2- to 4-hour) vascular assessment, as well as in study IV and V), verapamil at 10, 30, and 100 ug/min (endothelium- and NO independent vasodilator that does not release t-PA) was infused at the end of the study protocol.
Blood Flow Measurement
Forearm blood flow (FBF) was measured in infused and non infused arms by venous occlusion plethysmography with mercury in–silicone elastomer strain gauges. The difference in forearm volume results in a linear increase in forearm volume over time, which is proportional to arterial blood inflow (figure 7). Supine heart rate and blood pressure in the non- infused arm were monitored at intervals throughout each study with a semi automated, non-invasive, oscillometric sphygmomanometer.

SUBJECTS AND METHODS
41
Figure 7. Typical blood flow recording from non-infused and infused arms with infusion of intra-brachial bradykinin during venous plethysmography study.
Venous sampling and laboratory assays studies III-V
Forearm Venous Sampling
Venous cannulas (17 gauges) were inserted into large subcutaneous veins of the antecubital fossae of both arms. Blood (10 mL) was withdrawn simultaneously from each arm at baseline and during infusion of each dose of bradykinin and collected into acidified buffered citrate (Stabilyte tubes, Biopool International) for tissue plasminogen activator, t-PA, assays and into citrate (BD Vacutainer) for plasminogen activator inhibitor type 1 (PAI-1) assays.
Sample Preparation Samples were kept on ice before being centrifuged at 2,000g for 30 minutes at 4°C. Platelet-free plasma was decanted and stored at -80°C before assay. Plasma t-PA and PAI-1 antigen concentrations were determined by ELISAs (TintElize t-PA, Biopool EIA; Coaliza PAI-1, Chromogenix AB). Hematocrite was determined by capillary tube centrifugation at baseline and during infusion of bradykinin at 1000 pmol/min. Blood samples were taken immediately before and at 2, 6 and 24 hours after exposure and analyzed for total cells, differential cell counts, and platelets by an auto analyzer.
Fibrinolytic and Inflammatory/Oxidative stress Assays
Plasma interleukin-6 (IL-6), soluble p-selectin, soluble intracellular adhesion molecule-1, (ICAM-1) and tumour necrosis factor-α (TNF- α) were measured with commercially available ELISAs (Quantikine, R&D Systems). Plasma immunoreactive big endothelin (ET)-1 and ET-1 concentrations were measured according to an acetic acid extraction technique by use of a

SUBJECTS AND METHODS
42
modified commercial radioimmunoassay with rabbit anti-human big ET-1 or ET-1 (Peninsula Laboratories Europe). Serum C-reactive protein (CRP) concentrations were measured with an immunonephelometric assay (Behring BN II nephelometer). Plasma nitrite was measured using high performance liquid chromatography based on the methods by Misko TP (130). Total anti-oxidant capacity of plasma was measured in Trolox-equivelents as previously described (131). Briefly, this assay estimates the anti-oxidant capacity of plasma compared to a standard antioxidant Trolox (6–hydroxy–2, 5, 7, 8–tetramethylchroman–2–carboxylic acid). Trolox is a water-soluble derivative of vitamin E with potent antioxidant properties. The Trolox Equivalent Antioxidant Capacity (TEAC) assay is based on the scavenging of the 2,2'-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid)
(ABTS) radical. Loss of the ABTS radical induced by plasma is measured by a sensitive electron paramagnetic resonance technique and related to that induced by 1.0 mmol/L of Trolox standard giving the TEAC value.
Electron paramagnetic resonance (EPR) of diesel particulates
EPR was used to establish radical generation from diesel particulate. Diesel particulate (300μg) was collected on Teflon filters (PTFE Membrane Disc Filter, Pall Corporation, Pall Norden AB, Lund, Sweden) during clinical exposures. To remove particulate matter, filters were vortexed in 2 mL detergent (0.5% Tween20) and particle aggregates broken down by sonication (30 min). 100 μl suspension (particle concentration = 150μg/mL) was withdrawn and incubated with the spin-trap, Tempone-H (1 mmol/L) immediately before the initial measurement. A blank (unexposed) filter was treated in the same way to act as a control. Pyrogallol (100 μmol/L) was used as positive controls to generate superoxide radicals. In some samples superoxide dismutase (SOD; 500 U/mL) was added to scavenge superoxide generated in solution.
Samples were kept at 37oC throughout and measurements were taken at t=1, 20, 40 and 60 min by drawing 50 μl of sample into a capillary tube (Scientific Laboratory Ltd, Coatbridge, UK) and sealing with a plug of soft sealant (Cristaseal, VWR International, Lutterworth, UK). An X-band EPR machine (Magnettech MS-200, Berlin, Germany) was used with the following parameters: microwave frequency, 9.3-9.55 Hz; microwave power, 20 mW;
modulation frequency, 100 kHz; modulation amplitude, 1500 mG; center field, 3365 G; sweep width, 50 G; sweep time, 30 s; number of passes, 1; receiver gain, 1x101. The intensity scale on all graphs is an arbitrary scale base upon the area under the curve of the first derivative traces generated.
All agents were made freshly before experiment and dissolved in Hank’s balanced salt solution (Sigma, Poole, UK) except stock solutions of diesel suspension (dissolved in 0.5% Tween 20) and Tempone-H (dissolved in 0.01 mol/L EDTA). All compounds were purchased from Sigma-Aldrich (Poole, UK) with the exception of Tempone-H (Alexis/Axxora, Nottingham, UK).
Plethysmographic Data Analysis
Plethysmographic data were extracted from the Chart™ data files and forearm blood flows were calculated for individual venous occlusion cuff inflations by use of a template spreadsheet (Excel 2002; Microsoft Corporation, USA). Recordings from the first 60 seconds after wrist cuff inflation were not used because of the variability in blood flow that this incurs

SUBJECTS AND METHODS
43
[(124)]. Usually, the last five flow recordings in each 3-minute measurement period were calculated and averaged for each arm.
Reproducibility of Plethysmographic Data A single operator undertook analysis of all data collected during the forearm plethysmography study. Forearm blood flow responses are reported as absolute blood flow responses (mL/100 mL tissue/min) in the infused and non-infused arm. Previous work has assessed intra-subject variability and determined the coefficient of variation for forearm blood flow across resting conditions and a range of agonists to be between 24-27% (132)
Electrocardiographic recordings
Electrocardiographic recordings were analysed using the Reynolds Medical Pathfinder Digital 700 Series Analysis System (Delmar Reynolds, United Kingdom). ST-segment deviation was calculated by comparing the ST-segment during each 15-min exercise test with the average ST-segment for the 15-min immediately prior to the start of the exposure. The ST-segment amplitude was determined at the J-point plus 80 ms. The ischemic burden during each exercise test was determined as the product of the change in ST-segment amplitude and the duration of exercise. Leads II, V2, and V5 were selected a priori for ST-segment analysis to reflect separate regions of myocardium. Maximum ST-depression and ischemic burden were determined for these leads individually and as a composite.
Symptoms
In studies I-V, thorough all exposures, both air and diesel exhaust, symptoms were registered using a modified Borg`s scale. All registrations started when the participants just had entered the exposure chamber and thereafter at 30 minutes interval until the end of exposure (133). The modified scale consisted of a questionnaire of the following: headache, dizziness, nausea, tiredness, chest pain, coughing, and difficulty breathing, eye irritation, nose irritation, and unpleasant smell, bad taste in the mouth and throat irritation.
Data analysis and statistics
Study I
Wilcoxon´s nonparametric signed-rank test for paired observations to compare data on cells and soluble fluid phase mediators after air and diesel exhaust exposures. Paired t-test was used to assess lung function data. The percentage of neutrophils in induced sputum following diesel exposure was considered primary endpoint.
A p-value <0.05 was considered significant.

SUBJECTS AND METHODS
44
Study II
Data analysis and statistics were reported performed using a repeated measure ANOVA. Data are presented as Mean ± SEM. Statistical significance was taken at P<0.05.
Study III, IV and V
Plethysmographic data were analyzed as described previously (134). The estimated net release of t-PA antigen was defined as the product of the infused forearm plasma flow (based on the mean hematocrite and the infused FBF) and the concentration difference between the infused ([t-PA]Inf) and non-infused arms ([t-PA]Non-Inf) (134):
FBF x (1-Hct) x ([t-PA]Inf - [t-PA]Non-Inf)
Continuous variables are reported as mean ± SEM. Statistical analyses were performed with GraphPad Prism (Graph Pad Software) by ANOVA with repeated measures and 2-tailed Student t test, where appropriate. The area under the curve was calculated for the estimated net release of t-PA during the forearm study period. Statistical significance was taken at P<0.05.

45
MAIN RESULTS
STUDY I In this study, 12 elderly subjects with mild-moderate COPD (GOLD) fulfilled the study and were exposed to diesel exhaust at a PM10 concentration of 300μg/m3 vs. filtered air during one hour. Within the observation time of 18 hours, no diesel exhaust induced effects were found on lung function or airway inflammation, as determined by inflammatory cells or mediators in induced sputum.
STUDY II In this study, 15 elderly subjects with mild-moderate COPD (GOLD) were exposed to diesel exhaust at a PM10 concentration of 300μg/m3 vs. filtered air during one hour. Blood samples were drawn at before, 6 and 24 hours after the exposures and this study gives no support to the hypothesis that short-term exposure to diesel nanoparticles is associated with systemic inflammation, activation of blood coagulation, endothelial dysfunction or lung epithelial injury in at-risk individuals with COPD.
STUDY III In this double-blind, randomized, cross-over study, 30 healthy men were exposed to diluted diesel exhaust (300 μg/m3 particulate concentration) or air for 1 hour during intermittent exercise. Bilateral forearm blood flow and inflammatory factors were measured before and during unilateral intrabrachial infusion of vasodilators. bradykinin, acetylcholine, sodium nitroprusside and verapamil infusions 2 and 6 hours after exposure. There were no differences in resting forearm blood flow or inflammatory markers after exposure to diesel exhaust or air. Although there was a dose-dependent increase in blood flow with each vasodilator (P<0.0001 for all), this response was attenuated with bradykinin (P<0.05), acetylcholine (P<0.05), and sodium nitroprusside (P<0.001) infusions 2 hours after exposure to diesel exhaust, which persisted at 6 hours. Bradykinin caused a dose-dependent increase in plasma t-PA concentrations and net t-PA release in the infused arm (P<0.001 for both) that was suppressed (P<0.05, area under the curve decreased by 35%) following exposure to diesel exhaust.
STUDY IV In this study, fifteen healthy men were exposed to diesel exhaust (particulate concentration, 300 µg/m3) or filtered air for one hour in a double-blind randomised crossover study. Twenty-four hours following exposure, bilateral forearm blood flow, inflammation and fibrinolytic markers were measured before and during unilateral intra-brachial bradykinin, acetylcholine, sodium nitro-prusside and verapamil infusions.
Resting forearm blood flow, blood pressure and systemic inflammatory markers were similar 24 hours following either exposure. Following exposure to diesel exhaust, forearm vasodilatation (P<0.001 for all) was reduced with acetylcholine (P=0.01) and appeared to be attenuated with bradykinin (P=0.08). In contrast, there were no differences in either endothelial-independent vasodilatation or acute plasma tissue plasminogen activator (t-PA) release.

