Embed Size (px)
Citation preview

Biochimica et Biophysica Acta 1835 (2013) 194–210
Contents lists available at SciVerse ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r .com/ locate /bbacan
Review
Targeting LKB1 signaling in cancer
S.E. Korsse, M.P. Peppelenbosch, W. van Veelen ⁎Dept. of Gastroenterology and Hepatology, Erasmus Medical University Center, Rotterdam, The Netherlands
⁎ Corresponding author at: Dept. of Gastroenterology33322; fax: +31 10 70 32793.
E-mail address: [email protected] (W. van
0304-419X/$ – see front matter © 2012 Elsevier B.V. Alhttp://dx.doi.org/10.1016/j.bbcan.2012.12.006
a b s t r a c t
a r t i c l e i n f oArticle history:Received 14 August 2012Received in revised form 18 December 2012Accepted 20 December 2012Available online 31 December 2012
Keywords:LKB1Peutz–Jeghers syndromeSporadic cancerRapamycinMetforminmTOR
The serine/threonine kinase LKB1 is a master kinase involved in cellular responses such as energy metabo-lism, cell polarity and cell growth. LKB1 regulates these crucial cellular responses mainly via AMPK/mTOR sig-naling. Germ-line mutations in LKB1 are associated with the predisposition of the Peutz–Jeghers syndrome inwhich patients develop gastrointestinal hamartomas and have an enormously increased risk for developinggastrointestinal, breast and gynecological cancers. In addition, somatic inactivation of LKB1 has been associ-ated with sporadic cancers such as lung cancer. The exact mechanisms of LKB1-mediated tumor suppressionremain so far unidentified; however, the inability to activate AMPK and the resulting mTOR hyperactivationhas been detected in PJS-associated lesions. Therefore, targeting LKB1 in cancer is nowmainly focusing on theactivation of AMPK and inactivation of mTOR. Preclinical in vitro and in vivo studies show encouraging resultsregarding these approaches, which have even progressed to the initiation of a few clinical trials. In this re-view, we describe the functions, regulation and downstream signaling of LKB1, and its role in hereditaryand sporadic cancers. In addition, we provide an overview of several AMPK activators, mTOR inhibitors andadditional mechanisms to target LKB1 signaling, and describe the effect of these compounds on cancercells. Overall, we will explain the current strategies attempting to find a way of treating LKB1-associatedcancer.
© 2012 Elsevier B.V. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1951.1. The Peutz–Jeghers syndrome . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1951.2. Gastrointestinal hamartomas in PJS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1951.3. Cancer risk in PJS patients . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1951.4. The tumor suppressor gene LKB1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 195
2. LKB1 in sporadic cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1962.1. Colorectal cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1962.2. Pancreatic cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1972.3. Breast cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1972.4. Lung cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
3. LKB1 signaling and function . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1973.1. The LKB1 protein . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1973.2. LKB1 activation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1983.3. LKB1 in cell polarity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1983.4. LKB1 in energy metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1983.5. LKB1 in cell growth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 198
4. Targeting LKB1 signaling in cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1994.1. Metformin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1994.2. Rapamycin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2014.3. Combining mTOR inhibitors with other specific pathway inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2024.4. Other options to treat PJS-associated tumors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202
and Hepatology, Erasmus MC-Room L-463, 's Gravendijkwal 230, 3015 CE Rotterdam, The Netherlands. Tel.: +31 10 70
Veelen).
l rights reserved.
195S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
5. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202List of abbreviations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203Conflict of interest . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
1. Introduction
1.1. The Peutz–Jeghers syndrome
In 1921, theDutch physician Johannes Peutzwas the first to describethe combination of intestinal polyps, pigmentedmucocutaneous lesionsand heredity [1]. In 1949, Harold Jeghers specified the description ofthe syndrome [2], and in 1954 the eponym “Peutz–Jeghers syndrome”(PJS) was first used. PJS is a rare autosomal dominant disorder with aprevalence of 1 in 8300–280,000 [3]. The PJS-associatedmucocutaneoushyperpigmentation is predominantly apparent on the lips, oral mucosa,face and/or hands at young ages, and can diminish or disappear inadulthood. This pigmentation has unknown clinical implications but itcan be used in the diagnosis of PJS.
1.2. Gastrointestinal hamartomas in PJS
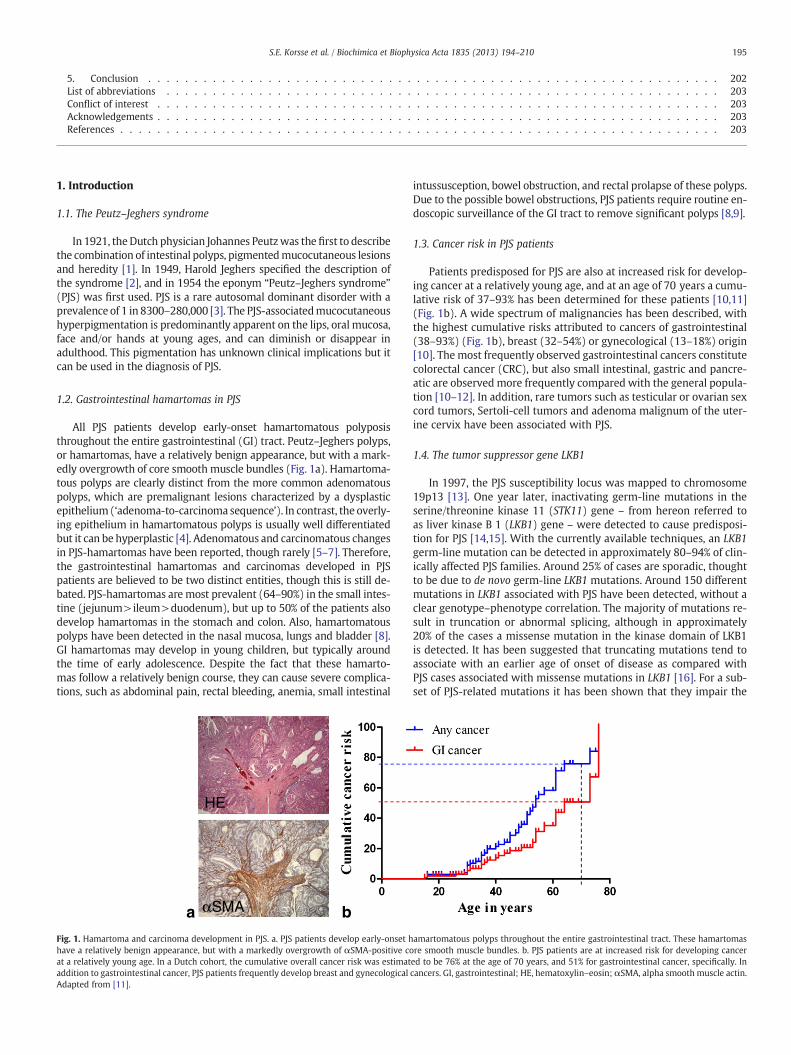
All PJS patients develop early-onset hamartomatous polyposisthroughout the entire gastrointestinal (GI) tract. Peutz–Jeghers polyps,or hamartomas, have a relatively benign appearance, but with a mark-edly overgrowth of core smooth muscle bundles (Fig. 1a). Hamartoma-tous polyps are clearly distinct from the more common adenomatouspolyps, which are premalignant lesions characterized by a dysplasticepithelium(‘adenoma-to-carcinoma sequence’). In contrast, the overly-ing epithelium in hamartomatous polyps is usually well differentiatedbut it can be hyperplastic [4]. Adenomatous and carcinomatous changesin PJS-hamartomas have been reported, though rarely [5–7]. Therefore,the gastrointestinal hamartomas and carcinomas developed in PJSpatients are believed to be two distinct entities, though this is still de-bated. PJS-hamartomas are most prevalent (64–90%) in the small intes-tine (jejunum>ileum>duodenum), but up to 50% of the patients alsodevelop hamartomas in the stomach and colon. Also, hamartomatouspolyps have been detected in the nasal mucosa, lungs and bladder [8].GI hamartomas may develop in young children, but typically aroundthe time of early adolescence. Despite the fact that these hamarto-mas follow a relatively benign course, they can cause severe complica-tions, such as abdominal pain, rectal bleeding, anemia, small intestinal
HE
αSMA ba
Fig. 1. Hamartoma and carcinoma development in PJS. a. PJS patients develop early-onset hhave a relatively benign appearance, but with a markedly overgrowth of αSMA-positive coat a relatively young age. In a Dutch cohort, the cumulative overall cancer risk was estimataddition to gastrointestinal cancer, PJS patients frequently develop breast and gynecologicalAdapted from [11].
intussusception, bowel obstruction, and rectal prolapse of these polyps.Due to the possible bowel obstructions, PJS patients require routine en-doscopic surveillance of the GI tract to remove significant polyps [8,9].
1.3. Cancer risk in PJS patients
Patients predisposed for PJS are also at increased risk for develop-ing cancer at a relatively young age, and at an age of 70 years a cumu-lative risk of 37–93% has been determined for these patients [10,11](Fig. 1b). A wide spectrum of malignancies has been described, withthe highest cumulative risks attributed to cancers of gastrointestinal(38–93%) (Fig. 1b), breast (32–54%) or gynecological (13–18%) origin[10]. The most frequently observed gastrointestinal cancers constitutecolorectal cancer (CRC), but also small intestinal, gastric and pancre-atic are observed more frequently compared with the general popula-tion [10–12]. In addition, rare tumors such as testicular or ovarian sexcord tumors, Sertoli-cell tumors and adenoma malignum of the uter-ine cervix have been associated with PJS.
1.4. The tumor suppressor gene LKB1
In 1997, the PJS susceptibility locus was mapped to chromosome19p13 [13]. One year later, inactivating germ-line mutations in theserine/threonine kinase 11 (STK11) gene – from hereon referred toas liver kinase B 1 (LKB1) gene – were detected to cause predisposi-tion for PJS [14,15]. With the currently available techniques, an LKB1germ-line mutation can be detected in approximately 80–94% of clin-ically affected PJS families. Around 25% of cases are sporadic, thoughtto be due to de novo germ-line LKB1 mutations. Around 150 differentmutations in LKB1 associated with PJS have been detected, without aclear genotype–phenotype correlation. The majority of mutations re-sult in truncation or abnormal splicing, although in approximately20% of the cases a missense mutation in the kinase domain of LKB1is detected. It has been suggested that truncating mutations tend toassociate with an earlier age of onset of disease as compared withPJS cases associated with missense mutations in LKB1 [16]. For a sub-set of PJS-related mutations it has been shown that they impair the
amartomatous polyps throughout the entire gastrointestinal tract. These hamartomasre smooth muscle bundles. b. PJS patients are at increased risk for developing cancered to be 76% at the age of 70 years, and 51% for gastrointestinal cancer, specifically. Incancers. GI, gastrointestinal; HE, hematoxylin–eosin; αSMA, alpha smooth muscle actin.
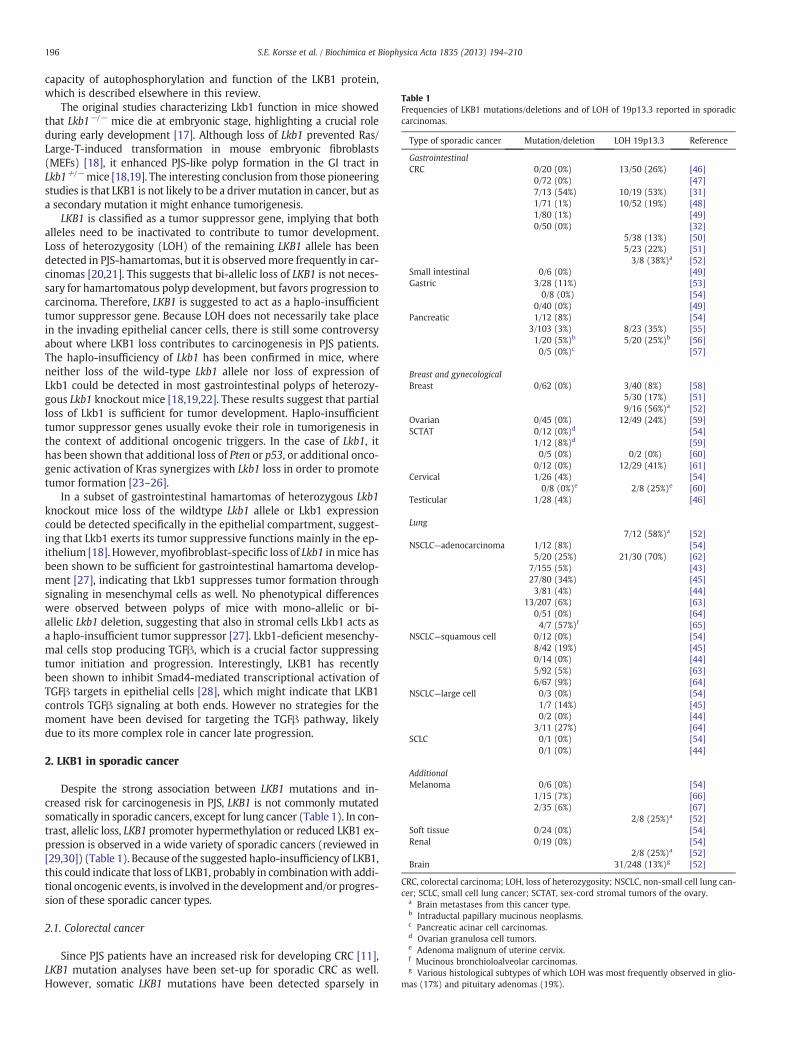
Table 1Frequencies of LKB1 mutations/deletions and of LOH of 19p13.3 reported in sporadiccarcinomas.
Type of sporadic cancer Mutation/deletion LOH 19p13.3 Reference
GastrointestinalCRC 0/20 (0%) 13/50 (26%) [46]
0/72 (0%) [47]7/13 (54%) 10/19 (53%) [31]1/71 (1%) 10/52 (19%) [48]1/80 (1%) [49]0/50 (0%) [32]
5/38 (13%) [50]5/23 (22%) [51]3/8 (38%)a [52]
Small intestinal 0/6 (0%) [49]Gastric 3/28 (11%) [53]
0/8 (0%) [54]0/40 (0%) [49]
Pancreatic 1/12 (8%) [54]3/103 (3%) 8/23 (35%) [55]1/20 (5%)b 5/20 (25%)b [56]0/5 (0%)c [57]
Breast and gynecologicalBreast 0/62 (0%) 3/40 (8%) [58]
5/30 (17%) [51]9/16 (56%)a [52]
Ovarian 0/45 (0%) 12/49 (24%) [59]SCTAT 0/12 (0%)d [54]
1/12 (8%)d [59]0/5 (0%) 0/2 (0%) [60]
0/12 (0%) 12/29 (41%) [61]Cervical 1/26 (4%) [54]
0/8 (0%)e 2/8 (25%)e [60]Testicular 1/28 (4%) [46]
Lung7/12 (58%)a [52]
NSCLC—adenocarcinoma 1/12 (8%) [54]5/20 (25%) 21/30 (70%) [62]
7/155 (5%) [43]27/80 (34%) [45]3/81 (4%) [44]
13/207 (6%) [63]0/51 (0%) [64]4/7 (57%)f [65]
NSCLC—squamous cell 0/12 (0%) [54]8/42 (19%) [45]0/14 (0%) [44]5/92 (5%) [63]6/67 (9%) [64]
NSCLC—large cell 0/3 (0%) [54]1/7 (14%) [45]0/2 (0%) [44]
3/11 (27%) [64]SCLC 0/1 (0%) [54]
0/1 (0%) [44]
AdditionalMelanoma 0/6 (0%) [54]
1/15 (7%) [66]2/35 (6%) [67]
2/8 (25%)a [52]Soft tissue 0/24 (0%) [54]Renal 0/19 (0%) [54]
2/8 (25%)a [52]Brain 31/248 (13%)g [52]
CRC, colorectal carcinoma; LOH, loss of heterozygosity; NSCLC, non-small cell lung can-cer; SCLC, small cell lung cancer; SCTAT, sex-cord stromal tumors of the ovary.
a Brain metastases from this cancer type.b Intraductal papillary mucinous neoplasms.c Pancreatic acinar cell carcinomas.d Ovarian granulosa cell tumors.e Adenoma malignum of uterine cervix.f Mucinous bronchioloalveolar carcinomas.g Various histological subtypes of which LOH was most frequently observed in glio-
mas (17%) and pituitary adenomas (19%).
196 S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
capacity of autophosphorylation and function of the LKB1 protein,which is described elsewhere in this review.
The original studies characterizing Lkb1 function in mice showedthat Lkb1−/− mice die at embryonic stage, highlighting a crucial roleduring early development [17]. Although loss of Lkb1 prevented Ras/Large-T-induced transformation in mouse embryonic fibroblasts(MEFs) [18], it enhanced PJS-like polyp formation in the GI tract inLkb1+/−mice [18,19]. The interesting conclusion from those pioneeringstudies is that LKB1 is not likely to be a driver mutation in cancer, but asa secondary mutation it might enhance tumorigenesis.
LKB1 is classified as a tumor suppressor gene, implying that bothalleles need to be inactivated to contribute to tumor development.Loss of heterozygosity (LOH) of the remaining LKB1 allele has beendetected in PJS-hamartomas, but it is observedmore frequently in car-cinomas [20,21]. This suggests that bi-allelic loss of LKB1 is not neces-sary for hamartomatous polyp development, but favors progression tocarcinoma. Therefore, LKB1 is suggested to act as a haplo-insufficienttumor suppressor gene. Because LOH does not necessarily take placein the invading epithelial cancer cells, there is still some controversyabout where LKB1 loss contributes to carcinogenesis in PJS patients.The haplo-insufficiency of Lkb1 has been confirmed in mice, whereneither loss of the wild-type Lkb1 allele nor loss of expression ofLkb1 could be detected in most gastrointestinal polyps of heterozy-gous Lkb1 knockout mice [18,19,22]. These results suggest that partialloss of Lkb1 is sufficient for tumor development. Haplo-insufficienttumor suppressor genes usually evoke their role in tumorigenesis inthe context of additional oncogenic triggers. In the case of Lkb1, ithas been shown that additional loss of Pten or p53, or additional onco-genic activation of Kras synergizes with Lkb1 loss in order to promotetumor formation [23–26].
In a subset of gastrointestinal hamartomas of heterozygous Lkb1knockout mice loss of the wildtype Lkb1 allele or Lkb1 expressioncould be detected specifically in the epithelial compartment, suggest-ing that Lkb1 exerts its tumor suppressive functions mainly in the ep-ithelium [18]. However,myofibroblast-specific loss of Lkb1 inmice hasbeen shown to be sufficient for gastrointestinal hamartoma develop-ment [27], indicating that Lkb1 suppresses tumor formation throughsignaling in mesenchymal cells as well. No phenotypical differenceswere observed between polyps of mice with mono-allelic or bi-allelic Lkb1 deletion, suggesting that also in stromal cells Lkb1 acts asa haplo-insufficient tumor suppressor [27]. Lkb1-deficient mesenchy-mal cells stop producing TGFβ, which is a crucial factor suppressingtumor initiation and progression. Interestingly, LKB1 has recentlybeen shown to inhibit Smad4-mediated transcriptional activation ofTGFβ targets in epithelial cells [28], which might indicate that LKB1controls TGFβ signaling at both ends. However no strategies for themoment have been devised for targeting the TGFβ pathway, likelydue to its more complex role in cancer late progression.
2. LKB1 in sporadic cancer
Despite the strong association between LKB1 mutations and in-creased risk for carcinogenesis in PJS, LKB1 is not commonly mutatedsomatically in sporadic cancers, except for lung cancer (Table 1). In con-trast, allelic loss, LKB1 promoter hypermethylation or reduced LKB1 ex-pression is observed in a wide variety of sporadic cancers (reviewed in[29,30]) (Table 1). Because of the suggested haplo-insufficiency of LKB1,this could indicate that loss of LKB1, probably in combinationwith addi-tional oncogenic events, is involved in the development and/or progres-sion of these sporadic cancer types.
2.1. Colorectal cancer
Since PJS patients have an increased risk for developing CRC [11],LKB1 mutation analyses have been set-up for sporadic CRC as well.However, somatic LKB1 mutations have been detected sparsely in

