Embed Size (px)
DESCRIPTION
I nummer 4 av Information från Läkemedelsverket kan du bland annat läsa om fotosensibilaserande läkemedel, försälningsstatestik av receptfria läkemedel. Du kan även läsa om den positiva utvecklingen av läkemedel mot HIV samt om inrapporterade biverkningar under 2011.
Citation preview

www.lakemedelsverket.se
Information frånLäkemedelsverketÅrgång 23 • nummer 4 • juni 2012
Nya regler för kliniska prövningar Den 1 februari trädde Läkemedelsverkets nya föreskrifter om kliniska läkemedelsprövningar på människor i kraft.
sid 15
approvals • authorisation • clinical trials • communication • competence • cosmetics • dialogue • directives • efficacy • environment • evaluation • guidelines • harmonisation • health economics • herbals • homeopathics • information • inspection laboratory analysis • market surveillance • medicinal products • medical devices • narcotics • public health • quality • registration • regulations • reliability • risk/benefit • safety • scientific • stan-dardisation • transparency • vigilance • approvals • authorisation • clinical trials • communication • competence • cosmetics • dialogue • directives • efficacy • environment • evaluation • guidelines • harmonisation • health eco-nomics • herbals • homeopathics • information • inspection • laboratory analysis • market surveillance • medicinal products • medical devices • narcotics • public health • quality • registration • regulations • reliability •
TLV informerarsid 39Nya läkemedelAmeluz (5-aminolevulin syra)Dexdor (dexmedetomidin)Plenadren (hydrokortison)Remicade (infliximab) – ny indikation
sid 30
Fler läkemedel mot sällsynta sjukdomar Förordningen om särläkemedel från 2000 har medfört en stark tillväxt på området och vi har idag fler godkända läkemedel för sällsynta sjukdomar.
sid 17 Inrapporterade biverkningar 2011 Under 2011 tog Läkemedelsverket emot 4 919 rapporter om biverkningar från den svenska hälso- och sjukvården och 597 från allmänheten.
sid 21
Fotosensibiliserande läkemedelDe flesta personer har inga större problem med överkänslighet mot solljus. Men i kombination med vissa läkemedel kan solen orsaka hudreaktioner.
sid 4
Positiv utveckling av läkemedel mot HIV Tillgången till effektiva behandlingsalternativ vid HIV-infektion har på kort tid radikalt förändrat förutsättningarna för utveckling av nya preparat.
sid 11
Försäljningen av receptfria läkemedel För att studera hur försäljningen av receptfria läkemedel påverkats av apoteksreformen har Läkemedelsverket analyserat försäljningsstatistik.
sid 6

Inom hälso- och sjukvården vet vi hur viktigt det är med utveckling och förändring. Nya forskningsrön, rapporter och utredningar hjälper oss att skapa en effektivare vård och en mer rationell läkemedelsanvändning. Men på samma sätt som förändringar är en förutsättning för utveckling är det lika viktigt att förändringar följs upp och utvärderas. Fick vi de förbättringar som var avsikten? Eller har förändringarna inneburit baksidor som vi behöver ta ställning till?
En stor förändring för Sveriges apotek, läkemedels- konsumenter och andra aktörer inom läkemedelsområdet var omregleringen av apoteksmarknaden som genomfördes 2009. Samtidigt med denna blev det också möjligt att köpa vissa receptfria läkemedel i butik utanför apotek. Nu, drygt två år senare, har Läkemedelsverket följt upp hur försälj-ningen av receptfria läkemedel eventuellt påverkats av denna förändring.
En uppenbar följd av förändringen är naturligtvis att konsumenten lättare får tillgång till vissa läkemedel för egenvård. Uppföljningen visar att det framför allt är läke-medel mot smärta, värk och feber samt näsdroppar som säljs i butik utanför apotek. Man kan konstatera att den totala försäljningen av paracetamol har ökat med drygt tre procent inom egenvården. Likaså har försäljningen av näsdroppar ökat. Redovisningen av försäljningen på substansnivå är en del av den totala uppföljningen som vi genomför inom ramen för projektet ” Säker användning av receptfria läke-medel – status efter omregleringen av apoteksmarknaden”. Läkemedelverket avser bland annat att följa försäljningen och utvecklingen av de frågor som allmänheten ställer om receptfria läkemedel fram till slutet av 2013. Mer om detta kan du läsa på sidan 6.
En annan viktig förändring, som vi också berättar om i detta nummer, var den nya förordning om särläkemedel som tillkom i EU 2000 för att stimulera läkemedelsföreta-gen att utveckla läkemedel för patienter med ovanliga sjuk-doms-tillstånd. En uppföljning av vad denna nya förordning inneburit visar på en konstant uppåtgående kurva. Från att ha haft en handfull läkemedel för sällsynta sjukdomar har vi idag nära 70 godkända särläkemedel på marknaden, och betydligt fler substanser är klassificerade som potentiella produkter. En betydande förändring med andra ord!
Här på Läkemedelsverket är vi också mitt uppe i ett stort förändringsarbete. Vi ser bland annat över vår organisation i syfte att bli mer tydliga i vårt uppdrag, både internt men också utåt gentemot sjukvård och allmänhet. I maj tog vi också del av Stefan Carlssons utredning om statens styrning av vård och omsorg – en utredning som kan innebära stora förändringar för oss och många andra myndigheter. Syftet med utredningen har varit att få till en tydligare nationell samordning och att skapa en bättre helhetsbild för mot- tagarna av myndigheternas tjänster och tillsyn. Vi välkomnar att en översyn görs med målet att få ett bättre helhets- perspektiv i vården.
Vi vet inte ännu hur regeringen kommer att hantera ut-redningen och dess förslag och inte heller vilka beslut som kommer att tas. Tills vi vet det kommer vi att fortsätta vårt uppdrag att verka för en bättre folk- och djurhälsa, genom att följa läkemedlens hela livscykel alltifrån utveckling och kliniska prövningar, till hur de används.
Önskar er alla en glad sommar!
Information från LäkemedelsverketBox 26, 751 03 UppsalaTelefon 018-17 46 00Telefax 018-54 85 66E-post: [email protected]
Ansvarig utgivare: Christina Rångemark Åkerman
Redaktion: Kristina Bergström, Malika Hadrati, Christina Hambn, Martina Tedenborg och Pernilla Örtqvist.Ytterligare exemplar kan rekvireras från: Läkemedelsanvändning, Medicinsk information
ISSN 1101-7104 Tryck: Elanders Sverige AB, 2012
Har du ändrat adress? Vill du ha tidningen till en ny adress ber vi dig skicka både din nya och gamla adress till oss via e-post eller brev.
2 • InformatIon fr ån L äkemedeLsverket 4 : 2012
LedarsIda
Christina Åkerman Generaldirektör
Förändringar leder oss framåt