MAIN RESULTS
46
STUDY V In this double blind randomized cross-over study, 20 men with prior myocardial infarction were exposed to dilute diesel exhaust (300 µg/m3) or filtered air during periods of rest and mild exercise in a controlled exposure facility. During the exposure, myocardial ischemia was quantified by ST-segment analysis using continuous 12-lead electrocardiography. Six hours following exposure, vascular vasomotor and fibrinolytic function was assessed by means of intra-arterial agonist infusions.
During both exposures, heart rate increased with exercise (P<0.001 for both) to a similar extent (P=ns; diesel exhaust versus filtered air). Exercise induced ST-segment depression was present in all patients but there was a greater increase in ischemic burden during exposure to diesel exhaust (-22±4 versus -8±6mVs, P<0.001). Exposure to diesel exhaust did not aggravate pre-existing vasomotor dysfunction, but did reduce acute endothelial tissue plasminogen activator release (P<0.05; area under the curve decreased by 35%). See figure 8 next page.
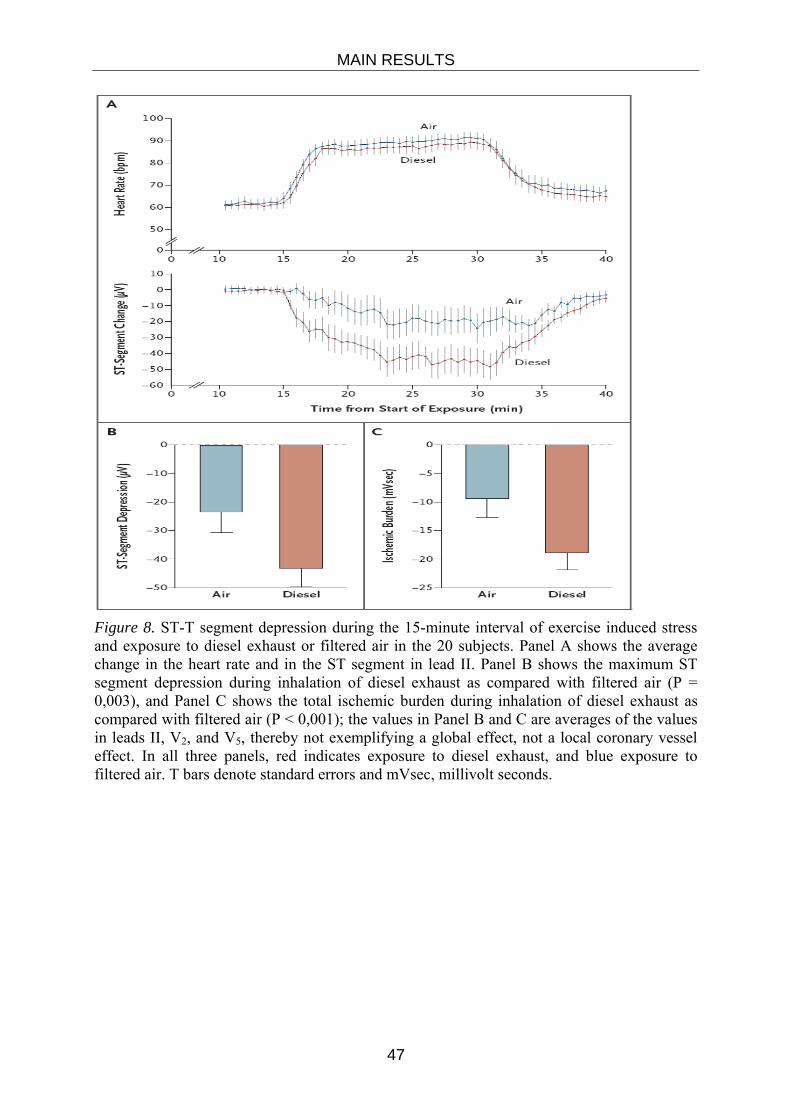
MAIN RESULTS
47
Figure 8. ST-T segment depression during the 15-minute interval of exercise induced stress and exposure to diesel exhaust or filtered air in the 20 subjects. Panel A shows the average change in the heart rate and in the ST segment in lead II. Panel B shows the maximum ST segment depression during inhalation of diesel exhaust as compared with filtered air (P = 0,003), and Panel C shows the total ischemic burden during inhalation of diesel exhaust as compared with filtered air (P < 0,001); the values in Panel B and C are averages of the values in leads II, V2, and V5, thereby not exemplifying a global effect, not a local coronary vessel effect. In all three panels, red indicates exposure to diesel exhaust, and blue exposure to filtered air. T bars denote standard errors and mVsec, millivolt seconds.

48

49
DISCUSSION
STUDIES I AND II Particulate matter air pollution is well known to be associated with increased morbidity and mortality in respiratory and cardiovascular conditions (8, 46, 135, 136).
In a series of experimental diesel engine exhaust studies we have characterised the airway inflammatory and bronchoconstrictive effects in healthy and asthmatic subjects (109, 120, 137). Since COPD patients also have been indicated to be a susceptible group to PM air pollution (138-140), it was of importance to address the effects of DE in this population. Another sensitive group is people with pre-existing cardiovascular disease due to their tendency to develop myocardial infarctions, stroke and even death following exposure to peaks in ambient PM pollution, as well as exposure in traffic situations (46, 52). Consequently, we set out also to investigate diesel exhaust effects in a group of subjects with proven coronary disease.
Diesel exhaust effects in COPD patients COPD is recognised as an airway inflammatory disease with a systemic involvement, including a long range of co-morbidities substantially contributing to the morbidity and burden of this common condition in today’s society (39). The systemic inflammation in COPD subjects is reflected by elevated levels of blood markers such as IL-6, TNF-α and CRP, which in some studies have been associated with disease severity (38, 141). These markers represent cardiovascular risk markers and may form a link to the common engagement of these organ systems in COPD.
The subjects in the current study represented a selected and relatively homogenous group of individuals with moderate COPD (FEV1/FVC<70%, 50 %< FEV1<80% predicted) according to GOLD. Prior to the study, they had undergone thorough medical examination in order to minimize the risk of co-morbidities such as ischemic coronary heart disease, arrhythmias and diabetes mellitus, which would act as an increased risk to the subjects and, furthermore, make the results difficult to interpret. No one was treated with corticosteroids (oral or inhaled) within 8 weeks prior to or during the study. They did not have chronic bronchitis, or a history of frequent exacerbations. This group of COPD patients had poor spirometric reversibility following administration of inhaled beta-2-agonists.
As this was the first DE-exposure study in COPD patients, we examined the airways with a non-invasive method. Sputum induction was performed in order to determine if exposure to diesel exhaust would induce an enhanced airway inflammation
Discussion of Results Within the observation time of 24 hours, DE exposure did not cause any deterioration in lung function or any increase in inflammatory cells or mediators in induced sputum. Neither could any signs of systemic inflammation, hyper-coagulability, endothelial dysfunction or lung epithelial integrity dysfunction be found in peripheral blood.

DISCUSSION
50
Lung function
In the present study we were not able to detect any lung function changes is the investigated group of COPD patients. We have previously demonstrated that exposure to DE caused a minor bronchoconstriction shown as an increase in airway resistance but not in FEV1 in healthy (142). Retrospectively, it would have been useful to perform a more detailed investigation of lung function changes in the COPD patients, but limited resources made this impossible. It cannot be excluded that other subgroups of COPD patients could have responded even on parameters such as FEV1 and vital capacity.
Blood samples
In recent years the view of COPD has gradually changed with the systemic components being increasingly in focus. From the time when the COPD study was planned, studies have revealed that IL-6 and TNF-alpha in peripheral blood are associated with severity, exacerbations and cardiovascular morbidity in COPD (38, 141). These markers were not addressed at the time of the study, but instead the downstream classical inflammatory marker CRP was measured using a high sensitivity assay. Nevertheless, CRP was unchanged from a normal baseline of 1-2 µg/ml, which was twice the concentration compared to healthy age-matched controls. However, whilst epidemiological studies have shown an association between high ambient air pollution exposures and increases in CRP (51), this could not be detected here.
We found no changes in the selected parameters of systemic inflammation within the studied time frame. Fibrinogen is a general marker for systemic inflammation as well as contributing to blood viscosity, coagulation and platelet aggregation (143). Increased fibrinogen has been reported in association with air pollution exposure in the classic Seaton study, leading to the systemic inflammation hypothesis related to air pollution exposure (144, 145) and has later been supported in some, but not in other studies (146-148). Increased fibrinogen concentrations have also been found in COPD subjects suffering from acute exacerbations (149). In the present material the fibrinogen concentration was approximately 50% higher in the COPD group in comparison with healthy aged matched controls, but was not further increased within the investigated time frame up to 24 hours similar to what was seen for CRP.
An affected coagulation process could potentially be detected as increased concentrations of D-dimer and Prothrombin fragment 1-2 since they are released into plasma at the end of the coagulation cascade, thus indicating of low grade blood coagulation. To reflect endothelial function we chose to determine vWF. This protein is strongly involved in vascular haemostasis and is increased in the acute coronary syndrome (150) by mediating platelet aggregation and adhesion on the injured vessel. Thus increased levels of vWF would indicate endothelial dysfunction and could be associated to acute events via increased thrombus developments. However, within the investigated time-frame no changes were detected in D-dimer, Prothrombin fragment 1-2 or vWF concentrations, indicating that there were no detectable systemic effects in this group of COPD patients following exposure to DE.
We also measured clara cell protein (CC16) in peripheral blood as determinant of pulmonary epithelial damage in terms of the. This is a pneumoprotein, i.e. a protein in principle only produced in the airways and leakage into the circulation is dependent on the permeability of the bronchial epithelium. Healthy participants in previous studies of ozone exposures have shown increases of CC16 in peripheral blood, indicating damage to the lung epithelial integrity (151). In contrast, no signs of DE-induced impaired lung epithelial integrity were

DISCUSSION
51
found in these COPD patients. This finding is in line with data from healthy subjects, in which no diesel exhaust-induced changes in serum CC16 concentrations were detected (Törnqvist ERS 2007).
Induced sputum
The presently investigated COPD patients showed no evident signs of a chronic airway inflammation and exposure to DE did not induce a neutrophil recruitment to the airways. There were neither any increases in the neutrophil chemoattractant IL-8 nor in neutrophilic activation as indicated by myeloperoxidase secretion found in the induced sputum. No increases in any other inflammatory cell numbers were detected.
Neutrophil counts in induced sputum are usually raised in COPD subjects, as compared to normal subjects (152). Exacerbations commonly increase mucus production as well as the inflammatory cell numbers (153). Inhalation of aged DE particles has previously been shown to increase induced sputum neutrophils in one study in healthy volunteers (67). This was confirmed following exposures of fresh DE in healthy subjects in where a significant increase was found in the presence of sputum PMNs together with increases in the concentration of IL-6 and methyl-histamine, 6 hours following DE exposure (120). However, using an identical study protocol, we have demonstrated a similar lack of DE induced airway inflammatory responses in asthmatics as in the present group of COPD patients (109).
In the present DE exposure study in COPD subjects, we chose induced sputum as a safe and non-invasive means to study airway inflammation, as induced sputum has been shown to be useful when addressing airway inflammation (109, 120, 154, 155). Although we were not able to demonstrate any airway inflammation in this COPD group following DE exposure, it can be argued that another time point for sampling would have given another result might an option to investigate in forthcoming studies. It is also possible that a differed selection of soluble components could have yielded a better sensitivity. A drawback of using induced sputum is the relatively small amount of supernatant available, which requires prioritisation of what inflammatory markers to measure. This was the case despite using commercially available high sensitivity assays requiring small amounts of fluid. Bronchoscopy is another option previously employed in other groups of subjects after DE exposure (69, 137). Bronchoscopy allows for more extensive sampling at different airway levels, but is an invasive procedure and may as such be less applicable in an elderly population.
The studied COPD population was selected for safety purposes as any unnecessary risks should not be taken in this first diesel exhaust study in COPD patients. It is quite likely that other groups of COPD patients could be more sensitive to diesel exhaust. Therefore we are not able to draw any general conclusion from this COPD population.
COPD patients with more extensive sputum production, more frequent exacerbations, and more rapid decline in lung function as well as those with co-morbidities could well be at higher risk of worsening in association with exposure to higher levels of PM pollution (156). Furthermore, in order to restrict confounding factors, smokers and patients treated with inhaled corticosteroids were not included in this study, but may be important groups to investigate at a later stage. COPD patients with eosinophilic airway inflammation have also attracted special interest as regards to pathophysiological mechanisms, disease progression as well as response to medication (157).
Exacerbations in COPD are often associated with increased mucus production and subsequent bacterial infection. It is interesting that Pourazar et al have demonstrated an increased
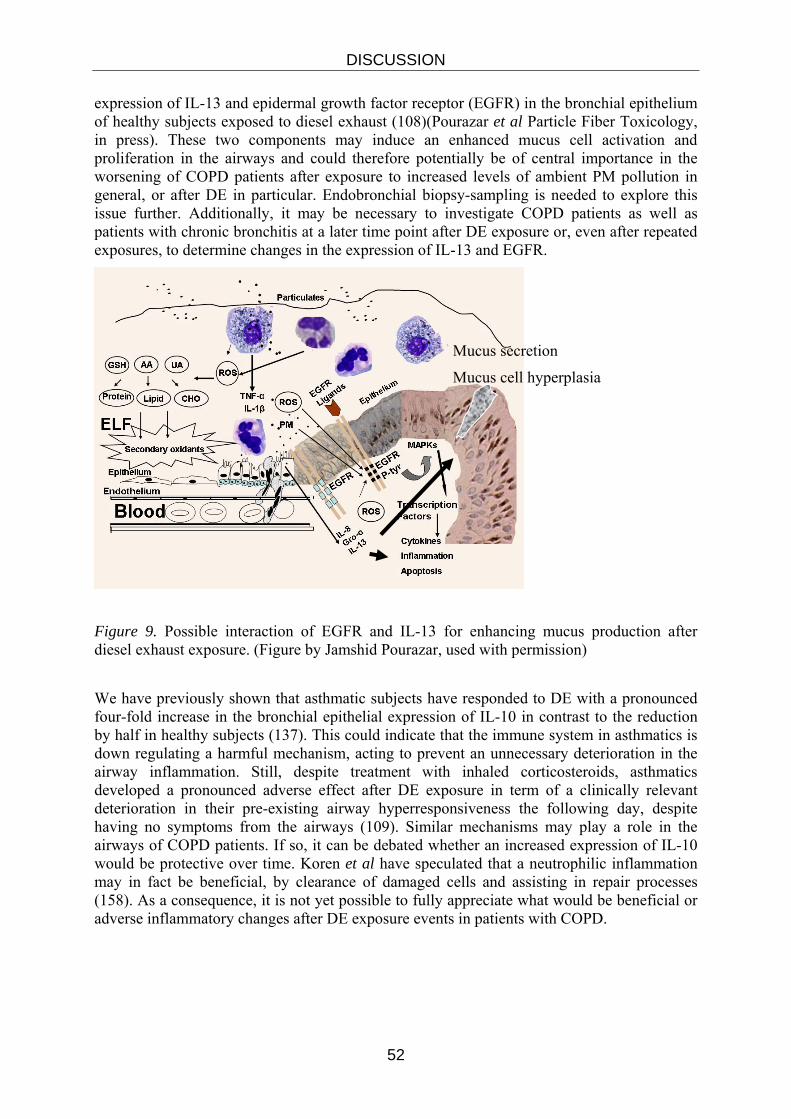
DISCUSSION
52
expression of IL-13 and epidermal growth factor receptor (EGFR) in the bronchial epithelium of healthy subjects exposed to diesel exhaust (108)(Pourazar et al Particle Fiber Toxicology, in press). These two components may induce an enhanced mucus cell activation and proliferation in the airways and could therefore potentially be of central importance in the worsening of COPD patients after exposure to increased levels of ambient PM pollution in general, or after DE in particular. Endobronchial biopsy-sampling is needed to explore this issue further. Additionally, it may be necessary to investigate COPD patients as well as patients with chronic bronchitis at a later time point after DE exposure or, even after repeated exposures, to determine changes in the expression of IL-13 and EGFR.
Figure 9. Possible interaction of EGFR and IL-13 for enhancing mucus production after diesel exhaust exposure. (Figure by Jamshid Pourazar, used with permission)
We have previously shown that asthmatic subjects have responded to DE with a pronounced four-fold increase in the bronchial epithelial expression of IL-10 in contrast to the reduction by half in healthy subjects (137). This could indicate that the immune system in asthmatics is down regulating a harmful mechanism, acting to prevent an unnecessary deterioration in the airway inflammation. Still, despite treatment with inhaled corticosteroids, asthmatics developed a pronounced adverse effect after DE exposure in term of a clinically relevant deterioration in their pre-existing airway hyperresponsiveness the following day, despite having no symptoms from the airways (109). Similar mechanisms may play a role in the airways of COPD patients. If so, it can be debated whether an increased expression of IL-10 would be protective over time. Koren et al have speculated that a neutrophilic inflammation may in fact be beneficial, by clearance of damaged cells and assisting in repair processes (158). As a consequence, it is not yet possible to fully appreciate what would be beneficial or adverse inflammatory changes after DE exposure events in patients with COPD.
Mucus secretion
Mucus cell hyperplasia