kinase domain1 43349 309 PP P PP P P P F
Ser31 Ser325 Thr363Thr402
Thr185Thr189 Thr336
Ser428Cys430
? ? ATM
PKAp90RSK
PKCζ
38-43
P
P
F
Autophosphorylation site
Phosphorylation site
Farnesylation site
Nuclear localization signal
Fig. 2. Schematic diagram of human LKB1. The human LKB1 protein constitutes 433amino acids. Amino acids 38–43 form a nuclear localization signal domain. Aminoacids 49–309 form the serine/threonine kinase domain. Four autophosphorylationsites have been identified, i.e. Thr185, Thr189, Thr336 and Thr402. Four phosphoryla-tion sites for other kinases have been identified, i.e. Ser31, Ser325, Thr363 and Ser428.Cys430 is a farnesylation site.
197S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
these carcinomas (Table 1). In contrast, one study reported amutationfrequency of 54% in carcinomas in the left-sided colon [31]. Thereforeit was suggested that LKB1 plays an important role in left-sided coloncancer carcinogenesis; however, in a later study no somatic LKB1mutations could be detected in 50 left-sided colon carcinomas [32](Table 1). LOH of 19p13.3 has been detected more frequently in spo-radic CRC with frequencies ranging from 13% to 53% (Table 1). Thissuggests that reduced levels of LKB1 may contribute to the develop-ment of sporadic CRC, though not through mutational inactivation.Lkb1-deficient mice did not develop intestinal carcinomas, nor didthe hamartomatous polyps progress to a more malignant histologicalphenotype when p53was additionally deleted [25,26]. Therefore, it re-mains elusive how loss of LKB1 contributes to colorectal carcinogenesis.
2.2. Pancreatic cancer
PJS patients have an increased risk for developing pancreatic can-cer, but somatic LKB1 mutations have been detected only in a smallproportion of sporadic pancreatic carcinomas (Table 1). In addition,a low frequency of reduced LKB1 expression has been observed inthese carcinomas [33]. However, reintroduction of LKB1 expressionin LKB1-silenced pancreatic cancer cells induced apoptosis, suggestingthat LKB1 suppresses pancreatic cancer cell survival in vitro [34]. Dele-tion of Lkb1 in murine pancreatic epithelial cells of the islets, ducts,and acini resulted in the development of ductal metaplasia andcystadenomas, but no formation of pancreatic intraepithelial neopla-sia (PanIN) or adenocarcinomas [24,35]. Introduction of an oncogenicKrasmutation in heterozygous Lkb1 knockout mice resulted in forma-tion of pancreatic ductal adenocarcinomas, indicating that Lkb1 is ahaplo-insufficient tumor suppressor and cooperates with Kras in thedevelopment of pancreatic cancer in mice [24].
2.3. Breast cancer
Somatic mutations in LKB1 and LOH of 19p13 are not observed fre-quently in primary sporadic breast carcinomas; however, LOH wasfrequently detected in brain metastases from breast carcinomas(Table 1). In addition, it has been shown that LOH of the LKB1 locusin primary breast carcinomas increased significantly as the tumorsprogressed to poorer histological grade, and that low LKB1 expressionin breast carcinomas is associated with poorer histological grade,presence of lymph node metastasis and a shorter overall survival[36–38]. In line with these observations, it has been shown thatLKB1 suppresses breast cancer cell migration and invasion in vitro,and tumor growth, microvessel density and lung metastasis in vivo[39,40]. Together, this suggests that loss of LKB1 is a late event inbreast cancer and promotes breast cancer progression.
2.4. Lung cancer
Despite the fact that lung cancer is not clearly associated with PJS,LKB1 is the third most frequently mutated gene in sporadic non-smallcell lung carcinomas (NSCLC). Though the proportion of missensemu-tations seems to be higher among the somatic mutations in sporadiccancer (45%) than among the germ-line mutations in PJS patients(21%), the mutations detected in NSCLC comprise mainly nonsensemutations or a combination of indels or large intragenic deletions onone LKB1 allele – resulting in truncation of the protein – plus largechromosomal deletions on the other allele [30,41]. Somatic LKB1 mu-tations are most frequently detected in adenocarcinomas, but also inother histological subtypes of NSCLC (Table 1). Notably, LKB1 muta-tions are more frequently observed in NSCLC of Caucasians comparedwith East Asian populations (reviewed in [42]). In addition, LKB1mutations frequently coincide with KRAS mutations. NSCLC patientswith LKB1; KRAS compound mutations tend to have a poorer prog-nosis compared with patients with KRAS-mutated NSCLC without a
concomitant LKB1 mutation, suggesting that loss of LKB1 inducesmore aggressive tumor phenotypes [43,44]. This is also shown inmice where double mutant Lkb1; Kras mice develop more aggressivetumors with higher tumor multiplicity, multiple NSCLC histologiesand more frequent metastasis [45].
Overall, loss of LKB1 is involved in cancers associated with PJS aswell as with a variety of sporadic malignancies, where it is associatedwith tumor progression. Therefore, LKB1 and its downstream sig-naling pathways suit a perfect target for therapeutic intervention ofthese cancers.
3. LKB1 signaling and function
3.1. The LKB1 protein
LKB1 encodes a 48 kDa protein, LKB1, which is ubiquitouslyexpressed in adult as well as fetal tissues, particularly in pancreas,liver, testes and skeletal muscles [14,15]. It contains an evolutionaryconserved serine/threonine kinase domain (Fig. 2), in which the vastmajority of PJS-associated missense mutations are located, result-ing in impaired kinase activity and cell growth suppressive capacity[15,68–70]. In addition to the mutations detected in the kinase do-main, a substantial proportion of PJS-associated mutation as well asmutations detected in sporadic cancers is located in the C-terminusof LKB1. Thesemutations have been shown not to impair kinase activ-ity or its ability to promote growth arrest [71]. In this C-terminalregion of LKB1, some posttranslational modification sites have beenidentified. First, four autophosphorylation sites have been described,i.e. threonine (Thr) 185, Thr189, Thr336 and Thr402 [72,73] (Fig. 2).Autophosphorylation of Thr336 seems not to affect its catalytic ac-tivity nor its cellular localization; however, it may inhibit the cellgrowth suppressive capacity of LKB1 [73]. In addition, four phosphor-ylation sites have been identified, i.e. serine (Ser) 31, Ser325, Thr363(mouse Thr366) and Ser428 (mouse Ser431) [73–75] (Fig. 2).Thr363 is phosphorylated by ATM in response to ionizing radiation[76]. Phosphorylation of Ser428 by cAMP-dependent kinase (PKA),p90RSK, and protein kinase Czeta (PKCζ) is not affecting its catalyticactivity, but it is essential for LKB1 to inhibit cell growth [74,75,77].The kinases for Ser31 and Ser325 have so far not been identified. Final-ly, a prenylation site, i.e. Cys430 (mouse Cys433), has been identified[75] (Fig. 2). Since it has been shown that farnesylation at Cys430 isnot essential for LKB1 to suppress cell growth, the functional relevanceof this prenylation is not yet understood [74].

STRAD
LKB1
MO
25
MARKBRSK
AMPK
mTOR
p70S6K/4E-BPsHIF-1α/VEGF
ATG13/ULK1/2
P21P27
P53
HMGCRACCGS PI3K/AKT
PTEN
Protein synthesis
PhoGTPasesPrimary cilium/mTOR
PhosphoinositideWnt/GSK3β
198 S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
3.2. LKB1 activation
LKB1 contains a nuclear localization signal domain which is likelyto be the reason that LKB1 is normally localized in the nucleus [78](Figs. 2, 3). Because LKB1 lacks a nuclear export domain of its own, itneeds interaction with other proteins in order to be actively exportedout of the nucleus. Activation of LKB1 is therefore associated with itstranslocation to the cytoplasm which is induced upon formation of aheterotrimer with the STE20-related adaptor (STRADα) and scaffold-ingmouse 25 (MO25) proteins [68,72] (Fig. 3). By facilitating the bind-ing of exportins to LKB1 and acting as a competitor for importin-α/β,STRADα prevents nuclear re-localization of LKB1.MO25merely servesas a stabilizer of the LKB1–STRAD interaction [68]. When in complexwith STRADα andMO25 and located in the cytoplasm, LKB1 phosphor-ylates and activates kinases of the AMP-activated kinase (AMPK) family,i.e. AMPKα1, AMPKα2, NUAK1, NUAK2, SIK1, SIK2, QSK, MARK1,MARK2, MARK3, MARK4, BRSK1, BRSK2 and SNRK [79–81] (Fig. 3). No-tably, for several PJS-associated mutations, it has been shown that theresulting LKB1 mutants are retained in the nucleus [15,68–70].
Isoprenoid synthesis Fatty acid synthesis Glycogen synthesis
Energy metabolism
Epithelial andneuronal polarity
Polarized cell migration Asymmetric cell division
Cell polarity
AngiogenesisAutophagyCell cycle
Apoptosis/survival
Cell growth
Fig. 4. LKB1 signaling and function. Via several downstream mediators, LKB1 regulatescrucial cellular processes involved in cell polarity, energy metabolism and cell growth.In addition, LKB1 regulates hematopoietic stem cell homeostasis. Since this seems to beindependent of AMPK/mTOR signaling, the underlying molecular mechanisms are yetto be identified.
3.3. LKB1 in cell polarity
Originally, the Caenorhabditis elegans and Drosophila melanogastercounterparts of LKB1, Par-4 and dLkb1 respectively, have been shownto play major roles in cellular polarity [82,83]. LKB1 induces epithelial-cell polarization through sorting of apical and basolateral mem-brane proteins, formation of epithelial junctions and reorganizationof the actin cytoskeleton, which is at least in part mediated via acti-vation of AMPKα [84–89]. In addition to activation of AMPKα, LKB1regulates polarization through the activation of MARK isoforms(mammalian counterparts of C. elegans and D. melanogaster Par-1),which play a remarkable role in microtubule skeleton organization[90,91]. Through activation of BRSK (or SAD) proteins, LKB1 regu-lates neuronal migration and axonal outgrowth [92,93]. A role forC. elegans and D. melanogaster Par-4/dLkb1 in asymmetric position-ing of the mitotic spindle and cytoplasmic determinants during mi-tosis has been established [94].
Inmammalian cells, LKB1 has recently been shown to be involved inepithelial polarization by controlling Rho GTPases, primary cilium,phosphoinositide and Wnt/GSK3β signaling [95–98]. More specifically,LKB1 is involved in centrosome positioning, lumen initiation andbrush border formation during epithelial morphogenesis [96,97].Together this shows that LKB1 acts as a master kinase regulatingneuronal and epithelial polarization, polarized cell migration andasymmetric cell division (Fig. 4). Given the role of LKB1 in these at-tributions, normal LKB1 function is required for proper functionand proliferation of epithelial tissues. Defects in LKB1 may thereforeresult in tissue disorganization associated with cancer [99–101]. No-tably, PJS-associated mutations as well as mutations detected in spo-radic cancers located in the C-terminus of LKB1 have been shown toimpair cellular polarity [71].
AMPKα1, AMPKα2MARK1, MARK2, MARK3, MARK4BRSK1, BRSK2 SIK1, SIK2 NUAK1, NUAK2 QSKSNRK
cytoplasmnucleus
STRAD
LKB1
MO
25
STRAD
LKB1
MO25
P
Fig. 3. LKB1 activation. Lacking a nuclear export signal of its own, LKB1 resides in thenucleus. Upon binding to STRADα and MO25, LKB1 is exported to the cytoplasm andbecomes catalytically active. In the cytoplasm, LKB1 has been shown to phosphorylatethe 14 serine/threonine kinases of the AMPK family.
3.4. LKB1 in energy metabolism
In addition to the regulation of cellular polarity, AMPK signalingcontrols lipid and glucose metabolism (Fig. 4). In circumstances ofenergy stress due to either excessive ATP consumption, or reducedaerobic ATP production, e.g. in the case of hypoxia, cellular AMP/ATPratios increase. This is sensed by AMPKγ which binds AMP, leadingto the complex formation of AMPKα, -β and -γ subunits [102–104].Threonine residue 172 in the activation loop of AMPKα is now ac-cessible to be phosphorylated by LKB1 [105–107]. Activated AMPKcontrols metabolic processes such as isoprenoid, fatty acid and glyco-gen synthesis via regulation of downstream targets such as HMG-CoA reductase, acetyl CoA carboxylase (ACC) and glycogen synthase[108–110]. Thus, by suppressing energy-consuming processes onthe one hand, and enhancing energy gaining pathways on the other,AMPK activation by LKB1 aids in restoration of the cellular energystatus.
3.5. LKB1 in cell growth
Moreover, LKB1 has been associated with cell growth control viamultiple different signaling pathways (Fig. 4). One such major down-stream pathway of LKB1/AMPK is TSC/mTOR signaling [111]. Activat-ed AMPK phosphorylates TSC2, thereby activating the TSC1:TSC2complex [112], which in turn regulates the activity of the mTORC1, acomplex consisting of mTOR, raptor and mLST8. The activated TSC1:TSC2 complex, which expresses GTPase activity towards Rheb, asmall G-protein that promotes mTORC1 activity when GTP-bound, in-duces conversion of active GTP-bound Rheb to inactive GDP-boundRhebwhich subsequently results in inhibition of mTORC1 [113]. In ad-dition to inhibiting mTORC1 via phosphorylation of TSC2, AMPK di-rectly phosphorylates raptor resulting in the inhibition of mTORC1[114]. mTORC1 plays a key role in protein translation by phosphorylat-ing and activating the ribosomal protein S6 kinase (S6K), and through

NH
H2NHN N
NHCH3
CH3
OH
CH3
CH3
OO
OO
O
O
O
HOH3C
CH3
CH3
CH3
CH3H3C
O
OH
OH3C
H3C
ON
ba
cSTRAD
LKB1
MO
25 AMPK
mTOR
Raptor
mL
ST
8
TSC1
TSC2P P
Metformin Rapamycin
FKBP12
AMP:ATP
Fig. 5. Metformin and rapamycin target LKB1 signaling. a. Chemical structure metfor-min. b. Chemical structure rapamycin (sirolimus). c. Both metformin and rapamycintarget the LKB1/AMPK/mTOR pathway. Metformin induces activation of AMPK viaupregulation of cellular AMP:ATP ratios, resulting in inhibition of mTOR. Rapamycinis a direct mTOR inhibitor. By binding to FKBP12 it induces dissociation of the mTORcomplex c (mTORC1) consisting of mTOR, raptor, and mLST8.
199S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
the inhibition of eukaryotic initiation factor 4E binding proteins(4E-BPs) [115]. In addition, mTORC1 activation stimulates angiogene-sis by stabilizing hypoxia-inducible factor 1α (HIF-1α) in conditionsof hypoxia [116]. Furthermore, mTORC1 activity inhibits autophagyvia phosphorylation of ATG13 and ULK1/2 [117,118]. Thus, LKB1 regu-latesmTOR signaling to control cell growth via different pathways andcellular responses. However, recently, it has been shown that LKB1 isnecessary to maintain hematopoietic stem cell (HSC) homeostasismainly independent of AMPK and mTOR [119–121], indicating thatalso other molecular pathways are involved in LKB1-regulated cellgrowth (Fig. 4).
LKB1 is also involved in cell cycle regulation and apoptosis. Re-introduction of LKB1 expression in cells lacking LKB1 induces cellcycle arrest in G1 [37,122–127], while knocking down LKB1 expres-sion triggers cell cycle progression from G1 to S phase [128]. Notably,reintroduction of LKB1 catalytic deficient mutants in CRC cells couldnot induce cell cycle arrest, but it even upregulated expression of thecell cycle inducer cyclin D [127]. LKB1-induced cell cycle arrest is me-diated by upregulation of the cell cycle inhibitors p21 and/or p27[37,122–125,127,129]. In addition, Lkb1-mediated tumor promotionhas been shown to be mediated by suppression of p21-dependentgrowth arrest in vivo [24]. LKB1-mediated cell cycle regulation hasbeen shown to be regulated via p53-dependent and p53-independentmechanisms [122,123,126,127,129,130]. Additionally, LKB1 has beenshown to interact with and phosphorylate p53, and mediatep53-induced apoptosis [122,131,132], while in Drosophila dLkb1induces p53-independent apoptosis via activation of the JNK pathway[133]. In mice, compound loss of p53 and Lkb1 accelerates onset andincreased incidence of polyps, indicating that Lkb1 and p53 cooperatein tumor development [25,26]. PJS-associated LKB1 mutants havebeen shown to diminish p53 activity [134]. Thus, LKB1 suppresses cellcycle progression and induces apoptosis most likely via suppression ofp21, though the role of p53 in these processes remains contradictory(Fig. 4).
PJS and Cowden syndrome are both hereditary hamartomatouspolyposis syndromes. In Cowden syndrome, patients are affected bya germ-line mutation in phosphatase and tensin homologue (PTEN), aphosphatase involved in the PI3K/AKT survival pathway. Althoughthese syndromes overlap in their clinical characteristics, they alsohave distinct features [135]. Several studies in cells and in micehave shown that LKB1 acts upstream of PTEN (Fig. 4). In fact, LKB1has been shown to interact with and phosphorylate PTEN, whichwas disrupted by introducing PJS-associated mutations in LKB1[136,137]. In addition, LKB1 induces expression and nuclear exportof PTEN resulting in reduced PI3K/AKT signaling and apoptosis[124,137,138]. This LKB1-mediated nuclear export of PTEN has beenshown to be independent of AMPK/mTOR signaling [138]. In vivo,loss of Lkb1 cooperates with Pten loss to accelerate tumorigenesis[23,139]. The tumorigenesis in these compound mutant mice wasshown to be, at least in part, mediated by mTOR signaling since treat-ment with mTOR inhibitors reduced tumor formation [139,140].
These studies reveal a pleiotropic role for LKB1 in cell polarity, en-ergy metabolism, and cell growth, processes that are deregulated incancer. Most of these processes are mediated via AMPK/mTOR signal-ing, suggesting that this major downstream pathway is a suitable can-didate to target for therapy against LKB1-associated cancer. However,other responses induced by loss of LKB1 have been shown to be inde-pendent of AMPK and/or mTOR signaling, suggesting that the tumorsuppression functions of LKB1 are, at least in part, also mediated viaother downstream effectors.
4. Targeting LKB1 signaling in cancer
Since various cancers have been associated with impaired AMPKactivation and/or mTOR inhibition, whether or not triggered by loss ofLKB1, AMPK/mTOR signaling has been suggested to serve a suitable
target for cancer treatment. Both pharmacological AMPK activators(metformin) andmTOR inhibitors (rapamycin and its analogs sirolimus,everolimus and temsirolimus) are available and used in clinical settings.In addition to these clinically approved drugs, additional compoundsaffecting LKB1 signaling have been identified and are being tested inpreclinical settings for their efficacy as anti-cancer agents. Since the dis-covery of LKB1 as the causative gene for PJS, and its signaling routes reg-ulating cell growth, several studies have been focusing on targetingLKB1/AMPK/mTOR signaling in order to treat PJS-associated tumors.
4.1. Metformin
In the 1970s, metformin, a biguanide (Fig. 5a), was approved bythe Food and Drugs Administration (FDA) for the treatment of Dia-betes Mellitus type 2 (DM2) in Europe, and in 1995 in the USA.Since then, millions of persons are using metformin, which has beenshown to increase overall survival and prevent macrovascular com-plications in DM2 patients [141]. Metformin is also used successfullyin polycystic ovarian syndrome (PCOS) and the management of themetabolic syndrome [142,143]. The efficacy of metformin in thesemetabolic disorders is attributed to the potential of metformin to re-duce hepatic gluconeogenesis and improve insulin sensitivity [144].Metformin has been shown to be an activator of AMPK, which isone of the master regulators of these metabolic processes (Fig. 5c)[145]. The interest in metformin as an anti-cancer drug arose whenpopulation studies showed that metformin use is associated with asignificant reduction of neoplasms in general, and of breast, pancreat-ic and prostate cancers in particular [144,146,147]. Therefore, metfor-min might also serve as an anti-tumor agent in cancer prevention andtreatment.
The anti-tumoral effect of metformin could partly be explained byits action in improving blood glucose and insulin levels [148]. Howev-er, metformin induces more direct anti-tumor responses in cancercells as well. Several in vitro studies have shown that metformin treat-ment inhibits cell growth, induces apoptosis, and reduces migrationand invasion in a variety of human cancer cell lines (Table 2). Theanti-proliferative effects have been shown to be mediated via inhibi-tion of insulin-like growth factor-1 (IGF-1) signaling [149–152], butalso via suppression of genes inducing cell cycle S- and M-phases[153–155]. The apoptosis-inducing effects of metformin have been