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 3
Fotosensibiliserande läkemedel ................................. 4Hur har försäljningen av receptfria läkemedel utvecklats sedan apoteksreformen? .......................... 6Den positiva utvecklingen av läkemedel mot HIV-infektion – implikationer för studiedesign och fortsatt läkemedelsutveckling .............................. 11Nya föreskrifter för kliniska läkemedelsprövningar ............................................... 15Ny förordning har gett oss fler läkemedel mot sällsynta sjukdomar ........................................... 17Samma aktiva substans men olika information i produktresumé (SmPC) och bipacksedel .............. 18Nitroglycerin Recip indraget på grund av stabilitetsproblem ................................................. 19Tillfällig brist på Betapred injektionsvätska ............ 20Pradaxa – tydligare information till patienter och förskrivare för att minska blödningsrisken ....... 20Ändrade rekommendationer om vaccination mot röda hund hos nyförlösta .................................. 21Inrapporterade biverkningar 2011 från hälso- och sjukvården samt allmänheten ............................ 21Allmänhetens inrapporterade misstänkta läkemedelsbiverkningar 2011 ................................... 26Q-Med upphör med Macrolane för behandling av bröst ...................................................................... 29Äldre Evab-patientsängar kan störa elektronisk utrustning ................................................................. 29
Tandvårds- och läkemedels- förmånsverket informerarTLV informerar ......................................................... 39
Observanda
Innehåll
Biverkningsblanketter
Biverkningsblankett .................................................. 42Biverkningsblankett, djur ......................................... 44Anmälan/Rapport Medicinteknisk avvikelse .......... 45
Nya läkemedel
Ameluz (5-aminolevulinsyra) .................................. 30Dexdor (dexmedetomidin) ...................................... 33Plenadren (hydrokortison) ....................................... 35Remicade (infliximab) – ny indikation .................... 37
Tidigare utgivna nummer
Tidigare utgivna nummer ......................................... 48
Av professor Olle Larkö, Sahlgrenska Universitets-sjukhuset, Hud- och könskliniken, 413 45 Göteborg.
Solen kan orsaka hudreaktioner, framför allt i kombina-tion med vissa läkemedel. I artikeln ges en översikt över solrelaterade hudreaktioner.
De flesta personer har inga större problem med överkänslig-het mot solljus. Den individuella känsligheten är dock stor eftersom vi har olika pigmentering. Inom dermatologin talar man om olika hudtyper där typ I representerar ljus-/rödhå-riga personer och typ VI personer som är mörka i huden. De flesta som har sitt ursprung i Sverige har hudtyp II–III. Normalt krävs cirka 30–45 minuter i solen mitt på dagen för att bli röd 24 timmar senare. Maximal rodnad i huden in-träffar 8–24 timmar efter solbelysning.
Solljus innehåller alla möjliga våglängder men i detta sammanhang är det framför allt det ultravioletta området som är intressant. Det indelas i UVA, UVB och UVC. UVA är våglängdsområdet 315–400 nm, UVB 280–315 nm och UVC våglängder kortare än 290 nm. I praktiken kan man bortse från våglängderna kortare än 290 nm eftersom de filtreras bort av ozonlagret. UVB ger erytem. Både UVB och UVA ger brunhet efter solexposition. UVB anses ge i första hand skivepitelcancer i huden medan malignt mela-nom sannolikt induceras av både UVA och UVB.
De flesta interaktioner mellan solljus och läkemedelsom-rådet sker inom UVA. Tyvärr är detta våglängdsområde svårare att skydda sig mot än UVB vilket gör att solskydds-medel endast har begränsad effekt på läkemedelsinducerad solljusöverkänslighet.
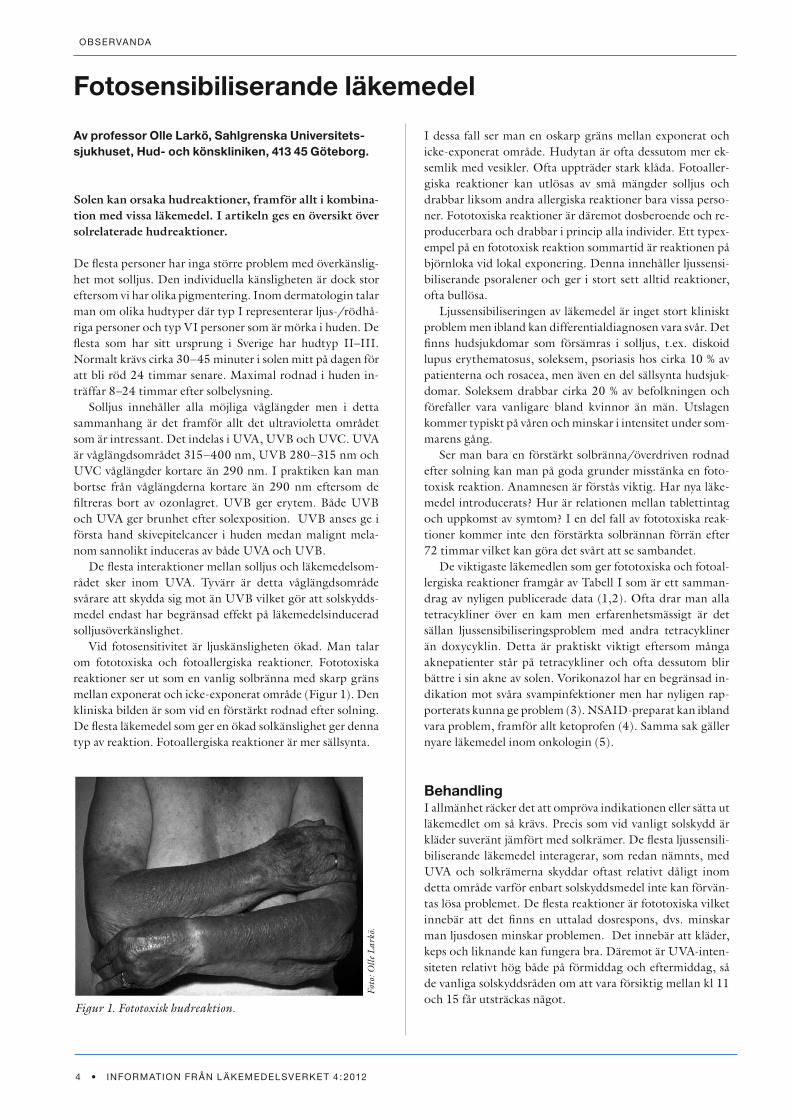
Vid fotosensitivitet är ljuskänsligheten ökad. Man talar om fototoxiska och fotoallergiska reaktioner. Fototoxiska reaktioner ser ut som en vanlig solbränna med skarp gräns mellan exponerat och icke-exponerat område (Figur 1). Den kliniska bilden är som vid en förstärkt rodnad efter solning. De flesta läkemedel som ger en ökad solkänslighet ger denna typ av reaktion. Fotoallergiska reaktioner är mer sällsynta.
I dessa fall ser man en oskarp gräns mellan exponerat och icke-exponerat område. Hudytan är ofta dessutom mer ek-semlik med vesikler. Ofta uppträder stark klåda. Fotoaller-giska reaktioner kan utlösas av små mängder solljus och drabbar liksom andra allergiska reaktioner bara vissa perso-ner. Fototoxiska reaktioner är däremot dosberoende och re-producerbara och drabbar i princip alla individer. Ett typex-empel på en fototoxisk reaktion sommartid är reaktionen på björnloka vid lokal exponering. Denna innehåller ljussensi-biliserande psoralener och ger i stort sett alltid reaktioner, ofta bullösa.
Ljussensibiliseringen av läkemedel är inget stort kliniskt problem men ibland kan differentialdiagnosen vara svår. Det finns hudsjukdomar som försämras i solljus, t.ex. diskoid lupus erythematosus, soleksem, psoriasis hos cirka 10 % av patienterna och rosacea, men även en del sällsynta hudsjuk-domar. Soleksem drabbar cirka 20 % av befolkningen och förefaller vara vanligare bland kvinnor än män. Utslagen kommer typiskt på våren och minskar i intensitet under som-marens gång.
Ser man bara en förstärkt solbränna/överdriven rodnad efter solning kan man på goda grunder misstänka en foto-toxisk reaktion. Anamnesen är förstås viktig. Har nya läke-medel introducerats? Hur är relationen mellan tablettintag och uppkomst av symtom? I en del fall av fototoxiska reak-tioner kommer inte den förstärkta solbrännan förrän efter 72 timmar vilket kan göra det svårt att se sambandet.
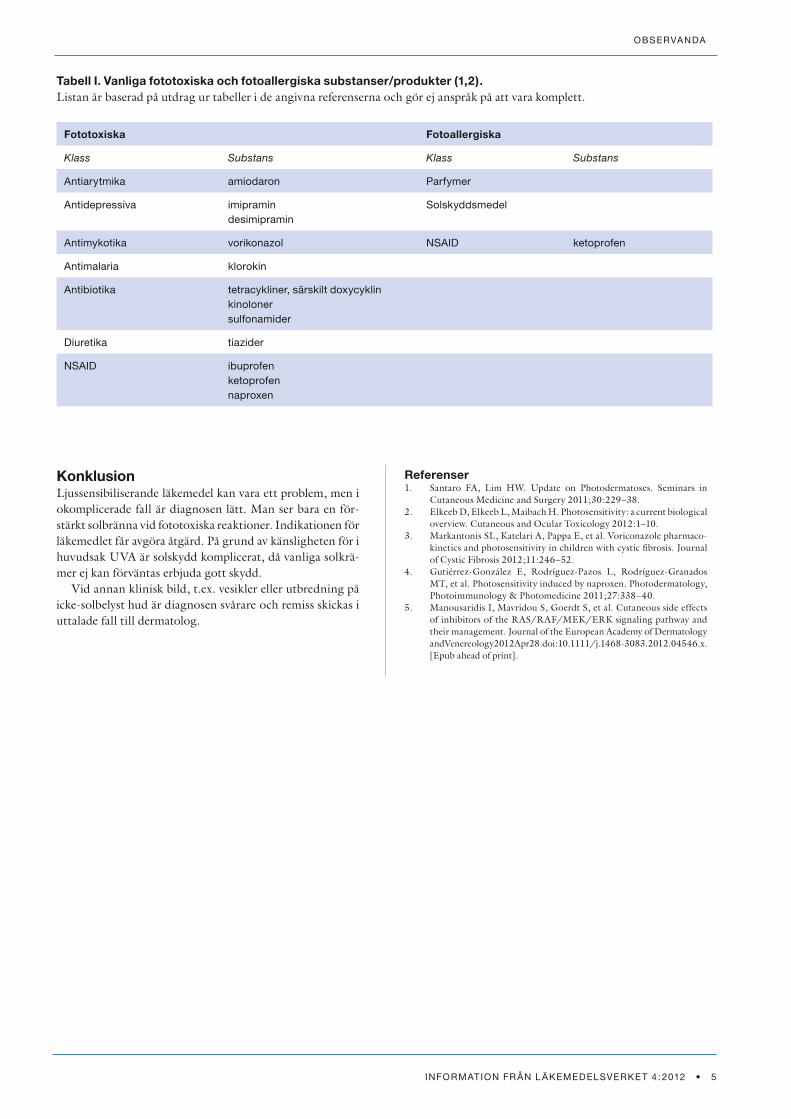
De viktigaste läkemedlen som ger fototoxiska och fotoal-lergiska reaktioner framgår av Tabell I som är ett samman-drag av nyligen publicerade data (1,2). Ofta drar man alla tetracykliner över en kam men erfarenhetsmässigt är det sällan ljussensibiliseringsproblem med andra tetracykliner än doxycyklin. Detta är praktiskt viktigt eftersom många aknepatienter står på tetracykliner och ofta dessutom blir bättre i sin akne av solen. Vorikonazol har en begränsad in-dikation mot svåra svampinfektioner men har nyligen rap-porterats kunna ge problem (3). NSAID-preparat kan ibland vara problem, framför allt ketoprofen (4). Samma sak gäller nyare läkemedel inom onkologin (5).
BehandlingI allmänhet räcker det att ompröva indikationen eller sätta ut läkemedlet om så krävs. Precis som vid vanligt solskydd är kläder suveränt jämfört med solkrämer. De flesta ljussensili-biliserande läkemedel interagerar, som redan nämnts, med UVA och solkrämerna skyddar oftast relativt dåligt inom detta område varför enbart solskyddsmedel inte kan förvän-tas lösa problemet. De flesta reaktioner är fototoxiska vilket innebär att det finns en uttalad dosrespons, dvs. minskar man ljusdosen minskar problemen. Det innebär att kläder, keps och liknande kan fungera bra. Däremot är UVA-inten-siteten relativt hög både på förmiddag och eftermiddag, så de vanliga solskyddsråden om att vara försiktig mellan kl 11 och 15 får utsträckas något.
4 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Figur 1. Fototoxisk hudreaktion.
Fotosensibiliserande läkemedel
Foto
: Olle
Lar
kö.
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 5
observanda
Tabell I. Vanliga fototoxiska och fotoallergiska substanser/produkter (1,2).Listan är baserad på utdrag ur tabeller i de angivna referenserna och gör ej anspråk på att vara komplett.
Fototoxiska Fotoallergiska
Klass Substans Klass Substans
antiarytmika amiodaron Parfymer
antidepressiva imipramindesimipramin
solskyddsmedel
antimykotika vorikonazol nsaId ketoprofen
antimalaria klorokin
antibiotika tetracykliner, särskilt doxycyklinkinolonersulfonamider
diuretika tiazider
nsaId ibuprofenketoprofennaproxen
KonklusionLjussensibiliserande läkemedel kan vara ett problem, men i okomplicerade fall är diagnosen lätt. Man ser bara en för-stärkt solbränna vid fototoxiska reaktioner. Indikationen för läkemedlet får avgöra åtgärd. På grund av känsligheten för i huvudsak UVA är solskydd komplicerat, då vanliga solkrä-mer ej kan förväntas erbjuda gott skydd.
Vid annan klinisk bild, t.ex. vesikler eller utbredning på icke-solbelyst hud är diagnosen svårare och remiss skickas i uttalade fall till dermatolog.
Referenser1. Santaro FA, Lim HW. Update on Photodermatoses. Seminars in
Cutaneous Medicine and Surgery 2011;30:229–38.2. Elkeeb D, Elkeeb L, Maibach H. Photosensitivity: a current biological
overview. Cutaneous and Ocular Toxicology 2012:1–10.3. Markantonis SL, Katelari A, Pappa E, et al. Voriconazole pharmaco-
kinetics and photosensitivity in children with cystic fibrosis. Journal of Cystic Fibrosis 2012;11:246–52.
4. Gutiérrez-González E, Rodríguez-Pazos L, Rodríguez-Granados MT, et al. Photosensitivity induced by naproxen. Photodermatology, Photoimmunology & Photomedicine 2011;27:338–40.
5. Manousaridis I, Mavridou S, Goerdt S, et al. Cutaneous side effects of inhibitors of the RAS/RAF/MEK/ERK signaling pathway and their management. Journal of the European Academy of Dermatology and Venereology 2012 Apr 28. doi: 10.1111/j.1468-3083.2012.04546.x. [Epub ahead of print].