DISCUSSION
53
Conclusions from diesel exhaust exposures in COPD The results indicate that inhalation of diesel exhaust as described in studies I and II, does not induce systemic inflammation, hyper-coagulability, endothelial dysfunction or signs of damaged airway epithelium integrity as reflected in peripheral blood samples from subjects with stable and moderate COPD. Neither was any acute lung function decline nor acute airway inflammation detected in induced sputum.
STUDIES III-V
Diesel exhaust effects in Cardiovascular patients Despite improved air quality during the last 50 years, there are still negative health effects (8, 53) below the present levels of air pollution (159). Individuals with established cardiovascular disease seem to be more vulnerable to these effects (17). However, the underlying detailed mechanisms of the air pollution-associated adverse cardiovascular health effects need to be clarified.
Using a well validated and powerful design we have combined two complementary experimental set ups in order to address important mechanisms that were considered to play a role in explaining the associations between PM air pollution and cardiovascular disease. We demonstrated that DE PM air pollution negatively affected two important and complementary cardiovascular risk factors. In young healthy subjects DE exposure reduced vasomotor response and impaired endogenous fibrinolysis, along with signs of a mild systemic inflammation. Furthermore, in patients with stable coronary heart disease a three-fold increase in ST-T segment depression was seen following DE exposure as compared to air. This could be caused of a systemic vascular oxidative stress indicating an early induction and upregulation of anti oxidative defence mechanisms as previously shown in toxicological studies (Baeza-Squiban, Bonvallot et al. 1999; Nel, Diaz-Sanchez et al. 2001; Donaldson, Stone et al. 2003; Xia, Kovochich et al. 2007).
Vasomotor and fibrinolytic function
Observational as well as epidemiological and clinical studies propose associations between exposure to air pollution and worsening of angina symptoms, (160) exacerbation of induced myocardial ischemia, (161) as well as triggering acute myocardial infarction (162). Many of these effects are suggested to be mediated through direct effects on the vasculature.
Of importance to present studies was the timing between traffic exposure and increased incidence of myocardial infarction as demonstrated by Peters et al. They demonstrated that exposure to traffic, e.g. inside a car or a bus or cycling at a road side, was associated with increased myocardial infarctions with peak incidences at 1-2, 6-7, 16-17 and more than 24 hours after exposure (52) (figure 10). The time points for vascular assessments in studies III-V were therefore chosen in order to mirror the effects described in this “real-life” study.
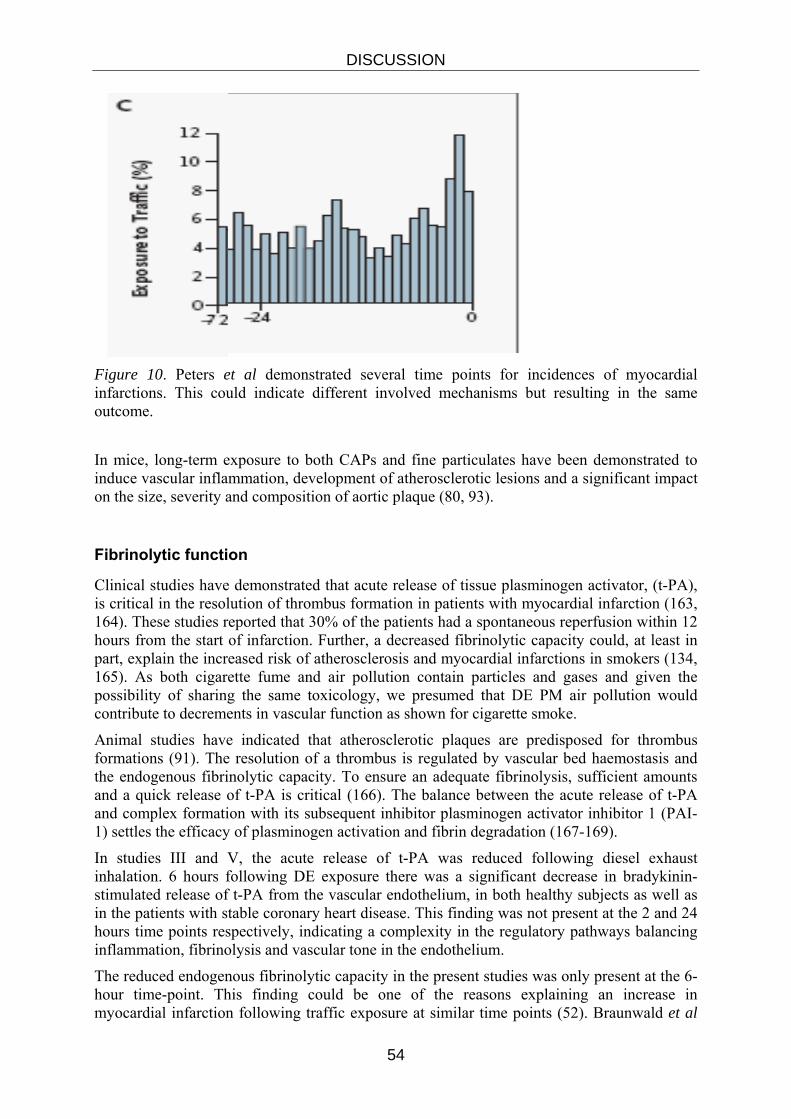
DISCUSSION
54
Figure 10. Peters et al demonstrated several time points for incidences of myocardial infarctions. This could indicate different involved mechanisms but resulting in the same outcome.
In mice, long-term exposure to both CAPs and fine particulates have been demonstrated to induce vascular inflammation, development of atherosclerotic lesions and a significant impact on the size, severity and composition of aortic plaque (80, 93).
Fibrinolytic function
Clinical studies have demonstrated that acute release of tissue plasminogen activator, (t-PA), is critical in the resolution of thrombus formation in patients with myocardial infarction (163, 164). These studies reported that 30% of the patients had a spontaneous reperfusion within 12 hours from the start of infarction. Further, a decreased fibrinolytic capacity could, at least in part, explain the increased risk of atherosclerosis and myocardial infarctions in smokers (134, 165). As both cigarette fume and air pollution contain particles and gases and given the possibility of sharing the same toxicology, we presumed that DE PM air pollution would contribute to decrements in vascular function as shown for cigarette smoke.
Animal studies have indicated that atherosclerotic plaques are predisposed for thrombus formations (91). The resolution of a thrombus is regulated by vascular bed haemostasis and the endogenous fibrinolytic capacity. To ensure an adequate fibrinolysis, sufficient amounts and a quick release of t-PA is critical (166). The balance between the acute release of t-PA and complex formation with its subsequent inhibitor plasminogen activator inhibitor 1 (PAI-1) settles the efficacy of plasminogen activation and fibrin degradation (167-169).
In studies III and V, the acute release of t-PA was reduced following diesel exhaust inhalation. 6 hours following DE exposure there was a significant decrease in bradykinin-stimulated release of t-PA from the vascular endothelium, in both healthy subjects as well as in the patients with stable coronary heart disease. This finding was not present at the 2 and 24 hours time points respectively, indicating a complexity in the regulatory pathways balancing inflammation, fibrinolysis and vascular tone in the endothelium.
The reduced endogenous fibrinolytic capacity in the present studies was only present at the 6-hour time-point. This finding could be one of the reasons explaining an increase in myocardial infarction following traffic exposure at similar time points (52). Braunwald et al

DISCUSSION
55
demonstrated a time-dependent pattern of t-PA release during myocardial infarctions (163). The different time points of increases in myocardial infarctions following traffic exposure, as shown by Peters et al, indicate different mechanisms resulting in the same outcome, a myocardial infarction. Interestingly, in study V a reduced endogenous fibrinolysis was demonstrated in patients with “cured” coronary atherosclerosis, despite full secondary prevention, including aspirin, statins and beta blockers. However, based on the present finding, it cannot be excluded that patients with more severe endothelial dysfunction, coronary atherosclerotic disease and no secondary prevention would experience an even more impaired fibrinolytic dysfunction and thus experience an increased risk for acute cardiovascular events.
Vasomotor function
Several experimental animal and human studies have demonstrated changes in vascular tone following air pollution challenges. In a rat model, Sun et al showed that long-term exposure to low concentration of PM2.5 altered vasomotor tone and demonstrated the development of endothelial dysfunction with increased atherosclerotic burden (93). One important measurement of endothelial dysfunction is reduced vasomotor function, which was demonstrated in the brachial artery by Brooks et al. They showed an acute impairment of vasodilatation following a combined air pollution exposure of Caps and ozone (100). Both studies reported air pollution-induced enhancement of vasoconstriction and reduced endothelium-dependent vasodilatation. Vasoconstriction or inability of vasodilatation is a clear risk factor of ischemia in periods of an increased blood supply demand and could be of major importance if this occurs in the coronary circulation. During physical or mental activity, there is an enhanced demand of oxygen supply to the heart. If this cannot be satisfactorily achieved, a myocardial infarction may occur. Impaired endothelium dependent and independent vasomotor function in the forearm vascular bed has been associated with an increased risk of acute cardiovascular events (170, 171).