Table 2Metformin treatment-induced cellular responses in cancer cell lines.
Cancer cell type Inhibiting cellgrowth
Inducingapoptosis
Inhibitingmigration/invasion
Reference
Breasta √ √ √ [150,154,155,157,160,203–206]
Pancreas √ √ √ [151,152,207,208]Colonb √ [171,209]Stomach √ [210]Lungc √ √ [149,156]Ovary √ [159]Prostate √ √ [158,171]Endometrium √ [211]Leukemiad √ [212]
a Part of the actions have been detected in triple-negative breast cancer cells.b These actions have been detected in P53-deficient colon cancer cells specifically.c Non-small cell lung cancer cells.d BCR-ABL-positive chronic myeloid leukemia cells.
200 S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
reported to be p53-dependent, mediated by downregulation of theBcl-2 protein family, targeting ERK and STAT3 signaling, and throughactivation of the JNK/p38MAPK pathway [156–160].
In addition to these anti-proliferative and pro-apoptotic effectsof metformin in vitro, metformin treatment reduces growth of vari-ous tumor types and/or delays tumor onset in animal models(Table 3). Contradictory effects on angiogenesis and metastasishave been reported in xenograft mouse models (Table 3). Metformintreatment reduced tumor growth, microvessel density and metasta-sis in ovarian cancer cell tumors, while it promoted tumor growthand the angiogenic phenotype in ERα-negative breast cancer cell tu-mors [161,162] (Table 3). In another high-energy diet fed xenograftmouse model, metformin did reduce growth of primary triple-negative breast cancer cell tumors, but did not affect metastasis[163] (Table 3). These results suggest that the effects of metforminon tumor progression depend on the tumor type and/or additionalcircumstances such as hormonal and metabolic status. Notably,metformin potentiates the effects of chemotherapeutic agents, suchas cisplatin, paclitaxel, doxorubicin and gemcitabine, suggestingthat metformin serves a potential adjuvant in conventional chemo-therapy [161,164–169].
Most of the in vitro and in vivo anti-tumoral actions of metforminhave been associated with AMPK activation and/or reduction in
Table 3Metformin treatment in animal models.
Tumor type Model
Pancreas Nu/nu mice xenograft MiaPaca-2 cells, Panc-1 cellsNu/nu mice xenograft LNCaP cellsSyrian golden hamsters+BOP+high-fat diet
Colon/intestinal Nu/nu mice xenograft HCT116 cellsa
C57/BL6 mice xenograft MC38 cells+high-energy dietBalb/c mice+AOMApc(Min/+) mice
Breast Nu/nu mice xenograft MDA-MB-231 cellsb
Nu/nu mice xenograft MDA-MB-435 cellsc
Balb/c mice xenograft 66cl4 cellsb+high-energy dietSprague–Dawley rats+NMUHER2/neu transgenic mice
Ovarian Nu/nu mice xenograft A2780 cells
Gastric Nu/nu mice xenograft MKN74 cellsLung A/J mice+NKKMelanoma C57/BL6 mice xenograft B16 cellsVariousd Lkb1fl/+; Pten+/− mice
AOM, azoxymethane; BOP, N-nitrosobis-(2-oxopropyl)amine; NKK, 4-(methylnitrosamino)a P53-deficient.b Triple-negative.c ERα-negative.d Intestinal polyps, lymphomas, pheochromocytomas, and prostate, breast, and pancreat
mTOR activity [158,170–173]. However, other studies specifically re-port AMPK-independent mechanisms of action of metformin as well[174–176]. Even though AMPK is activated upon metformin treat-ment, AMPK is not the direct target of metformin. Metformin is sug-gested to directly target complex I of the mitochondrial respiratorychain which evokes a rise in cellular AMP:ATP ratios [177,178]. Thissubsequently potentiates AMPK to be phosphorylated and activatedby LKB1. Interestingly, metformin has been shown to induce LKB1 cy-tosolic translocation [134,179]. Contradictory results are reportedabout the requirement of LKB1 in metformin-induced AMPK acti-vation and subsequent effects on metabolism in cells [23,175,179–183]. However, in women with PCOS single nucleotide polymor-phisms in LKB1were associated with a significantly reduced responseto metformin treatment [184]. Moreover, metformin failed to inhibitcell growth in cells lacking LKB1, indicating that LKB1 is required formetformin-induced growth inhibitory effects [23,175]. Therefore, itis proposed that metformin is not a suitable drug for the treatmentof tumors showing bi-allelic loss of LKB1 and/or loss of LKB1 expres-sion, as is often the case in PJS patients. In these patients, metforminmight, however, be a useful drug to prevent hamartoma and carcino-ma development and outgrowth, although studies testing this inLkb1-mouse models and in PJS patients have not yet been performed.
Although scarce, clinical evidence for anti-cancer effects ofmetforminhave been reported for sporadic cancer. Metformin use suppressed co-lonic epithelial proliferation and formation of rectal aberrant crypt foci,an early feature of CRC, in non-diabetic patients [170]. More recently,anti-proliferative effects of metformin use in non-diabetic women withoperable invasive breast cancer have been described [185]. All epidemi-ologic, preclinical and clinical evidence has led to the design of a numberof prospective clinical trials investigating metformin therapy for cancerin the neoadjuvant and adjuvant (in combination with standard chemo-therapy or hormone therapy) settings (www.clinicaltrials.gov). Themajority of studies concern breast, prostate, and pancreatic cancer, inline with previous observations of population studies. Two phase IItrials investigate the use of metformin as chemopreventive agent inpre-malignant conditions, i.e. Barrett esophagus (NCT01447927) andcolorectal adenomas (NCT01312467). In addition, one phase III trial iscurrently recruiting participants (NCT01101438), evaluating the effectsof metformin on early-stage breast cancer outcomes, including recur-rence and death. Results of these trials are awaited.
In addition tometformin, other compounds such as TZDs [186], statins[187] and D942 [188] have been shown to activate AMPK indirectly by
Effect of metformin treatment Reference
Reduced tumor growth [152]Reduced tumor growth [213]Reduced tumor incidence [214]Reduced tumor growth [209]Reduced tumor growth [215]Reduced tumor growth (mild) [216]Decreased incidence of large (>2 mm) adenomas [217]Reduced tumor growth [203]Increased tumor growth, increased angiogenesis [162]Reduced primary tumor growth, no effect on metastasis [163]Delayed tumor onset [218]Decreased tumor incidence and size, delayed tumor onset [219]Reduced tumor growth, and microvessel density,and metastatic nodules
[161]
Reduced tumor growth [210]Reduced tumor growth [220]Reduced tumor growth [174]Delayed tumor onset [23]
-1-(3-pyridyl)-1-butanone (tobacco carcinogen); NMU, N-methyl-N-nitrosourea.
ic carcinomas.

201S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
inhibitingmitochondrial ATP production, thereby increasing the cellularAMP/ATP ratio. Also natural polyphenols such as berberine and resver-atrol have been identified as indirect activators of AMPK [189,190].AICAR is another indirect AMPK activator which, after uptake by thecells, is converted to ZMP, an AMP mimetic that binds to the AMPKγsubunit [191,192]. Novel direct AMPK activators such as A-769662[193] and PT1 [194] have been identified, of which A-769662 hasbeen shown to target AMPK subunit β1 [195] while PT1 activates bothα1 and α2 subunits by reducing its auto-inhibition [194]. Preclinicalstudies have shown that these AMPK activators can inhibit tumor cellgrowth [23,196–202].
4.2. Rapamycin
In 1999, rapamycin, a macrolide (Fig. 5b) was approved as animmuno-suppressant by the FDA, and used in organ transplantationto prevent allograft rejection [221]. Rapamycin specifically inhibitsmTOR signaling by binding to the cytosolic FK-binding protein-12(FKBP-12). This rapamycin–FKBP-12 complex binds to the mTORprotein resulting in the dissociation of mTORC1 (Fig. 5c) [222]. Be-cause of the discovery of hyperactivated mTOR signaling in a varietyof human cancers, rapamycin got attention for its putative efficacy ininhibiting cancer cell growth. Numerous studies have revealed thatrapamycin inhibits growth of several cancer cell types such as breast,pancreas, prostate, kidney and lung in vitro, and reduces tumorgrowth and formation of metastases in tumor xenograft animalmodels [223–227]. Notably, rapamycin has been shown to increasethe sensitivity for chemotherapy (gemcitabine) and radiotherapy invitro and in vivo [228–231] Additional in vivo studies demonstratedthat treatment with rapamycin reduces tumor growth in differentgenetically engineered mouse models that spontaneously develop tu-mors (Table 4). In these models, mTOR signaling was activated by de-letion of e.g. Lkb1, Tsc1 or Tsc2, Nf1, or Pten, but also by overexpressionof Akt or Neu/ErbB2 (Table 4). Since these genetic events are commonevents in sporadic cancers as well as in cancers associated with hered-itary hamartoma syndromes such as PJS, tuberous sclerosis complex(TSC), neurofibromatosis (NF) and Cowden's Disease, rapamycin hasbeen suggested to serve as an effective anti-tumor drug in those disor-ders [232].
Clinical trials have been set-up to test the efficacy of rapamycin incancer patients, and in particular for patients with renal cell carcinoma
Table 4Genetically engineered tumor mouse models in which rapamycin treatment reduced tumo
Model Tum
Lkb1+/− mice PJSApcΔ716 mice InteApc(Min/+) mice InteBasal colonic crypt cell-specific Apc−/− mice (Adeno-Cre)a Discis-Nf1+/−:p53−/− mice MaTsc2+/− mice RenENU-treated Tsc2+/− mice RenPten+/− mice PheEndometrial-specific Lkb1−/− mice (Sprr2f-Cre) InvMISIIR-TAg transgenic mice OvaOvarian-specific Apc−/−; Pten−/− mice (Adeno-Cre)b OvaPten+/−; TRβPV/PV mice FollBladder-specific Pten−/−; p53−/− mice (Adeno-Cre)c InvProstate-specific Pten−/− mice (Pb-Cre4) ProPb-Akt1 transgenic mice ProMMTV-NeuYD transgenic mice MaMMTV-NeuYD; VEGF transgenic mice MaMMTV-c-Neu/ErbB2 transgenic mice MaMMTV-PyMT transgenic mice MaBK5-ErbB2 transgenic mice Gal
ENU, N-ethyl-N-nitrosourea; PJS, Peutz–Jeghers syndrome.a Adeno-Cre delivery in distal colon.b Adeno-Cre delivery in ovarian bursae.c Adeno-Cre delivery in bladder lumen.
(RCC), TSC or pancreatic neuroendocrine tumors (NETs). In an in-ternational randomized phase III trial, the efficacy of temsirolimusto treat RCC has been compared with conventional interferon(IFN)α treatment (NCT00065468). Temsirolimus superiorly improvedprogression-free and overall survival in these patients [233]. A ran-domized placebo-controlled phase III trial documented that the use ofeverolimus in patients with advanced RCC after progression onsunitinib and/or sorafenib stabilized tumor progression, and improvedprogression-free survival with acceptable tolerability (RECORD-1,NCT00410124) [234,235]. In two nonrandomized, open-label trials(NCT00457808 and NCT00490789), it has been shown that sirolimustreatment of patients with TSC or sporadic lymphangioleiomyomatosis(LAM) resulted in regression of angiomyolipomas; however, these tu-mors tended to increase in volume after the therapy was stopped[236–238]. In a prospective, open-label phase I/II trial (NCT00411619),everolimus treatment of patients with subependymal giant cell astro-cytomas (SEGA) was associated with marked reduction in tumor vol-ume and seizure frequency [239]. A multicenter phase II trial(NCT00126672) showed that sirolimus treatment of TSC patients in-duced regression of kidney and liver angiomyolipomas, and SEGAs[240]. Two phase II trials showed that everolimus, both alone and com-bined with octreotide long-acting release (LAR), improved disease con-trol in patients with advanced NETs [241,242]. Two placebo-controlledphase III trials have been published investigating everolimus plusoctreotide LAR versus octreotide LAR alone in patients with advancedcarcinoids (RADIANT-2, NCT00412061) and everolimus monotherapyfor advanced pancreatic NET (RADIANT-3, NCT00510068). In both stud-ies everolimus improved progression-free survivalwith a low rate of se-vere adverse events [243,244]. Both temsirolimus (in May 2007) andeverolimus (in March 2009) have been approved by the FDA for thetreatment of advanced RCC after failure of treatment with sunitinib orsorafenib. In October 2010, the FDA approved the use of everolimus totreat subependymal giant cell astrocytomas in individuals with TSCwho require treatment but are not candidates for surgery. In May2011, everolimus has also been approved to treat patientswith progres-sive NETs of the pancreas that are unresectable, locally advanced ormetastatic.
Clinical studies to test the efficacy of mTOR inhibiting drugs inother cancers such as lung, breast, and CRC are currently ongoing.Besides the relatively high frequency of LKB1 mutations in sporadicNSCLC, mutations in EGFR and activation of AKT are frequently
r growth.
or type Reference
-like gastrointestinal polyps [261–264]stinal polyps [265]stinal polyps [266]tal colon tumors [267]lignant peripheral nerve sheath tumors [268]al cystadenomas and liver hemangiomas [269,270]al cystadenomas [271]ochromocytomas and endometrial hyperplasia [272]asive endometrial carcinomas [273]rian tumors [274]rian endometrioid adenocarcinomas [275]icular thyroid carcinomas [276]asive bladder carcinomas [277]state tumors [278]state tumors [279]mmary tumors [280]mmary tumors [280]mmary tumors [281]mmary tumors [282]lbladder tumors [283]

202 S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
detected in these tumors as well [245,246]. Though all these events re-sult in increased mTOR activity, the results of clinically inhibitingmTOR in NSCLC are so far not promising. Combined everolimus/gefitinib therapy in patients with advanced NSCLC led to a partial re-sponse in only 13% of the patients (8/62, of which two patients hadmutations in KRAS) [247]. The LKB1 mutation status in these patientswas not reported. As described in this review, loss of LKB1 is detectedin breast carcinomas; however, loss of PTEN and mutations in PIK3CAhas been detected more frequently. These genetic events all result inaberrant mTOR signaling, and are associated with resistance to hor-monal therapy [248,249]. In a randomized placebo-controlled phaseIII trial (BOLERO-2, NCT00863655), postmenopausal women withhormone-receptor positive advanced breast tumors, resistant to endo-crine therapy alone, were treated with everolimus and an aromataseinhibitor, which improved their progression-free survival [250]. Thisstudy indicates that inhibiting mTOR signaling sensitizes breast carci-nomas to hormonal therapy in human patients. Also for advanced CRC,in which loss of LKB1 occurs infrequently but mTOR hyperactivationhas been reported more commonly [251,252], inhibition of mTORwith rapamycin analogs has been proposed for reversion of chemo-therapy resistance. Several phase I and phase II trials are currently in-vestigating this hypothesis by adding everolimus or temsirolimus tochemotherapy treatment regimens (www.clinicaltrials.gov). Further-more, results of a phase II trial for the treatment of refractory meta-static CRC in 50 patients with a combination of bevacizumab andsirolimus were published last year [253]. Unfortunately, the combina-tion of drugs showed modest activity on the disease and considerableside effects.
In addition to the approved rapalogs sirolimus, temsirolimus andeverolimus, the efficacy of a fourth rapalog, i.e. ridaforolimus, is cur-rently being tested in phase III clinical trials. Besides these mTORC1inhibitors, dual mTORC1 and mTORC2 inhibitors such as AZD8055[140,254,255]; PP242 [256]; OSI-027 [244]; WAY-600; WYE-687;WYE-354 [257]; WYE-132 [258]; KU-0063794 [259]; and X-387[260] have been developed and are currently being tested for their ef-ficacy as anti-tumor agents.
4.3. Combining mTOR inhibitors with other specific pathway inhibitors
Rapamycin has been shown to be most effective in cells that rely onAKT signaling for their proliferation and tumor growth, whereas cellswhich depend on other signaling pathways for their growth are resis-tant to rapamycin [224,284,285]. Tumors that are responsive to rapa-mycin may develop resistance when alternative survival pathways,such as the mitogen-activated extracellular kinase (MAPK/ERK) signal-ing pathway, become activated [267,284]. Combination of rapamycinwith other specific pathway inhibitors could solve this problem of resis-tance. Notably, rapamycin has been shown to synergize with specificinhibitors of e.g. the EGFR/ErbB2, MEK/ERK and Hedgehog signalingin vitro and in vivo [286–295].
In rapamycin-sensitive as well as rapamycin-resistant tumors, in-hibition of mTOR by rapamycin treatment induces a feedback loopresulting in upregulation of PI3K/AKT signaling [296,297]. Therefore,blocking of both PI3K/AKT signaling and mTOR signaling by combin-ing rapamycin with specific PI3K inhibitors (LY294002, Wortmannin,ZSTK474) has been shown to be more effective in tumor-growth inhi-bition than rapamycin alone [297–300]. Recently, a new inhibitor hasbeen identified targeting both mTOR and PI3K, i.e. NVP-BEZ235 [301].NVP-BEZ235 is an imidazo-quinoline derivative, which can bind to theATP-binding cleft of both PI3K and mTOR [301]. Several in vitro and invivo studies have shown that this dual inhibitor induces apoptosis andcell cycle arrest in a wide variety of tumor-cell types, and in most ofthese cases, the anti-tumor efficacy of NVP-BEZ235 was greater com-pared with that of rapamycin (e.g. [298,301–308]). Even in chemo-resistant tumor cells, or tumor cells which have been shown difficultto treat, dual PI3K/mTOR inhibition by NVP-BEZ235 efficiently inhibits
tumor-cell growth, e.g. in KRAS-mutant NSCLC and in breast cancersresistant to ErbB2 inhibitors [309–314]. In addition, NVP-BEZ235enhances sensitivity to chemotherapy and radiotherapy [315–318].In some cases, combining NVP-BEZ235 with MEK or RAF inhibitorsshowed an increased efficacy in inhibiting tumor-cell growth, sug-gesting that some cancers require inhibition of both PI3K/mTOR andMEK/ERK signaling pathways for efficient anti-cancer treatment[296,319–322]. NVP-BEZ235 has entered phase I/II clinical studiesfor patients with advanced breast, renal, endometrial and other solidcancers (www.clinicaltrials.gov).
In addition to the NVP-BEZ235 inhibitor, novel dual PI3K/mTOR in-hibitors such as PI-103 [323–325], PF-04691502 [326,327], GDC-0980[328,329], NVP-BGT226 [330–332], GSK2126458 [333], and PKI-402[334,335] have been developed and are currently under investigationfor their anti-tumor activities.
4.4. Other options to treat PJS-associated tumors
Since germ-line LKB1 mutations predispose to PJS, LKB1/AMPK/mTOR signaling has been proposed as a suitable target for treatmentof PJS-associated hamartomas and carcinomas. In addition to thepreclinical studies using metformin and rapamycin to inhibit tumorcell growth in vitro and in vivo as described in this review, other op-tions for targeted treatment have been suggested. In tumors ofLkb1+/− mice as well as in tumors of PJS patients, elevated levels ofcyclooxygenase-2 (COX-2) have been detected [336,337]. Inhibitionof COX-2 with celecoxib, a non-steroidal anti-inflammatory drug, inLkb1+/− mice reduced tumor burden and was associated with de-creased vascularity [338]. In addition, 2 of 6 PJS patients respondedwell to celecoxib treatment as they showed reduced gastric polyposis[338], suggesting that inhibition of COX-2 serves a suitable strategy totreat PJS-associated lesions.
Recently, the SRC protein has been identified as a target of LKB1 inan integrative genomic and proteomic approach [339]. In this study,Src was shown to be activated in Lkb1-deficient Kras-mutated murineprimary and metastatic lung tumors, which was validated in humanlung carcinoma samples. Inhibition of Src by Dasatinib did not affecttumor growth, but it restored the sensitivity to PI3K/mTOR/MEK inhi-bition using NVP-BEZ235 and AZD2644 [339]. These results suggestthat inhibition of SRC, whether or not in combination with other spe-cific inhibitors, might be a suitable approach for treatment of PJS pa-tients as well, although the activity status of SRC in PJS-associatedlesions has to be determined yet.
5. Conclusion
As described in this review, loss of LKB1 is involved in a variety ofhuman cancers, both in sporadic cancers and in cancers associatedwith PJS. LKB1 signaling has been attributed to a wide diversity of bi-ological processes, involving a multitude of downstream mediators.These biological processes are known to be essential and deregulatedin tumors, indicating the relevance of LKB1 loss in cancer. However,since only little is known about the biological consequences of LKB1loss in carcinomas and the molecular mechanisms underlying thesebiological consequences, the exact tumor suppressor functions ofLKB1 are yet to be elucidated. So far, most efforts to target LKB1 signal-ing in cancer have focused on activating AMPK and inhibiting mTOR.However, since activation of AMPK might require intact LKB1, andsince it has been shown that some molecular and cellular responsesof LKB1 loss are independent of AMPK and/or mTOR activity, targetingthese twomediatorsmight not be efficient in treating LKB1-associatedcancer. More insight into the tumor suppressor functions and mediatingmolecular mechanisms of LKB1 will likely uncover additional and/ornovel proteins and pathways to target, in order to treat LKB1-associatedcancer. Nevertheless, hyperactivation of mTOR downstream of LKB1 isalso mediated via other major signaling pathways such as the PI3K/AKT