Den 1 november 2009 blev det möjligt att köpa vissa receptfria läkemedel i butiker utanför apotek. Regel-ändringen medförde att konsumenterna fick enklare tillgång till vissa läkemedel avsedda för egenvård. För att studera om användningsmönstret har påverkats av den ökade tillgången har Läkemedelsverket bl.a. analy-serat försäljningsstatistiken för ett antal läkemedels-substanser som används inom egenvård.
InledningEftersom även receptfria läkemedel kan orsaka problem som leder till ökad vårdkonsumtion och miljöbelastning är det av betydelse att veta hur mycket som säljs av varje substans. Dessa uppgifter saknas dock i den offentliga statistiken. Genom att studera hur försäljningen av olika substanser ut-vecklas över tid är det rimligt att anta att man erhåller kun-skap om eventuella förändringar i konsumtions- och miljö-belastningsmönstret.
När ett läkemedel ändrar legal status och blir mer till-gängligt för allmänheten kan en viss förskjutning ske vad avser inköpskälla. Det som tidigare förskrevs och användes inom vården eller köptes mot recept på apotek, kanske nu köps utan recept på apotek eller i allmän handel. En annan möjlig utveckling är att den totala försäljningen ökar utan att någon förskjutning sker mellan inköpskällorna, och vi får en ökad användning av de aktuella preparaten. För att få en heltäckande bild och för att fånga eventuella övervältrings-effekter bör försäljningen studeras i alla förekommande försäljningskanaler.
I butiker utanför apotek säljs framförallt läkemedel mot smärta/värk/feber samt näsdroppar. Läkemedel mot allergi säljs i mycket liten omfattning i dessa butiker och även för-säljningen av nikotinersättningsmedel sker i mycket stor ut-sträckning fortfarande via apotek. I butiker utanför apotek får dessa läkemedel endast säljas till personer över 18 år, till skillnad från på apotek där det inte finns någon ålders-gräns.
SyfteDetta projekt syftar till att studera och analysera hur försälj-ningen av receptfria läkemedel på apotek och hos andra ak-törer utvecklas. Försäljningen analyseras som mängd försålda gram alternativt milligram av respektive substans.
MetodData samlas in från Apotekens Service AB:s register över försäljning av receptfria läkemedel i butik utanför apotek samt från Concise (registret över läkemedel sålda på apotek, för egenvård, mot recept, i slutenvård eller mot rekvisition). Dessa data analyseras och en omräkning görs till antal gram eller milligram av respektive substans utifrån innehållet av aktiv substans per förpackning.
Enligt Apotekens Service AB rapporterar nu cirka 90 % av försäljningsställena in sin försäljningsstatistik. Alla stora kedjor som t.ex. ICA och Coop rapporterar in sina resultat. Under första delen av 2010 var motsvarande siffra runt 70 %.
De substanser som analyserats i denna studie är:paracetamol, ATC-kod N02BE01: lätta analgetika och •antipyretikaacetylsalicylsyra inklusive kombinationer, ATC-kod •N02BA01 och N02BA51: lätta analgetika och anti-pyretika ibuprofen, ATC-kod M01AE01: antiinflammatoriska •och antireumatiska medeloximetazolin, ATC-kod R01AA05: avsvällande •behandling vid nässjukdomarxylometazolin inklusive kombinationer, ATC-kod •R01AA07 och R01AB06: avsvällande behandling vid nässjukdomarcetirizin, ATC-kod R06AE07: antihistaminer för •systemiskt brukloratadin, ATC-kod R06AX02: antihistaminer för •systemiskt bruknikotin, ATC-kod N07BA01: medel vid nikotin- •beroende.
ResultatVissa förpackningsstorlekar av dessa läkemedel får endast säljas på apotek och vissa är dessutom receptbelagda. Beräkningarna avser läkemedel som säljs på apoteken (recept och egenvård) samt de läkemedel som säljs i butik utanför apotek.
Figur 1 visar att egenvårdsförsäljningen av ibuprofen på apotek har minskat. Minskningen för antalet sålda gram när man jämför kvartal 1-2010 och kvartal 4-2011 är 5,8 %. Inom butik utanför apotek har en fördubbling av försälj-ningen (101 %) skett. Den totala egenvårdsförsäljningen av ibuprofen är 16 % större kvartal 4-2011 jämfört med kvartal 1-2010.
Figur 2 visar att egenvårdsförsäljningen av acetylsalicyl-syra inklusive kombinationer på apoteken har minskat. Minskningen för antalet sålda gram är 8,5 % mellan kvartal 1-2010 och kvartal 4-2011. Under samma tid har försälj-ningen inom butik utanför apotek ökat med 92 % och nu säljs det mer acetylsalicylsyra inklusive kombinationer i butik utanför apotek än på apotek. Den totala försäljningen av acetylsalicylsyra inklusive kombinationer utan recept har ökat med 31 %.
Figur 3 visar att egenvårdsförsäljningen av paracetamol via apotek har minskat. Minskningen för antalet sålda gram är 9,3 % mellan kvartal 1-2010 och kvartal 4-2011. Under samma tid har försäljningen inom butik utanför apotek ökat med 44 %. Den totala försäljningen av paracetamol utan recept har ökat med 3,1 %.
6 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Hur har försäljningen av receptfria läkemedel utvecklats sedan apoteksreformen?

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 7
observanda
apotekbutik utanför apotektotalt
25 000 000
20 000 000
15 000 000
10 000 000
5 000 000
02010 kv 1 2010 kv 2 2010 kv 3 2010 kv 4 2011 kv 1 2011 kv 2 2011 kv 3 2011 kv 4
Fö
rsä
ljnin
g a
v ib
up
rofe
n (g
)
Figur 1. Antal sålda gram ibuprofen 2010–2011 i egenvård på apotek och i butik utanför apotek.
Figur 2. Antal sålda gram acetylsalicylsyra inklusive kombinationer 2010–2011 i egenvård på apotek och i butik utanför apotek.
apotekbutik utanför apotektotalt
2010 kv 1 2010 kv 2 2010 kv 3 2010 kv 4 2011 kv 1 2011 kv 2 2011 kv 3 2011 kv 4
40 000 000
35 000 000
30 000 000
25 000 000
20 000 000
15 000 000
10 000 000
5 000 000
Fö
rsä
ljnin
g a
v p
ara
ce
tam
ol (
g)
Figur 3. Antal sålda gram paracetamol 2010–2011 i egenvård på apotek och i butik utanför apotek.
apotekbutik utanför apotektotalt
2010 kv 1 2010 kv 2 2010 kv 3 2010 kv 4 2011 kv 1 2011 kv 2 2011 kv 3 2011 kv 4
14 000 000
12 000 000
10 000 000
8 000 000
6 000 000
4 000 000
2 000 000
Fö
rsä
ljnin
g a
v ac
ety
lsa
licyl
syra
(g
)

8 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Figur 4. Antal sålda milligram xylometazolin inklusive kombinationer 2010–2011 i egenvård på apotek och i butik utanför apotek.
Figur 5. Försäljningen av nikotin till kund 2010–2011 i egenvård på apotek och i butik utanför apotek i kronor.
2010 kv 1 2010 kv 2 2010 kv 3 2010 kv 4 2011 kv 1 2011 kv 2 2011 kv 3 2011 kv 4
apotekbutik utanför apotektotalt
16 000 000
14 000 000
12 000 000
10 000 000
8 000 000
6 000 000
4 000 000
2 000 000
Fö
rsä
ljnin
g a
v xy
lom
eta
zolin
(m
g)
2010 kv 1 2010 kv 2 2010 kv 3 2010 kv 4 2011 kv 1 2011 kv 2 2011 kv 3 2011 kv 4
apotekbutik utanför apotektotalt
200 000 000
180 000 000
160 000 000
140 000 000
120 000 000
100 000 000
80 000 000
60 000 000
40 000 000
20 000 000
Fö
rsä
ljnin
g a
v n
iko
tin
(SE
K)
Även egenvårdsförsäljningen via apotek av xylometazolin inklusive kombinationer har minskat, vilket framgår av Figur 4. Minskningen av antalet sålda milligram är 6,9 % mellan kvartal 1-2010 och kvartal 4-2011. Under samma tid har försäljningen i butik utanför apotek ökat med 75 %. Den totala försäljningen av xylometazolin inklusive kombinatio-ner utan recept har ökat med 11 %.
För nikotin (Figur 5) har försäljningsvärdet för apoteken ökat med 14 % och i butik utanför apotek med 17 %. Den totala egenvårdsförsäljningen har ökat med 15 %.
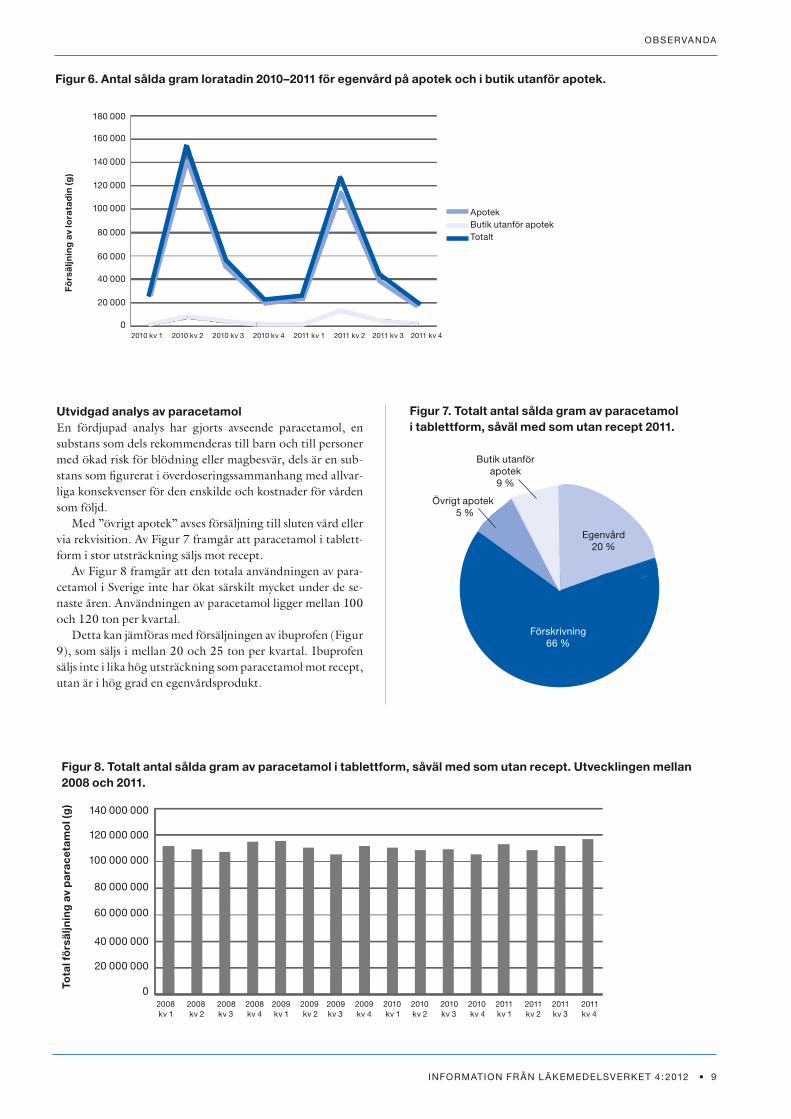
För loratadin (Figur 6) försvåras analysen av att försälj-ningen är säsongsbunden. Man kan konstatera att loratadin
säljs i liten omfattning i butik utanför apotek, men att en viss ökning av försäljningen i butik utanför apotek har skett mel-lan 2010 och 2011 och att en viss minskning av försälj-ningen på apotek har skett.
I butik utanför apotek säljs framför allt läkemedel mot smärta/värk/feber samt näsdroppar.
Läkemedel mot allergier säljs endast i begränsad omfatt-ning inom butik utanför apotek. Även nikotinförsäljningen sker i mycket stor utsträckning fortfarande via apotek.
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 9
observanda
Figur 6. Antal sålda gram loratadin 2010–2011 för egenvård på apotek och i butik utanför apotek.
2010 kv 1 2010 kv 2 2010 kv 3 2010 kv 4 2011 kv 1 2011 kv 2 2011 kv 3 2011 kv 4
apotekbutik utanför apotektotalt
180 000
160 000
140 000
120 000
100 000
80 000
60 000
40 000
20 000
0
Fö
rsä
ljnin
g a
v lo
rata
din
(g
)
Utvidgad analys av paracetamolEn fördjupad analys har gjorts avseende paracetamol, en substans som dels rekommenderas till barn och till personer med ökad risk för blödning eller magbesvär, dels är en sub-stans som figurerat i överdoseringssammanhang med allvar-liga konsekvenser för den enskilde och kostnader för vården som följd.
Med ”övrigt apotek” avses försäljning till sluten vård eller via rekvisition. Av Figur 7 framgår att paracetamol i tablett-form i stor utsträckning säljs mot recept.
Av Figur 8 framgår att den totala användningen av para-cetamol i Sverige inte har ökat särskilt mycket under de se-naste åren. Användningen av paracetamol ligger mellan 100 och 120 ton per kvartal.
Detta kan jämföras med försäljningen av ibuprofen (Figur 9), som säljs i mellan 20 och 25 ton per kvartal. Ibuprofen säljs inte i lika hög utsträckning som paracetamol mot recept, utan är i hög grad en egenvårdsprodukt.
Figur 7. Totalt antal sålda gram av paracetamol i tablettform, såväl med som utan recept 2011.
egenvård20 %
förskrivning66 %
butik utanför apotek
9 %
Övrigt apotek5 %
Tota
l fö
rsä
ljnin
g a
v p
ara
cet
am
ol (
g)
2008 2008 2008 2008 2009 2009 2009 2009 2010 2010 2010 2010 2011 2011 2011 2011 kv 1 kv 2 kv 3 kv 4 kv 1 kv 2 kv 3 kv 4 kv 1 kv 2 kv 3 kv 4 kv 1 kv 2 kv 3 kv 4
140 000 000
120 000 000
100 000 000
80 000 000
60 000 000
40 000 000
20 000 000
0
Figur 8. Totalt antal sålda gram av paracetamol i tablettform, såväl med som utan recept. Utvecklingen mellan 2008 och 2011.