DISCUSSION
56
Figure 11. Hypothesis of diesel exhaust-induced effects on endothelial function. Both vascular endothelial dependent and independent vasomotor functions were deteriorated following DEPs exposures indicating decreased NO-bioavailability caused by an oxidative stress (Adapted from Mills et al 2007) The vasodilators acetylcholine (Ach) and bradykinin (BK) are endothelial dependent as they both need pre-existing nitrogen monoxide synthase (eNOS) to exert their vasodilatation effect. Acetylcholine and bradykinin act on membrane bound G-protein coupled receptors on the vascular endothelium stimulating calcium influx and NO-synthetase activation, which increases the NO concentrations in the adjacent smooth muscle cells, thereby dilating the vessels and increasing blood flow. Sodium-nitroprusside (SNP) is a nitro-glycerine derivate, donating NO directly to the smooth muscle, and thereby not dependent of any pre-existing NOS in the endothelial layer. Verapamil (VP) is both endothelial independent and NO-independent in blocking calcium channels, thereby relaxing smooth muscle directly. Due to their physical (nanometer diameter) and chemical properties (hydrocarbons and transitional metals), diesel exhaust particles (DEP) are highly oxidative. DEPs may penetrate into the smooth muscle, thereby inducing oxidative stress. As a consequence, different reactive oxygen species, such as superoxide radical (O2-.) and peroxynitrit (OONO.) are produced. Peroxynitrit inhibits the activity of second messengers in smooth muscle cells, e.g. soluble guanylyl cyclase (172) and cGMP-dependent protein kinase (173) deteriorating both endothelial dependent and endothelial independent NO mediated vasodilatation. Impaired endothelium dependent and independent vasomotor function in the forearm vascular bed has been associated with an increased risk of acute cardiovascular events (170, 171). In the present studies, we have therefore employed the golden standard method of forearm plethysmography in order to address both endothelial dependent and endothelial independent vasodilatation (124, 127).
Both endothelial dependent and independent vasomotor dysfunction was demonstrated in young and healthy individuals. Following DE exposure, an acute impairment in vasomotor function occurred as early as 2 hours after the exposure with a persistent pattern at 6 hours. At 24 hours after DE exposure only a selectively impaired endothelial-dependent vasodilatation was present. (There was a significant decrease in vasodilatation function of acetylcholine following diesel exhaust exposure, but only borderline significance for the vasodilator bradykinin). In contrast, the endothelium independent nitric oxide (NO)-donor sodium-nitroprusside and the calcium blocker verapamil did not induce a vasodilatation at that later time-point. All vasodilators used in the present studies, except for verapamil, are exerting

DISCUSSION
57
their effects via NO pathways. Acetylcholine and bradykinin release NO synthase within the endothelial wall, thereby inducing the increase of NO into the smooth vascular wall which relaxes and subsequently dilates the vessel. Sodium-nitroprusside is independent of the endothelial NO supply since it donates NO directly into the smooth muscles. Verapamil is acting directly on smooth muscle calcium receptors without any effects on NO-related pathways. Taken together, the results from studies III and IV, indicate a diesel exhaust-induced reduction in NO-bioavailability as responsible for the vasomotor dysfunction, at least in part. DE is believed to induce oxidative stress in the vasculature which consumes NO and decrease vascular wall smooth muscle relaxation, something of special importance in physical activity.
Diesel exhaust particles cause oxidative stress through their physical as well as their chemical properties (19). The amount of NO within the vascular wall is thought to be consumed following oxidative stress and inflammation (84). Oxidative stress occur when the balance between antioxidants and oxidants is disturbed, thereby forming reactive oxidant species (ROS), e.g. superoxide radical (O2-.) and peroxynitrite (OONO.). One hypothesis that has been put forward is that superoxide radicals can bind to NO forming peroxynitrite and thus reducing the bioavailability of NO and as a consequence affect vascular tone.
A recent hypothesis when addressing the mechanisms behind the impaired vasomotor function following diesel exhaust exposure is the role of endothelin-1 (ET-1). ET-1 is a very potent vasoconstrictor, mainly released by the vascular endothelium in response to stress. Following a controlled exposure to diesel exhaust (particulate concentration 200 µg/m3), plasma ET-1 concentrations have been shown to be significantly elevated at 3 hours (Peretz et al ATS 2007). The local release of endothelin-1 therefore offers a plausible mechanism for the impairment of vascular tone seen after diesel exhaust exposure. This may be even more important in those with underlying cardiovascular disease. In contrast, in study III we were not been able to demonstrate any increased ET-1 or Big ET-1 (precursor of ET-1). Neither was systemic blood pressure increased after diesel exposure, which would have been expected if endothelin had been of significant importance. However, further studies regarding the relevance of endothelin-1 in the diesel exhaust-induced vasomotor dysfunction are warranted.
Oxidative stress
It has been suggested that diesel exhaust particles (DEP) exert their systemic effects indirectly through pulmonary-derived inflammatory-generated cytokines or directly through the possible translocation into the systemic circulation (69, 93). In previous DE studies on airway responses we have demonstrated that respiratory inflammation is mediated by enhanced cytokine activity suggested to be regulated through the augmentation of redox sensitive transcription factor activity associated with increased MAPkinase activation (108)(Pourazar et al Particle Fiber Toxicology, in press). It has been postulated that DEP-associated oxidative stress is mediated by transitional metals, poly-aromatic hydrocarbons (PAHs) including quinones (174, 175) ROS may be generated by DE and cause respiratory cell necrosis and increased release of cytokines. Moreover, studies on bronchial endothelial cells following PM instillation of diesel particles containing quinones, demonstrate damage to mitochondria by oxidative stress within the “energy-factories of the cells” (176). If this may occur in the peripheral vasculature remains to be explored. Phenanthraquinones belong to the great hydrocarbon family and travels with other compounds on the DEP surface. Studies have suggested phenanthraquinones to be involved in DEP-induced oxidative stress in the vasculature and might in part explain the impairment of vasodilatation seen after diesel exhaust exposure, by reducing endothelial nitric oxide synthase (eNOS) activity

DISCUSSION
58
(177).Furthermore, an impaired vasodilatation may also be due to phenanthraquinones interference with oxidative stress-mediated signal transduction and capability to interact with NADPH-cytochrome-P450-reductase, leading to an overproduction of ROS (178).
Study IV demonstrated that diesel exhaust particles had oxidative properties already before any action in human tissue. Electron paramagnetic resonance (EPR) is a method that in this study was used to establish radical generation from diesel particulates.
When incubating DEP with superoxide dismutase (SOD) the antioxidant defence pattern was reduced, indicating an involvement of SOD in the defence reactions against oxidative stress. Moreover, at 24 hour after DE exposure there was an increase in Trolox Equivalent Antioxidant Capacity (TEAC) which is the total antioxidative capacity (131).
In the studies in this thesis we have demonstrated that the particles generated by a diesel engine produce oxidative particles. We have also reported a time-related pattern of both vasomotor dysfunction and fibrinolytic impairment. In the time-kinetic study (study IV), we showed mild systemic inflammation 24 hour after diesel exposure. Given these results, there are probably several different mechanisms involved in the development of important air pollution-induced cardiovascular events such as myocardial infarction. These include two important and complementary risk factors of vascular endothelial dysfunction; decreased vasomotor response and impaired endogenous fibrinolytic capacity. The early findings of deteriorated endothelial dysfunction could represent signs of an acute oxidative stress on the vascular endothelium, most probably induced by a direct systemic particulate matter effect. However, it needs to be confirmed whether this response is due to a translocation of the nanoparticulate components of the diesel exhaust PM or its chemical properties. The sustained vasomotor response, still apparent at 24 hours, was present along with signs of a mild systemic inflammatory response as well as an upregulated antioxidant defence. Therefore, it cannot be excluded that a systemic vascular inflammation induces the activation of inflammatory cells and, hence, an endogenous production of ROS, which in turn activates the antioxidant defences.
Systemic inflammation
We have previously demonstrated both pulmonary and systemic inflammation in young, healthy volunteers (69). In study IV, we could show a mild systemic inflammation following DE exposure noted as significant increases in TNF-α, soluble-p-selectin and IL-6 in peripheral blood at 24 hours. These are pro-inflammatory markers/cytokines involved in establishing systemic inflammatory cell recruitment. In contrast, no increase was found in the levels of ultra sensitive-CRP at any time point after the DE exposure.
In patients with cardiovascular disease (study V), no signs of a DE-induced increase in inflammatory blood cell counts, cytokines or in CRP were found. This could at least in part be explained by their secondary preventive medication, i.e. statins and aspirin, which both have been suggested to have anti-inflammatory properties. Furthermore, it has been implied that statins are capable of reducing systemic inflammation in patients with acute and chronic coronary syndromes (179). Therefore, it cannot be excluded that treatment with statins and aspirin are of importance in explaining the absence of signs of an enhanced systemic inflammation. This lack of a systemic response was found despite demonstrating impaired fibrinolytic capacity, an important risk factor of acute cardiovascular events in this patient group. However, it cannot be excluded that patients with more severe atherosclerosis and/or without secondary medical prevention would have experienced a measurable systemic inflammatory response after diesel exhaust exposure.

DISCUSSION
59
Autonomic function
In the Framingham study, heart rate variability (HRV) has been suggested to be associated with an increased risk of cardiovascular morbidity and mortality in healthy individuals (58). HRV is relevant in air pollution studies, as air pollution episodes have been associated with cardiac arrhythmias, a well known risk factor of sudden death. Peters et al found an association between fine particulates and episodes of defibrillations in patients with implantable cardiac defibrillators (ICD) (61). Dockery et al conducted a similar study but reported a lag time from time of exposure to defibrillation of nearly three days (62). It has also been demonstrated that increased levels of PM2.5 were associated with lower cardiac autonomic control, suggesting a possible mechanistic link between PM and cardiovascular disease mortality (60, 98). A plausible causative mechanism between air pollution and dysfunction of cardiovascular autonomic function is decreased vagal tone, causing increased resting heart rate (180). Moreover, systemic blood pressure has been measured and discussed in terms of autonomic function. Urch et al demonstrated an immediate increase in blood pressure as well as arterial vasoconstriction following a combined air pollution exposure to ozone and CAPs (181).
In the present studies, no significant changes were found in pulse rate or systemic blood pressure after DE exposure compared to filtered air. Neither were there any changes in HRV or signs of arrhythmia present. However, when the men with a stable coronary heart disease (study V) started their first mild physical exercise during DE exposure, an immediate increase in ST-T depression was shown as compared to during air exposure. The three-fold, but non-symptomatic increase in exercise induced ST-T segment depression was apparent despite no significant changes in heart rate or blood pressure. See figure 12 next page.
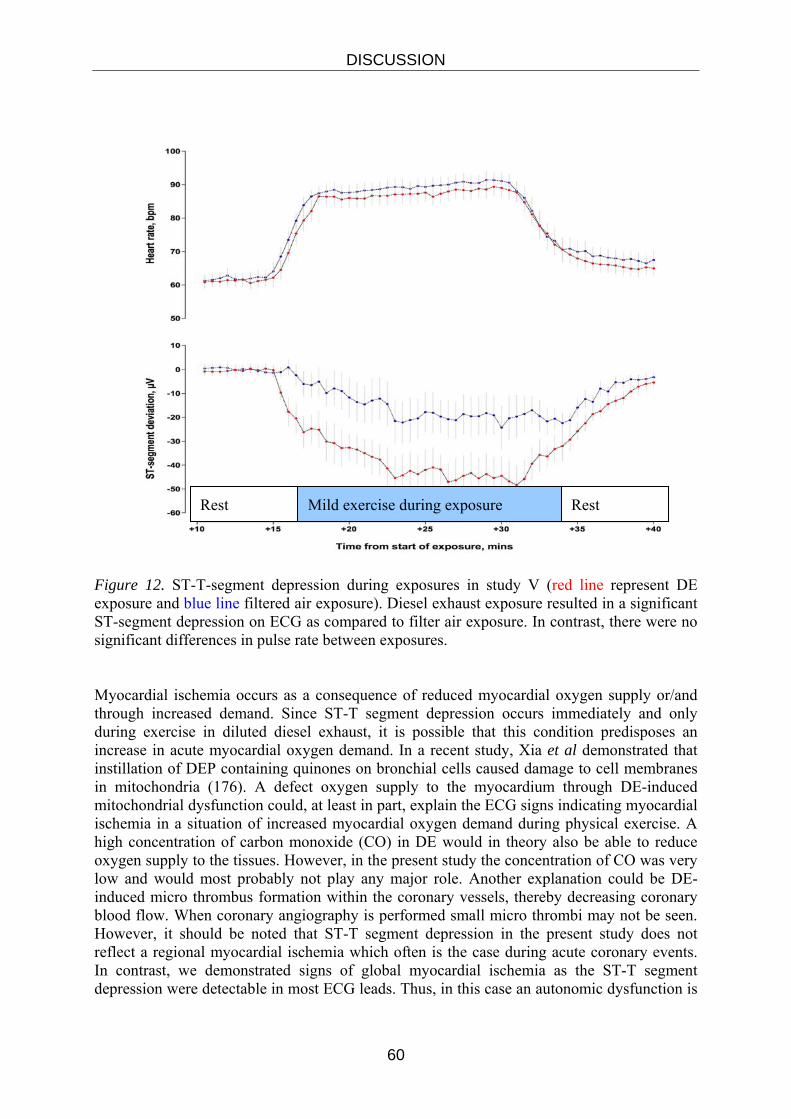
DISCUSSION
60
Figure 12. ST-T-segment depression during exposures in study V (red line represent DE exposure and blue line filtered air exposure). Diesel exhaust exposure resulted in a significant ST-segment depression on ECG as compared to filter air exposure. In contrast, there were no significant differences in pulse rate between exposures. Myocardial ischemia occurs as a consequence of reduced myocardial oxygen supply or/and through increased demand. Since ST-T segment depression occurs immediately and only during exercise in diluted diesel exhaust, it is possible that this condition predisposes an increase in acute myocardial oxygen demand. In a recent study, Xia et al demonstrated that instillation of DEP containing quinones on bronchial cells caused damage to cell membranes in mitochondria (176). A defect oxygen supply to the myocardium through DE-induced mitochondrial dysfunction could, at least in part, explain the ECG signs indicating myocardial ischemia in a situation of increased myocardial oxygen demand during physical exercise. A high concentration of carbon monoxide (CO) in DE would in theory also be able to reduce oxygen supply to the tissues. However, in the present study the concentration of CO was very low and would most probably not play any major role. Another explanation could be DE-induced micro thrombus formation within the coronary vessels, thereby decreasing coronary blood flow. When coronary angiography is performed small micro thrombi may not be seen. However, it should be noted that ST-T segment depression in the present study does not reflect a regional myocardial ischemia which often is the case during acute coronary events. In contrast, we demonstrated signs of global myocardial ischemia as the ST-T segment depression were detectable in most ECG leads. Thus, in this case an autonomic dysfunction is
Rest Mild exercise during exposure Rest