203S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
survival pathway, which is frequently aberrantly activated in cancer.More insights into the genes and pathways deregulated in tumors andessential for their growthwill provide novel strategies in order to devel-op therapeutic agents specifically targeting these pathways. In particu-lar, a combination of these specific agents, and/or a combination ofthese agents with conventional hormonal, chemotherapy and radio-therapy are most likely to result in efficient anti-cancer regimens with-out inducing therapy resistance.
List of abbreviations4E-BPs eukaryotic initiation factor 4E binding proteinsACC acetyl CoA carboxylaseAMPK AMP-activated kinaseCOX-2 cyclooxygenase-2CRC colorectal cancerDM2 Diabetes Mellitus type 2FDA Food and Drugs AdministrationFKBP-12 FK-binding protein-12GI gastrointestinalHIF-1α hypoxia-inducible factor 1 alphaHSC hematopoietic stem cellIFN interferonIGF-1 insulin-like growth factor-1LAM lymphangioleiomyomatosisLAR long-acting releaseLKB1 liver kinase B 1LOH loss of heterozygosityMEFs mouse embryonic fibroblastsMO25 scaffolding mouse 25NETs neuroendocrine tumorsNSCLC non-small cell lung carcinomasPCOS polycystic ovarian syndromePJS Peutz–Jeghers syndromePKA cAMP-dependent kinasePKCζ protein kinase C zetaPTEN phosphatase and tensin homologueRCC renal cell carcinomaSEGA subependymal giant cell astrocytomasSer serineSTK11 serine/threonine kinase 11STRADα STE20-related adaptor alphaThr threonineTSC tuberous sclerosis complex
Conflict of interest
The authors do not have any conflicts of interest to declare.
Acknowledgements
This study was supported via ALW Grant #81702002 to MPP.
References
[1] J.L.A. Peutz, Over een zeer merkwaardige, gecombineerde familiaire polyposis vande slijmvliezen van den tractus intestinalis met die van de neuskeelholte engepaardmet eigenaardige pigmentaties van huid en slijmvliezen, Ned. Maandschr.v. Geneesk. 10 (1921) 134–146.
[2] H. Jeghers, K.V. Mc, K.H. Katz, Generalized intestinal polyposis and melaninspots of the oral mucosa, lips and digits; a syndrome of diagnostic significance,N. Engl. J. Med. 241 (1949) 1031–1036.
[3] N.M. Lindor, M.L. McMaster, C.J. Lindor, M.H. Greene, Concise handbook of famil-ial cancer susceptibility syndromes— second edition, J. Natl. Cancer Inst. Monogr.(2008) 1–93.
[4] F.T. Bosman, F. Carneiro, R.H. Hruban, N.D. Theise,WHO Classification of Tumoursof the Digestive System, vol. 3, IARC Press, Lyon, 2010, p. 417.
[5] A.R. Latchford, K. Neale, R.K. Phillips, S.K. Clark, Peutz–Jeghers syndrome: in-triguing suggestion of gastrointestinal cancer prevention from surveillance,Dis. Colon Rectum 54 (2011) 1547–1551.
[6] K. Hizawa, M. Iida, T. Matsumoto, N. Kohrogi, T. Yao, M. Fujishima, Neoplastictransformation arising in Peutz–Jeghers polyposis, Dis. Colon Rectum 36 (1993)953–957.
[7] M.M. Entius, A.M. Westerman, F.M. Giardiello, M.L. van Velthuysen, M.M. Polak,R.J. Slebos, J.H. Wilson, S.R. Hamilton, G.J. Offerhaus, Peutz–Jeghers polyps, dys-plasia, and K-ras codon 12 mutations, Gut 41 (1997) 320–322.
[8] I.P. Tomlinson, R.S. Houlston, Peutz–Jeghers syndrome, J. Med. Genet. 34 (1997)1007–1011.
[9] M.G. van Lier, E.M.Mathus-Vliegen, A.Wagner,M.E. van Leerdam, E.J. Kuipers, Highcumulative risk of intussusception in patients with Peutz–Jeghers syndrome: timeto update surveillance guidelines? Am. J. Gastroenterol. 106 (2011) 940–945.
[10] M.G. van Lier, A. Wagner, E.M. Mathus-Vliegen, E.J. Kuipers, E.W. Steyerberg, M.E.van Leerdam, High cancer risk in Peutz–Jeghers syndrome: a systematic reviewand surveillance recommendations, Am. J. Gastroenterol. 105 (2010) 1258–1264,(author reply 1265).
[11] M.G. van Lier, A.M. Westerman, A. Wagner, C.W. Looman, J.H. Wilson, F.W. deRooij, V.E. Lemmens, E.J. Kuipers, E.M. Mathus-Vliegen, M.E. van Leerdam,High cancer risk and increased mortality in patients with Peutz–Jeghers syn-drome, Gut 60 (2011) 141–147.
[12] F.M. Giardiello, S.B. Welsh, S.R. Hamilton, G.J. Offerhaus, A.M. Gittelsohn, S.V.Booker, A.J. Krush, J.H. Yardley, G.D. Luk, Increased risk of cancer in the Peutz–Jeghers syndrome, N. Engl. J. Med. 316 (1987) 1511–1514.
[13] A. Hemminki, I. Tomlinson, D. Markie, H. Jarvinen, P. Sistonen, A.M. Bjorkqvist, S.Knuutila, R. Salovaara, W. Bodmer, D. Shibata, A. de la Chapelle, L.A. Aaltonen,Localization of a susceptibility locus for Peutz–Jeghers syndrome to 19p usingcomparative genomic hybridization and targeted linkage analysis, Nat. Genet.15 (1997) 87–90.
[14] A. Hemminki, D. Markie, I. Tomlinson, E. Avizienyte, S. Roth, A. Loukola, G.Bignell, W. Warren, M. Aminoff, P. Hoglund, H. Jarvinen, P. Kristo, K. Pelin, M.Ridanpaa, R. Salovaara, T. Toro, W. Bodmer, S. Olschwang, A.S. Olsen, M.R.Stratton, A. de la Chapelle, L.A. Aaltonen, A serine/threonine kinase gene defec-tive in Peutz–Jeghers syndrome, Nature 391 (1998) 184–187.
[15] D.E. Jenne, H. Reimann, J. Nezu, W. Friedel, S. Loff, R. Jeschke, O. Muller, W. Back,M. Zimmer, Peutz–Jeghers syndrome is caused by mutations in a novel serinethreonine kinase, Nat. Genet. 18 (1998) 38–43.
[16] C.I. Amos, M.B. Keitheri-Cheteri, M. Sabripour, C. Wei, T.J. McGarrity, M.F. Seldin, L.Nations, P.M. Lynch, H.H. Fidder, E. Friedman, M.L. Frazier, Genotype–phenotypecorrelations in Peutz–Jeghers syndrome, J. Med. Genet. 41 (2004) 327–333.
[17] A. Ylikorkala, D.J. Rossi, N. Korsisaari, K. Luukko, K. Alitalo, M. Henkemeyer, T.P.Makela, Vascular abnormalities and deregulation of VEGF in Lkb1-deficientmice, Science 293 (2001) 1323–1326.
[18] N. Bardeesy, M. Sinha, A.F. Hezel, S. Signoretti, N.A. Hathaway, N.E. Sharpless, M.Loda, D.R. Carrasco, R.A. DePinho, Loss of the Lkb1 tumour suppressor provokes in-testinal polyposis but resistance to transformation, Nature 419 (2002) 162–167.
[19] H. Miyoshi, M. Nakau, T.O. Ishikawa, M.F. Seldin, M. Oshima, M.M. Taketo, Gas-trointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice,Cancer Res. 62 (2002) 2261–2266.
[20] M.M. Entius, J.J. Keller, A.M. Westerman, B.P. van Rees, M.L. van Velthuysen, A.F.de Goeij, J.H. Wilson, F.M. Giardiello, G.J. Offerhaus, Molecular genetic alter-ations in hamartomatous polyps and carcinomas of patients with Peutz–Jegherssyndrome, J. Clin. Pathol. 54 (2001) 126–131.
[21] S.B. Gruber, M.M. Entius, G.M. Petersen, S.J. Laken, P.A. Longo, R. Boyer, A.M.Levin, U.J. Mujumdar, J.M. Trent, K.W. Kinzler, B. Vogelstein, S.R. Hamilton,M.H. Polymeropoulos, G.J. Offerhaus, F.M. Giardiello, Pathogenesis of adenocar-cinoma in Peutz–Jeghers syndrome, Cancer Res. 58 (1998) 5267–5270.
[22] K. Jishage, J. Nezu, Y. Kawase, T. Iwata, M. Watanabe, A. Miyoshi, A. Ose, K. Habu,T. Kake, N. Kamada, O. Ueda, M. Kinoshita, D.E. Jenne, M. Shimane, H. Suzuki,Role of Lkb1, the causative gene of Peutz–Jegher's syndrome, in embryogenesisand polyposis, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 8903–8908.
[23] X. Huang, S. Wullschleger, N. Shpiro, V.A. McGuire, K. Sakamoto, Y.L. Woods, W.McBurnie, S. Fleming,D.R. Alessi, Important role of the LKB1–AMPKpathway in sup-pressing tumorigenesis in PTEN-deficient mice, Biochem. J. 412 (2008) 211–221.
[24] J.P. Morton, N.B. Jamieson, S.A. Karim, D. Athineos, R.A. Ridgway, C. Nixon, C.J.McKay, R. Carter, V.G. Brunton, M.C. Frame, A. Ashworth, K.A. Oien, T.R. Evans,O.J. Sansom, LKB1 haploinsufficiency cooperates with Kras to promote pancreat-ic cancer through suppression of p21-dependent growth arrest, Gastroenterolo-gy 139 (2010) 586–597, (597 e581-586).
[25] H. Takeda, H. Miyoshi, Y. Kojima, M. Oshima, M.M. Taketo, Accelerated onsets ofgastric hamartomas and hepatic adenomas/carcinomas in Lkb1+/− p53−/−
compound mutant mice, Oncogene 25 (2006) 1816–1820.[26] C. Wei, C.I. Amos, L.C. Stephens, I. Campos, J.M. Deng, R.R. Behringer, A. Rashid,
M.L. Frazier, Mutation of Lkb1 and p53 genes exert a cooperative effect on tu-morigenesis, Cancer Res. 65 (2005) 11297–11303.
[27] P. Katajisto, K. Vaahtomeri, N. Ekman, E. Ventela, A. Ristimaki, N. Bardeesy, R.Feil, R.A. DePinho, T.P. Makela, LKB1 signaling in mesenchymal cells requiredfor suppression of gastrointestinal polyposis, Nat. Genet. 40 (2008) 455–459.
[28] A. Moren, E. Raja, C.H. Heldin, A. Moustakas, Negative regulation of TGFbeta sig-naling by the kinase LKB1 and the scaffolding protein LIP1, J. Biol. Chem. 286(2011) 341–353.
[29] J.L. Herrmann, Y. Byekova, C.A. Elmets, M. Athar, Liver kinase B1 (LKB1) in thepathogenesis of epithelial cancers, Cancer Lett. 306 (2011) 1–9.
[30] M. Sanchez-Cespedes, A role for LKB1 gene in human cancer beyond the Peutz–Jeghers syndrome, Oncogene 26 (2007) 7825–7832.

204 S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
[31] S.M. Dong, K.M. Kim, S.Y. Kim, M.S. Shin, E.Y. Na, S.H. Lee, W.S. Park, N.J. Yoo, J.J.Jang, C.Y. Yoon, J.W. Kim, Y.M. Yang, S.H. Kim, C.S. Kim, J.Y. Lee, Frequent somaticmutations in serine/threonine kinase 11/Peutz–Jeghers syndrome gene inleft-sided colon cancer, Cancer Res. 58 (1998) 3787–3790.
[32] V. Launonen, E. Avizienyte, A. Loukola, P. Laiho, R. Salovaara, H. Jarvinen, J.P.Mecklin, A. Oku, M. Shimane, H.C. Kim, J.C. Kim, J. Nezu, L.A. Aaltonen, No evi-dence of Peutz–Jeghers syndrome gene LKB1 involvement in left-sided colorec-tal carcinomas, Cancer Res. 60 (2000) 546–548.
[33] F. Sahin, A. Maitra, P. Argani, N. Sato, N. Maehara, E. Montgomery, M. Goggins,R.H. Hruban, G.H. Su, Loss of Stk11/Lkb1 expression in pancreatic and biliaryneoplasms, Mod. Pathol. 16 (2003) 686–691.
[34] S. Qanungo, S. Haldar, A. Basu, Restoration of silenced Peutz–Jeghers syndromegene, LKB1, induces apoptosis in pancreatic carcinoma cells, Neoplasia 5 (2003)367–374.
[35] A.F. Hezel, S. Gurumurthy, Z. Granot, A. Swisa, G.C. Chu, G. Bailey, Y. Dor, N.Bardeesy, R.A. Depinho, Pancreatic LKB1 deletion leads to acinar polarity defectsand cystic neoplasms, Mol. Cell. Biol. 28 (2008) 2414–2425.
[36] H. Fenton, B. Carlile, E.A. Montgomery, H. Carraway, J. Herman, F. Sahin, G.H. Su, P.Argani, LKB1 protein expression in human breast cancer, Appl. Immunohistochem.Mol. Morphol. 14 (2006) 146–153.
[37] Z. Shen, X.F. Wen, F. Lan, Z.Z. Shen, Z.M. Shao, The tumor suppressor gene LKB1 isassociated with prognosis in human breast carcinoma, Clin. Cancer Res. 8 (2002)2085–2090.
[38] T.L. Yang, Y.R. Su, C.S. Huang, J.C. Yu, Y.L. Lo, P.E. Wu, C.Y. Shen, High-resolution19p13.2-13.3 allelotyping of breast carcinomas demonstrates frequent loss ofheterozygosity, Genes Chromosom. Cancer 41 (2004) 250–256.
[39] L. Taliaferro-Smith, A. Nagalingam, D. Zhong, W. Zhou, N.K. Saxena, D. Sharma,LKB1 is required for adiponectin-mediated modulation of AMPK–S6K axis andinhibition of migration and invasion of breast cancer cells, Oncogene 28 (2009)2621–2633.
[40] Z.G. Zhuang, G.H. Di, Z.Z. Shen, J. Ding, Z.M. Shao, Enhanced expression of LKB1in breast cancer cells attenuates angiogenesis, invasion, and metastatic poten-tial, Mol. Cancer Res. 4 (2006) 843–849.
[41] V. Launonen, Mutations in the human LKB1/STK11 gene, Hum. Mutat. 26 (2005)291–297.
[42] Y. Gao, G. Ge, H. Ji, LKB1 in lung cancerigenesis: a serine/threonine kinase astumor suppressor, Protein Cell 2 (2011) 99–107.
[43] S. Matsumoto, R. Iwakawa, K. Takahashi, T. Kohno, Y. Nakanishi, Y. Matsuno, K.Suzuki, M. Nakamoto, E. Shimizu, J.D. Minna, J. Yokota, Prevalence and specific-ity of LKB1 genetic alterations in lung cancers, Oncogene 26 (2007) 5911–5918.
[44] R. Onozato, T. Kosaka, H. Achiwa, H. Kuwano, T. Takahashi, Y. Yatabe, T.Mitsudomi, LKB1 gene mutations in Japanese lung cancer patients, Cancer Sci.98 (2007) 1747–1751.
[45] H. Ji, M.R. Ramsey, D.N. Hayes, C. Fan, K. McNamara, P. Kozlowski, C. Torrice, M.C.Wu, T. Shimamura, S.A. Perera, M.C. Liang, D. Cai, G.N. Naumov, L. Bao, C.M.Contreras, D. Li, L. Chen, J. Krishnamurthy, J. Koivunen, L.R. Chirieac, R.F. Padera,R.T. Bronson, N.I. Lindeman, D.C. Christiani, X. Lin, G.I. Shapiro, P.A. Janne, B.E.Johnson, M. Meyerson, D.J. Kwiatkowski, D.H. Castrillon, N. Bardeesy, N.E.Sharpless, K.K. Wong, LKB1 modulates lung cancer differentiation and metasta-sis, Nature 448 (2007) 807–810.
[46] E. Avizienyte, S. Roth, A. Loukola, A. Hemminki, R.A. Lothe, A.E. Stenwig, S.D.Fossa, R. Salovaara, L.A. Aaltonen, Somatic mutations in LKB1 are rare in sporadiccolorectal and testicular tumors, Cancer Res. 58 (1998) 2087–2090.
[47] Z.J. Wang, F. Taylor, M. Churchman, G. Norbury, I. Tomlinson, Genetic pathwaysof colorectal carcinogenesis rarely involve the PTEN and LKB1 genes outside theinherited hamartoma syndromes, Am. J. Pathol. 153 (1998) 363–366.
[48] N. Resta, C. Simone, C. Mareni, M. Montera, M. Gentile, F. Susca, R. Gristina, S.Pozzi, L. Bertario, P. Bufo, N. Carlomagno, M. Ingrosso, F.P. Rossini, R. Tenconi,G. Guanti, STK11 mutations in Peutz–Jeghers syndrome and sporadic colon can-cer, Cancer Res. 58 (1998) 4799–4801.
[49] H. Nakagawa, K. Koyama, S. Nakamori, M. Kameyama, S. Imaoka, M. Monden, Y.Nakamura, Frameshift mutation of the STK11 gene in a sporadic gastrointestinalcancer with microsatellite instability, Jpn. J. Cancer Res. 90 (1999) 633–637.
[50] J. Trojan, A. Brieger, J. Raedle, M. Esteller, S. Zeuzem, 5′-CpG island methylationof the LKB1/STK11 promoter and allelic loss at chromosome 19p13.3 in sporadiccolorectal cancer, Gut 47 (2000) 272–276.
[51] L.F. Forster, S. Defres, D.R. Goudie, D.U. Baty, F.A. Carey, An investigation of thePeutz–Jeghers gene (LKB1) in sporadic breast and colon cancers, J. Clin. Pathol.53 (2000) 791–793.
[52] S.B. Sobottka, M. Haase, G. Fitze, M. Hahn, H.K. Schackert, G. Schackert, Frequentloss of heterozygosity at the 19p13.3 locus without LKB1/STK11 mutations inhuman carcinoma metastases to the brain, J. Neurooncol. 49 (2000) 187–195.
[53] W.S. Park, Y.W. Moon, Y.M. Yang, Y.S. Kim, Y.D. Kim, B.G. Fuller, A.O. Vortmeyer,F. Fogt, I.A. Lubensky, Z. Zhuang, Mutations of the STK11 gene in sporadic gastriccarcinoma, Int. J. Oncol. 13 (1998) 601–604.
[54] E. Avizienyte, A. Loukola, S. Roth, A. Hemminki, M. Tarkkanen, R. Salovaara, J. Arola,R. Butzow, K. Husgafvel-Pursiainen, A. Kokkola, H. Jarvinen, L.A. Aaltonen, LKB1 so-matic mutations in sporadic tumors, Am. J. Pathol. 154 (1999) 677–681.
[55] G.H. Su, R.H. Hruban, R.K. Bansal, G.S. Bova, D.J. Tang, M.C. Shekher, A.M.Westerman, M.M. Entius, M. Goggins, C.J. Yeo, S.E. Kern, Germline and somaticmutations of the STK11/LKB1 Peutz–Jeghers gene in pancreatic and biliary can-cers, Am. J. Pathol. 154 (1999) 1835–1840.
[56] N. Sato, C. Rosty, M. Jansen, N. Fukushima, T. Ueki, C.J. Yeo, J.L. Cameron, C.A.Iacobuzio-Donahue, R.H. Hruban, M. Goggins, STK11/LKB1 Peutz–Jeghers geneinactivation in intraductal papillary-mucinous neoplasms of the pancreas, Am.J. Pathol. 159 (2001) 2017–2022.
[57] R.F. de Wilde, N.A. Ottenhof, M. Jansen, F.H. Morsink, W.W. de Leng, G.J.Offerhaus, L.A. Brosens, Analysis of LKB1 mutations and other molecular alter-ations in pancreatic acinar cell carcinoma, Mod. Pathol. 24 (2011) 1229–1236.
[58] G.R. Bignell, R. Barfoot, S. Seal, N. Collins, W.Warren, M.R. Stratton, Low frequen-cy of somatic mutations in the LKB1/Peutz–Jeghers syndrome gene in sporadicbreast cancer, Cancer Res. 58 (1998) 1384–1386.
[59] Z.J. Wang, M. Churchman, I.G. Campbell, W.H. Xu, Z.Y. Yan, W.G. McCluggage, W.D.Foulkes, I.P. Tomlinson, Allele loss and mutation screen at the Peutz–Jeghers(LKB1) locus (19p13.3) in sporadic ovarian tumours, Br. J. Cancer 80 (1999) 70–72.
[60] D.C. Connolly, H. Katabuchi, W.A. Cliby, K.R. Cho, Somatic mutations in theSTK11/LKB1 gene are uncommon in rare gynecological tumor types associatedwith Peutz–Jegher's syndrome, Am. J. Pathol. 156 (2000) 339–345.
[61] N. Kato, M. Romero, L. Catasus, J. Prat, The STK11/LKB1 Peutz–Jegher gene is notinvolved in the pathogenesis of sporadic sex cord-stromal tumors, although lossof heterozygosity at 19p13.3 indicates other gene alteration in these tumors,Hum. Pathol. 35 (2004) 1101–1104.
[62] M. Sanchez-Cespedes, P. Parrella, M. Esteller, S. Nomoto, B. Trink, J.M. Engles,W.H. Westra, J.G. Herman, D. Sidransky, Inactivation of LKB1/STK11 is a commonevent in adenocarcinomas of the lung, Cancer Res. 62 (2002) 3659–3662.
[63] J.P. Koivunen, J. Kim, J. Lee, A.M. Rogers, J.O. Park, X. Zhao, K. Naoki, I. Okamoto,K. Nakagawa, B.Y. Yeap, M. Meyerson, K.K. Wong, W.G. Richards, D.J. Sugarbaker,B.E. Johnson, P.A. Janne, Mutations in the LKB1 tumour suppressor are frequent-ly detected in tumours from Caucasian but not Asian lung cancer patients, Br. J.Cancer 99 (2008) 245–252.
[64] M. Strazisar, V.Mlakar, T. Rott, D. Glavac, Somatic alterations of the serine/threoninekinase LKB1 gene in squamous cell (SCC) and large cell (LCC) lung carcinoma, Can-cer Investig. 27 (2009) 407–416.
[65] A. Osoegawa, T. Kometani, K. Nosaki, K. Ondo, M. Hamatake, F. Hirai, T. Seto, K.Sugio, Y. Ichinose, LKB1 mutations frequently detected in mucinous bronchio-loalveolar carcinoma, Jpn. J. Clin. Oncol. 41 (2011) 1132–1137.
[66] A. Rowan, V. Bataille, R. MacKie, E. Healy, D. Bicknell, W. Bodmer, I. Tomlinson,Somatic mutations in the Peutz–Jeghers (LKB1/STKII) gene in sporadic malig-nant melanomas, J. Investig. Dermatol. 112 (1999) 509–511.
[67] P. Guldberg, P. thor Straten, V. Ahrenkiel, T. Seremet, A.F. Kirkin, J. Zeuthen, So-matic mutation of the Peutz–Jeghers syndrome gene, LKB1/STK11, in malignantmelanoma, Oncogene 18 (1999) 1777–1780.
[68] J. Boudeau, A.F. Baas, M. Deak, N.A. Morrice, A. Kieloch, M. Schutkowski, A.R.Prescott, H.C. Clevers, D.R. Alessi, MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm,EMBO J. 22 (2003) 5102–5114.
[69] H. Mehenni, C. Gehrig, J. Nezu, A. Oku, M. Shimane, C. Rossier, N. Guex, J.L.Blouin, H.S. Scott, S.E. Antonarakis, Loss of LKB1 kinase activity in Peutz–Jegherssyndrome, and evidence for allelic and locus heterogeneity, Am. J. Hum. Genet.63 (1998) 1641–1650.
[70] A. Ylikorkala, E. Avizienyte, I.P. Tomlinson, M. Tiainen, S. Roth, A. Loukola, A.Hemminki, M. Johansson, P. Sistonen, D. Markie, K. Neale, R. Phillips, P. Zauber,T. Twama, J. Sampson, H. Jarvinen, T.P. Makela, L.A. Aaltonen, Mutations and im-paired function of LKB1 in familial and non-familial Peutz–Jeghers syndromeand a sporadic testicular cancer, Hum. Mol. Genet. 8 (1999) 45–51.
[71] C. Forcet, S. Etienne-Manneville, H. Gaude, L. Fournier, S. Debilly, M. Salmi, A. Baas,S. Olschwang, H. Clevers,M. Billaud, Functional analysis of Peutz–Jeghersmutationsreveals that the LKB1 C-terminal region exerts a crucial role in regulating both theAMPK pathway and the cell polarity, Hum. Mol. Genet. 14 (2005) 1283–1292.
[72] A.F. Baas, J. Boudeau, G.P. Sapkota, L. Smit, R. Medema, N.A. Morrice, D.R. Alessi,H.C. Clevers, Activation of the tumour suppressor kinase LKB1 by the STE20-likepseudokinase STRAD, EMBO J. 22 (2003) 3062–3072.
[73] G.P. Sapkota, J. Boudeau, M. Deak, A. Kieloch, N. Morrice, D.R. Alessi, Identifica-tion and characterization of four novel phosphorylation sites (Ser31, Ser325,Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz–Jeghers cancer syndrome, Biochem. J. 362 (2002) 481–490.
[74] G.P. Sapkota, A. Kieloch, J.M. Lizcano, S. Lain, J.S. Arthur, M.R. Williams, N.Morrice, M. Deak, D.R. Alessi, Phosphorylation of the protein kinase mutatedin Peutz–Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) andcAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essen-tial for LKB1 to suppress cell growth, J. Biol. Chem. 276 (2001) 19469–19482.
[75] S.P. Collins, J.L. Reoma, D.M. Gamm, M.D. Uhler, LKB1, a novel serine/threonineprotein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo, Biochem. J. 345 (Pt 3)(2000) 673–680.
[76] G.P. Sapkota, M. Deak, A. Kieloch, N. Morrice, A.A. Goodarzi, C. Smythe, Y. Shiloh,S.P. Lees-Miller, D.R. Alessi, Ionizing radiation induces ataxia telangiectasia mu-tated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366,Biochem. J. 368 (2002) 507–516.
[77] P. Song, Z. Xie, Y. Wu, J. Xu, Y. Dong, M.H. Zou, Protein kinase Czeta-dependentLKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosisin endothelial cells, J. Biol. Chem. 283 (2008) 12446–12455.
[78] D.P. Smith, J. Spicer, A. Smith, S. Swift, A. Ashworth, The mouse Peutz–Jeghers syn-drome gene Lkb1 encodes a nuclear protein kinase, Hum. Mol. Genet. 8 (1999)1479–1485.
[79] S.A. Hawley, J. Boudeau, J.L. Reid, K.J. Mustard, L. Udd, T.P. Makela, D.R. Alessi,D.G. Hardie, Complexes between the LKB1 tumor suppressor, STRAD alpha/betaand MO25 alpha/beta are upstream kinases in the AMP-activated protein kinasecascade, J. Biol. 2 (2003) 28.
[80] M. Jaleel, A. McBride, J.M. Lizcano, M. Deak, R. Toth, N.A. Morrice, D.R. Alessi,Identification of the sucrose non-fermenting related kinase SNRK, as a novelLKB1 substrate, FEBS Lett. 579 (2005) 1417–1423.