10 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Figur 9. Antal sålda gram av ibuprofen, såväl med som utan recept under 2011.
SammanfattningIntressant att notera är att efter omregleringen av apoteksmarknaden
sker 16 % av all försäljning av läkemedel för egenvård, räknat i antal kronor, i butik utanför apotek,•säljs paracetamol räknat i gram mest mot recept (66%), men substansen ligger även i topp inom butik utanför apotek,•har den totala försäljningen av paracetamol räknat i gram ökat med 3,1 % inom egenvård och butik utanför apotek,•har den totala försäljningen räknat i milligram av näsdroppar innehållande xylometazolin ökat med 11 % (apotek och •butik utanför apotek)säljs medel mot rökning liksom medel mot pollenallergier mest via apotek.•
kvartal 1 2009 kvartal 1 2010 kvartal 2 2010 kvartal 3 2010 kvartal 4 2010
egenvård (apotek)
apotek övrigt (recept, rekv. m.m.)
butik utanför apotek
totalt antal sålda gram ibuprofen
25 000 000
20 000 000
15 000 000
10 000 000
5 000 000
0
Tota
l fö
rsä
ljnin
g a
v ib
up
rofe
n (g
)
Det har gått drygt 30 år sedan HIV upptäcktes, och 25 år sedan det första läkemedlet godkändes. Idag finns sex klas-ser av specifikt verkande läkemedel, som sinsemellan angri-per olika delar av virusets livscykel. Tillgången till effektiva behandlingsalternativ har på kort tid radikalt förändrat förutsättningarna för utveckling av nya preparat.
Den kliniska upptäckten av AIDS gjordes 1981 då man i Kalifornien noterade ett kluster av homosexuella män med diagnostiserad pneumocystis-pneumoni, men som saknade de vanliga riskfaktorerna för denna opportunistiska infek-tion. Ett par år senare, 1983, publicerade två forskargrupper (Montagnier och Gallo) oberoende av varandra att man hade isolerat orsaken, HIV-viruset. Luc Montagnier och Françoise Barré-Sinoussi fick 2008 nobelpriset i medicin för upp- täckten. I ett tidigt skede rådde en stor optimism avseende möjligheten att inom kort skapa ett vaccin, vilket inte blev fallet. Nu 30 år senare finns fortfarande inget vaccin, och cirka 30 miljoner människor beräknas ha avlidit i AIDS.
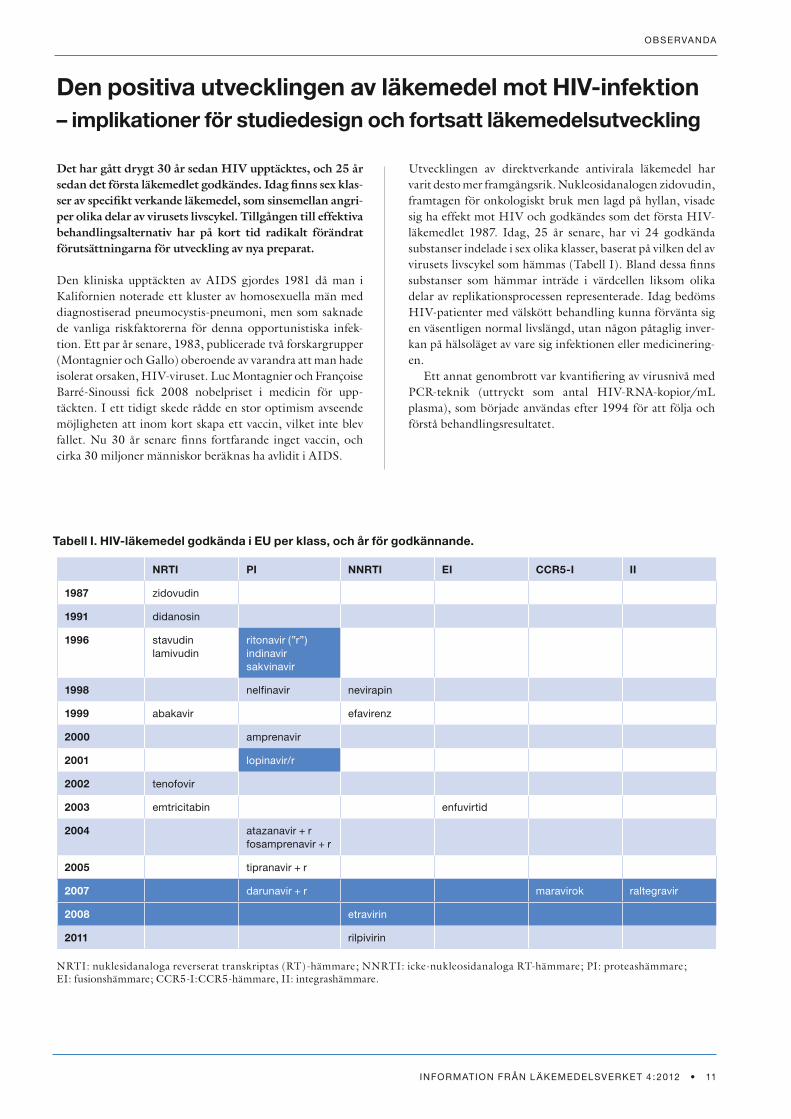
Utvecklingen av direktverkande antivirala läkemedel har varit desto mer framgångsrik. Nukleosidanalogen zidovudin, framtagen för onkologiskt bruk men lagd på hyllan, visade sig ha effekt mot HIV och godkändes som det första HIV-läkemedlet 1987. Idag, 25 år senare, har vi 24 godkända substanser indelade i sex olika klasser, baserat på vilken del av virusets livscykel som hämmas (Tabell I). Bland dessa finns substanser som hämmar inträde i värdcellen liksom olika delar av replikationsprocessen representerade. Idag bedöms HIV-patienter med välskött behandling kunna förvänta sig en väsentligen normal livslängd, utan någon påtaglig inver-kan på hälsoläget av vare sig infektionen eller medicinering-en.
Ett annat genombrott var kvantifiering av virusnivå med PCR-teknik (uttryckt som antal HIV-RNA-kopior/mL plasma), som började användas efter 1994 för att följa och förstå behandlingsresultatet.
NRTI PI NNRTI EI CCR5-I II
1987 zidovudin
1991 didanosin
1996 stavudinlamivudin
ritonavir (”r”)indinavirsakvinavir
1998 nelfinavir nevirapin
1999 abakavir efavirenz
2000 amprenavir
2001 lopinavir/r
2002 tenofovir
2003 emtricitabin enfuvirtid
2004 atazanavir + rfosamprenavir + r
2005 tipranavir + r
2007 darunavir + r maravirok raltegravir
2008 etravirin
2011 rilpivirin
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 11
observanda
Tabell I. HIV-läkemedel godkända i EU per klass, och år för godkännande.
Den positiva utvecklingen av läkemedel mot HIV-infektion – implikationer för studiedesign och fortsatt läkemedelsutveckling
NRTI: nuklesidanaloga reverserat transkriptas (RT)-hämmare; NNRTI: icke-nukleosidanaloga RT-hämmare; PI: proteashämmare; EI: fusionshämmare; CCR5-I:CCR5-hämmare, II: integrashämmare.