DISCUSSION
61
less probable, as no pulse rate or blood pressure differences were detected between the two different exposures. The finding of an increased ST-T segment depression was found in a selected patient cohort of elderly men with a stable coronary heart disease. This should be taken into consideration when interpreting the present results and comparing to the real life situation. However, large epidemiological studies demonstrate that women respond in a similar way as men following long-term exposure to ambient fine particulate air pollution (56). In study V, 20 male patients with previous myocardial infarction participated. None of them had clinical symptom of cardiac failure or detectable arrhythmia or signs of myocardial ischemia (> 1mm ST-T depression) at the pre-study exercise test. All had suffered a myocardial infarction in the past and had been investigated with coronary angiography which had demonstrated coronary atherosclerosis and all had eventually been successfully treated with PCI. They were all in a completely stable condition taking all usual medication including aspirin, statins and beta blockers for prevention of secondary cardiac events.
Despite a known coronary heart disease, the patients in study V represented a selected “healthy” patient cohort, not totally representative for “the average man in the street”. Study V is to be considered as a safety study. Therefore, the actual study was designed not to expose the patients to any theoretical risk of an adverse cardiovascular event during the study. If a group of patients with less controlled coronary heart disease, i.e. present symptoms of angina, heart failure, important co-morbidities or without adequate medical treatment would have been investigated, it is likely that the consequences of a DE exposure would have been more pronounced.

62

63
FINAL COMMENTS
The present thesis aimed to explore mechanisms which could explain the worsening of cardiovascular disease and COPD by particulate matter air pollution. We have previously described the airway inflammation cascade from EGFR, kinase and transcription factor activation along with downstream cytokine production and inflammatory cell recruitment after diesel engine exhaust exposure. Asthmatic subjects have been shown to demonstrate both similarities and differences in their response to diesel exhaust as compared with healthy subjects. This could be related to the already established asthmatic airway inflammation. Since it is evident that PM air pollution is related to cardiovascular disease in terms of myocardial infarctions and stroke, it is of major importance to address the mechanistic aspects of the adverse health effects reported in epidemiological studies. The pivotal study by Peters and co-workers in NEJM 2004 showed strong associations between exposure to traffic and myocardial infarction and also revealed a time course with several peaks of increased risk that suggested different underlying mechanisms contributing at various time points. We therefore chose to utilise a number of highly sensitive and well validated methods to address cardiovascular mechanisms known to be associated to increased risk of ischemic heart and cerebrovascular events. This led us to discover that short-term exposure to diluted diesel exhaust adversely affected two important, highly relevant and complementary aspects of vascular function in healthy subjects; the regulation of vascular tone and endogenous fibrinolysis. Interestingly, it was found that the identified vascular events corresponded to the time course of increased risk for myocardial infarctions, reported by Peters’s group. We therefore proceeded with an investigation of heart patients. It was of paramount importance not to take overdue risks when designing such a study and selecting the patient population. Out of a vast number of coronary patients, we included individuals who had received a successful PCI treatment for coronary heart disease and consequently had no coronary stenoses, were asymptomatic and on full preventive medication including beta-blockers, statins and aspirin. This group had a proven coronary heart disease, but was at less risk for an adverse cardiovascular event than the average elderly population. We confirmed that patients with coronary heart disease had an impaired endogenous fibrinolysis, in terms of reduced t-PA release and activity after diesel exhaust, of similar magnitude as in healthy subjects. The reduction in t-PA by approximately 30% would substantially increase the risk for a coronary thombosis due to a progressive high grade coronary stenosis or an atherosclerotic plaque rupture. A considerable proportion of the population has significant coronary stenoses and may therefore be at risk when exposed to diesel exhaust in traffic. The efficacy of the patients’ extensive medication to prevent adverse coronary events

FINAL COMMENTS
64
was not specifically addressed in the present study. Yet, it was evident that beta-blockers, aspirin and statins were unable to abolish the global ST-T segment depression that emerged rapidly during exercise in a diesel exhaust atmosphere, as opposed to filtered air. Despite only a pulse increase to around 90 beats per minute, the ECG changes were present in all individuals with previously proven but PCI-treated coronary disease. A pulse rate at or above this level is probably very common in people in hectic traffic and may therefore represent a risk both in individuals at some level of physical exercise in traffic environment or driving a vehicle. In this thesis, the role of the endothelium in the impaired vascular function is highlighted. The detailed mechanisms of this dysfunction are currently under further exploration, as is the mode whereby inhaled diesel exhaust does not only induce airway inflammatory events but also a systemic response. From the endobronchial biopsy explorative research by Jamshid Pourazar, it is indicated that even the superficial vasculature in the bronchi are affected by diesel exhaust. It is still unclear whether the particles themselves translocate from the lungs to the blood and enter the vascular wall and myocardium. This could occur through particles entering the blood stream or being transported by phagocytic cells. Both mechanisms have been indicated in animal studies, while studies in humans have not yet confirmed transalveolar transport. Another alternative is that inflammatory signals, either cell mediated or via soluble components, could cause the systemic vascular events. It has been suggested that diesel exhaust particles exert their toxic effects both to their size and chemical composition. Nanoparticles appear more toxic than coarse particles and additional chemical components on diesel particles such as PAHs have been indicated to cause disturbances in cell functions, which at least in part are mediated through oxidative stress. Nel and co-workers have indicated quinones to be of particular interest. Their oxidative capacity has been shown to result in mitochondrial dysfunction, which together with microvascular dysfunction could potentially explain some of the observed cardiovascular events. The “COPD epidemic” means a major burden for the individual, the health care system and the society in general. Individuals with COPD have been indicated to be susceptible to particulate matter air pollution. In the first investigation of COPD subjects, we included stable ex-smoking patients without any history of exacerbations or co-morbidities. Due to safety precautions, we also selected non-invasive procedures when addressing potential worsening of the condition as a result the exposures. We were not able to demonstrate any effects suggesting worsening of COPD after diesel exhaust exposure. This does not preclude that COPD patients may be sensitive to diesel exhaust or other PM pollutants, but suggests that other potentially more sensitive subgroups of COPD-patients should be investigated in forthcoming studies. The panel of methods may also be extended to address both respiratory and cardiovascular endpoints In conclusion, this thesis has confirmed the link between diesel exposure and the previously described adverse cardiovascular events related to traffic exposure. It

FINAL COMMENTS
65
also showed that COPD patients may safely be studied after diesel exhaust exposure and suggests complementary sub-groups and techniques to be utilised when addressing this important group.

66

67
CONCLUSIONS It is concluded that:
These exposure studies support the epidemiological evidence of an association between particulate matter pollution and adverse health effects in humans and suggest mechanisms for the air pollution-induced events in the cardiovascular system.
Exposure to diesel exhaust at high ambient concentrations did not induce a deterioration of lung function or an increased airway inflammation in individuals with stable and moderately severe COPD.
Diesel exhaust exposure was not associated with systemic inflammation, hypercoagulability, endothelial dysfunction or deteriorated lung epithelial integrity as determined in peripheral blood in the investigated population with moderate and stable COPD.
In healthy subjects, inhalation of diluted diesel exhaust deteriorated two important, highly relevant and complementary aspects of vascular function; the regulation of vascular tone and endogenous fibrinolysis.
Studies of the time kinetics of the vascular and systemic effects of exposure to diesel exhaust indicated a selective and persistent impairment of endothelium-dependent vasodilatation along with a systemic inflammatory response for at least 24 hours after exposure.
Short-term exposure to diluted diesel exhaust increased signs of exercise-induced myocardial ischemia in terms of greater ST-segment depression on ECG along with impaired endogenous fibrinolytic capacity in men with stable coronary heart disease.

68
ACKNOWLEDGEMENTS Writing this thesis is the end of my period as a PhD-student. I have been a PhD-student for many years and it has been one of the busiest times in my life. My wish is to thank everyone that has helped me in any way but it is impossible to name them all. However, I would like to express my sincere gratitude to those who have been closest and contributed the most: Associate Professor Anders Blomberg, my main supervisor in research and roommate at several international conferences. At congresses it has been good to share room with someone who has the same diurnal rhythm. Thank you for being patient and supporting as well as pushy when needed. Thank you for your excellent capacity in forming my thoughts into readable English as well as keeping things together. Professor Thomas Sandström, my co-tutor. Thank you for your enthusiasm and valuable support. Without your exceptional capacity to build bridges and contact-nets worldwide as well as back home, the contents of this thesis would probably have looked totally different. Thank you for all help with writing up this thesis, and discussing dog-matters. Associate Professor Ellinor Ädelroth, who have helped me a lot during writing this thesis as well as and with the COPD manuscript. I am most grateful for the feed-back that you have given me. Thank you for support, fruitful discussions and sharing your great scientific experience. Dr Nick Mills, my friend, who has been so important in introducing new research technique to Umeå, as well as during writing the papers. Thank you for all your efforts and skilled work. It has been a privilege to work with you and to share all the joyful moments in Edinburgh, Umeå as well as in the ski-slopes in Åre and elsewhere. Thank you for everything. Professor David Newby, the cardiovascular oracle. Thank you for providing such an excellent research knowledge and research platform, and your kind support in different phases of this collaborative project between the University of Edinburgh and Umeå. My collaborators at the Department of Medicine, Dr Manuel Gonzalez and Associate Professor Stefan Söderberg. Thank you for excellent collaboration when introducing the forearm plethysmography technique to Umeå. Special thanks to Manuel who shared room with me during periods in Edinburgh when we learnt the plethysmography technique from Nick Mills et al, and for all the hard labour during the vascular studies. It has also been a pleasure to watch nice football games in your home. Real Madrid is quite a decent football team after all. Ken Donaldson, Simon Robinson, Bill McNee, Nick Boon and Kareen Darnley for excellent collaboration and contributions to the development of this research field. Cedric Hermans and Lesley Desmyter for sharing your tremendous expertise in the field of coagulation. My clinical tutor Associate Professor Karl Franklin, thank you for many nice discussions of clinical matter as well as the importance of having meaningful hobbies. You have really meant a lot to me. Thank you for everything. Dr Ragnberth Helleday, Head of the Department of Respiratory Medicine and Allergy, for providing excellent research conditions and for helping me with the layout of this thesis. Dr Eva Norrman my college and Deputy Head of the Department of Respiratory Medicine and Allergy, for always being there, when I needed support. My colleagues and the staff at the clinical division of the Department of Respiratory Medicine and Allergy in Umeå, for all your support.

69
Dr Jenny Bosson, my colleague and always positive research friend who also happily volunteered to be my “toastress” at my dissertation party. Dr Jamshid Pourazar, who is so skilled and always kind and helpful. Thank you for sorting the amounts of desired sampling in these papers as well as taking care of me at the research lab during several summer scholar ships. Research nurses Annika Johansson, Frida Holmström, Helena Bogseth, Margot Johansson and Veronika Sjögren for co-ordinating the studies and for your exceptional capacity in sorting out problems before, during and after the studies. Ann-Britt Lundström, Maj-Cari Ledin, Maria Sehlstedt, Ester Roos-Engstrand for your excellent technical assistance during the studies. Hans, Jocke, Tage and Stefan at Svensk Maskinprovning, SMP, for the brilliant technical support concerning everything around the diesel exhaust setup. Lena Åström, our great administrator who keeps “everything” in order. Thank you also for giving me and my family access to your lovely sport house in Grönfjäll several times. My research-collaborators at the Department of Respiratory Medicine and Allergy in Umeå, Annelie Behndig, Fredrik Valham, Magnus Lundbäck and Stefan Barath for many joyful moments and all memories from national and international meetings. Marie Eriksson for helping me understanding that the “numbers are telling stories”. My parents, Rolf and Ulla Törnqvist who raised me and taught me that nothing is really impossible. My brother Göran and his family, Lena and their sons Carl-Johan and Fredrik. Thank for many great full moments during camping life. My sister Gunilla and her family, Lars and their children Linn and Johan. My sister Görel and her family, Leif-Thomas and their children Johanna and Viktor. My good friend Per and his wife Karin and their children Amanda, Kajsa, Sofia and Andreas. Thank you for being such good friends. All the positive and enthusiastic volunteers, who participated in my projects. Thanks to everybody that I have forgot to mention. Uschigårdens Kenzo, who has taught me so much about dogs. Integrity is not only for two- leg creatures. Joacim and Marcus, my two clever sons, who are my pride and source of happiness. There are not words enough to express the joy you have given and are giving me. Thank you for being you. Anette, my dear, beautiful and beloved wife, who has never stopped believing in me. You are the most supporting and understanding wife a man can ever wish. I love you eternally. The studies in this thesis were generously supported by grants from the Swedish Heart Lung Foundation, the Heart and Lung Associations in Sollefteå, Dorotea, Härnösand and Örnsköldsvik, the British Heart Foundation, the Swedish Research Council for Environment, Agricultural sciences and Spatial planning, Front Research, the County of Västerbotten, Umeå University, Sweden and the Swedish Air pollution programme: the Swedish Emission Research Program).