205S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
[81] J.M. Lizcano, O. Goransson, R. Toth, M. Deak, N.A. Morrice, J. Boudeau, S.A.Hawley, L. Udd, T.P. Makela, D.G. Hardie, D.R. Alessi, LKB1 is a master kinasethat activates 13 kinases of the AMPK subfamily, including MARK/PAR-1, EMBOJ. 23 (2004) 833–843.
[82] S.G. Martin, D. St Johnston, A role for Drosophila LKB1 in anterior–posterior axisformation and epithelial polarity, Nature 421 (2003) 379–384.
[83] J.L. Watts, D.G. Morton, J. Bestman, K.J. Kemphues, The C. elegans par-4 gene en-codes a putative serine-threonine kinase required for establishing embryonicasymmetry, Development 127 (2000) 1467–1475.
[84] A.F. Baas, J. Kuipers, N.N. van der Wel, E. Batlle, H.K. Koerten, P.J. Peters, H.C.Clevers, Complete polarization of single intestinal epithelial cells upon activa-tion of LKB1 by STRAD, Cell 116 (2004) 457–466.
[85] C.R. Cowan, A.A. Hyman, Acto-myosin reorganization and PAR polarity in C.elegans, Development 134 (2007) 1035–1043.
[86] J.H. Lee, H. Koh, M. Kim, Y. Kim, S.Y. Lee, R.E. Karess, S.H. Lee, M. Shong, J.M. Kim,J. Kim, J. Chung, Energy-dependent regulation of cell structure by AMP-activatedprotein kinase, Nature 447 (2007) 1017–1020.
[87] L. Zhang, J. Li, L.H. Young, M.J. Caplan, AMP-activated protein kinase regulates theassembly of epithelial tight junctions, Proc. Natl. Acad. Sci. U. S. A. 103 (2006)17272–17277.
[88] S. Zhang, K. Schafer-Hales, F.R. Khuri, W. Zhou, P.M. Vertino, A.I. Marcus, Thetumor suppressor LKB1 regulates lung cancer cell polarity by mediating cdc42recruitment and activity, Cancer Res. 68 (2008) 740–748.
[89] B. Zheng, L.C. Cantley, Regulation of epithelial tight junction assembly and disas-sembly by AMP-activated protein kinase, Proc. Natl. Acad. Sci. U. S. A. 104 (2007)819–822.
[90] G. Drewes, A. Ebneth, E.M. Mandelkow, MAPs, MARKs and microtubule dynam-ics, Trends Biochem. Sci. 23 (1998) 307–311.
[91] Y. Kojima,H.Miyoshi, H.C. Clevers,M. Oshima,M. Aoki,M.M. Taketo, Suppression oftubulin polymerization by the LKB1-microtubule-associated protein/microtubuleaffinity-regulating kinase signaling, J. Biol. Chem. 282 (2007) 23532–23540.
[92] A.P. Barnes, B.N. Lilley, Y.A. Pan, L.J. Plummer, A.W. Powell, A.N. Raines, J.R.Sanes, F. Polleux, LKB1 and SAD kinases define a pathway required for the polar-ization of cortical neurons, Cell 129 (2007) 549–563.
[93] M. Kishi, Y.A. Pan, J.G. Crump, J.R. Sanes, Mammalian SAD kinases are requiredfor neuronal polarization, Science 307 (2005) 929–932.
[94] J. Betschinger, J.A. Knoblich, Dare to be different: asymmetric cell division inDrosophila, C. elegans and vertebrates, Curr. Biol. 14 (2004) R674–R685.
[95] C. Boehlke, F. Kotsis, V. Patel, S. Braeg, H. Voelker, S. Bredt, T. Beyer, H. Janusch, C.Hamann, M. Godel, K. Muller, M. Herbst, M. Hornung, M. Doerken, M. Kottgen, R.Nitschke, P. Igarashi, G. Walz, E.W. Kuehn, Primary cilia regulate mTORC1 activ-ity and cell size through Lkb1, Nat. Cell Biol. 12 (2010) 1115–1122.
[96] M. Gloerich, J.P. ten Klooster, M.J. Vliem, T. Koorman, F.J. Zwartkruis, H. Clevers,J.L. Bos, Rap2A links intestinal cell polarity to brush border formation, Nat. CellBiol. 14 (2012) 793–801.
[97] A.E. Rodriguez-Fraticelli, M. Auzan, M.A. Alonso, M. Bornens, F. Martin-Belmonte,Cell confinement controls centrosome positioning and lumen initiation duringepithelial morphogenesis, J. Cell Biol. 198 (2012) 1011–1023.
[98] O. Ossipova, N. Bardeesy, R.A. DePinho, J.B. Green, LKB1 (XEEK1) regulates Wntsignalling in vertebrate development, Nat. Cell Biol. 5 (2003) 889–894.
[99] F. Martin-Belmonte, M. Perez-Moreno, Epithelial cell polarity, stem cells andcancer, Nat. Rev. Cancer 12 (2012) 23–38.
[100] M. Jansen, J.P. Ten Klooster, G.J. Offerhaus, H. Clevers, LKB1 and AMPK family sig-naling: the intimate link between cell polarity and energy metabolism, Physiol.Rev. 89 (2009) 777–798.
[101] L.M. McCaffrey, I.G. Macara, Epithelial organization, cell polarity and tumorigen-esis, Trends Cell Biol. 21 (2011) 727–735.
[102] J.M. Corton, J.G. Gillespie, D.G. Hardie, Role of the AMP-activated protein kinasein the cellular stress response, Curr. Biol. 4 (1994) 315–324.
[103] D.G. Hardie, Minireview: the AMP-activated protein kinase cascade: the keysensor of cellular energy status, Endocrinology 144 (2003) 5179–5183.
[104] A. Woods, P.C. Cheung, F.C. Smith, M.D. Davison, J. Scott, R.K. Beri, D. Carling, Char-acterization of AMP-activated protein kinase beta and gamma subunits. Assemblyof the heterotrimeric complex in vitro, J. Biol. Chem. 271 (1996) 10282–10290.
[105] S.A. Hawley, M. Davison, A. Woods, S.P. Davies, R.K. Beri, D. Carling, D.G. Hardie,Characterization of the AMP-activated protein kinase from rat liver and identifica-tion of threonine 172 as the major site at which it phosphorylates AMP-activatedprotein kinase, J. Biol. Chem. 271 (1996) 27879–27887.
[106] S.P. Hong, F.C. Leiper, A. Woods, D. Carling, M. Carlson, Activation of yeast Snf1and mammalian AMP-activated protein kinase by upstream kinases, Proc. Natl.Acad. Sci. U. S. A. 100 (2003) 8839–8843.
[107] R.J. Shaw, M. Kosmatka, N. Bardeesy, R.L. Hurley, L.A. Witters, R.A. DePinho, L.C.Cantley, The tumor suppressor LKB1 kinase directly activates AMP-activated ki-nase and regulates apoptosis in response to energy stress, Proc. Natl. Acad. Sci.U. S. A. 101 (2004) 3329–3335.
[108] D. Carling, P.R. Clarke, V.A. Zammit, D.G. Hardie, Purification and characteriza-tion of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxyl-ase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities, Eur.J. Biochem. 186 (1989) 129–136.
[109] D. Carling, D.G. Hardie, The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphory-lase kinase, Biochim. Biophys. Acta 1012 (1989) 81–86.
[110] A.T. Sim, D.G. Hardie, The low activity of acetyl-CoA carboxylase in basal andglucagon-stimulated hepatocytes is due to phosphorylation by the AMP-activatedprotein kinase and not cyclic AMP-dependent protein kinase, FEBS Lett. 233(1988) 294–298.
[111] R.J. Shaw, N. Bardeesy, B.D. Manning, L. Lopez, M. Kosmatka, R.A. DePinho, L.C.Cantley, The LKB1 tumor suppressor negatively regulates mTOR signaling, Can-cer Cell 6 (2004) 91–99.
[112] K. Inoki, T. Zhu, K.L. Guan, TSC2 mediates cellular energy response to control cellgrowth and survival, Cell 115 (2003) 577–590.
[113] Y. Zhang, X. Gao, L.J. Saucedo, B. Ru, B.A. Edgar, D. Pan, Rheb is a direct target of thetuberous sclerosis tumour suppressor proteins, Nat. Cell Biol. 5 (2003) 578–581.
[114] D.M. Gwinn, D.B. Shackelford, D.F. Egan, M.M. Mihaylova, A. Mery, D.S. Vasquez,B.E. Turk, R.J. Shaw, AMPK phosphorylation of raptor mediates a metaboliccheckpoint, Mol. Cell 30 (2008) 214–226.
[115] K. Hara, K. Yonezawa, M.T. Kozlowski, T. Sugimoto, K. Andrabi, Q.P. Weng, M.Kasuga, I. Nishimoto, J. Avruch, Regulation of eIF-4E BP1 phosphorylation bymTOR, J. Biol. Chem. 272 (1997) 26457–26463.
[116] C.C. Hudson, M. Liu, G.G. Chiang, D.M. Otterness, D.C. Loomis, F. Kaper, A.J.Giaccia, R.T. Abraham, Regulation of hypoxia-inducible factor 1alpha expressionand function by the mammalian target of rapamycin, Mol. Cell. Biol. 22 (2002)7004–7014.
[117] N. Hosokawa, T. Hara, T. Kaizuka, C. Kishi, A. Takamura, Y. Miura, S. Iemura, T.Natsume, K. Takehana, N. Yamada, J.L. Guan, N. Oshiro, N. Mizushima, Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex requiredfor autophagy, Mol. Biol. Cell 20 (2009) 1981–1991.
[118] C.H. Jung, C.B. Jun, S.H. Ro, Y.M. Kim, N.M. Otto, J. Cao, M. Kundu, D.H. Kim,ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy ma-chinery, Mol. Biol. Cell 20 (2009) 1992–2003.
[119] B. Gan, J. Hu, S. Jiang, Y. Liu, E. Sahin, L. Zhuang, E. Fletcher-Sananikone, S. Colla,Y.A. Wang, L. Chin, R.A. Depinho, Lkb1 regulates quiescence and metabolic ho-meostasis of haematopoietic stem cells, Nature 468 (2010) 701–704.
[120] S. Gurumurthy, S.Z. Xie, B. Alagesan, J. Kim, R.Z. Yusuf, B. Saez, A. Tzatsos, F.Ozsolak, P. Milos, F. Ferrari, P.J. Park, O.S. Shirihai, D.T. Scadden, N. Bardeesy,The Lkb1 metabolic sensor maintains haematopoietic stem cell survival, Nature468 (2010) 659–663.
[121] D. Nakada, T.L. Saunders, S.J. Morrison, Lkb1 regulates cell cycle and energy me-tabolism in haematopoietic stem cells, Nature 468 (2010) 653–658.
[122] P.Y. Zeng, S.L. Berger, LKB1 is recruited to the p21/WAF1 promoter by p53 tomediate transcriptional activation, Cancer Res. 66 (2006) 10701–10708.
[123] M. Tiainen, K. Vaahtomeri, A. Ylikorkala, T.P. Makela, Growth arrest by the LKB1tumor suppressor: induction of p21(WAF1/CIP1), Hum. Mol. Genet. 11 (2002)1497–1504.
[124] A.I. Jimenez, P. Fernandez, O. Dominguez, A. Dopazo,M. Sanchez-Cespedes, Growthand molecular profile of lung cancer cells expressing ectopic LKB1: down-regulation of the phosphatidylinositol 3′-phosphate kinase/PTEN pathway, CancerRes. 63 (2003) 1382–1388.
[125] X. Xie, Z. Wang, Y. Chen, Association of LKB1 with a WD-repeat protein WDR6 isimplicated in cell growth arrest and p27(Kip1) induction, Mol. Cell. Biochem.301 (2007) 115–122.
[126] S. Gurumurthy, A.F. Hezel, E. Sahin, J.H. Berger, M.W. Bosenberg, N. Bardeesy,LKB1 deficiency sensitizes mice to carcinogen-induced tumorigenesis, CancerRes. 68 (2008) 55–63.
[127] K.D. Scott, S. Nath-Sain, M.D. Agnew, P.A. Marignani, LKB1 catalytically deficientmutants enhance cyclin D1 expression, Cancer Res. 67 (2007) 5622–5627.
[128] X. Liang, P. Wang, Q. Gao, T. Xiang, X. Tao, Endogenous LKB1 knockdown accel-erates G(1)/S transition through p53 and p16 pathways, Cancer Biol. Ther. 9(2010) 156–160.
[129] T. Setogawa, S. Shinozaki-Yabana, T. Masuda, K. Matsuura, T. Akiyama, Thetumor suppressor LKB1 induces p21 expression in collaboration with LMO4,GATA-6, and Ldb1, Biochem. Biophys. Res. Commun. 343 (2006) 1186–1190.
[130] J. Liang, S.H. Shao, Z.X. Xu, B. Hennessy, Z. Ding, M. Larrea, S. Kondo, D.J. Dumont,J.U. Gutterman, C.L. Walker, J.M. Slingerland, G.B. Mills, The energy sensingLKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the deci-sion to enter autophagy or apoptosis, Nat. Cell Biol. 9 (2007) 218–224.
[131] H. Cheng, P. Liu, Z.C. Wang, L. Zou, S. Santiago, V. Garbitt, O.V. Gjoerup, J.D.Iglehart, A. Miron, A.L. Richardson, W.C. Hahn, J.J. Zhao, SIK1 couples LKB1 top53-dependent anoikis and suppresses metastasis, Sci. Signal. 2 (2009) ra35.
[132] P. Karuman, O. Gozani, R.D. Odze, X.C. Zhou, H. Zhu, R. Shaw, T.P. Brien, C.D.Bozzuto, D. Ooi, L.C. Cantley, J. Yuan, The Peutz–Jegher gene product LKB1 is amediator of p53-dependent cell death, Mol. Cell 7 (2001) 1307–1319.
[133] J.H. Lee, H. Koh, M. Kim, J. Park, S.Y. Lee, S. Lee, J. Chung, JNK pathway mediatesapoptotic cell death induced by tumor suppressor LKB1 in Drosophila, Cell DeathDiffer. 13 (2006) 1110–1122.
[134] S.S. Deepa, L. Zhou, J. Ryu, C. Wang, X. Mao, C. Li, N. Zhang, N. Musi, R.A.DeFronzo, F. Liu, L.Q. Dong, APPL1 mediates adiponectin-induced LKB1 cytosoliclocalization through the PP2A-PKCzeta signaling pathway, Mol. Endocrinol. 25(2011) 1773–1785.
[135] J.A. Hobert, C. Eng, PTEN hamartoma tumor syndrome: an overview, Genet. Med.11 (2009) 687–694.
[136] H. Mehenni, N. Lin-Marq, K. Buchet-Poyau, A. Reymond, M.A. Collart, D. Picard,S.E. Antonarakis, LKB1 interacts with and phosphorylates PTEN: a functionallink between two proteins involved in cancer predisposing syndromes, Hum.Mol. Genet. 14 (2005) 2209–2219.
[137] P. Song, Y.Wu, J. Xu, Z. Xie, Y. Dong,M. Zhang,M.H. Zou, Reactive nitrogen species in-duced by hyperglycemia suppresses Akt signaling and triggers apoptosis by up-regulating phosphatase PTEN (phosphatase and tensin homologue deleted onchromosome 10) in an LKB1-dependentmanner, Circulation 116 (2007) 1585–1595.
[138] L. Liu, X. Du, J. Nie, A novel de novo mutation in LKB1 gene in a Chinese PeutzJeghers syndrome patient significantly diminished p53 activity, Clin. Res. Hepatol.Gastroenterol. 35 (2011) 221–226.