12 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
HIV-behandling och resistensutvecklingLäkemedelsresistens är av central betydelse inom HIV- behandling, och måste därför förklaras i viss detalj i den aktu-ella diskussionen. HIV har en hög spontan mutationsbenä-genhet, vilket innebär att det i varje obehandlad HIV- infekterad individ finns en enorm mängd olika så kallade kvasispecies representerade. Grovt räknat finns därmed virion (en enstaka viruspartikel) med alla tänkbara singelmutationer och likaledes sannolikt alla tänkbara kombinationer av dub-belmutationer representerade, redan innan någon behand-ling mot HIV har påbörjats. Chansen, eller risken, att tre specifika mutationer skall hittas inom ett och samma virion i den obehandlade individen är däremot extremt låg.
För flertalet HIV-läkemedel finns det singelmutationer som medför kraftigt nedsatt effekt (upp till 100-faldigt). Idag är det därför lätt att förstå varför monoterapi med zido-vudin hade en blygsam och snabbt övergående effekt, och varför kombinationsbehandling med två nukleosidanaloger inte heller gav varaktig effekt – resistent virus selekterades fram. Inom var och en av de sex läkemedelsklasserna (i den mån det finns flera läkemedel i klassen) råder hög grad av korsresistens. Däremot förekommer inte korsresistens mel-lan klasserna.
Att korsresistens uppstår inom läkemedelsklasserna men inte emellan dem är bakgrunden till framgångskonceptet inom behandling av HIV-infektion: trippelbehandling, kombinationsterapi med tre läkemedel med olika resistens-mönster. Strategin kom i bruk efter 1996, i samband med godkännandet av läkemedel i en andra klass (proteashäm-marna), och innebar starten på en helt ny era inom HIV-behandlingen.
Genotypisk resistensbestämning, där mutationer associe-rade med nedsatt läkemedelseffekt snabbt kan påvisas med PCR-teknik, blev tillgänglig 1991, och har haft mycket stor betydelse både för läkemedelsutvecklingen och i den kliniska vardagen. Sverige var ett av de första länderna att introdu-cera dessa tekniker i kliniskt bruk.
Selektion av resistenta stammar, och därefter ackumule-rande resistensutveckling sker i närvaro av läkemedel när full virussuppression inte uppnås med behandlingen. Vid full-god viruskontroll (definierat som icke detekterbara virusni-våer med ovan nämnda metod) ses däremot inte någon fortsatt resistensutveckling. HIV tillhör gruppen retrovirus och bygger därmed in sitt genom i värdcellens. För cellin-träde måste HIV binda till ett par olika receptorer, CD4-receptorn och en co-receptor. Detta är anledningen till att just vissa celltyper i immunsystemet (framför allt CD4-posi-tiva T-hjälparceller, ”T-celler”, och makrofager) infekteras. Sådana celler kan ha mycket lång livslängd (så kallade min-nesceller), varför uppkommen resistens i praktiken finns lagrad för överskådlig framtid, och man måste alltså ta hän-syn till den i kommande behandling.
Proteashämmarna, för närvarande ett särfall ur resistenssynpunktEn kort passus bör också ägnas proteashämmargruppen inför kommande resonemang. Proteashämmarna skiljer sig i resistenshänseende från övriga läkemedelsklasser. Enstaka
(singel-) mutationer hos viruset medför bara måttligt ned-satt känslighet för dessa preparat (< femfaldigt nedsatt känslighet). Istället krävs en kombination av ett flertal mu-tationer i proteasgenen för att påtagligt nedsatt känslighet (> 10-faldig) ska uppstå. En hög dosering kan således poten-tiellt sett ge bra effekt mot virus som utvecklat en viss grad av resistens mot substansklassen. För den tidigare obehand-lade patienten medför detta att en hög exponering för pro-teashämmare ger en effektiv barriär mot resistensutveckling. Behandlingen medför då låg risk för resistensutveckling, även vid bristande grad av följsamhet.
Utveckling av läkemedel med påtagligt högre resistens-barriär är av stort kliniskt värde, även inom de andra läke-medelsklasserna, och kan vara på väg att lyckas för integras-hämmarna.
Ritonavir för ökad biotillgänglighetEtt effektivt sätt att kraftigt öka koncentrationen (ökat upptag, minskad nedbrytning) av proteashämmare är samadministrering av ritonavir i låg dos, vilket kallas ”boostring”. Ritonavir var den första HIV-proteashämma-ren som godkändes, och gavs som sådan i en dosering om 1 200 mg per dygn med en mycket besvärlig biverknings-profil. När ritonavir används i boostrande syfte, är man inte ute efter substansens antivirala effekt. Istället utnyttjas att ritonavir redan i låg dosering är en mycket potent hämmare av CYP-3A4, den huvudsakliga nedbrytningsvägen för proteashämmarna. Dosen är då 100–200 mg per dygn, bero-ende på vilken proteashämmare som används. I dessa låga doser är biverkningarna också ett betydligt mindre problem.
Sedan lopinavir/r godkändes 2001(med lågdos ritonavir, ”r”, i fast kombination) har sådan boostring utnyttjats för nya preparat i klassen, vilket framgår av Tabell I. Näst intill alla patienter som idag bär på virus resistent mot proteas-hämmare (nedsatt effekt) ådrog sig i praktiken denna resis-tens under behandlingssvikt med icke boostrad proteashäm-mare. Hos patienter som redan från början behandlas med boostrad proteashämmare (som nu är standard), är utveck-ling av resistens mycket ovanlig, även för patienter som inte har förmågan att ta behandlingen med optimal följsamhet.
Ett behandlingslandskap i förändringVid en titt i Tabell I kan man, utifrån tidigare resonemang, peka ut tre perioder i läkemedelsutvecklingen som kanske haft särskilt stor betydelse för den allt bättre kliniska situationen:
Åren 1996/1997, då trippelbehandling medförde det •stora genombrottet och snabbt blev klinisk praxis efter godkännandet av proteashämmarna. Efter 2001, då konceptet med boostrad proteashäm-•mare har haft en påtaglig effekt. Åren 2007/2008, när flera läkemedel godkändes vilket •möjliggjorde framgångsrik behandling (dvs. fullgod virussuppression) även av patienter som påbörjat HIV-terapi under tidigt skede och på grund av icke-optimala behandlingar ådragit sig avancerad resistens:
– Darunavir/r är en proteashämmare med påtagligt god effekt även vid uttalad proteashämmarresistens.
– Raltegavir (integrashämmare) och maravirok (CCR5-hämmare) är de första substanserna inom två helt nya klasser.
– Etravirin kan vara effektivt även hos många patienter som tidigare sviktat med andra NNRTI (nevirapin eller efavirenz).
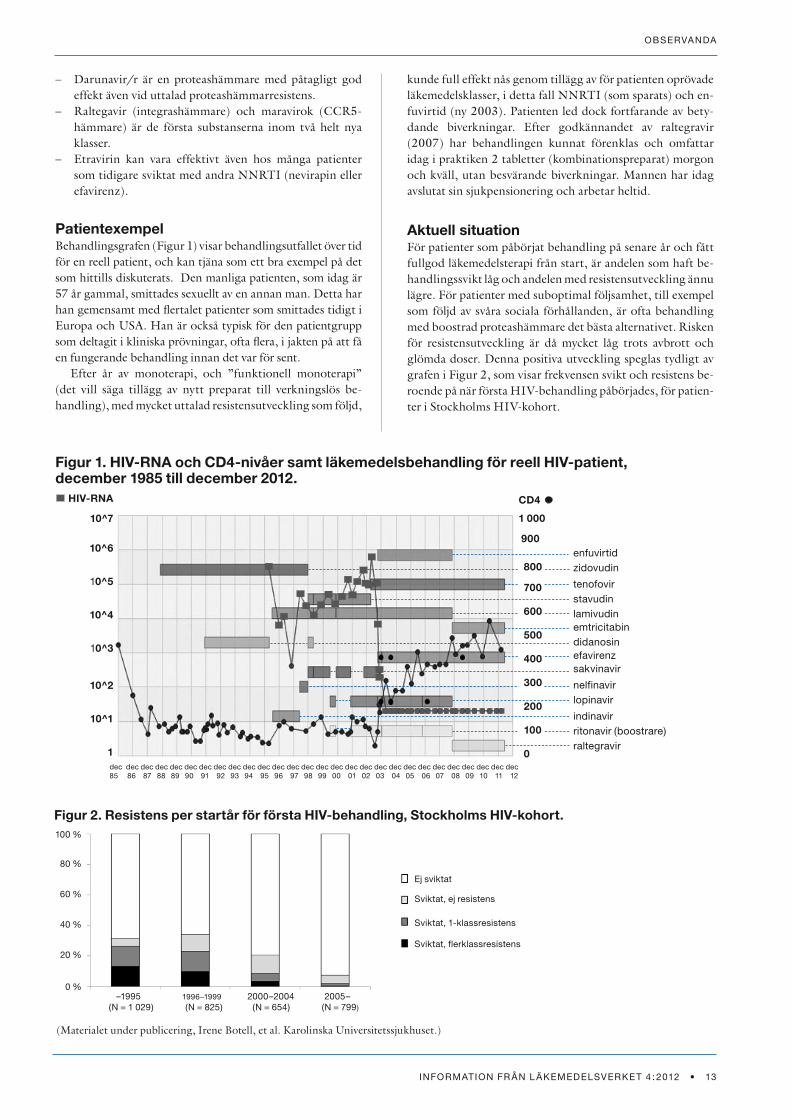
PatientexempelBehandlingsgrafen (Figur 1) visar behandlingsutfallet över tid för en reell patient, och kan tjäna som ett bra exempel på det som hittills diskuterats. Den manliga patienten, som idag är 57 år gammal, smittades sexuellt av en annan man. Detta har han gemensamt med flertalet patienter som smittades tidigt i Europa och USA. Han är också typisk för den patientgrupp som deltagit i kliniska prövningar, ofta flera, i jakten på att få en fungerande behandling innan det var för sent.
Efter år av monoterapi, och ”funktionell monoterapi” (det vill säga tillägg av nytt preparat till verkningslös be-handling), med mycket uttalad resistensutveckling som följd,
kunde full effekt nås genom tillägg av för patienten oprövade läkemedelsklasser, i detta fall NNRTI (som sparats) och en-fuvirtid (ny 2003). Patienten led dock fortfarande av bety-dande biverkningar. Efter godkännandet av raltegravir (2007) har behandlingen kunnat förenklas och omfattar idag i praktiken 2 tabletter (kombinationspreparat) morgon och kväll, utan besvärande biverkningar. Mannen har idag avslutat sin sjukpensionering och arbetar heltid.
Aktuell situationFör patienter som påbörjat behandling på senare år och fått fullgod läkemedelsterapi från start, är andelen som haft be-handlingssvikt låg och andelen med resistensutveckling ännu lägre. För patienter med suboptimal följsamhet, till exempel som följd av svåra sociala förhållanden, är ofta behandling med boostrad proteashämmare det bästa alternativet. Risken för resistensutveckling är då mycket låg trots avbrott och glömda doser. Denna positiva utveckling speglas tydligt av grafen i Figur 2, som visar frekvensen svikt och resistens be-roende på när första HIV-behandling påbörjades, för patien-ter i Stockholms HIV-kohort.
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 13
observanda
Figur 1. HIV-RNA och CD4-nivåer samt läkemedelsbehandling för reell HIV-patient, december 1985 till december 2012.
Figur 2. Resistens per startår för första HIV-behandling, Stockholms HIV-kohort.
(Materialet under publicering, Irene Botell, et al. Karolinska Universitetssjukhuset.)
100 %
80 %
60 %
40 %
20 %
0 % –1995 1996–1999 2000–2004 2005–(n = 1 029) (n = 825) (n = 654) (n = 799)
ej sviktat
sviktat, ej resistens
sviktat, 1-klassresistens
sviktat, flerklassresistens
enfuvirtidzidovudin
tenofovirstavudinlamivudinemtricitabindidanosinefavirenzsakvinavir
nelfinavirlopinavir
indinavirritonavir (boostrare)raltegravir
CD4
900
800
700
600
500
400
300
200
100
0
HIV-RNA
10^7
10^6
10^5
10^4
10^3
10^2
10^1
1dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec dec85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 00 01 02 03 04 05 06 07 08 09 10 11 12
1 000