70

71
REFERENCES R
1. Nemery B, Hoet PH, Nemmar A. The Meuse Valley fog of 1930: an air pollution disaster. Lancet. 2001 Mar 3;357(9257):704-8. 2. Health effects of outdoor air pollution. Committee of the Environmental and Occupational Health Assembly of the American Thoracic Society. Am J Respir Crit Care Med. 1996;153(1):3-50. 3. Bell ML, Davis DL. Reassessment of the lethal London fog of 1952: novel indicators of acute and chronic consequences of acute exposure to air pollution. Environ Health Perspect. 2001 Jun;109 Suppl 3:389-94. 4. Davis DL, Bell ML, Fletcher T. A look back at the London smog of 1952 and the half century since. Environ Health Perspect. 2002 Dec;110(12):A734-5. 5. Pemberton J, Goldberg C. Air pollution and bronchitis. Br Med J. 1954 Sep 4;2(4887):567-70. 6. Lave LB, Seskin EP. Air pollution and human health. Science. 1970 Aug 21;169(947):723-33. 7. Holland WW, Bennett AE, Cameron IR, Florey CV, Leeder SR, Schilling RS, et al. Health effects of particulate pollution: reappraising the evidence. Am J Epidemiol. 1979 Nov;110(5):527-659. 8. Dockery DW, Pope CA, Xu XP, Spengler JD, Ware JH, Fay ME, et al. An association between air-pollution and mortality in 6 united-states cities. New England Journal Of Medicine. 1993;329:1753-9. 9. Pope CA, III, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, et al. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA. 2002;287(9):1132-41. 10. Krzyzanowski M, Vandenberg J, Stieb D. Perspectives on air quality policy issues in Europe and North America. J Toxicol Environ Health A. 2005 Jul 9-23;68(13-14):1057-61. 11. EU CAFECP-EC. Working Group on Particulate Matter. Second Position Paper on Particulate Matter; December 20, 2004. 12. Forsberg B, Hansson HC, Johansson C, Areskoug H, Persson K, Jarvholm B. Comparative health impact assessment of local and regional particulate air pollutants in Scandinavia. Ambio. 2005 Feb;34(1):11-9. 13. Kunzli N, Kaiser R, Medina S, Studnicka M, Chanel O, Filliger P, et al. Public-health impact of outdoor and traffic-related air pollution: a European assessment. Lancet. 2000;356(9232):795-801. 14. Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL. Fine particulate air pollution and mortality in 20 U.S. cities, 1987-1994. N Engl J Med. 2000;343(24):1742-9. 15. Laden F, Neas LM, Dockery DW, Schwartz J. Association of fine particulate matter from different sources with daily mortality in six U.S. cities. Environ Health Perspect. 2000;108(10):941-7.

REFERENCES
72
16. Le Tertre A, Medina S, Samoli E, Forsberg B, Michelozzi P, Boumghar A, et al. Short-term effects of particulate air pollution on cardiovascular diseases in eight European cities. Journal of Epidemiology and Community Health. 2002;56(10):773-9. 17. Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, et al. Air Pollution and Cardiovascular Disease: A Statement for Healthcare Professionals From the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation. 2004;109(21):2655-71. 18. Churg A, Brauer M. Human lung parenchyma retains PM2.5. Am J Respir Crit Care Med. 1997;155(6):2109-11. 19. Donaldson K, Tran L, Jimenez L, Duffin R, Newby D, Mills N, et al. Combustion-derived nanoparticles: A review of their toxicology following inhalation exposure. Particle and Fibre Toxicology. 2005;2(1):10. 20. Brunekreef B, Forsberg B. Epidemiological evidence of effects of coarse airborne particles on health. Eur Respir J. 2005 Aug;26(2):309-18. 21. Sydbom A, Blomberg A, Parnia S, Stenfors N, Sandstrom T, Dahlen SE. Health effects of diesel exhaust emissions. Eur Respir J. 2001;17(4):733-46. 22. Pino P, Walter T, Oyarzun M, Villegas R, Romieu I. Fine particulate matter and wheezing illnesses in the first year of life. Epidemiology. 2004 Nov;15(6):702-8. 23. Lacasana M, Esplugues A, Ballester F. Exposure to ambient air pollution and prenatal and early childhood health effects. Eur J Epidemiol. 2005;20(2):183-99. 24. Huynh M, Woodruff TJ, Parker JD, Schoendorf KC. Relationships between air pollution and preterm birth in California. Paediatr Perinat Epidemiol. 2006 Nov;20(6):454-61. 25. Bateson TF, Schwartz J. Children's response to air pollutants. J Toxicol Environ Health A. 2008 Feb;71(3):238-43. 26. Gauderman WJ, Avol E, Gilliland F, Vora H, Thomas D, Berhane K, et al. The effect of air pollution on lung development from 10 to 18 years of age. N Engl J Med. 2004 Sep 9;351(11):1057-67. 27. Rojas-Martinez R, Perez-Padilla R, Olaiz-Fernandez G, Mendoza-Alvarado L, Moreno-Macias H, Fortoul T, et al. Lung function growth in children with long-term exposure to air pollutants in Mexico City. Am J Respir Crit Care Med. 2007 Aug 15;176(4):377-84. 28. Trenga CA, Sullivan JH, Schildcrout JS, Shepherd KP, Shapiro GG, Liu LJ, et al. Effect of particulate air pollution on lung function in adult and pediatric subjects in a Seattle panel study. Chest. 2006 Jun;129(6):1614-22. 29. Kulkarni N, Pierse N, Rushton L, Grigg J. Carbon in Airway Macrophages and Lung Function in Children. The New England Journal of Medicine. 2006;355(1):21-30. 30. von Klot S, Wolke G, Tuch T, Heinrich J, Dockery DW, Schwartz J, et al. Increased asthma medication use in association with ambient fine and ultrafine particles. Eur Respir J. 2002;20(3):691-702. 31. Halonen JI, Lanki T, Yli-Tuomi T, Kulmala M, Tiittanen P, Pekkanen J. Urban Air Pollution And Asthma And Copd Hospital Emergency Room Visits. Thorax. 2008 Feb 11. 32. Ko FW, Tam W, Wong TW, Lai CK, Wong GW, Leung TF, et al. Effects of air pollution on asthma hospitalization rates in different age groups in Hong Kong. Clin Exp Allergy. 2007 Sep;37(9):1312-9. 33. Chalupa DC, Morrow PE, Oberdorster G, Utell MJ, Frampton MW. Ultrafine particle deposition in subjects with asthma. Environ Health Perspect. 2004 Jun;112(8):879-82.

REFERENCES
73
34. Penard-Morand C, Charpin D, Raherison C, Kopferschmitt C, Caillaud D, Lavaud F, et al. Long-term exposure to background air pollution related to respiratory and allergic health in schoolchildren. Clin Exp Allergy. 2005 Oct;35(10):1279-87. 35. Boezen HM, Vonk JM, van der Zee SC, Gerritsen J, Hoek G, Brunekreef B, et al. Susceptibility to air pollution in elderly males and females. Eur Respir J. 2005 Jun;25(6):1018-24. 36. McCreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, et al. Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med. 2007 Dec 6;357(23):2348-58. 37. Sin DD, Man SF. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003;107(11):1514-9. 38. Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004 Jul;59(7):574-80. 39. Calverley PM, Scott S. Is airway inflammation in chronic obstructive pulmonary disease (COPD) a risk factor for cardiovascular events? Copd. 2006 Dec;3(4):233-42. 40. Atkinson RW, Anderson HR, Sunyer J, Ayres J, Baccini M, Vonk JM, et al. Acute effects of particulate air pollution on respiratory admissions: results from APHEA 2 project. Air Pollution and Health: a European Approach. Am J RespirCrit Care Med. 2001;164(10 Pt 1):1860-6. 41. Schikowski T, Sugiri D, Ranft U, Gehring U, Heinrich J, Wichmann HE, et al. Long-term air pollution exposure and living close to busy roads are associated with COPD in women. Respir Res. 2005;6:152. 42. Hart JE, Laden F, Schenker MB, Garshick E. Chronic obstructive pulmonary disease mortality in diesel-exposed railroad workers. Environ Health Perspect. 2006 Jul;114(7):1013-7. 43. Ulvestad B, Bakke B, Eduard W, Kongerud J, Lund MB. Cumulative exposure to dust causes accelerated decline in lung function in tunnel workers. Occup Environ Med. 2001 Oct;58(10):663-9. 44. Ulvestad B, Bakke B, Melbostad E, Fuglerud P, Kongerud J, Lund MB. Increased risk of obstructive pulmonary disease in tunnel workers. Thorax. 2000 Apr;55(4):277-82. 45. Ulvestad B, Lund MB. [Increased risk of chronic obstructive pulmonary disease among tunnel construction workers]. Tidsskr Nor Laegeforen. 2003 Aug 28;123(16):2292-5. 46. Hoek G, Brunekreef B, Goldbohm S, Fischer P, van den Brandt PA. Association between mortality and indicators of traffic-related air pollution in the Netherlands: a cohort study. The Lancet. 2002;360(9341):1203-9. 47. Rosenlund M, Berglind N, Pershagen G, Hallqvist J, Jonson T, Bellander T. Long-term exposure to urban air pollution and myocardial infarction. Epidemiology. 2006 Jul;17(4):383-90. 48. Brook RD, Brook JR, Rajagopalan S. Air pollution: the "Heart" of the problem. [Review] [55 refs]. Current Hypertension Reports. 2003;5(1):32-9. 49. Routledge HC, Ayres JG, Townend JN. Why cardiologists should be interested in air pollution. Heart. 2003;89(12):1383-8. 50. Peters A, Dockery DW, Muller JE, Mittleman MA. Increased particulate air pollution and the triggering of myocardial infarction. Circulation. 2001;103(23):2810-5.

REFERENCES
74
51. Peters A, Frohlich M, Doring A, Immervoll T, Wichmann HE, Hutchinson WL, et al. Particulate air pollution is associated with an acute phase response in men; results from the MONICA-Augsburg Study. Eur Heart J. 2001;22(14):1198-204. 52. Peters A, von Klot S, Heier M, Trentinaglia I, Hormann A, Wichmann HE, et al. Exposure to Traffic and the Onset of Myocardial Infarction. The New England Journal of Medicine. 2004;351(17):1721-30. 53. Pope CA, III, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, et al. Cardiovascular Mortality and Long-Term Exposure to Particulate Air Pollution: Epidemiological Evidence of General Pathophysiological Pathways of Disease. Circulation. 2004;109(1):71-7. 54. von Klot S, Peters A, Aalto P, Bellander T, Berglind N, D'Ippoliti D, et al. Ambient Air Pollution Is Associated With Increased Risk of Hospital Cardiac Readmissions of Myocardial Infarction Survivors in Five European Cities. Circulation. 2005;112(20):3073-9. 55. Hoek G, Brunekreef B, Fischer P, van Wijnen J. The association between air pollution and heart failure, arrhythmia, embolism, thrombosis, and other cardiovascular causes of death in a time series study. Epidemiology. 2001;12(3):355-7. 56. Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, et al. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med. 2007 Feb 1;356(5):447-58. 57. Kunzli N, Jerrett M, Mack WJ, Beckerman B, LaBree L, Gilliland F, et al. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect. 2005 Feb;113(2):201-6. 58. Tsuji H, Larson MG, Venditti FJ, Jr., Manders ES, Evans JC, Feldman CL, et al. Impact of reduced heart rate variability on risk for cardiac events. The Framingham Heart Study. Circulation. 1996 Dec 1;94(11):2850-5. 59. Kleiger RE, Miller JP, Bigger JT, Jr., Moss AJ. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol. 1987 Feb 1;59(4):256-62. 60. Liao D, Creason J, Shy C, Williams R, Watts R, Zweidinger R. Daily variation of particulate air pollution and poor cardiac autonomic control in the elderly. Environmental Health Perspectives. 1999;107(7):521-5. 61. Peters A, Liu E, Verrier RL, Schwartz J, Gold DR, Mittleman M, et al. Air pollution and incidence of cardiac arrhythmia [see comments]. Epidemiology. 2000;11(1):11-7. 62. Dockery DW, Luttmann-Gibson H, Rich DQ, Link MS, Mittleman MA, Gold DR, et al. Association of air pollution with increased incidence of ventricular tachyarrhythmias recorded by implanted cardioverter defibrillators. Environ Health Perspect. 2005;113(6):670-4. 63. Auchincloss AH, Roux AV, Dvonch JT, Brown PL, Barr RG, Daviglus ML, et al. Associations between Recent Exposure to Ambient Fine Particulate Matter and Blood Pressure in the Multi-Ethnic Study of Atherosclerosis (MESA). Environ Health Perspect. 2008 Apr;116(4):486-91. 64. Godlee F. Air pollution: II--Road traffic and modern industry. BMJ. 1991;303(6816):1539-43. 65. Salvi SUND, Blomberg ANDE, Rudell BERT, Kelly FRAN, Sandstrom THOM, Holgate STEP, et al. Acute Inflammatory Responses in the Airways and Peripheral Blood After Short-Term Exposure to Diesel Exhaust in Healthy Human Volunteers. American Journal of Respiratory and Critical Care Medicine. 1999;159(3):702-9.