206 S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
[139] B.Y. Shorning, D. Griffiths, A.R. Clarke, Lkb1 and Pten synergise to suppressmTOR-mediated tumorigenesis and epithelial-mesenchymal transition in themouse bladder, PLoS One 6 (2011) e16209.
[140] J.M. Garcia-Martinez, S. Wullschleger, G. Preston, S. Guichard, S. Fleming, D.R.Alessi, S.L. Duce, Effect of PI3K- and mTOR-specific inhibitors on spontaneousB-cell follicular lymphomas in PTEN/LKB1-deficient mice, Br. J. Cancer 104(2011) 1116–1125.
[141] United Kingdom Prospective Diabetes Study (UKPDS), 13: Relative efficacyof randomly allocated diet, sulphonylurea, insulin, or metformin in patientswith newly diagnosed non-insulin dependent diabetes followed for threeyears, BMJ 310 (1995) 83–88.
[142] E. Diamanti-Kandarakis, F. Economou, S. Palimeri, C. Christakou, Metformin inpolycystic ovary syndrome, Ann. N. Y. Acad. Sci. 1205 (2010) 192–198.
[143] C. Bianchi, G. Penno, F. Romero, S. Del Prato, R. Miccoli, Treating the metabolicsyndrome, Expert. Rev. Cardiovasc. Ther. 5 (2007) 491–506.
[144] B. Viollet, B. Guigas, N. Sanz Garcia, J. Leclerc, M. Foretz, F. Andreelli, Cellular andmolecular mechanisms of metformin: an overview, Clin. Sci. (Lond.) 122 (2012)253–270.
[145] G. Zhou, R. Myers, Y. Li, Y. Chen, X. Shen, J. Fenyk-Melody, M. Wu, J. Ventre, T.Doebber, N. Fujii, N. Musi, M.F. Hirshman, L.J. Goodyear, D.E. Moller, Role ofAMP-activated protein kinase in mechanism of metformin action, J. Clin. Invest.108 (2001) 1167–1174.
[146] N. Papanas, E. Maltezos, D.P. Mikhailidis, Metformin and cancer: licence to heal?Expert Opin. Investig. Drugs 19 (2010) 913–917.
[147] J. Li, B.N. Zhang, J.H. Fan, Y. Pang, P. Zhang, S.L. Wang, S. Zheng, B. Zhang, H.J.Yang, X.M. Xie, Z.H. Tang, H. Li, J.Y. Li, J.J. He, Y.L. Qiao, A nation-wide multicenter10-year (1999–2008) retrospective clinical epidemiological study of femalebreast cancer in China, BMC Cancer 11 (2011) 364.
[148] P.J. Goodwin, K.I. Pritchard, M. Ennis, M. Clemons, M. Graham, I.G. Fantus,Insulin-lowering effects of metformin in women with early breast cancer, Clin.Breast Cancer 8 (2008) 501–505.
[149] B. Salani, S. Maffioli, M. Hamoudane, A. Parodi, S. Ravera, M. Passalacqua, A.Alama, M. Nhiri, R. Cordera, D. Maggi, Caveolin-1 is essential for metformin in-hibitory effect on IGF1 action in non-small-cell lung cancer cells, FASEB J. 26(2012) 788–798.
[150] B. Liu, Z. Fan, S.M. Edgerton, X. Yang, S.E. Lind, A.D. Thor, Potent anti-proliferativeeffects of metformin on trastuzumab-resistant breast cancer cells via inhibitionof erbB2/IGF-1 receptor interactions, Cell Cycle 10 (2011) 2959–2966.
[151] E. Rozengurt, J. Sinnett-Smith, K. Kisfalvi, Crosstalk between insulin/insulin-likegrowth factor-1 receptors and G protein-coupled receptor signaling systems: anovel target for the antidiabetic drug metformin in pancreatic cancer, Clin. Can-cer Res. 16 (2010) 2505–2511.
[152] K. Kisfalvi, G. Eibl, J. Sinnett-Smith, E. Rozengurt, Metformin disrupts crosstalkbetween G protein-coupled receptor and insulin receptor signaling systemsand inhibits pancreatic cancer growth, Cancer Res. 69 (2009) 6539–6545.
[153] C. Oliveras-Ferraros, A. Vazquez-Martin, J.A. Menendez, Genome-wide inhibito-ry impact of the AMPK activator metformin on [kinesins, tubulins, histones, au-roras and polo-like kinases] M-phase cell cycle genes in human breast cancercells, Cell Cycle 8 (2009) 1633–1636.
[154] I.N. Alimova, B. Liu, Z. Fan, S.M. Edgerton, T. Dillon, S.E. Lind, A.D. Thor, Metfor-min inhibits breast cancer cell growth, colony formation and induces cell cyclearrest in vitro, Cell Cycle 8 (2009) 909–915.
[155] Y. Zhuang, W.K. Miskimins, Cell cycle arrest in metformin treated breast cancercells involves activation of AMPK, downregulation of cyclin D1, and requiresp27Kip1 or p21Cip1, J. Mol. Signal. 3 (2008) 18.
[156] N.Wu, C. Gu, H. Gu, H. Hu, Y. Han, Q. Li, Metformin induces apoptosis of lung can-cer cells through activating JNK/p38 MAPK pathway and GADD153, Neoplasma58 (2011) 482–490.
[157] A. Malki, A. Youssef, Antidiabetic drug metformin induces apoptosis in humanMCF breast cancer via targeting ERK signaling, Oncol. Res. 19 (2011) 275–285.
[158] I. Ben Sahra, K. Laurent, S. Giuliano, F. Larbret, G. Ponzio, P. Gounon, Y. LeMarchand-Brustel, S. Giorgetti-Peraldi, M. Cormont, C. Bertolotto, M. Deckert,P. Auberger, J.F. Tanti, F. Bost, Targeting cancer cell metabolism: the combinationof metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostatecancer cells, Cancer Res. 70 (2010) 2465–2475.
[159] A. Yasmeen, M.C. Beauchamp, E. Piura, E. Segal, M. Pollak, W.H. Gotlieb, Induc-tion of apoptosis by metformin in epithelial ovarian cancer: involvement ofthe Bcl-2 family proteins, Gynecol. Oncol. 121 (2011) 492–498.
[160] X.S. Deng, S. Wang, A. Deng, B. Liu, S.M. Edgerton, S.E. Lind, R. Wahdan-Alaswad,A.D. Thor, Metformin targets Stat3 to inhibit cell growth and induce apoptosis intriple-negative breast cancers, Cell Cycle 11 (2012) 367–376.
[161] R. Rattan, R.P. Graham, J.L. Maguire, S. Giri, V. Shridhar, Metformin suppressesovarian cancer growth and metastasis with enhancement of cisplatin cytotoxic-ity in vivo, Neoplasia 13 (2011) 483–491.
[162] K.N. Phoenix, F. Vumbaca, K.P. Claffey, Therapeutic metformin/AMPK activationpromotes the angiogenic phenotype in the ERalpha negative MDA-MB-435breast cancer model, Breast Cancer Res. Treat. 113 (2009) 101–111.
[163] K.N. Phoenix, F. Vumbaca, M.M. Fox, R. Evans, K.P. Claffey, Dietary energy avail-ability affects primary and metastatic breast cancer and metformin efficacy,Breast Cancer Res. Treat. 123 (2010) 333–344.
[164] G. Chen, S. Xu, K. Renko, M. Derwahl, Metformin inhibits growth of thyroid carcino-ma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the ef-fect of chemotherapeutic agents, J. Clin. Endocrinol. Metab. 97 (2012) E510–520.
[165] H.A. Hirsch, D. Iliopoulos, P.N. Tsichlis, K. Struhl, Metformin selectively targetscancer stem cells, and acts together with chemotherapy to block tumor growthand prolong remission, Cancer Res. 69 (2009) 7507–7511.
[166] R.K. Hanna, C. Zhou, K.M. Malloy, P.A. Gehrig, V.L. Bae-Jump, Metformin potenti-ates the effects of paclitaxel in endometrial cancer cells through inhibition of cellproliferation and modulation of the mTOR pathway, Gynecol. Oncol. 125 (2012)458–469.
[167] T. Kawanami, S. Takiguchi, N. Ikeda, A. Funakoshi, A humanized anti-IGF-1Rmono-clonal antibody (R1507) and/or metformin, Oncol. Rep. 27 (2012) 867–872.
[168] G.Z. Rocha, M.M. Dias, E.R. Ropelle, F. Osorio-Costa, F.A. Rossato, A.E. Vercesi, M.J.Saad, J.B. Carvalheira, Metformin amplifies chemotherapy-induced AMPK acti-vation and antitumoral growth, Clin. Cancer Res. 17 (2011) 3993–4005.
[169] D. Iliopoulos, H.A. Hirsch, K. Struhl, Metformin decreases the dose of chemother-apy for prolonging tumor remission in mouse xenografts involving multiplecancer cell types, Cancer Res. 71 (2011) 3196–3201.
[170] K. Hosono, H. Endo, H. Takahashi, M. Sugiyama, E. Sakai, T. Uchiyama, K. Suzuki,H. Iida, Y. Sakamoto, K. Yoneda, T. Koide, C. Tokoro, Y. Abe, M. Inamori, H.Nakagama, A. Nakajima, Metformin suppresses colorectal aberrant crypt fociin a short-term clinical trial, Cancer Prev. Res. (Phila) 3 (2010) 1077–1083.
[171] M. Zakikhani, R.J. Dowling, N. Sonenberg, M.N. Pollak, The effects of adiponectinand metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase, Cancer Prev. Res. (Phila) 1 (2008) 369–375.
[172] L. Tosca, C. Rame, C. Chabrolle, S. Tesseraud, J. Dupont, Metformin decreasesIGF1-induced cell proliferation and protein synthesis through AMP-activated pro-tein kinase in cultured bovine granulosa cells, Reproduction 139 (2010) 409–418.
[173] K.A. Brown, N.I. Hunger, M. Docanto, E.R. Simpson, Metformin inhibits aroma-tase expression in human breast adipose stromal cells via stimulation ofAMP-activated protein kinase, Breast Cancer Res. Treat. 123 (2010) 591–596.
[174] K. Janjetovic, L. Harhaji-Trajkovic, M. Misirkic-Marjanovic, L. Vucicevic, D.Stevanovic, N. Zogovic, M. Sumarac-Dumanovic, D. Micic, V. Trajkovic, In vitro andin vivo anti-melanoma action ofmetformin, Eur. J. Pharmacol. 668 (2011) 373–382.
[175] R. Rattan, S. Giri, L.C. Hartmann, V. Shridhar, Metformin attenuates ovariancancer cell growth in an AMP-kinase dispensable manner, J. Cell. Mol. Med. 15(2011) 166–178.
[176] K. Janjetovic, L. Vucicevic, M. Misirkic, U. Vilimanovich, G. Tovilovic, N. Zogovic,Z. Nikolic, S. Jovanovic, V. Bumbasirevic, V. Trajkovic, L. Harhaji-Trajkovic, Met-formin reduces cisplatin-mediated apoptotic death of cancer cells throughAMPK-independent activation of Akt, Eur. J. Pharmacol. 651 (2011) 41–50.
[177] M.Y. El-Mir, V. Nogueira, E. Fontaine, N. Averet, M. Rigoulet, X. Leverve,Dimethylbiguanide inhibits cell respiration via an indirect effect targeted onthe respiratory chain complex I, J. Biol. Chem. 275 (2000) 223–228.
[178] M.R. Owen, E. Doran, A.P. Halestrap, Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratorychain, Biochem. J. 348 (Pt 3) (2000) 607–614.
[179] Z. Xie, Y. Dong, R. Scholz, D. Neumann, M.H. Zou, Phosphorylation of LKB1 at ser-ine 428 by protein kinase C-zeta is required for metformin-enhanced activationof the AMP-activated protein kinase in endothelial cells, Circulation 117 (2008)952–962.
[180] R.J. Shaw, K.A. Lamia, D. Vasquez, S.H. Koo, N. Bardeesy, R.A. Depinho, M.Montminy, L.C. Cantley, The kinase LKB1 mediates glucose homeostasis inliver and therapeutic effects of metformin, Science 310 (2005) 1642–1646.
[181] R.J. Dowling, M. Zakikhani, I.G. Fantus, M. Pollak, N. Sonenberg, Metformin in-hibits mammalian target of rapamycin-dependent translation initiation inbreast cancer cells, Cancer Res. 67 (2007) 10804–10812.
[182] J. Ouyang, R.A. Parakhia, R.S. Ochs, Metformin activates AMP kinase through in-hibition of AMP deaminase, J. Biol. Chem. 286 (2011) 1–11.
[183] M. Foretz, S. Hebrard, J. Leclerc, E. Zarrinpashneh, M. Soty, G. Mithieux, K.Sakamoto, F. Andreelli, B. Viollet, Metformin inhibits hepatic gluconeogenesisin mice independently of the LKB1/AMPK pathway via a decrease in hepatic en-ergy state, J. Clin. Invest. 120 (2010) 2355–2369.
[184] R.S. Legro, H.X. Barnhart, W.D. Schlaff, B.R. Carr, M.P. Diamond, S.A. Carson, M.P.Steinkampf, C. Coutifaris, P.G. McGovern, N.A. Cataldo, G.G. Gosman, J.E. Nestler,L.C. Giudice, K.G. Ewens, R.S. Spielman, P.C. Leppert, E.R. Myers, Ovulatory re-sponse to treatment of polycystic ovary syndrome is associated with a polymor-phism in the STK11 gene, J. Clin. Endocrinol. Metab. 93 (2008) 792–800.
[185] S. Hadad, T. Iwamoto, L. Jordan, C. Purdie, S. Bray, L. Baker, G. Jellema, S. Deharo,D.G. Hardie, L. Pusztai, S. Moulder-Thompson, J.A. Dewar, A.M. Thompson, Evi-dence for biological effects of metformin in operable breast cancer: apre-operative, window-of-opportunity, randomized trial, Breast Cancer Res.Treat. 128 (2011) 783–794.
[186] N.K. LeBrasseur, M. Kelly, T.S. Tsao, S.R. Farmer, A.K. Saha, N.B. Ruderman, E.Tomas, Thiazolidinediones can rapidly activate AMP-activated protein ki-nase in mammalian tissues, Am. J. Physiol. Endocrinol. Metab. 291 (2006)E175–E181.
[187] W. Sun, T.S. Lee, M. Zhu, C. Gu, Y. Wang, Y. Zhu, J.Y. Shyy, Statins activateAMP-activated protein kinase in vitro and in vivo, Circulation 114 (2006)2655–2662.
[188] T. Kosaka, R. Okuyama, W. Sun, T. Ogata, J. Harada, K. Araki, M. Izumi, T. Yoshida,A. Okuno, T. Fujiwara, J. Ohsumi, K. Ichikawa, Identification of molecular targetof AMP-activated protein kinase activator by affinity purification and mass spec-trometry, Anal. Chem. 77 (2005) 2050–2055.
[189] M. Zang, S. Xu, K.A. Maitland-Toolan, A. Zuccollo, X. Hou, B. Jiang, M. Wierzbicki,T.J. Verbeuren, R.A. Cohen, Polyphenols stimulate AMP-activated protein kinase,lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice, Diabetes 55 (2006) 2180–2191.
[190] J.M. Brusq, N. Ancellin, P. Grondin, R. Guillard, S. Martin, Y. Saintillan, M.Issandou, Inhibition of lipid synthesis through activation of AMP kinase: an ad-ditional mechanism for the hypolipidemic effects of berberine, J. Lipid Res. 47(2006) 1281–1288.

207S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
[191] J.E. Sullivan, K.J. Brocklehurst, A.E.Marley, F. Carey, D. Carling, R.K. Beri, Inhibition oflipolysis and lipogenesis in isolated rat adipocyteswith AICAR, a cell-permeable ac-tivator of AMP-activated protein kinase, FEBS Lett. 353 (1994) 33–36.
[192] J.M. Corton, J.G. Gillespie, S.A. Hawley, D.G. Hardie, 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activatedprotein kinase in intact cells? Eur. J. Biochem. 229 (1995) 558–565.
[193] B. Cool, B. Zinker, W. Chiou, L. Kifle, N. Cao, M. Perham, R. Dickinson, A. Adler, G.Gagne, R. Iyengar, G. Zhao, K. Marsh, P. Kym, P. Jung, H.S. Camp, E. Frevert, Iden-tification and characterization of a small molecule AMPK activator that treatskey components of type 2 diabetes and the metabolic syndrome, Cell Metab. 3(2006) 403–416.
[194] T. Pang, Z.S. Zhang, M. Gu, B.Y. Qiu, L.F. Yu, P.R. Cao, W. Shao, M.B. Su, J.Y. Li, F.J.Nan, J. Li, Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells, J. Biol. Chem. 283 (2008) 16051–16060.
[195] J.W. Scott, B.J. van Denderen, S.B. Jorgensen, J.E. Honeyman, G.R. Steinberg, J.S.Oakhill, T.J. Iseli, A. Koay, P.R. Gooley, D. Stapleton, B.E. Kemp, Thienopyridonedrugs are selective activators of AMP-activated protein kinase beta1-containingcomplexes, Chem. Biol. 15 (2008) 1220–1230.
[196] X. Xiang, A.K. Saha, R. Wen, N.B. Ruderman, Z. Luo, AMP-activated protein kinaseactivators can inhibit the growth of prostate cancer cells by multiple mecha-nisms, Biochem. Biophys. Res. Commun. 321 (2004) 161–167.
[197] S. Han, J. Roman, Rosiglitazone suppresses human lung carcinoma cell growththrough PPARgamma-dependent and PPARgamma-independent signal path-ways, Mol. Cancer Ther. 5 (2006) 430–437.
[198] P.M. Yang, Y.L. Liu, Y.C. Lin, C.T. Shun, M.S. Wu, C.C. Chen, Inhibition of autophagyenhances anticancer effects of atorvastatin in digestive malignancies, CancerRes. 70 (2010) 7699–7709.
[199] J. Woodard, S. Joshi, B. Viollet, N. Hay, L.C. Platanias, AMPK as a therapeutic tar-get in renal cell carcinoma, Cancer Biol. Ther. 10 (2010) 1168–1177.
[200] H.S. Kim, M.J. Kim, E.J. Kim, Y. Yang, M.S. Lee, J.S. Lim, Berberine-induced AMPK ac-tivation inhibits themetastatic potential ofmelanoma cells via reduction of ERK ac-tivity and COX-2 protein expression, Biochem. Pharmacol. 83 (2012) 385–394.
[201] J.T. Hwang, D.W. Kwak, S.K. Lin, H.M. Kim, Y.M. Kim, O.J. Park, Resveratrol in-duces apoptosis in chemoresistant cancer cells via modulation of AMPK signal-ing pathway, Ann. N. Y. Acad. Sci. 1095 (2007) 441–448.
[202] P. Baumann, S. Mandl-Weber, B. Emmerich, C. Straka, R. Schmidmaier, Activa-tion of adenosine monophosphate activated protein kinase inhibits growth ofmultiple myeloma cells, Exp. Cell Res. 313 (2007) 3592–3603.
[203] B. Liu, Z. Fan, S.M. Edgerton, X.S. Deng, I.N. Alimova, S.E. Lind, A.D. Thor, Metfor-min induces unique biological and molecular responses in triple negative breastcancer cells, Cell Cycle 8 (2009) 2031–2040.
[204] M. Zakikhani, R. Dowling, I.G. Fantus, N. Sonenberg, M. Pollak, Metformin is anAMP kinase-dependent growth inhibitor for breast cancer cells, Cancer Res. 66(2006) 10269–10273.
[205] Y. Zhuang, W.K. Miskimins, Metformin induces both caspase-dependent andpoly(ADP-ribose) polymerase-dependent cell death in breast cancer cells, Mol.Cancer Res. 9 (2011) 603–615.
[206] A. Vazquez-Martin, C. Oliveras-Ferraros, S. Cufi, S. Del Barco, B. Martin-Castillo,E. Lopez-Bonet, J.A. Menendez, The anti-diabetic drug metformin suppressesthe metastasis-associated protein CD24 in MDA-MB-468 triple-negative breastcancer cells, Oncol. Rep. 25 (2011) 135–140.
[207] L.W. Wang, Z.S. Li, D.W. Zou, Z.D. Jin, J. Gao, G.M. Xu, Metformin induces apopto-sis of pancreatic cancer cells, World J. Gastroenterol. 14 (2008) 7192–7198.
[208] B. Bao, Z. Wang, S. Ali, A. Ahmad, A.S. Azmi, S.H. Sarkar, S. Banerjee, D. Kong, Y. Li,S. Thakur, F.H. Sarkar, Metformin inhibits cell proliferation, migration and inva-sion by attenuating CSC function mediated by deregulating miRNAs in pancreat-ic cancer cells, Cancer Prev. Res. (Phila) 5 (2012) 355–364.
[209] M. Buzzai, R.G. Jones, R.K. Amaravadi, J.J. Lum, R.J. DeBerardinis, F. Zhao, B.Viollet, C.B. Thompson, Systemic treatment with the antidiabetic drug metfor-min selectively impairs p53-deficient tumor cell growth, Cancer Res. 67(2007) 6745–6752.
[210] K. Kato, J. Gong, H. Iwama, A. Kitanaka, J. Tani, H. Miyoshi, K. Nomura, S. Mimura,M. Kobayashi, Y. Aritomo, H. Kobara, H. Mori, T. Himoto, K. Okano, Y. Suzuki, K.Murao, T. Masaki, The antidiabetic drug metformin inhibits gastric cancer cellproliferation in vitro and in vivo, Mol. Cancer Ther. 11 (2012) 549–560.
[211] B.K. Tan, R. Adya, J. Chen, H. Lehnert, L.J. Sant Cassia, H.S. Randeva, Metformintreatment exerts antiinvasive and antimetastatic effects in human endometrialcarcinoma cells, J. Clin. Endocrinol. Metab. 96 (2011) 808–816.
[212] E. Vakana, J.K. Altman, H. Glaser, N.J. Donato, L.C. Platanias, Antileukemic effectsof AMPK activators on BCR-ABL-expressing cells, Blood 118 (2011) 6399–6402.
[213] I. Ben Sahra, K. Laurent, A. Loubat, S. Giorgetti-Peraldi, P. Colosetti, P. Auberger,J.F. Tanti, Y. Le Marchand-Brustel, F. Bost, The antidiabetic drug metformin ex-erts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1level, Oncogene 27 (2008) 3576–3586.
[214] M.B. Schneider, H. Matsuzaki, J. Haorah, A. Ulrich, J. Standop, X.Z. Ding, T.E.Adrian, P.M. Pour, Prevention of pancreatic cancer induction in hamsters bymetformin, Gastroenterology 120 (2001) 1263–1270.
[215] C. Algire, L. Amrein, M. Zakikhani, L. Panasci, M. Pollak, Metformin blocks thestimulative effect of a high-energy diet on colon carcinoma growth in vivo andis associated with reduced expression of fatty acid synthase, Endocr. Relat. Can-cer 17 (2010) 351–360.
[216] K. Hosono, H. Endo, H. Takahashi, M. Sugiyama, T. Uchiyama, K. Suzuki, Y. Nozaki,K. Yoneda, K. Fujita, M. Yoneda, M. Inamori, A. Tomatsu, T. Chihara, K. Shimpo, H.Nakagama, A. Nakajima, Metformin suppresses azoxymethane-induced colorec-tal aberrant crypt foci by activating AMP-activated protein kinase, Mol. Carcinog.49 (2010) 662–671.
[217] A. Tomimoto, H. Endo, M. Sugiyama, T. Fujisawa, K. Hosono, H. Takahashi, N.Nakajima, Y. Nagashima, K. Wada, H. Nakagama, A. Nakajima, Metformin sup-presses intestinal polyp growth in ApcMin/+ mice, Cancer Sci. 99 (2008)2136–2141.
[218] B. Bojkova, P. Orendas, M. Garajova, M. Kassayova, V. Kutna, E. Ahlersova, I.Ahlers, Metformin in chemically-induced mammary carcinogenesis in rats,Neoplasma 56 (2009) 269–274.
[219] V.N. Anisimov, L.M. Berstein, P.A. Egormin, T.S. Piskunova, I.G. Popovich, M.A.Zabezhinski, I.G. Kovalenko, T.E. Poroshina, A.V. Semenchenko, M. Provinciali,F. Re, C. Franceschi, Effect of metformin on life span and on the developmentof spontaneous mammary tumors in HER-2/neu transgenic mice, Exp. Gerontol.40 (2005) 685–693.
[220] R.M. Memmott, J.R. Mercado, C.R. Maier, S. Kawabata, S.D. Fox, P.A. Dennis, Met-formin prevents tobacco carcinogen-induced lung tumorigenesis, Cancer Prev.Res. (Phila) 3 (2010) 1066–1076.
[221] X. Yu, P. Carpenter, C. Anasetti, Advances in transplantation tolerance, Lancet357 (2001) 1959–1963.
[222] J. Chung, C.J. Kuo, G.R. Crabtree, J. Blenis, Rapamycin-FKBP specifically blocksgrowth-dependent activation of and signaling by the 70 kd S6 protein kinases,Cell 69 (1992) 1227–1236.
[223] T. Asano, Y. Yao, J. Zhu, D. Li, J.L. Abbruzzese, S.A. Reddy, The rapamycin analogCCI-779 is a potent inhibitor of pancreatic cancer cell proliferation, Biochem.Biophys. Res. Commun. 331 (2005) 295–302.
[224] K. Yu, L. Toral-Barza, C. Discafani, W.G. Zhang, J. Skotnicki, P. Frost, J.J. Gibbons,mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor,in preclinical models of breast cancer, Endocr. Relat. Cancer 8 (2001) 249–258.
[225] G.V. Thomas, C. Tran, I.K. Mellinghoff, D.S. Welsbie, E. Chan, B. Fueger, J. Czernin,C.L. Sawyers, Hypoxia-inducible factor determines sensitivity to inhibitors ofmTOR in kidney cancer, Nat. Med. 12 (2006) 122–127.
[226] L. Wu, D.C. Birle, I.F. Tannock, Effects of the mammalian target of rapamycin in-hibitor CCI-779 used alone or with chemotherapy on human prostate cancercells and xenografts, Cancer Res. 65 (2005) 2825–2831.
[227] D.J. Boffa, F. Luan, D. Thomas, H. Yang, V.K. Sharma, M. Lagman, M. Suthanthiran,Rapamycin inhibits the growth and metastatic progression of non-small celllung cancer, Clin. Cancer Res. 10 (2004) 293–300.
[228] D. Ito, K. Fujimoto, T. Mori, K. Kami, M. Koizumi, E. Toyoda, Y. Kawaguchi, R.Doi, In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabinein xenograft models of human pancreatic cancer, Int. J. Cancer 118 (2006)2337–2343.
[229] T. Okada, T. Sawada, K. Kubota, Rapamycin enhances the anti-tumor effect ofgemcitabine in pancreatic cancer cells, Hepatogastroenterology 54 (2007)2129–2133.
[230] P.C. Manegold, C. Paringer, U. Kulka, K. Krimmel, M.E. Eichhorn, R. Wilkowski,K.W. Jauch, M. Guba, C.J. Bruns, Antiangiogenic therapy with mammalian targetof rapamycin inhibitor RAD001 (Everolimus) increases radiosensitivity in solidcancer, Clin. Cancer Res. 14 (2008) 892–900.
[231] S. Paglin, N.Y. Lee, C. Nakar, M. Fitzgerald, J. Plotkin, B. Deuel, N. Hackett, M.McMahill, E. Sphicas, N. Lampen, J. Yahalom, Rapamycin-sensitive pathway reg-ulates mitochondrial membrane potential, autophagy, and survival in irradiatedMCF-7 cells, Cancer Res. 65 (2005) 11061–11070.
[232] W. van Veelen, S.E. Korsse, L. van de Laar, M.P. Peppelenbosch, The long andwinding road to rational treatment of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling, Oncogene 30 (2011) 2289–2303.
[233] G. Hudes, M. Carducci, P. Tomczak, J. Dutcher, R. Figlin, A. Kapoor, E. Staroslawska,J. Sosman, D. McDermott, I. Bodrogi, Z. Kovacevic, V. Lesovoy, I.G. Schmidt-Wolf,O. Barbarash, E. Gokmen, T. O'Toole, S. Lustgarten, L. Moore, R.J. Motzer,Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma, N.Engl. J. Med. 356 (2007) 2271–2281.
[234] R.J. Motzer, B. Escudier, S. Oudard, T.E. Hutson, C. Porta, S. Bracarda, V. Grunwald,J.A. Thompson, R.A. Figlin, N. Hollaender, G. Urbanowitz, W.J. Berg, A. Kay, D.Lebwohl, A. Ravaud, Efficacy of everolimus in advanced renal cell carcinoma: adouble-blind, randomised, placebo-controlled phase III trial, Lancet 372 (2008)449–456.
[235] R.J. Motzer, B. Escudier, S. Oudard, T.E. Hutson, C. Porta, S. Bracarda, V. Grunwald,J.A. Thompson, R.A. Figlin, N. Hollaender, A. Kay, A. Ravaud, Phase 3 trial ofeverolimus for metastatic renal cell carcinoma: final results and analysis ofprognostic factors, Cancer 116 (2010) 4256–4265.
[236] J.J. Bissler, F.X. McCormack, L.R. Young, J.M. Elwing, G. Chuck, J.M. Leonard, V.J.Schmithorst, T. Laor, A.S. Brody, J. Bean, S. Salisbury, D.N. Franz, Sirolimus forangiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis,N. Engl. J. Med. 358 (2008) 140–151.
[237] D.M. Davies, S.R. Johnson, A.E. Tattersfield, J.C. Kingswood, J.A. Cox, D.L.McCartney, T. Doyle, F. Elmslie, A. Saggar, P.J. de Vries, J.R. Sampson, Sirolimustherapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis, N. Engl.J. Med. 358 (2008) 200–203.
[238] D.M. Davies, P.J. de Vries, S.R. Johnson, D.L. McCartney, J.A. Cox, A.L. Serra, P.C.Watson, C.J. Howe, T. Doyle, K. Pointon, J.J. Cross, A.E. Tattersfield, J.C. Kingswood,J.R. Sampson, Sirolimus therapy for angiomyolipoma in tuberous sclerosis andsporadic lymphangioleiomyomatosis: a phase 2 trial, Clin. Cancer Res. 17 (2011)4071–4081.
[239] D.A. Krueger, M.M. Care, K. Holland, K. Agricola, C. Tudor, P. Mangeshkar, K.A.Wilson, A. Byars, T. Sahmoud, D.N. Franz, Everolimus for subependymal giant-cellastrocytomas in tuberous sclerosis, N. Engl. J. Med. 363 (2010) 1801–1811.
[240] S.L. Dabora, D.N. Franz, S. Ashwal, A. Sagalowsky, F.J. DiMario Jr., D. Miles, D.Cutler, D. Krueger, R.N. Uppot, R. Rabenou, S. Camposano, J. Paolini, F. Fennessy,N. Lee, C. Woodrum, J. Manola, J. Garber, E.A. Thiele, Multicenter phase 2 trial of