Tidigare fanns således ett stort antal patienter med sviktande behandlingar och resistens i olika omfattning. Behovet av nya läkemedel var stort, och det fanns goda möjligheter att snabbt rekrytera alltifrån tidigare obehandlade och patienter med måttlig resistens till patienter med mycket uttalad resis-tens till kliniska prövningar. De senare årens framgångar, inte minst under perioden 2007/2008, har som framgår helt förändrat detta patientpanorama.
I dagsläget är sviktande behandling oftast en följd av bristande följsamhet, men utan att någon relevant resistens kan påvisas. Dessa patienter lämpar sig sällan för kliniska studier. Patienter med god följsamhet och utan mycket avancerad resistens har idag fullgoda behandlingsresultat med redan godkända läkemedel, och är således inte heller självklart aktuella för studier. Lite tillspetsat kan man säga att de patienter som nu kan vara aktuella att rekrytera till studier är de obehandlade (utan tidigare behandling) å ena sidan, och ett tämligen begränsat antal patienter med mycket avancerad resistens, som inte får behandlingskontroll med tillgängliga läkemedel trots fullgod följsamhet (”be-gränsade möjligheter”), å andra sidan. Läget utgör således en skarp kontrast till situationen som rådde för några år sedan, då en betydligt större andel patienter stod på svik-tande behandlingar och uppvisade resistens av varierande grad.
Riktlinjer för läkemedelsutvecklingEuropeiska läkemedelsmyndigheten (EMA) och Food and Drug Administration (FDA) i USA utarbetar regulatoriska riktlinjer som beskriver vad som krävs inom ett utvecklings-program – typ av studier, antal patienter etc. – för nygod-kännande av läkemedel i EU/USA. Riktlinjerna skall vara utformade på ett sådant sätt att kliniska studier skall ge god möjlighet att utvärdera effekt och säkerhet av det nya läke-medlet, utan att studiepatienternas säkerhet åsidosätts.
EMAs riktlinjer reviderades senast 2007/2008, med ut-gångspunkt från den situation som rådde då och baserat på de kliniska studier som genomfördes åren innan. Innan denna revision hade flertalet nya läkemedel i första hand studerats på patienter med sviktande behandling, ofta med avancerad resistens (patienter av den typ som gavs exempel på ovan). Därefter gjordes studier på patienter med mindre avancerad resistens eller utan tidigare behandling. Studierna i sviktande patienter var kontrollerade, antingen gentemot placebo eller mot bästa tillgängliga aktiva läkemedel ur samma klass. Vid revisionen 2007/2008 ville man i första hand undvika att patienter med avancerad resistens och problem att hitta en fungerande behandling skulle delta i studier där de kunde lottas till placebo – med en oacceptabel risk för fortsatt resistensutveckling som följd. Dessa (nu gällande) riktlinjer förordade därför att studier på patienter med behandlingssvikt skulle utföras på patienter utan alltför avancerad resistens, där optimerad bakgrundsbehandling medför en så pass hög aktivitet att en randomiserad studie är acceptabel ur patientetisk synvinkel. Patienter med begrän-sade möjligheter skulle istället ges möjlighet att erhålla det nya läkemedlet inom ramen för expanded access/compas-sionate use.
I ljuset av utvecklingen som pågick tämligen parallellt, dis-kuterat tidigare, står det nu klart att detta förhållningssätt inte var en framkomlig väg. Den patientgrupp som kunde ingå i studier med önskad design kunde efterhand i praktiken svårligen rekryteras. De aktuella riktlinjerna måste därför anses hämma en möjlig fortsatt utveckling av läkemedel till patienter med uttalade resistensproblem – den grupp som fortfarande har ett stort behov av nya behandlingsalternativ.
Genomgripande revidering pågårRiktlinjerna för industrin genomgår nu en genomgripande förändring, med följande grundupplägg:
Hittills har studier på patienter med behandlingssvikt (och resistensproblematik mot godkända läkemedel) krävts för att få indikation för behandling av sådana ”behand-lingserfarna” patienter, även för nytt läkemedel ur ny klass. Med den nya versionen avses att det första läkemedlet i en ny klass (vilket innebär att resistens mot läkemedlet inte förekommer) kan få obegränsad indikation på basis av studier på tidigare obehandlade patienter. Ingen vet om, och i så fall när, det kommer någon ny klass av direktver-kande HIV-läkemedel.
Den fortsatta utvecklingen inom befintliga klasser pågår. För ett sådant läkemedel, som inte bara är avsett för patienter utan resistens mot preparatklassen, sätts med den nya revi-sionen fokus på en rimlig studiedesign för patienter med avancerad resistensproblematik (dvs. begränsade möjlighe-ter). Sådana patienter är möjliga att rekrytera, om än i relativt begränsat antal, och de har ett stort behov av nya läkemedel. Nu föreslås att studierna skall omfatta en kort period (i storleksordningen 14 dagar) av ”funktionell monoterapi”, där det nya läkemedlet jämförs med placebo, som tillägg till sviktande behandling där läkemedelsklassen i fråga ingår. Efter denna period ska studien vara enarmad och alla patien-ter ska få aktiv substans.
Effekten kommer i dessa studier att utvärderas med avse-ende på grad av virussänkning som uppnås under den funktionella monoterapin (primär endpoint), liksom hur bestående effekten förefaller vara under fortsatt optimerad behandling under 24–48 veckor (sekundär endpoint). Den enarmade studiedesignen, som nu anses vara den enda möj-liga för denna patientgrupp, medför klara inskränkningar i möjligheten att utvärdera effektens varaktighet och läke-medlets säkerhet. Parallellt med nämnda studiemodell krävs därför även en traditionell kontrollerad studie på tidigare obehandlade patienter, även om detta inte skulle vara den primära målgruppen för det nya läkemedlet. Därmed garan-teras inte minst att säkerheten kan utvärderas på ett adekvat sätt, det vill säga i randomiserat upplägg mot lämpligt refe-rensläkemedel.
Det tar tid att revidera riktlinjer, och ofta har redan något läkemedel följt den tänkta, kommande linjen med stöd från vetenskaplig och regulatorisk rådgivning. Detta är fallet i den aktuella revisionen, där en ny integrashämmare med effekt även på integrashämmarresistent virus i hög grad följer det upplägg som diskuterats ovan. Detta ger förhoppningar om att de nya riktlinjerna ska vara förenliga med fortsatt utveckling av nya HIV-läkemedel, trots en alltmer komplex situation.
14 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 15
observanda
Nya föreskrifter för kliniska läkemedelsprövningar
Den 1 februari 2012 trädde Läkemedelsverkets före-skrifter (LVFS 2011:19) om kliniska prövningar på människor i kraft. Samtidigt publicerades en vägledning vars syfte är att beskriva och tolka innehållet i gällande regelverk. Vägledningen kommer att kompletteras efter hand. Både föreskrifterna och vägledningen finns tillgängliga via Läkemedelsverkets webbplats www.lakemedelsverket.se
BakgrundI samband med att LVFS 2011:19 trädde i kraft upphävdes de gamla föreskrifterna om kliniska läkemedelsprövningar (LVFS 2003:6). De utgjorde i huvudsak Sveriges anpassning till det EU-gemensamma direktivet för kliniska läkemedelspröv-ningar som infördes den 1 maj 2004. Föreskrifterna har där-efter uppdaterats, bland annat för att tillgodose GCP-direkti-vet från 2005 och den pediatriska lagstiftningen från 2006. Behovet av en mer omfattande översyn av föreskrifterna har vuxit i takt med ytterligare lagändringar, omregleringen av apoteksmarknaden och ändrade europeiska riktlinjer.
Generella ändringarFrågor som regleras av överordnad lagstiftning har ersatts av upplysningsparagrafer som hänvisar till den aktuella lagstift-ningen för att undvika dubbelreglering. De allmänna råden har lyfts ut från föreskriften och ersatts av en separat vägledning.
Prövarens ansvarI de nya föreskrifterna har det förtydligats att legitimerad läkare eller legitimerad tandläkare alltid ansvarar för medi-cinska beslut och medicinsk vård. I vägledningen har det förtydligats att samtycke endast i undantagsfall får inhämtas av annan hälso- och sjukvårdspersonal än legitimerad läkare/tandläkare. Förutsättningen är att Etikprövningsnämnden har godkänt undantaget efter bedömning av samtyckespro-cedurens lämplighet.
Distribution och hantering av prövnings-läkemedelSedan lagen (2009:366) om handel med läkemedel trädde i kraft har förutsättningarna för läkemedelshanteringen i kliniska läkemedelsprövningar förändrats. Enligt lagen gäl-ler olika bestämmelser för distribution av prövningsläke-medel till prövningsställen inom sjukhus respektive till prövningsställen utanför sjukhus. De nya föreskrifterna inne-håller mer detaljerade bestämmelser om hantering och distribution av prövningsläkemedel. Ytterligare förtydligan-den ges i vägledningen.
AnsökningsförfarandetEnligt de nya föreskrifterna ska ansökan till Läkemedels-verket sändas in elektroniskt.
Läkemedelsverkets och Etikprövnings-nämndens skilda rollerDe nya föreskrifterna har tydligare anpassats till de delvis skilda roller som Läkemedelsverket och Etikprövningsnämn-den har i bedömningen av kliniska läkemedelsprövningar.
Prövarnas och prövningsställenas lämplighet bedöms som huvudregel inte längre av Läkemedelsverket. Som ett led i granskningen av ansökan om klinisk läkemedelsprövning inspekterar Läkemedelsverket dock även fortsättningsvis prövningsställen där ett nytt läkemedel som bedöms kunna medföra hög risk för första gången ges till människa. Inspek-tionen utförs för att säkerställa att prövningsstället är ända-målsenligt.
Inte heller proceduren för inhämtande av samtycke be-döms som huvudregel längre av Läkemedelsverket. Detta innebär att patientinformation och samtyckesblankett i fortsättningen inte ingår i den dokumentation som ska läm-nas till Läkemedelsverket.
Ny terminologiMed de nya föreskrifterna införs ett par nya termer:
Icke-prövningsläkemedel• beskriver andra läkemedel än prövningsläkemedel som enligt protokollet ingår i prövningen.Prövningsställe• har införts i definitionsavsnittet.Incident • ersätter oönskad händelse. Biverkning• ersätter oönskad reaktion.I definitionen av begreppet incident har ordet oönskad •ersatts av ordet ogynnsam för att få bättre överensstäm-melse mellan definitionerna av incident och biverkning.
Väsentliga ändringar och avslutande av prövningenOväsentliga ändringar och ändringar som endast är väsent-liga för Etikprövningsnämndens bedömning får införas utan godkännande av Läkemedelsverket. De ska inte skickas till Läkemedelsverket för kännedom. Den ändrade dokumenta-tionen ska istället ingå vid kommande ansökningar om vä-sentliga ändringar. Tidigare meddelanden från Läkemedels-verket om eventuella invändningar mot väsentliga ändringar ersätts av tillståndsbeslut. Om prövningen fortfarande pågår i andra länder behöver Läkemedelsverket inte informeras när prövningen avslutas i Sverige. Läkemedelsverket ska endast informeras när prövningen avslutas i sin helhet.

16 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
SäkerhetsuppföljningRapportering av misstänkta allvarliga oförutsedda biverk-ningar till Läkemedelsverket ska ske i den europeiska bi-verkningsdatabasen EudraVigilance, om inte annat över-enskommits. I vägledningen har det förtydligats att årliga säkerhetsrapporter ska lämnas till Läkemedelsverket så länge prövningen pågår i Sverige, det vill säga innan end-of-trial-definitionen har uppfyllts i Sverige, samt att det rekommen-derade formatet för dessa är ICH E2F (Development Safety Update Reports).
Överträdelser och avvikelserI de nya föreskrifterna har det förtydligats att sponsor är skyldig att omedelbart informera Läkemedelsverket om överträdelser av gällande regelverk eller avvikelser från prövningsprotokollet har gjorts som väsentligen kan påverka försökspersonernas integritet eller säkerhet eller prövning-ens vetenskapliga värde.
Erratum – Läkemedelsbehandling vid inflammatorisk tarmsjukdomI ”Läkemedelsbehandling vid inflammatorisk tarmsjukdom”, publicerad i Information från Läkemedelsverket i april 2012, har överläkare Judith Bruchfeld, Infektionskliniken, Karolinska Universitetssjukhuset, Solna, bidragit med värde-full bakgrundsinformation.
På sidan 18 i behandlingsrekommendationen, under avsnittet ”Tuberkulosscreening och profylax”, ska följande tillägg göras:
Screening och behandling av LTBI (latent tuberculosis infection) bör utföras av behandlande läkare med vana vid detta eller av tbc-specialist.
För närvarande finns det ingen referensmetod för diagnostik av LTBI. I diagnostiken av LTBI ingår därför en sam-manvägd bedömning av en strukturerad anamnes avseende ökad risk for tbc-exponering eller tidigare genomgången och adekvat behandlad latent eller aktiv tbc, symtomscreening, kliniskt status, röntgen av lungor samt resultat av testning avseende immunreaktivitet mot tbc-antigen.
För ytterligare bakgrund till bedömning av tbc hos patienter som behandlas med TNF-hämmare hänvisas till motsva-rande avsnitt om screening och profylax av tuberkulos i Läkemedelsverkets rekommendationer om läkemedelsbehandling av psoriasis (juni 2011) samt till den behandlingsrekommendationens bakgrundsdokument ”Tuberkulos – screening och profylax”.
Bakgrundsdokument och behandlingsrekommendationer finns tillgängliga via Läkemedelsverkets webbplats, www.lakemedelsverket.se, under fliken Hälso- & sjukvård och vidare Behandlingsrekommendationer.

År 2000 fick vi en förordning om särläkemedel (orphan medicinal products) som tillkom för att stimulera läke-medelsföretagen att utveckla läkemedel för patienter med ovanliga sjukdomstillstånd. Den nya förordningen har inneburit en stark tillväxt på området och att vi fått fler godkända läkemedel för sällsynta sjukdomar.
Särläkemedel (orphan medicinal products) är läkemedel som är avsedda för allvarliga, livshotande eller kroniska sjukdoms-tillstånd som förekommer hos högst 5/10 000 individer inom EU och där det inte finns några behandlingar alls, alternativt att det nya läkemedlet förväntas innebära en bety-dande fördel för patienterna jämfört med tillgänglig behand-ling. Många patienter med dessa sällsynta sjukdomar saknar tillgång till läkemedel på grund av att utvecklings- och marknadsföringskostnaderna blir väldigt höga i förhållande till den vinst läkemedelsföretagen kan göra på de aktuella preparaten. Med anledning av detta tillkom en ny förordning om särläkemedel inom EU som antogs av EU-kommissionen i april 2000. Syftet med förordningen var att underlätta och stimulera läkemedelsföretagen att utveckla särläkemedel.
I samband med detta bildades COMP (Committee for Orphan Medicinal Products) som bland annat har till uppgift att bedöma klassificering av särläkemedel, utforma riktlinjer, vara rådgivande till kommissionen i policy-frågor och svara för internationellt samarbete med patientgrupper, forskare/hälso- och sjukvård och regulatoriska myndigheter utanför EU.
– Den nya förordningen blev en grindöppnare för den här typen av läkemedel. Från att ha haft en handfull läkemedel för sällsynta sjukdomar har vi nu nära 70 godkända särläke-medel på marknaden och vi har nyligen klassificerat den tu-sende substansen ”potentiella produkter”, berättar Kerstin Westermark, senior expert på Läkemedelsverket som sedan 2006 är ordförande i COMP.
Administrativt och ekonomiskt stödFör företagen innebär förordningen att de får administra-tivt, vetenskapligt och ekonomiskt stöd, exempelvis genom befrielse från eller reduktion av regulatoriska avgifter. En av de största fördelarna för de lite större företagen är att de får tio års ensamrätt för sin produkt för den aktuella sjukdoms-indikationen. För mindre företag eller enskilda forskare är gratis regulatorisk rådgivning en mycket betydelsefull sti-mulansåtgärd.
– Idag kan en ensam akademiker med en idé till nytt läke-medel göra en gratis ansökan om att få en substans klassifice-rad som ett särläkemedel, vilket innebär en större chans att substansen köps upp av ett större företag som sedan kan ut-veckla läkemedlet, säger Kerstin Westermark.
Samma krav på kvalitetGodkännandet av särläkemedel går enligt samma procedur som för andra läkemedel och samma krav ställs på visad ef-fekt och säkerhet.
– Den enda skillnaden är att man av förklarliga skäl inte kan kräva studier på lika många patienter. I vissa fall sker godkännandet ”under exceptional circumstances” eftersom det inte är möjligt att få en lika omfattande klinisk dokumen-tation som för vanliga sjukdomar. Då ställs istället krav på företagen att göra uppföljningsstudier efter godkännandet.
Ovanliga cancerformer vanligaste indikationen40 % av de ansökningar som kommer in för klassificering är indicerade för sällsynta cancerformer. Glivec är ett av de mest betydelsefulla särläkemedel som godkänts. Andra exempel på särläkemedel är olika enzymersättningsmedel för medfödda metabola sjukdomar, cystisk fibros och olika blodsjukdomar.
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 17
observanda
samtliga nummer av Information från Läkemedelsverket 2001–2012 finns på
www.lakemedelsverket.se
Ny förordning har gett oss fler läkemedel mot sällsynta sjukdomar