REFERENCES
75
66. Rudell B, Sandstrom T, Hammarstrom U, Ledin ML, Horstedt P, Stjernberg N. Evaluation of an exposure setup for studying effects of diesel exhaust in humans. Int Arch Occup Environ Health. 1994;66(2):77-83. 67. Nightingale JA, Maggs RICH, Cullinan PAUL, Donnelly LE, Rogers DF, Kinnersley ROBE, et al. Airway Inflammation after Controlled Exposure to Diesel Exhaust Particulates. American Journal of Respiratory and Critical Care Medicine. 2000;162(1):161-6. 68. Blomberg A, Sainsbury C, Rudell B, Frew AJ, Holgate ST, Sandstrom T, et al. Nasal cavity lining fluid ascorbic acid concentration increases in healthy human volunteers following short term exposure to diesel exhaust. Free Radic Res. 1998;28(1):59-67. 69. Salvi S, Blomberg A, Rudell B, Kelly F, Sandstrom T, Holgate ST, et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159(3):702-9. 70. Ghio AJ, Huang YC. Exposure to Concentrated Ambient Particles (CAPs): A Review. Inhal Toxicol. 2004;16(1):53-9. 71. Seaton A, MacNee W, Donaldson K, Godden D. Particulate air-pollution and acute health-effects. Lancet. 1995;345:176-8. 72. Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, et al. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health A. 2002;65(20):1513-30. 73. Nadel JA. Role of neutrophil elastase in hypersecretion during COPD exacerbations, and proposed therapies. Chest. 2000 May;117(5 Suppl 2):386S-9S. 74. Nemmar A, Hoet PH, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, et al. Passage of inhaled particles into the blood circulation in humans. Circulation. 2002;105(4):411-4. 75. Nemmar A, Vanbilloen H, Hoylaerts MF, Hoet PH, Verbruggen A, Nemery B. Passage of intratracheally instilled ultrafine particles from the lung into the systemic circulation in hamster. Am J Respir Crit Care Med. 2001;164(9):1665-8. 76. Oberdorster G, Sharp Z, Atudorei V, Elder A, Gelein R, Lunts A, et al. Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J ToxicolEnviron Health A. 2002;65(20):1531-43. 77. Mills NL, Amin N, Robinson SD, Anand A, Davies J, Patel D, et al. Do Inhaled Carbon Nanoparticles Translocate Directly into the Circulation in Humans? American Journal of Respiratory and Critical Care Medicine. 2006;173(4):426-31. 78. Wiebert P, Sanchez-Crespo A, Seitz J, Falk R, Philipson K, Kreyling WG, et al. Negligible clearance of ultrafine particles retained in healthy and affected human lungs. European Respiratory Journal. 2006;28(2):286-90. 79. Guzman LA, Labhasetwar V, Song C, Jang Y, Lincoff AM, Levy R, et al. Local Intraluminal Infusion of Biodegradable Polymeric Nanoparticles: A Novel Approach for Prolonged Drug Delivery After Balloon Angioplasty. Circulation. 1996;94(6):1441-8. 80. Chen LC, Nadziejko C. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal Toxicol. 2005 Apr;17(4-5):217-24. 81. Oberdorster G, Sharp Z, Atudorei V, Elder A, Gelein R, Kreyling W, et al. Translocation of inhaled ultrafine particles to the brain. Inhal Toxicol. 2004 Jun;16(6-7):437-45. 82. Cruts B, van Etten L, Tornqvist H, Blomberg A, Sandstrom T, Mills NL, et al. Exposure to diesel exhaust induces changes in EEG in human volunteers. Part Fibre Toxicol. 2008 Mar 11;5(1):4.

REFERENCES
76
83. Fiorito S, Serafino A, Andreola F, Togna A, Togna G. Toxicity and biocompatibility of carbon nanoparticles. J Nanosci Nanotechnol. 2006 Mar;6(3):591-9. 84. Donaldson K, Stone V, Borm PJ, Jimenez LA, Gilmour PS, Schins RP, et al. Oxidative stress and calcium signalling in the adverse effects of environmental particles (PM10). Free Radic Biol Med. 2003;34(11):1369-82. 85. Donaldson K, Tran L, Jimenez LA, Duffin R, Newby DE, Mills N, et al. Combustion-derived nanoparticles: a review of their toxicology following inhalation exposure. Part Fibre Toxicol. 2005;2:10.:10. 86. Nel AE, Diaz-Sanchez D, Li N. The role of particulate pollutants in pulmonary inflammation and asthma: evidence for the involvement of organic chemicals and oxidative stress. Curr Opin Pulm Med. 2001;7(1):20-6. 87. Steerenberg PA, Withagen CE, Dormans JA, van Dalen WJ, van Loveren H, Casee FR. Adjuvant activity of various diesel exhaust and ambient particles in two allergic models. J Toxicol Environ Health A. 2003 Aug 8;66(15):1421-39. 88. Ma JY, Ma JK. The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2002 Nov;20(2):117-47. 89. Radomski A, Jurasz P, onso-Escolano D, Drews M, Morandi M, Malinski T, et al. Nanoparticle-induced platelet aggregation and vascular thrombosis. 2005;146(6):882-93. 90. Nemmar A, Hoylaerts MF, Hoet PH, Nemery B. Possible mechanisms of the cardiovascular effects of inhaled particles: systemic translocation and prothrombotic effects. Toxicol Lett. 2004 Apr 1;149(1-3):243-53. 91. Suwa T, Hogg JC, Quinlan KB, Ohgami A, Vincent R, van Eeden SF. Particulate air pollution induces progression of atherosclerosis. J Am Coll Cardiol. 2002;39(6):935-42. 92. Goto Y, Hogg JC, Shih CH, Ishii H, Vincent R, van Eeden SF. Exposure to ambient particles accelerates monocyte release from the bone marrow in atherosclerotic rabbits. AJP - Lung Cellular and Molecular Physiology. 2004:00425. 93. Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, et al. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294(23):3003-10. 94. Nemmar A, Hoet PHM, Vermylen J, Nemery B, Hoylaerts MF. Pharmacological Stabilization of Mast Cells Abrogates Late Thrombotic Events Induced by Diesel Exhaust Particles in Hamsters. Circulation. 2004:01. 95. Yokota S, Furuya M, Seki T, Marumo H, Ohara N, Kato A. Delayed exacerbation of acute myocardial ischemia/reperfusion-induced arrhythmia by tracheal instillation of diesel exhaust particles. Inhal Toxicol. 2004 May;16(5):319-31. 96. Campen MJ, Babu NS, Helms GA, Pett S, Wernly J, Mehran R, et al. Nonparticulate components of diesel exhaust promote constriction in coronary arteries from ApoE-/- mice. Toxicol Sci. 2005 Nov;88(1):95-102. 97. Ghio AJ, Hall A, Bassett MA, Cascio WE, Devlin RB. Exposure to concentrated ambient air particles alters hematologic indices in humans. Inhal Toxicol. 2003 Dec;15(14):1465-78. 98. Devlin RB, Ghio AJ, Kehrl H, Sanders G, Cascio W. Elderly humans exposed to concentrated air pollution particles have decreased heart rate variability. European Respiratory Journal - Supplement40:76s-80s. 2003.

REFERENCES
77
99. Pietropaoli AP, Frampton MW, Hyde RW, Morrow PE, Oberdorster G, Cox C, et al. Pulmonary function, diffusing capacity, and inflammation in healthy and asthmatic subjects exposed to ultrafine particles. Inhal Toxicol. 2004;16 Suppl 1:59-72. 100. Brook RD, Brook JR, Urch B, Vincent R, Rajagopalan S, Silverman F. Inhalation of fine particulate air pollution and ozone causes acute arterial vasoconstriction in healthy adults. Circulation. 2002;105(13):1534-6. 101. Larsson BM, Sehlstedt M, Grunewald J, Skold CM, Lundin A, Blomberg A, et al. Road tunnel air pollution induces bronchoalveolar inflammation in healthy subjects. Eur Respir J. 2007 Apr;29(4):699-705. 102. Svartengren M. Short-term exposure to air pollution in a road tunnel enhances the asthmatic response to allergen. European Respiratory Journal15(4):716-24. 2000. 103. Routledge HC, Manney S, Harrison RM, Ayres JG, Townend JN. Effect of inhaled sulphur dioxide and carbon particles on heart rate variability and markers of inflammation and coagulation in human subjects. Heart. 2006;92(2):220-7. 104. Rudell B, Ledin MC, Hammarstrom U, Stjernberg N, Lundback B, Sandstrom T. Effects on symptoms and lung function in humans experimentally exposed to diesel exhaust. Occup Environ Med. 1996;53(10):658-62. 105. Salvi S, Blomberg A, Rudell B, Kelly F, Sandstrom T, Holgate ST, et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999 Mar;159(3):702-9. 106. Stenfors N, Nordenhall C, Salvi SS, Mudway I, Soderberg M, Blomberg A, et al. Different airway inflammatory responses in asthmatic and healthy humans exposed to diesel. Eur Respir J. 2004 Jan;23(1):82-6. 107. Behndig AF, Mudway IS, Brown JL, Stenfors N, Helleday R, Duggan ST, et al. Airway antioxidant and inflammatory responses to diesel exhaust exposure in healthy humans. Eur Respir J. 2006 Feb;27(2):359-65. 108. Pourazar J, Mudway IS, Samet JM, Helleday R, Blomberg A, Wilson SJ, et al. Diesel exhaust activates redox-sensitive transcription factors and kinases in human airways. AJP - Lung Cellular and Molecular Physiology. 2005;289(5):L724-L30. 109. Nordenhall C, Pourazar J, Ledin MC, Levin JO, Sandstrom T, Adelroth E. Diesel exhaust enhances airway responsiveness in asthmatic subjects. Eur Respir J. 2001 May;17(5):909-15. 110. Gould T, Larson T, Stewart J, Kaufman JD, Slater D, McEwen N. A controlled inhalation diesel exhaust exposure facility with dynamic feedback control of PM concentration. Inhal Toxicol. 2008 Jan;20(1):49-52. 111. Peretz A, Kaufman JD, Trenga CA, Allen J, Carlsten C, Aulet MR, et al. Effects of diesel exhaust inhalation on heart rate variability in human volunteers. Environ Res. 2008 Mar 7. 112. Peretz A, Peck EC, Bammler TK, Beyer RP, Sullivan JH, Trenga CA, et al. Diesel exhaust inhalation and assessment of peripheral blood mononuclear cell gene transcription effects: an exploratory study of healthy human volunteers. Inhal Toxicol. 2007 Nov;19(14):1107-19. 113. Bateman ED, Feldman C, O'Brien J, Plit M, Joubert JR. Guideline for the management of chronic obstructive pulmonary disease (COPD): 2004 revision. S Afr Med J. 2004 Jul;94(7 Pt 2):559-75.