208 S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors re-gress and VEGF-D levels decrease, PLoS One 6 (2011) e23379.
[241] J.C. Yao, A.T. Phan, D.Z. Chang, R.A. Wolff, K. Hess, S. Gupta, C. Jacobs, J.E. Mares,A.N. Landgraf, A. Rashid, F. Meric-Bernstam, Efficacy of RAD001 (everolimus)and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tu-mors: results of a phase II study, J. Clin. Oncol. 26 (2008) 4311–4318.
[242] J.C. Yao, C. Lombard-Bohas, E. Baudin, L.K. Kvols, P. Rougier, P. Ruszniewski, S.Hoosen, J. St Peter, T. Haas, D. Lebwohl, E. Van Cutsem, M.H. Kulke, T.J. Hobday,T.M. O'Dorisio, M.H. Shah, G. Cadiot, G. Luppi, J.A. Posey, B. Wiedenmann, Dailyoral everolimus activity in patients with metastatic pancreatic neuroendocrinetumors after failure of cytotoxic chemotherapy: a phase II trial, J. Clin. Oncol.28 (2010) 69–76.
[243] M.E. Pavel, J.D. Hainsworth, E. Baudin, M. Peeters, D. Horsch, R.E. Winkler, J.Klimovsky, D. Lebwohl, V. Jehl, E.M. Wolin, K. Oberg, E. Van Cutsem, J.C. Yao,Everolimus plus octreotide long-acting repeatable for the treatment of advancedneuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): arandomised, placebo-controlled, phase 3 study, Lancet 378 (2011) 2005–2012.
[244] S.V. Bhagwat, P.C. Gokhale, A.P. Crew, A. Cooke, Y. Yao, C. Mantis, J. Kahler, J.Workman, M. Bittner, L. Dudkin, D.M. Epstein, N.W. Gibson, R. Wild, L.D.Arnold, P.J. Houghton, J.A. Pachter, Preclinical characterization of OSI-027, a po-tent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin,Mol. Cancer Ther. 10 (2011) 1394–1406.
[245] E. Conde, B. Angulo, M. Tang, M. Morente, J. Torres-Lanzas, A. Lopez-Encuentra,F. Lopez-Rios, M. Sanchez-Cespedes, Molecular context of the EGFR mutations:evidence for the activation of mTOR/S6K signaling, Clin. Cancer Res. 12 (2006)710–717.
[246] B.R. Balsara, J. Pei, Y. Mitsuuchi, R. Page, A. Klein-Szanto, H. Wang, M. Unger, J.R.Testa, Frequent activation of AKT in non-small cell lung carcinomas andpreneoplastic bronchial lesions, Carcinogenesis 25 (2004) 2053–2059.
[247] K.A. Price, C.G. Azzoli, L.M. Krug, M.C. Pietanza, N.A. Rizvi, W. Pao, M.G. Kris, G.J.Riely, R.T. Heelan, M.E. Arcila, V.A. Miller, Phase II trial of gefitinib and everolimusin advanced non-small cell lung cancer, J. Thorac. Oncol. 5 (2010) 1623–1629.
[248] P.P. Pandolfi, Breast cancer—loss of PTEN predicts resistance to treatment,N. Engl. J. Med. 351 (2004) 2337–2338.
[249] E. Tokunaga, Y. Kimura, K. Mashino, E. Oki, A. Kataoka, S. Ohno, M. Morita, Y.Kakeji, H. Baba, Y. Maehara, Activation of PI3K/Akt signaling and hormone resis-tance in breast cancer, Breast Cancer 13 (2006) 137–144.
[250] J. Baselga, M. Campone, M. Piccart, H.A. Burris III, H.S. Rugo, T. Sahmoud, S.Noguchi, M. Gnant, K.I. Pritchard, F. Lebrun, J.T. Beck, Y. Ito, D. Yardley, I.Deleu, A. Perez, T. Bachelot, L. Vittori, Z. Xu, P. Mukhopadhyay, D. Lebwohl,G.N. Hortobagyi, Everolimus in postmenopausal hormone-receptor-positive ad-vanced breast cancer, N. Engl. J. Med. 366 (2012) 520–529.
[251] H. Nozawa, T. Watanabe, H. Nagawa, Phosphorylation of ribosomal p70 S6 ki-nase and rapamycin sensitivity in human colorectal cancer, Cancer Lett. 251(2007) 105–113.
[252] Y.J. Zhang, Q. Dai, D.F. Sun, H. Xiong, X.Q. Tian, F.H. Gao, M.H. Xu, G.Q. Chen, Z.G.Han, J.Y. Fang, mTOR signaling pathway is a target for the treatment of colorectalcancer, Ann. Surg. Oncol. 16 (2009) 2617–2628.
[253] I. Altomare, J.C. Bendell, K.E. Bullock, H.E. Uronis, M.A. Morse, S.D. Hsu, S.Y. Zafar,G.C. Blobe, H. Pang, W. Honeycutt, L. Sutton, H.I. Hurwitz, A phase II trial ofbevacizumab plus everolimus for patients with refractory metastatic colorectalcancer, Oncologist 16 (2011) 1131–1137.
[254] C.M. Chresta, B.R. Davies, I. Hickson, T. Harding, S. Cosulich, S.E. Critchlow, J.P.Vincent, R. Ellston, D. Jones, P. Sini, D. James, Z. Howard, P. Dudley, G. Hughes,L. Smith, S. Maguire, M. Hummersone, K. Malagu, K. Menear, R. Jenkins, M.Jacobsen, G.C. Smith, S. Guichard, M. Pass, AZD8055 is a potent, selective, andorally bioavailable ATP-competitive mammalian target of rapamycin kinase in-hibitor with in vitro and in vivo antitumor activity, Cancer Res. 70 (2010)288–298.
[255] Q. Jiang, J.M.Weiss, T. Back, T. Chan, J.R. Ortaldo, S. Guichard, R.H.Wiltrout, mTORkinase inhibitor AZD8055 enhances the immunotherapeutic activity of an ago-nist CD40 antibody in cancer treatment, Cancer Res. 71 (2011) 4074–4084.
[256] M.R. Janes, J.J. Limon, L. So, J. Chen, R.J. Lim, M.A. Chavez, C. Vu, M.B. Lilly, S.Mallya, S.T. Ong, M. Konopleva, M.B. Martin, P. Ren, Y. Liu, C. Rommel, D.A.Fruman, Effective and selective targeting of leukemia cells using a TORC1/2 ki-nase inhibitor, Nat. Med. 16 (2010) 205–213.
[257] K. Yu, L. Toral-Barza, C. Shi, W.G. Zhang, J. Lucas, B. Shor, J. Kim, J. Verheijen, K.Curran, D.J. Malwitz, D.C. Cole, J. Ellingboe, S. Ayral-Kaloustian, T.S. Mansour,J.J. Gibbons, R.T. Abraham, P. Nowak, A. Zask, Biochemical, cellular, and in vivoactivity of novel ATP-competitive and selective inhibitors of the mammalian tar-get of rapamycin, Cancer Res. 69 (2009) 6232–6240.
[258] K. Yu, C. Shi, L. Toral-Barza, J. Lucas, B. Shor, J.E. Kim, W.G. Zhang, R. Mahoney, C.Gaydos, L. Tardio, S.K. Kim, R. Conant, K. Curran, J. Kaplan, J. Verheijen, S. Ayral-Kaloustian, T.S. Mansour, R.T. Abraham, A. Zask, J.J. Gibbons, Beyond rapalogtherapy: preclinical pharmacology and antitumor activity of WYE-125132, anATP-competitive and specific inhibitor of mTORC1 and mTORC2, Cancer Res. 70(2010) 621–631.
[259] J.M. Garcia-Martinez, J. Moran, R.G. Clarke, A. Gray, S.C. Cosulich, C.M. Chresta,D.R. Alessi, Ku-0063794 is a specific inhibitor of the mammalian target ofrapamycin (mTOR), Biochem. J. 421 (2009) 29–42.
[260] S.M. Chen, J.L. Liu, X. Wang, C. Liang, J. Ding, L.H. Meng, Inhibition of tumor cellgrowth, proliferation and migration by X-387, a novel active-site inhibitor ofmTOR, Biochem. Pharmacol. 83 (2012) 1183–1194.
[261] C. Wei, C.I. Amos, N. Zhang, X. Wang, A. Rashid, C.L. Walker, R.R. Behringer, M.L.Frazier, Suppression of Peutz–Jeghers polyposis by targeting mammalian targetof rapamycin signaling, Clin. Cancer Res. 14 (2008) 1167–1171.
[262] J. Robinson, C. Lai, A. Martin, E. Nye, I. Tomlinson, A. Silver, Oral rapamycin re-duces tumour burden and vascularization in Lkb1(+/−) mice, J. Pathol. 219(2009) 35–40.
[263] D.B. Shackelford, D.S. Vasquez, J. Corbeil, S. Wu, M. Leblanc, C.L. Wu, D.R. Vera, R.J.Shaw, mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mousemodel of Peutz–Jeghers syndrome, Proc. Natl. Acad. Sci. U. S. A. 106 (2009)11137–11142.
[264] C. Wei, C.I. Amos, N. Zhang, J. Zhu, X. Wang, M.L. Frazier, Chemopreventive effi-cacy of rapamycin on Peutz–Jeghers syndrome in a mouse model, Cancer Lett.277 (2009) 149–154.
[265] T. Fujishita, K. Aoki, H.A. Lane, M. Aoki, M.M. Taketo, Inhibition of the mTORC1pathway suppresses intestinal polyp formation and reduces mortality inApcDelta716 mice, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 13544–13549.
[266] G.E. Koehl, M. Spitzner, J. Ousingsawat, R. Schreiber, E.K. Geissler, K. Kunzelmann,Rapamycin inhibits oncogenic intestinal ion channels andneoplasia inAPC(Min/+)mice, Oncogene 29 (2010) 1553–1560.
[267] K.E. Hung, M.A. Maricevich, L.G. Richard, W.Y. Chen, M.P. Richardson, A. Kunin,R.T. Bronson, U. Mahmood, R. Kucherlapati, Development of a mouse modelfor sporadic and metastatic colon tumors and its use in assessing drug treat-ment, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 1565–1570.
[268] C.M. Johannessen, B.W. Johnson, S.M. Williams, A.W. Chan, E.E. Reczek, R.C.Lynch, M.J. Rioth, A. McClatchey, S. Ryeom, K. Cichowski, TORC1 is essential forNF1-associated malignancies, Curr. Biol. 18 (2008) 56–62.
[269] L. Lee, P. Sudentas, B. Donohue, K. Asrican, A.Worku, V.Walker, Y. Sun, K. Schmidt,M.S. Albert, N. El-Hashemite, A.S. Lader, H. Onda, H. Zhang, D.J. Kwiatkowski, S.L.Dabora, Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous scle-rosis mouse models, Genes Chromosom. Cancer 42 (2005) 213–227.
[270] N. Lee, C.L. Woodrum, A.M. Nobil, A.E. Rauktys, M.P. Messina, S.L. Dabora,Rapamycin weekly maintenance dosing and the potential efficacy of combina-tion sorafenib plus rapamycin but not atorvastatin or doxycycline in tuberoussclerosis preclinical models, BMC Pharmacol. 9 (2009) 8.
[271] K. Pollizzi, I. Malinowska-Kolodziej, M. Stumm, H. Lane, D. Kwiatkowski, Equiv-alent benefit of mTORC1 blockade and combined PI3K-mTOR blockade in amouse model of tuberous sclerosis, Mol. Cancer 8 (2009) 38.
[272] K. Podsypanina, R.T. Lee, C. Politis, I. Hennessy, A. Crane, J. Puc, M. Neshat, H.Wang, L. Yang, J. Gibbons, P. Frost, V. Dreisbach, J. Blenis, Z. Gaciong, P. Fisher,C. Sawyers, L. Hedrick-Ellenson, R. Parsons, An inhibitor of mTOR reduces neo-plasia and normalizes p70/S6 kinase activity in Pten+/−mice, Proc. Natl.Acad. Sci. U. S. A. 98 (2001) 10320–10325.
[273] C.M. Contreras, E.A. Akbay, T.D. Gallardo, J.M. Haynie, S. Sharma, O. Tagao, N.Bardeesy, M. Takahashi, J. Settleman, K.K. Wong, D.H. Castrillon, Lkb1 inactiva-tion is sufficient to drive endometrial cancers that are aggressive yet highly re-sponsive to mTOR inhibitor monotherapy, Dis. Model. Mech. 3 (2010) 181–193.
[274] S. Mabuchi, D.A. Altomare, D.C. Connolly, A. Klein-Szanto, S. Litwin, M.K. Hoelzle,H.H. Hensley, T.C. Hamilton, J.R. Testa, RAD001 (Everolimus) delays tumor onsetand progression in a transgenic mouse model of ovarian cancer, Cancer Res. 67(2007) 2408–2413.
[275] R. Wu, T.C. Hu, A. Rehemtulla, E.R. Fearon, K.R. Cho, Preclinical testing ofPI3K/AKT/mTOR signaling inhibitors in a mouse model of ovarian endometrioidadenocarcinoma, Clin. Cancer Res. 17 (2011) 7359–7372.
[276] C.J. Guigon, L. Fozzatti, C. Lu, M.C. Willingham, S.Y. Cheng, Inhibition of mTORC1signaling reduces tumor growth but does not prevent cancer progression in amouse model of thyroid cancer, Carcinogenesis 31 (2010) 1284–1291.
[277] A.M. Puzio-Kuter, M. Castillo-Martin, C.W. Kinkade, X. Wang, T.H. Shen, T. Matos,M.M. Shen, C. Cordon-Cardo, C. Abate-Shen, Inactivation of p53 and Pten pro-motes invasive bladder cancer, Genes Dev. 23 (2009) 675–680.
[278] W. Zhang, J. Zhu, C.L. Efferson, C. Ware, J. Tammam, M. Angagaw, J. Laskey, K.A.Bettano, S. Kasibhatla, J.F. Reilly, C. Sur, P.K. Majumder, Inhibition of tumorgrowth progression by antiandrogens and mTOR inhibitor in a Pten-deficientmouse model of prostate cancer, Cancer Res. 69 (2009) 7466–7472.
[279] P.K. Majumder, P.G. Febbo, R. Bikoff, R. Berger, Q. Xue, L.M. McMahon, J. Manola, J.Brugarolas, T.J. McDonnell, T.R. Golub, M. Loda, H.A. Lane, W.R. Sellers, mTOR inhibi-tion reverses Akt-dependent prostate intraepithelial neoplasia through regulation ofapoptotic and HIF-1-dependent pathways, Nat. Med. 10 (2004) 594–601.
[280] M. Liu, A. Howes, J. Lesperance, W.B. Stallcup, C.A. Hauser, K. Kadoya, R.G. Oshima,R.T. Abraham, Antitumor activity of rapamycin in a transgenic mouse model ofErbB2-dependent human breast cancer, Cancer Res. 65 (2005) 5325–5336.
[281] J.D. Mosley, J.T. Poirier, D.D. Seachrist, M.D. Landis, R.A. Keri, Rapamycin inhibitsmultiple stages of c-Neu/ErbB2 induced tumor progression in a transgenic mousemodel of HER2-positive breast cancer, Mol. Cancer Ther. 6 (2007) 2188–2197.
[282] X. Wang, Y. Zhan, L. Zhao, J. Alvarez, I. Chaudhary, B.B. Zhou, R.T. Abraham, G.Z.Feuerstein, Multimodal biomarker investigation on efficacy and mechanism ofaction for the mammalian target of rapamycin inhibitor, temsirolimus, in apreclinical mammary carcinoma OncoMouse model: a translational medicinestudy in support for early clinical development, J. Pharmacol. Exp. Ther. 339(2011) 421–429.
[283] Q. Wu, K. Kiguchi, T. Kawamoto, T. Ajiki, J. Traag, S. Carbajal, L. Ruffino, H.Thames, I. Wistuba, M. Thomas, K.M. Vasquez, J. DiGiovanni, Therapeutic effectof rapamycin on gallbladder cancer in a transgenic mouse model, Cancer Res.67 (2007) 3794–3800.
[284] D. Xing, S. Orsulic, A genetically defined mouse ovarian carcinoma model for themolecular characterization of pathway-targeted therapy and tumor resistance,Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 6936–6941.
[285] H.G. Wendel, A. Malina, Z. Zhao, L. Zender, S.C. Kogan, C. Cordon-Cardo, J.Pelletier, S.W. Lowe, Determinants of sensitivity and resistance to rapamycin-chemotherapy drug combinations in vivo, Cancer Res. 66 (2006) 7639–7646.