Läkemedelsverket får återkommande frågor om varför bipacksedlar och SmPC:er kan skilja mellan originallä-kemedel och generika och mellan generikaläkemedlen sinsemellan, och vad som kan göras åt dessa skillnader. Vi har tidigare informerat om orsaken till skillnaderna, men vi tar åter tillfället i akt att förklara situationen.
Det är viktigt att påpeka att det faktum att en och samma läkemedelssubstans kan ha olika indikationer inte behöver medföra några medicinska risker. Läkemedelsverket säker-ställer att nya generika är likvärdiga med originalpreparatet och att produktresumén inte är missledande på ett sätt som kan innebära risker för patienten. För läkemedel som be-dömts som utbytbara på apoteken föreligger ingen verklig skillnad i effekt eller biverkningar, även om informationen kan se olika ut.
Hur kan preparat som innehåller samma läkemedel ha olika indikationer?Det förekommer att nya generika av äldre läkemedelssub-stanser har fler indikationer än vad originalpreparatet har. I vissa fall kan det vara så att generika saknar någon indikation på grund av att originalets patent fortfarande gäller för den indikationen. Den vanligaste orsaken till skillnader i indika-tioner är dock att originalläkemedlet är godkänt i enskilda länder (nationella procedurer) i Europa och de indikationer som godkänts kan skilja sig avsevärt mellan olika länder. När sedan ansökan om godkännande av ett generikapreparat skickas in sker detta vanligen i en EU-gemensam procedur (antingen den så kallade decentrala eller den ömsesidiga godkännandeproceduren). Ansökan skickas då till ett av länderna som har originalpreparatet godkänt och detta land ansvarar för godkännandeproceduren (”referensland”). Om referenslandet tidigare har godkänt en originalprodukt med en vidare indikation än den vi har i Sverige, så är detta lands myndighet positivt till att godkänna den indikation som originalet redan har i landet. Sverige kan då som medlems-land ifrågasätta den vidare indikationen och efterfråga do-kumentationen för den. Referenslandet bedömer dokumen-tation för den vidare indikationen och övriga länder godkänner eller underkänner indikationen. Om det då är så att den vidare indikationen är godkänd i ett övervägande antal av medlemsländerna, och Sverige inte har mycket goda skäl att ifrågasätta den insända dokumentationen, kan Läke-medelsverket inte ensidigt hävda att indikationen för det nya generiska preparatet ska begränsas. Detta är i enlighet med den EU-gemensamma läkemedelslagstiftningen.
Ett exempel på ovanstående, där originalet och flera äldre generika har smalare indikationer än ett nyare generiskt preparat är azatioprin, där originalet Imurel, godkänt i
Sverige 1967 genom en nationell godkännandeprocedur, har indikationen:
”Transplantation. Systemisk lupus erytematosus då kortiko-steroider inte gett resultat eller då resultaten uppnåtts med doser, som förorsakat biverkningar. Hos patienter med biverk-ningar kan man med hjälp av azatioprinterapi minska den nödvändiga underhållsdosen av steroider.”
De äldre generikapreparaten har en indikation som är mycket lik det svenska originalets indikation medan däremot det senast godkända generikapreparatet Immunoprin (god-känt 2010) har en betydligt vidare indikation:
”Immunoprin är indicerat som immunosuppressiv be-handling som tillägg till immunosuppressiva preparat som är grundläggande i behandlingen.
Immunoprin är indicerat i kombination med andra im-munosuppressiva ämnen som profylax mot transplantatavstöt-ning hos patienter som fått allogena njure-, lever-, hjärt-, lung- eller pankreastransplantat.
Immunoprin används som en immunosuppressiv antime-tabolit antingen ensamt eller, vanligare, i kombination med andra substanser (vanligen kortikosteroider) och/eller proce-durer som påverkar immunsvaret. Terapeutisk effekt kan i-bland ses först efter veckor eller månader och innefatta en steroidbesparande effekt, och därigenom minska toxiciteten förknippad med hög dos och långvarig användning av korti-kosteroider.
Immunoprin är indicerat antingen ensamt eller i kombi-nation med kortikosteroider och/eller andra läkemedel och behandlingar i svåra fall av följande sjukdomar, hos patienter som inte tål steroider eller som är beroende av steroider hos vilka det terapeutiska svaret är otillräckligt trots behandling med höga doser steroider:
svår aktiv reumatoid artrit som inte kan kontrolleras •med mindre toxiska substanser (sjukdomsmodifierande antireumatiska läkemedel, DMARD)svår eller måttligt svår inflammatorisk tarmsjukdom •(Crohns sjukdom eller Ulcerös colit)systemisk lupus erytematosus•dermatomyosit och polymyosit•autoimmun kronisk aktiv hepatit•polyarteritis nodosa•autoimmun hemolytisk anemi•kronisk refraktär idiopatisk trombocytopen purpura”•
Att olika preparat med samma aktiva substans kan ha olika indikationer är förstås inte tillfredsställande. Läkemedels-verket har drivit denna fråga inom EU-samarbetet och lagt fram olika förslag till åtgärder. Att harmonisera produktre-
18 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Samma aktiva substans men olika information i produktresumé (SmPC) och bipacksedel

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 19
observanda
suméerna för originalläkemedlen, så att dessa blir lika i alla EU-länder, är ett sådant förslag och en kontinuerlig översyn pågår. Om originalläkemedlet hade samma produktresumé i alla berörda länder skulle även alla generika för detta läkeme-del erhålla samma informationstext i alla länder. I nuläget är det dock bara ett fåtal läkemedel per år som genomgår en sådan EU-gemensam harmoniseringsprocedur. I väntan på att informationen gås igenom kan läkemedelsmyndigheterna dock inte att vidta andra åtgärder än att informera förskri-vare och patienter om den rådande situationen.
De flesta nya originalläkemedel som godkänts efter 2008 och som utsätts för generikakonkurrens är antingen god-
kända via den centrala eller via den ömsesidiga proceduren, och har därmed en i alla länder gemensam informationstext. Generika till dessa nya läkemedel kommer därför också att få produktresuméer som är likalydande i hela EU. Problemet med olika indikationer för olika fabrikat av samma aktiva substans kommer dock att kvarstå för generika till äldre ori-ginalläkemedel som ännu inte fått sina produktresuméer harmoniserade. I de fall som indikationsskillnaden beror på ett ännu inte utgånget patent för originalläkemedlet så kan tillståndsinnehavaren för ett generikum ansöka om att få även den nya indikationen godkänd när originalets patent går ut.
Nitroglycerin Recip indraget på grund av stabilitetsproblemDen 24 april drogs kärlkrampsmedicinen Nitroglycerin Recip in från marknaden på grund av kvalitetsproblem. Läkemedlet beräknas vara indraget under cirka fyra månader. Patienter som för närvarande använder medlet rekommenderas att kontakta sjukvården för att få alter-nativt läkemedel utskrivet.
Företaget drog in alla tillverkningssatser av Nitroglycerin Recip den 25 april 2012. Orsaken var stabilitetsproblem som påvisades vid efterkontroll. Företaget beräknar att det tar cirka fyra månader att åtgärda kvalitetsproblemen så att produkten åter kan släppas ut på marknaden.
Nitroglycerin Recip är ett snabbverkande läkemedel som placeras under tungan och används för att behandla akuta attacker av kärlkramp. Läkemedlet är godkänt i styrkorna 0,25 och 0,5 mg.
Rekommendation till sjukvårdenNyinsättning I första hand rekommenderas någon av nitroglycerinspray-erna Glytrin eller Nitrolingual.
Ersättning för patienter som behandlas med Nitroglycerin RecipPatienter som behandlas med Nitroglycerin Recip bör kon-takta sin läkare för att få recept på annat läkemedel under den tid som bristsituationen kommer att bestå. Om möjligt bör patienterna istället få Glytrin eller Nitrolingual. Det är viktigt att patienterna får god information om hur produk-terna ska användas.
Ett annat, men mindre snabbverkande, nitroglycerinläkeme-del är Suscard buckaltablett (som liksom sprayerna innehåller glyceryltrinitrat) och som kan användas både förebyggande och för akut behandling. Tabletten löser dock inte upp sig lika snabbt som Nitroglycerin Recip och effekten uppnås därmed långsammare. Suscard finns dock för närvarande inte tillgängligt på marknaden.
För patienter där det är svårt att hitta en likvärdig be-handling med godkänt läkemedel finns det möjlighet att ansöka om licens för ett kortverkande, i Sverige ej registrerat, nitroglycrinpreparat. För anvisningar om licensförskrivning, se länk: www.lakemedelsverket.se
Rekommendation till patienterPatienter som använder Nitroglycerin Recip rekommenderas att kontakta sjukvården för att få alternativt läkemedel för-skrivet.

Under försommaren rådde det brist på injektionsvätska Betapred (betametason) 5 × 1 mL, vilket resulterade i att även lagret för förpackningsstorleken 1 × 5 mL nästan tog slut.
Företaget uppgav i slutet av maj att leverans av Betapred in-jektionsvätska förväntades tidigast i mitten av juni.
För de patienter som inte kan behandlas med Betapred i tablettform finns möjligheten att ansöka om licens för ett i
Sverige ej registrerat betametsasonpreparat i form av injek-tionsvätska.
För anvisningar om licensförskrivning, se Läkemedels-verkets webbplats www.lakemedelsverket.se
20 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Tillfällig brist på Betapred injektionsvätska
Pradaxa – tydligare information till patienter och förskrivare för att minska blödningsriskenTydligare vägledning bör ges till läkare och patienter för att minska den blödningsrisk som är förknippad med antikoagulantiabehandling. Det konstaterar EMAs (Eu-ropean Medicines Agency) vetenskapliga kommitté, CHMP, efter en utredning och föreslår därför att pro-duktinformationen uppdateras.
Pradaxa förskrivs för att förebygga stroke och systemisk emboli hos vuxna patienter som har genomgått elektiv total protesoperation i höft- eller knäled och till vuxna patienter med icke-valvulärt förmaksflimmer.
Förekomsten av blödningar med dödlig utgång har varit lägre efter marknadsföringen av Pradaxa jämfört med vad som observerades i de kliniska prövningar som genomfördes inför godkännandet av läkemedlet. CHMP konstaterar att nytta-riskbalansen för Pradaxa är fortsatt positiv och att lä-kemedlet är ett viktigt alternativ till andra antikoagulantia. Rådgivningen till läkare och patienter behöver dock uppda-teras och förstärkas för att ge tydligare vägledning för en bättre användning av läkemedlet.
Följande uppdateringar rekommenderas i produktinforma-tionen för Pradaxa:
Patienter ska akut söka läkarvård om de ramlar eller •skadar sig under pågående behandling, speciellt om de skadar huvudet, på grund av risk för blödning. Pradaxa ska inte användas tillsammans med andra •antikoagulantia, förutom under en period när behand-lingen ändras till eller från Pradaxa. Pradaxa ska inte användas hos patienter med skada eller •tillstånd som kan leda till en ökad risk för en större blödning.Njurfunktionen bör undersökas hos alla patienter •innan en behandling med Pradaxa inleds och upprepas om försämring av njurfunktionen kan misstänkas.
Sök läkemedelsfakta www.lakemedelsverket.se