REFERENCES
78
114. Hingorani AD, Cross J, Kharbanda RK, Mullen MJ, Bhagat K, Taylor M, et al. Acute systemic inflammation impairs endothelium-dependent dilatation in humans. Circulation. 2000 Aug 29;102(9):994-9. 115. Newby DE, Witherow FN, Wright RA, Bloomfield P, Ludlam CA, Boon NA, et al. Hypercholesterolaemia and lipid lowering treatment do not affect the acute endogenous fibrinolytic capacity in vivo. Heart. 2002 Jan;87(1):48-53. 116. Rusznak C. Airway response of asthmatic subjects to inhaled allergen after exposure to pollutants. Thorax51(11):1105-8. 1996. 117. Rudell B, Blomberg A, Helleday R, Ledin MC, Lundback B, Stjernberg N, et al. Bronchoalveolar inflammation after exposure to diesel exhaust: comparison between unfiltered and particle trap filtered exhaust. Occupational and Environmental Medicine. 1999;56(8):527-34. 118. Pin I, Gibson PG, Kolendowicz R, Girgis-Gabardo A, Denburg JA, Hargreave FE, et al. Use of induced sputum cell counts to investigate airway inflammation in asthma. Thorax. 1992 Jan;47(1):25-9. 119. Pizzichini MM, Popov TA, Efthimiadis A, Hussack P, Evans S, Pizzichini E, et al. Spontaneous and induced sputum to measure indices of airway inflammation in asthma. Am J Respir Crit Care Med. 1996 Oct;154(4 Pt 1):866-9. 120. Nordenhall C, Pourazar J, Blomberg A, Levin JO, Sandstrom T, Adelroth E. Airway inflammation following exposure to diesel exhaust: a study of time kinetics using induced sputum [In Process Citation]. Eur Respir J. 2000;15(6):1046-51. 121. Johnson AM, Ledue TB, Collins MF. Commutability of the CRM 470 C-reactive protein value in the Dade Behring N High Sensitivity CRP assay. Clin Chem Lab Med. 2003 Feb;41(2):177-82. 122. Wilson DB, Gard KM. Evaluation of an automated, latex-enhanced turbidimetric D-dimer test (advanced D-dimer) and usefulness in the exclusion of acute thromboembolic disease. Am J Clin Pathol. 2003 Dec;120(6):930-7. 123. Hermans C, Dong P, Robin M, Jadoul M, Bernard A, Bersten AD, et al. Determinants of serum levels of surfactant proteins A and B and Clara cell protein CC16. Biomarkers. 2003 Nov-Dec;8(6):461-71. 124. Benjamin N, Calver A, Collier J, Robinson B, Vallance P, Webb D. Measuring Forearm Blood Flow and Interpreting the Responses to Drugs and Mediators. Hypertension. 1995 May 1, 1995;25(5):918-23. 125. Anderson TJ, Uehata A, Gerhard MD, Meredith IT, Knab S, Delagrange D, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995 Nov 1;26(5):1235-41. 126. Vitalis TZ, Keicho N, Itabashi S, Hayashi S, Hogg JC. A model of latent adenovirus-5 infection in the guinea-pig (Cavia- porcellus). American Journal Of Respiratory Cell And Molecular Biology. 1996;14:225-31. 127. Webb DJ. The pharmacology of human blood vessels in vivo. J Vasc Res. 1995 Jan-Feb;32(1):2-15. 128. Walker HA, Jackson G, Ritter JM, Chowienczyk PJ. Assessment of forearm vasodilator responses to acetylcholine and albuterol by strain gauge plethysmography: reproducibility and influence of strain gauge placement. Br J Clin Pharmacol. 2001;51(3):225-9. 129. Wilkinson IB, Webb DJ. Venous occlusion plethysmography in cardiovascular research: methodology and clinical applications. Br J Clin Pharmacol. 2001 Dec;52(6):631-46.

REFERENCES
79
130. Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG. A fluorometric assay for the measurement of nitrite in biological samples. Anal Biochem. 1993 Oct;214(1):11-6. 131. Miller NJ, Rice-Evans C, Davies MJ, Gopinathan V, Milner A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin Sci (Lond). 1993 Apr;84(4):407-12. 132. Walker HA, Jackson G, Ritter JM, Chowienczyk PJ. Assessment of forearm vasodilator responses to acetylcholine and albuterol by strain gauge plethysmography: reproducibility and influence of strain gauge placement. Br J Clin Pharmacol. 2001 Mar;51(3):225-9. 133. Borg GA. Psychophysical bases of perceived exertion. Med Sci Sports Exerc. 1982;14(5):377-81. 134. Newby DE, Wright RA, Labinjoh C, Ludlam CA, Fox KA, Boon NA, et al. Endothelial dysfunction, impaired endogenous fibrinolysis, and cigarette smoking: a mechanism for arterial thrombosis and myocardial infarction. Circulation. 1999;99(11):1411-5. 135. Pope CA, III, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, et al. Cardiovascular Mortality and Long-Term Exposure to Particulate Air Pollution. Epidemiological Evidence of General Pathophysiological Pathways of Disease. Circulation. 2003:01. 136. Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, et al. Long-Term Exposure to Air Pollution and Incidence of Cardiovascular Events in Women. N Engl J Med. 2007 February 1, 2007;356(5):447-58. 137. Stenfors N, Nordenhall C, Salvi SS, Mudway I, Soderberg M, Blomberg A, et al. Different airway inflammatory responses in asthmatic and healthy humans exposed to diesel. Eur Respir J. 2004;23(1):82-6. 138. Thurston GD. A critical review of PM10-mortality time-series studies. J Expo Anal Environ Epidemiol. 1996;6(1):3-21. 139. Pope CA. Epidemiology of fine particulate air pollution and human health: biologic mechanisms and Who's at risk? Environ Health Perspect. 2000;108 Suppl 4:713-23. 140. Lagorio S, Forastiere F, Pistelli R, Iavarone I, Michelozzi P, Fano V, et al. Air pollution and lung function among susceptible adult subjects: a panel study. Environ Health. 2006;5:11. 141. Sin DD, Man SF. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003 Mar 25;107(11):1514-9. 142. Rudell B, Ledin MC, Hammarstrom U, Stjernberg N, Lundback B, Sandstrom T. Effects on symptoms and lung function in humans experimentally exposed to diesel exhaust. Occup Environ Med. 1996 Oct;53(10):658-62. 143. Kannel WB, McGee DL. Diabetes and cardiovascular risk factors: the Framingham study. Circulation. 1979 Jan;59(1):8-13. 144. Seaton A, MacNee W, Donaldson K, Godden D. Particulate air pollution and acute health effects. Lancet. 1995;345(8943):176-8. 145. Seaton A, Soutar CA, Crawford V, Elton R, McNerlan S, Cherrie J, et al. Particulate air pollution and the blood. Thorax. 1999;54(11):1027-32. 146. Ghio AJ, Kim C, Devlin RB. Concentrated ambient air particles induce mild pulmonary inflammation in healthy human volunteers. Am J Respir Crit Care Med. 2000;162(3 Pt 1):981-8. 147. Pekkanen J, Brunner EJ, Anderson HR, Tiittanen P, Atkinson RW. Daily concentrations of air pollution and plasma fibrinogen in London. Occupational & Environmental Medicine. 2000;57(12):818-22.

REFERENCES
80
148. Schwartz J. Air pollution and blood markers of cardiovascular risk. Environmental Health Perspectives. 2001;109:Suppl-9. 149. Wedzicha JA, Seemungal TA, MacCallum PK, Paul EA, Donaldson GC, Bhowmik A, et al. Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost. 2000 Aug;84(2):210-5. 150. Horvath B, Hegedus D, Szapary L, Marton Z, Alexy T, Koltai K, et al. [Investigation of von Willebrand-factor as a marker of endothelial dysfunction in atherosclerotic patients]. Orv Hetil. 2003 Dec 14;144(50):2471-6. 151. Blomberg A, Mudway I, Svensson M, Hagenbjork-Gustafsson A, Thomasson L, Helleday R, et al. Clara cell protein as a biomarker for ozone-induced lung injury in humans. Eur Respir J. 2003 Dec;22(6):883-8. 152. Keatings VM, Barnes PJ. Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjects. Am J Respir Crit Care Med. 1997 Feb;155(2):449-53. 153. Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 2000 Feb;55(2):114-20. 154. Jayaram L, Parameswaran K, Sears MR, Hargreave FE. Induced sputum cell counts: their usefulness in clinical practice. Eur Respir J. 2000 Jul;16(1):150-8. 155. Brightling CE. Clinical applications of induced sputum. Chest. 2006 May;129(5):1344-8. 156. Vestbo J, Prescott E, Lange P. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am J Respir Crit Care Med. 1996 May;153(5):1530-5. 157. Siva R, Green RH, Brightling CE, Shelley M, Hargadon B, McKenna S, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. Eur Respir J. 2007 May;29(5):906-13. 158. Koren H, O'Neill M. Experimental assessment of the influence of atmospheric pollutants on respiratory disease. Toxicol Lett. 1998 Dec 28;102-103:317-21. 159. Ware JH. Particulate Air Pollution and Mortality -- Clearing the Air. The New England Journal of Medicine. 2000;343(24):1798-9. 160. Hosseinpoor AR, Forouzanfar MH, Yunesian M, Asghari F, Naieni KH, Farhood D. Air pollution and hospitalization due to angina pectoris in Tehran, Iran: a time-series study. Environ Res. 2005 Sep;99(1):126-31. 161. Pekkanen J, Peters A, Hoek G, Tiittanen P, Brunekreef B, de Hartog J, et al. Particulate air pollution and risk of ST-segment depression during repeated submaximal exercise tests among subjects with coronary heart disease: the Exposure and Risk Assessment for Fine and Ultrafine Particles in Ambient Air (ULTRA) study. Circulation. 2002;106(8):933-8. 162. Peters A, Dockery DW, Muller JE, Mittleman MA. Increased particulate air pollution and the triggering of myocardial infarction. Circulation. 2001;103(23):2810-5. 163. Braunwald E. Coronary artery patency in patients with myocardial infarction. J Am Coll Cardiol. 1990 Dec;16(7):1550-2. 164. Van Lierde J, De Geest H, Verstraete M, Van de Werf F. Angiographic assessment of the infarct-related residual coronary stenosis after spontaneous or therapeutic thrombolysis. J Am Coll Cardiol. 1990 Dec;16(7):1545-9. 165. Newby DE, McLeod AL, Uren NG, Flint L, Ludlam CA, Webb DJ, et al. Impaired coronary tissue plasminogen activator release is associated with coronary atherosclerosis and

REFERENCES
81
cigarette smoking: direct link between endothelial dysfunction and atherothrombosis. Circulation. 2001;103(15):1936-41. 166. Harrod KS, Jaramillo RJ, Rosenberger CL, Wang SZ, Berger JA, McDonald JD, et al. Increased susceptibility to RSV infection by exposure to inhaled diesel engine emissions. Am J Respir Cell Mol Biol. 2003 Apr;28(4):451-63. 167. Emeis JJ. Regulation of the acute release of tissue-type plasminogen activator from the endothelium by coagulation activation products. Ann N Y Acad Sci. 1992 Dec 4;667:249-58. 168. Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem. 1982 Mar 25;257(6):2912-9. 169. Plow EF, Felez J, Miles LA. Cellular regulation of fibrinolysis. Thromb Haemost. 1991 Jul 12;66(1):32-6. 170. Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001 Nov 27;104(22):2673-8. 171. Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001 Apr;280(4):C719-41. 172. Mulsch A, Bauersachs J, Schafer A, Stasch JP, Kast R, Busse R. Effect of YC-1, an NO-independent, superoxide-sensitive stimulator of soluble guanylyl cyclase, on smooth muscle responsiveness to nitrovasodilators. Br J Pharmacol. 1997 Feb;120(4):681-9. 173. Oelze M, Mollnau H, Hoffmann N, Warnholtz A, Bodenschatz M, Smolenski A, et al. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signalling and endothelial dysfunction. Circ Res. 2000 Nov 24;87(11):999-1005. 174. Monks TJ, Hanzlik RP, Cohen GM, Ross D, Graham DG. Quinone chemistry and toxicity. Toxicol Appl Pharmacol. 1992 Jan;112(1):2-16. 175. Dellinger B, Pryor WA, Cueto R, Squadrito GL, Hegde V, Deutsch WA. Role of free radicals in the toxicity of airborne fine particulate matter. Chem Res Toxicol. 2001 Oct;14(10):1371-7. 176. Xia T, Korge P, Weiss JN, Li N, Venkatesen MI, Sioutas C, et al. Quinones and aromatic chemical compounds in particulate matter induce mitochondrial dysfunction: implications for ultrafine particle toxicity. Environ Health Perspect. 2004 Oct;112(14):1347-58. 177. Kumagai Y, Hayashi T, Miyauchi T, Endo A, Iguchi A, Kiriya-Sakai M, et al. Phenanthraquinone inhibits eNOS activity and suppresses vasorelaxation. Am J Physiol Regul Integr Comp Physiol. 2001 Jul;281(1):R25-30. 178. Kumagai T, Nakamura Y, Osawa T, Uchida K. Role of p38 mitogen-activated protein kinase in the 4-hydroxy-2-nonenal-induced cyclooxygenase-2 expression. Arch Biochem Biophys. 2002 Jan 15;397(2):240-5. 179. Kinlay S, Selwyn AP. Effects of statins on inflammation in patients with acute and chronic coronary syndromes. Am J Cardiol. 2003 Feb 20;91(4A):9B-13B. 180. Peters A, Perz S, Doring A, Stieber J, Koenig W, Wichmann HE. Increases in heart rate during an air pollution episode. American Journal of Epidemiology. 1999;150(10):1094-8. 181. Urch B, Silverman F, Corey P, Brook JR, Lukic KZ, Rajagopalan S, et al. Acute blood pressure responses in healthy adults during controlled air pollution exposures. Environ Health Perspect. 2005;113(8):1052-5.





