209S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
[286] E. Buck, A. Eyzaguirre, E. Brown, F. Petti, S. McCormack, J.D. Haley, K.K. Iwata,N.W. Gibson, G. Griffin, Rapamycin synergizes with the epidermal growth factorreceptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breasttumors, Mol. Cancer Ther. 5 (2006) 2676–2684.
[287] A. Azzariti, L. Porcelli, G. Gatti, A. Nicolin, A. Paradiso, Synergic antiproliferativeand antiangiogenic effects of EGFR and mTor inhibitors on pancreatic cancercells, Biochem. Pharmacol. 75 (2008) 1035–1044.
[288] M.T. Mueller, P.C. Hermann, J. Witthauer, B. Rubio-Viqueira, S.F. Leicht, S. Huber,J.W. Ellwart, M. Mustafa, P. Bartenstein, J.G. D'Haese, M.H. Schoenberg, F. Berger,K.W. Jauch, M. Hidalgo, C. Heeschen, Combined targeted treatment to eliminatetumorigenic cancer stem cells in human pancreatic cancer, Gastroenterology137 (2009) 1102–1113.
[289] Q. Chang, E. Chen, D.W. Hedley, Effects of combined inhibition of MEK andmTORon downstream signaling and tumor growth in pancreatic cancer xenograftmodels, Cancer Biol. Ther. 8 (2009) 1893–1901.
[290] R. Bianco, S. Garofalo, R. Rosa, V. Damiano, T. Gelardi, G. Daniele, R. Marciano, F.Ciardiello, G. Tortora, Inhibition of mTOR pathway by everolimus cooperateswith EGFR inhibitors in human tumours sensitive and resistant to anti-EGFRdrugs, Br. J. Cancer 98 (2008) 923–930.
[291] L.H. Wang, J.L. Chan, W. Li, Rapamycin together with herceptin significantly in-creased anti-tumor efficacy compared to either alone in ErbB2 over expressingbreast cancer cells, Int. J. Cancer 121 (2007) 157–164.
[292] R. Mi, J. Ma, D. Zhang, L. Li, H. Zhang, Efficacy of combined inhibition of mTORand ERK/MAPK pathways in treating a tuberous sclerosis complex cell model,J. Genet. Genomics 36 (2009) 355–361.
[293] S. Buonamici, J.Williams,M.Morrissey, A.Wang, R. Guo, A. Vattay, K. Hsiao, J. Yuan,J. Green, B. Ospina, Q. Yu, L. Ostrom, P. Fordjour, D.L. Anderson, J.E. Monahan, J.F.Kelleher, S. Peukert, S. Pan, X. Wu, S.M. Maira, C. Garcia-Echeverria, K.J. Briggs,D.N.Watkins, Y.M. Yao, C. Lengauer, M.Warmuth,W.R. Sellers, M. Dorsch, Interfer-ingwith resistance to smoothened antagonists by inhibition of the PI3K pathway inmedulloblastoma, Sci. Transl. Med. 2 (2010) 51ra70.
[294] H. Shi, X. Kong, A. Ribas, R.S. Lo, Combinatorial treatments that overcomePDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition, Can-cer Res. 71 (2011) 5067–5074.
[295] T. Shimizu, A.W. Tolcher, K.P. Papadopoulos, M. Beeram, D. Rasco, L.S. Smith, S.Gunn, L. Smetzer, T.A. Mays, B. Kaiser, M.J. Wick, C. Alvarez, A. Cavazos, G.L.Mangold, A. Patnaik, The clinical effect of the dual-targeting strategy involvingPI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer,Clin. Cancer Res. 18 (2012) 2316–2325.
[296] K. Zitzmann, J. Ruden, S. Brand, B. Goke, J. Lichtl, G. Spottl, C.J. Auernhammer,Compensatory activation of Akt in response to mTOR and Raf inhibitors — a ra-tionale for dual-targeted therapy approaches in neuroendocrine tumor disease,Cancer Lett. 295 (2010) 100–109.
[297] S. Banerjee, S.M. Gianino, F. Gao, U. Christians, D.H. Gutmann, Interpreting mam-malian target of rapamycin and cell growth inhibition in a geneticallyengineered mouse model of Nf1-deficient astrocytes, Mol. Cancer Ther. 10(2011) 279–291.
[298] R. Marone, D. Erhart, A.C. Mertz, T. Bohnacker, C. Schnell, V. Cmiljanovic, F.Stauffer, C. Garcia-Echeverria, B. Giese, S.M. Maira, M.P. Wymann, Targeting mel-anoma with dual phosphoinositide 3-kinase/mammalian target of rapamycin in-hibitors, Mol. Cancer Res. 7 (2009) 601–613.
[299] A.A. Elfiky, S.A. Aziz, P.J. Conrad, S. Siddiqui, W. Hackl, M. Maira, C.L. Robert, H.M.Kluger, Characterization and targeting of phosphatidylinositol-3 kinase (PI3K)and mammalian target of rapamycin (mTOR) in renal cell cancer, J. Transl.Med 9 (2011) 133.
[300] D.C. Cho, M.B. Cohen, D.J. Panka, M. Collins, M. Ghebremichael, M.B. Atkins, S.Signoretti, J.W. Mier, The efficacy of the novel dual PI3-kinase/mTOR inhibitorNVP-BEZ235 compared with rapamycin in renal cell carcinoma, Clin. CancerRes. 16 (2010) 3628–3638.
[301] S.M. Maira, F. Stauffer, J. Brueggen, P. Furet, C. Schnell, C. Fritsch, S. Brachmann, P.Chene, A. De Pover, K. Schoemaker, D. Fabbro, D. Gabriel, M. Simonen, L. Murphy,P. Finan, W. Sellers, C. Garcia-Echeverria, Identification and characterizationof NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity,Mol. Cancer Ther. 7 (2008) 1851–1863.
[302] P. Cao, S.M. Maira, C. Garcia-Echeverria, D.W. Hedley, Activity of a novel, dualPI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic can-cers grown as orthotopic xenografts, Br. J. Cancer 100 (2009) 1267–1276.
[303] D.W. McMillin, M. Ooi, J. Delmore, J. Negri, P. Hayden, N. Mitsiades, J. Jakubikova,S.M. Maira, C. Garcia-Echeverria, R. Schlossman, N.C. Munshi, P.G. Richardson,K.C. Anderson, C.S. Mitsiades, Antimyeloma activity of the orally bioavailabledual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitorNVP-BEZ235, Cancer Res. 69 (2009) 5835–5842.
[304] A.P. Bhatt, P.M. Bhende, S.H. Sin, D. Roy, D.P. Dittmer, B. Damania, Dual inhibitionof PI3K and mTOR inhibits autocrine and paracrine proliferative loops inPI3K/Akt/mTOR-addicted lymphomas, Blood 115 (2010) 4455–4463.
[305] F. Chiarini, C. Grimaldi, F. Ricci, P.L. Tazzari, C. Evangelisti, A. Ognibene, M.Battistelli, E. Falcieri, F. Melchionda, A. Pession, P. Pagliaro, J.A. McCubrey, A.M.Martelli, Activity of the novel dual phosphatidylinositol 3-kinase/mammaliantarget of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblasticleukemia, Cancer Res. 70 (2010) 8097–8107.
[306] J. Roper, M.P. Richardson, W.V. Wang, L.G. Richard, W. Chen, E.M. Coffee, M.J.Sinnamon, L. Lee, P.C. Chen, R.T. Bronson, E.S. Martin, K.E. Hung, The dualPI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a geneticallyengineered mouse model of PIK3CA wild-type colorectal cancer, PLoS One 6(2011) e25132.
[307] V.A. Herrera, E. Zeindl-Eberhart, A. Jung, R.M. Huber, A. Bergner, The dualPI3K/mTOR inhibitor BEZ235 is effective in lung cancer cell lines, Anticancer.Res. 31 (2011) 849–854.
[308] C. Santiskulvong, G.E. Konecny, M. Fekete, K.Y. Chen, A. Karam, D. Mulholland, C.Eng, H. Wu, M. Song, O. Dorigo, Dual targeting of phosphoinositide 3-kinase andmammalian target of rapamycin using NVP-BEZ235 as a novel therapeutic ap-proach in human ovarian carcinoma, Clin. Cancer Res. 17 (2011) 2373–2384.
[309] V. Serra, B. Markman, M. Scaltriti, P.J. Eichhorn, V. Valero, M. Guzman, M.L.Botero, E. Llonch, F. Atzori, S. Di Cosimo, M. Maira, C. Garcia-Echeverria, J.L.Parra, J. Arribas, J. Baselga, NVP-BEZ235, a dual PI3K/mTOR inhibitor, preventsPI3K signaling and inhibits the growth of cancer cells with activating PI3K mu-tations, Cancer Res. 68 (2008) 8022–8030.
[310] S.M. Brachmann, I. Hofmann, C. Schnell, C. Fritsch, S. Wee, H. Lane, S. Wang, C.Garcia-Echeverria, S.M. Maira, Specific apoptosis induction by the dualPI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breastcancer cells, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 22299–22304.
[311] C. Brunner-Kubath, W. Shabbir, V. Saferding, R. Wagner, C.F. Singer, P. Valent, W.Berger, B. Marian, C.C. Zielinski, M. Grusch, T.W. Grunt, The PI3 kinase/mTORblocker NVP-BEZ235 overrides resistance against irreversible ErbB inhibitorsin breast cancer cells, Breast Cancer Res. Treat. 129 (2011) 387–400.
[312] P.J. Eichhorn, M. Gili, M. Scaltriti, V. Serra, M. Guzman, W. Nijkamp, R.L.Beijersbergen, V. Valero, J. Seoane, R. Bernards, J. Baselga, Phosphatidylinositol3-kinase hyperactivation results in lapatinib resistance that is reversed by themTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235, Cancer Res. 68(2008) 9221–9230.
[313] G. Konstantinidou, E.A. Bey, A. Rabellino, K. Schuster, M.S. Maira, A.F. Gazdar, A.Amici, D.A. Boothman, P.P. Scaglioni, Dual phosphoinositide 3-kinase/mammaliantarget of rapamycin blockade is an effective radiosensitizing strategy for the treat-ment of non-small cell lung cancer harboring K-RAS mutations, Cancer Res. 69(2009) 7644–7652.
[314] A.C. Faber, D. Li, Y. Song, M.C. Liang, B.Y. Yeap, R.T. Bronson, E. Lifshits, Z. Chen,S.M. Maira, C. Garcia-Echeverria, K.K. Wong, J.A. Engelman, Differential induc-tion of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition,Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 19503–19508.
[315] C. Schult, M. Dahlhaus, A. Glass, K. Fischer, S. Lange, M. Freund, C. Junghanss, Thedual kinase inhibitor NVP-BEZ235 in combination with cytotoxic drugs exertsanti-proliferative activity towards acute lymphoblastic leukemia cells, Antican-cer. Res. 32 (2012) 463–474.
[316] E. Fokas, J.H. Im, S. Hill, S. Yameen, M. Stratford, J. Beech, W. Hackl, S.M. Maira,E.J. Bernhard, W.G. McKenna, R.J. Muschel, Dual inhibition of the PI3K/mTORpathway increases tumor radiosensitivity by normalizing tumor vasculature,Cancer Res. 72 (2012) 239–248.
[317] N. Awasthi, P.L. Yen, M.A. Schwarz, R.E. Schwarz, The efficacy of a novel, dualPI3K/mTOR inhibitor NVP-BEZ235 to enhance chemotherapy and antiangiogenicresponse in pancreatic cancer, J. Cell. Biochem. 113 (2012) 784–791.
[318] A. Dubrovska, J. Elliott, R.J. Salamone, S. Kim, L.J. Aimone, J.R. Walker, J. Watson,M. Sauveur-Michel, C. Garcia-Echeverria, C.Y. Cho, V.A. Reddy, P.G. Schultz, Com-bination therapy targeting both tumor-initiating and differentiated cellpopulations in prostate carcinoma, Clin. Cancer Res. 16 (2010) 5692–5702.
[319] J.A. Engelman, L. Chen, X. Tan, K. Crosby, A.R. Guimaraes, R. Upadhyay, M. Maira, K.McNamara, S.A. Perera, Y. Song, L.R. Chirieac, R. Kaur, A. Lightbown, J. Simendinger,T. Li, R.F. Padera, C. Garcia-Echeverria, R.Weissleder, U.Mahmood, L.C. Cantley, K.K.Wong, Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D andPIK3CA H1047R murine lung cancers, Nat. Med. 14 (2008) 1351–1356.
[320] J. Sunayama, K. Matsuda, A. Sato, K. Tachibana, K. Suzuki, Y. Narita, S. Shibui, K.Sakurada, T. Kayama, A. Tomiyama, C. Kitanaka, Crosstalk between the PI3K/mTORand MEK/ERK pathways involved in the maintenance of self-renewal and tumori-genicity of glioblastoma stem-like cells, Stem Cells 28 (2010) 1930–1939.
[321] N. Jin, T. Jiang, D.M. Rosen, B.D. Nelkin, D.W. Ball, Synergistic action of a RAFinhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer, Clin. Cancer Res.17 (2011) 6482–6489.
[322] D. Roulin, L. Waselle, A. Dormond-Meuwly, M. Dufour, N. Demartines, O.Dormond, Targeting renal cell carcinoma with NVP-BEZ235, a dual PI3K/mTORinhibitor, in combination with sorafenib, Mol. Cancer 10 (2011) 90.
[323] M.G. Kharas, M.R. Janes, V.M. Scarfone, M.B. Lilly, Z.A. Knight, K.M. Shokat, D.A.Fruman, Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dualPI3K/mTOR inhibitor prevents expansion of human BCR-ABL+leukemia cells,J. Clin. Invest. 118 (2008) 3038–3050.
[324] C.Y. Zou, K.D. Smith, Q.S. Zhu, J. Liu, I.E. McCutcheon, J.M. Slopis, F. Meric-Bernstam, Z. Peng, W.G. Bornmann, G.B. Mills, A.J. Lazar, R.E. Pollock, D. Lev,Dual targeting of AKT and mammalian target of rapamycin: a potential thera-peutic approach for malignant peripheral nerve sheath tumor, Mol. CancerTher. 8 (2009) 1157–1168.
[325] J. Werzowa, S. Koehrer, S. Strommer, D. Cejka, T. Fuereder, E. Zebedin, V.Wacheck, Vertical inhibition of the mTORC1/mTORC2/PI3K pathway shows syn-ergistic effects against melanoma in vitro and in vivo, J. Investig. Dermatol. 131(2011) 495–503.
[326] J. Yuan, P.P. Mehta, M.J. Yin, S. Sun, A. Zou, J. Chen, K. Rafidi, Z. Feng, J. Nickel, J.Engebretsen, J. Hallin, A. Blasina, E. Zhang, L. Nguyen, M. Sun, P.K. Vogt, A.McHarg, H. Cheng, J.G. Christensen, J.L. Kan, S. Bagrodia, PF-04691502, a potentand selective oral inhibitor of PI3K and mTOR kinases with antitumor activity,Mol. Cancer Ther. 10 (2011) 2189–2199.
[327] K.M. Kinross, D.V. Brown, M. Kleinschmidt, S. Jackson, J. Christensen, C.Cullinane, R.J. Hicks, R.W. Johnstone, G.A. McArthur, In vivo activity of combinedPI3K/mTOR and MEK inhibition in a Kras(G12D);Pten deletion mouse model ofovarian cancer, Mol. Cancer Ther. 10 (2011) 1440–1449.

210 S.E. Korsse et al. / Biochimica et Biophysica Acta 1835 (2013) 194–210
[328] D.P. Sutherlin, L. Bao, M. Berry, G. Castanedo, I. Chuckowree, J. Dotson, A. Folks, L.Friedman, R. Goldsmith, J. Gunzner, T. Heffron, J. Lesnick, C. Lewis, S. Mathieu, J.Murray, J. Nonomiya, J. Pang, N. Pegg, W.W. Prior, L. Rouge, L. Salphati, D.Sampath, Q. Tian, V. Tsui, N.C. Wan, S. Wang, B. Wei, C. Wiesmann, P. Wu, B.Y.Zhu, A. Olivero, Discovery of a potent, selective, and orally available class I phos-phatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) ki-nase inhibitor (GDC-0980) for the treatment of cancer, J. Med. Chem. 54(2011) 7579–7587.
[329] J.J. Wallin, K.A. Edgar, J. Guan, M. Berry, W.W. Prior, L. Lee, J.D. Lesnick, C. Lewis, J.Nonomiya, J. Pang, L. Salphati, A.G. Olivero, D.P. Sutherlin, C. O'Brien, J.M.Spoerke, S. Patel, L. Lensun, R. Kassees, L. Ross, M.R. Lackner, D. Sampath, M.Belvin, L.S. Friedman, GDC-0980 is a novel class I PI3K/mTOR kinase inhibitorwith robust activity in cancer models driven by the PI3K pathway, Mol. CancerTher. 10 (2011) 2426–2436.
[330] P. Baumann, L. Schneider, S. Mandl-Weber, F. Oduncu, R. Schmidmaier, Simulta-neous targeting of PI3K and mTOR with NVP-BGT226 is highly effective in mul-tiple myeloma, Anticancer Drugs 23 (2012) 131–138.
[331] K.Y. Chang, S.Y. Tsai, C.M. Wu, C.J. Yen, B.F. Chuang, J.Y. Chang, Novelphosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potentgrowth-inhibitory activity against human head and neck cancer cells in vitroand in vivo, Clin. Cancer Res. 17 (2011) 7116–7126.
[332] W. Glienke, L. Maute, J. Wicht, L. Bergmann, The dual PI3K/mTOR inhibitorNVP-BGT226 induces cell cycle arrest and regulates Survivin gene expressionin human pancreatic cancer cell lines, Tumour Biol. 33 (2011) 757–765.
[333] E. Leung, J.E. Kim, G.W. Rewcastle, G.J. Finlay, B.C. Baguley, Comparison of the ef-fects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells, Cancer Biol. Ther. 11 (2011) 938–946.
[334] C.M. Dehnhardt, A.M. Venkatesan, E. Delos Santos, Z. Chen, O. Santos, S.Ayral-Kaloustian, N. Brooijmans, R. Mallon, I. Hollander, L. Feldberg, J. Lucas, I.Chaudhary, K. Yu, J. Gibbons, R. Abraham, T.S. Mansour, Lead optimization ofN-3-substituted 7-morpholinotriazolopyrimidines as dual phosphoinositide3-kinase/mammalian target of rapamycin inhibitors: discovery of PKI-402,J. Med. Chem. 53 (2010) 798–810.
[335] R.Mallon, I. Hollander, L. Feldberg, J. Lucas, V. Soloveva, A. Venkatesan, C. Dehnhardt, E.Delos Santos, Z. Chen,O.Dos Santos, S. Ayral-Kaloustian, J. Gibbons, Antitumor efficacyprofile of PKI-402, a dual phosphatidylinositol 3-kinase/mammalian target ofrapamycin inhibitor, Mol. Cancer Ther. 9 (2010) 976–984.
[336] D.J. Rossi, A. Ylikorkala, N. Korsisaari, R. Salovaara, K. Luukko, V. Launonen, M.Henkemeyer, A. Ristimaki, L.A. Aaltonen, T.P. Makela, Induction of cyclooxygenase-2 in a mouse model of Peutz–Jeghers polyposis, Proc. Natl. Acad. Sci. U. S. A. 99(2002) 12327–12332.
[337] C. Wei, C.I. Amos, A. Rashid, M. Sabripour, L. Nations, T.J. McGarrity, M.L. Frazier,Correlation of staining for LKB1 and COX-2 in hamartomatous polyps and carci-nomas from patients with Peutz–Jeghers syndrome, J. Histochem. Cytochem. 51(2003) 1665–1672.
[338] L. Udd, P. Katajisto, D.J. Rossi, A. Lepisto, A.M. Lahesmaa, A. Ylikorkala, H.J.Jarvinen, A.P. Ristimaki, T.P. Makela, Suppression of Peutz–Jeghers polyposisby inhibition of cyclooxygenase-2, Gastroenterology 127 (2004) 1030–1037.
[339] J. Carretero, T. Shimamura, K. Rikova, A.L. Jackson, M.D. Wilkerson, C.L. Borgman,M.S. Buttarazzi, B.A. Sanofsky, K.L. McNamara, K.A. Brandstetter, Z.E. Walton, T.L.Gu, J.C. Silva, K. Crosby, G.I. Shapiro, S.M. Maira, H. Ji, D.H. Castrillon, C.F. Kim, C.Garcia-Echeverria, N. Bardeesy, N.E. Sharpless, N.D. Hayes, W.Y. Kim, J.A.Engelman, K.K. Wong, Integrative genomic and proteomic analyses identify tar-gets for Lkb1-deficient metastatic lung tumors, Cancer Cell 17 (2010) 547–559.





