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 21
observanda
Ändrade rekommendationer om vaccination mot röda hund hos nyförlösta
Kvinnor som saknar immunitet mot röda hund (rubella) har sedan många år erbjudits vaccination före graviditet eller efter förlossning. Med anledning av att monovalent vaccin mot röda hund inte längre finns tillgängligt i Sverige rekommenderas att istället använda kombina-tionsvacciner.
I Sverige har det tidigare funnits ett vaccin som enbart skyddade mot röda hund, men detta vaccin saluförs inte längre. Vaccinet har erbjudits till nyförlösta kvinnor vilka vid provtagning under graviditeten har visats sakna immunitet mot rubella.
De vacciner som idag finns tillgängliga i Sverige är MMR-VAXPRO och Priorix, som utöver skydd mot röda hund också ger skydd mot mässling och påssjuka (MPR-vaccin). Båda dessa kombinationsvacciner innehåller levande, försva-gat virus och kan ges till icke gravida rubella-negativa kvin-nor, antingen före graviditet eller efter förlossning, för att förebygga rubellasmitta och därmed minska risken för fos-terskador vid kommande graviditeter.
Även kvinnor som under graviditet visats ha dåligt skydd mot röda hund rekommenderas vaccination efter förlossningen.
Inrapporterade biverkningar 2011 från hälso- och sjukvården samt allmänheten
Varje år sammanställer Läkemedelsverket de biverkningar som hälso- och sjukvården samt allmänheten har rappor-terat till myndigheten. Under 2011 tog Läkemedelsverket emot 4 919 rapporter om biverkningar från den svenska hälso- och sjukvården och 597 från allmänheten.
Hälso- och sjukvårdens inrapporterade biverkningarFrån den svenska hälso- och sjukvården inkommer årligen biverkningsrapporter till Läkemedelsverket. Dessa rapporter matas in i SWEDIS, den svenska biverkningsdatabasen. Av dessa rapporter var 4 538 spontanrapporter och resten, 381 rapporter, kom från icke-interventionsstudier, till exempel registerstudier.
Varför behövs biverkningsrapporter?Vid godkännandet av nya läkemedel är endast de vanligaste biverkningarna kända. Kunskapen om mer ovanliga biverk-ningar är ofta mycket begränsad. En bra biverkningsrapporte-ring är därför av stor betydelse för att klargöra riskprofilen hos nya läkemedel när de kommit i normalt bruk av patienter.
Vad ska rapporteras?Den som bedriver verksamhet inom hälso- och sjukvården (läkare, tandläkare, sjuksköterskor) ska till Läkemedelsverket rapportera alla misstänkta biverkningar av nya läkemedel
som ej står upptagna som ”vanliga” eller ”mycket vanliga” biverkningar i produktinformationen. Vidare ska samtliga allvarliga biverkningar, samtliga oförutsedda biverkningar samt sådana biverkningar som verkar öka i frekvens snarast inrapporteras.
Hur görs en sambandsbedömning av biverkningsrapporter?Läkemedelsverket uppmanar hälso- och sjukvården att rap-portera redan vid misstanke om läkemedelsbiverkning. Det innebär att den som rapporterar in biverkningen inte behöver ha tagit ställning till om ett orsakssamband föreligger mellan läkemedlet och den rapporterade händelsen.
Inkomna biverkningsrapporter granskas av experter vid regionala biverkningscentra och Läkemedelsverket och en bedömning görs om det kan föreligga ett orsakssamband mellan läkemedlet och den aktuella händelsen. Från och med den 1 juli i år kommer all granskning av inkomna biverk-ningsrapporter att ske på Läkemedelsverket.
Viktiga faktorer att ta hänsyn till vid bedömningen är bland annat patientens sjukdom, samtidiga intag av andra läkemedel samt tidsförloppen för intaget och för händelsen. I de fall ett samband mellan läkemedlet och den uppkomna händelsen har bedömts vara troligt eller möjligt är detta inte detsamma som att ett orsakssamband mellan läkemedlet och den uppkomna biverkningen har säkerställts. I dessa fall kan ett orsakssamband inte uteslutas och behöver därför utredas vidare.

Hur ska uppgifterna från biverknings- databasen, Swedis, tolkas?Uppgifterna från SWEDIS kan alltså inte användas för att dra direkta slutsatser om ett visst läkemedel har orsakat en specifik biverkning eller orsakat ett dödsfall. Den rapporte-rade reaktionen kan naturligtvis vara orsakad av läkemedlet i fråga men kan även vara orsakad av andra läkemedel som patienten behandlas med, kan vara orsakad av patientens bakomliggande sjukdom eller ha uppkommit oberoende av läkemedlet.
Den enskilda biverkningsrapporten ska ses som en pus-selbit i ett mönster där man kan dra slutsatser om ett eventu-ellt samband med läkemedlet först efter analys av andra lik-nande rapporter, t.ex. från andra länder och från resultat i vetenskapliga undersökningar.
Rapporteringsmönstret 2011Rapporteringsmönstret är i princip oförändrat för år 2011 om vaccinrapporterna undantas. I Tabell I–IV redovisas hur rapporterna fördelar sig på allvarlighetsgrad, ålder/kön, ålder/kön vid dödsfall samt förlopp.
En allvarlig biverkning är en sådan som leder till döden, är livshotande, nödvändiggör sjukhusvård eller förlängd sjuk-husvård, leder till invalidisering eller medför missbildning.För dödsfall har sambandet mellan reaktionen och den fa-tala utgången bedömts som möjlig eller ej utesluten.
Tabell I. Fördelning på allvarlighetsgrad.
Rapporternas allvarlighetsgrad Antal rapporter
allvarliga – ej dödsfall 1 995
allvarliga – dödsfall 101
ej allvarliga 2 825
Liksom tidigare år tillhörde vaccinerna de mest frekvent rapporterade läkemedlen (Tabell V och VI). 184 rapporter rör Pandemrix, varav 101 rapporter gäller narkolepsi. För övriga vacciner visar det stora antalet rapporter en utbredd användning och frekvent rapportering av lokala reaktioner.
För åren 2009–2010 var andelen vaccinrapporter särskilt hög på grund av den intensiva Pandemrixvaccinationen.
Tabell VII redovisar vilka läkemedel som oftast ger upp-hov till rapporter om allvarliga biverkningar. Waran är det läkemedel som rapporterades mest frekvent. Viktigt att no-tera är att flertalet inkomna rapporter för Remicade och Enbrel har skett via registeruppföljning i sjukvården inom ramen för ett särskilt projekt för registeruppföljning (ARTIS) med målet att registrera samtliga allvarliga biverk-ningar. Det representerar därmed en ”intensifierad spontan-rapportering”.
Tabell VIII visar vilka yrkeskategorier som rapporterar biverkningar. Under året 2007 genomfördes generell sjuk-sköterskerapportering. Detta har ännu inte fått genomslag i statistiken. Antal rapporter från sjuksköterskor 2006–2011 ses i Tabell IX.
22 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Tabell II. Ålders- och könsfördelning för samtliga rapporter.
Ålder (år) Män % av total Kvinnor % av total Totalt %
0–20 501 10 543 11 1 047 21
21–40 208 4 329 7 537 11
41–60 400 8 613 12 1 013 21
61–80 747 15 924 19 1 672 34
81–100 260 5 385 8 647 13
okänd 2 0 1 0 3 0
totalt 2 118 43 2 795 57 4 919 100
Tabell III. Ålders- och könsfördelning för dödsfall.
Ålder (år) Män % av totalt antal dödsfall
Kvinnor % av totalt antal dödsfall
Totalt %
0–20 1 1 1 1 2 2
21–40 2 2 0 0 2 2
41–60 9 9 8 8 17 17
61–80 32 31 25 25 57 56
81–100 13 13 10 10 23 23
totalt 57 56 44 44 101 100
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 23
observanda
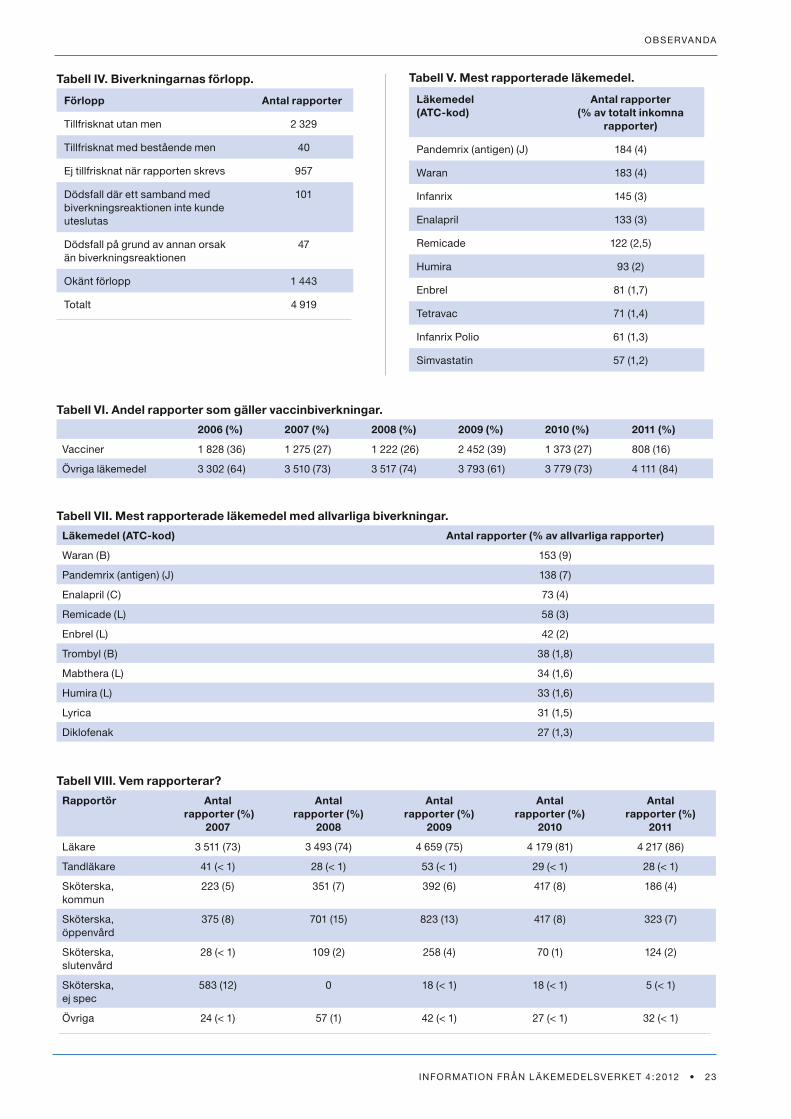
Tabell V. Mest rapporterade läkemedel.
Läkemedel(ATC-kod)
Antal rapporter(% av totalt inkomna
rapporter)
Pandemrix (antigen) (J) 184 (4)
Waran 183 (4)
Infanrix 145 (3)
enalapril 133 (3)
remicade 122 (2,5)
Humira 93 (2)
enbrel 81 (1,7)
tetravac 71 (1,4)
Infanrix Polio 61 (1,3)
simvastatin 57 (1,2)
Tabell VI. Andel rapporter som gäller vaccinbiverkningar.
2006 (%) 2007 (%) 2008 (%) 2009 (%) 2010 (%) 2011 (%)
vacciner 1 828 (36) 1 275 (27) 1 222 (26) 2 452 (39) 1 373 (27) 808 (16)
Övriga läkemedel 3 302 (64) 3 510 (73) 3 517 (74) 3 793 (61) 3 779 (73) 4 111 (84)
Tabell IV. Biverkningarnas förlopp.
Förlopp Antal rapporter
tillfrisknat utan men 2 329
tillfrisknat med bestående men 40
ej tillfrisknat när rapporten skrevs 957
dödsfall där ett samband med biverkningsreaktionen inte kunde uteslutas
101
dödsfall på grund av annan orsak än biverkningsreaktionen
47
okänt förlopp 1 443
totalt 4 919
Tabell VII. Mest rapporterade läkemedel med allvarliga biverkningar.
Läkemedel (ATC-kod) Antal rapporter (% av allvarliga rapporter)
Waran (b) 153 (9)
Pandemrix (antigen) (J) 138 (7)
enalapril (C) 73 (4)
remicade (L) 58 (3)
enbrel (L) 42 (2)
trombyl (b) 38 (1,8)
mabthera (L) 34 (1,6)
Humira (L) 33 (1,6)
Lyrica 31 (1,5)
diklofenak 27 (1,3)
Tabell VIII. Vem rapporterar?
Rapportör Antal rapporter (%)
2007
Antal rapporter (%)
2008
Antal rapporter (%)
2009
Antal rapporter (%)
2010
Antal rapporter (%)
2011
Läkare 3 511 (73) 3 493 (74) 4 659 (75) 4 179 (81) 4 217 (86)
tandläkare 41 (< 1) 28 (< 1) 53 (< 1) 29 (< 1) 28 (< 1)
sköterska, kommun
223 (5) 351 (7) 392 (6) 417 (8) 186 (4)
sköterska, öppenvård
375 (8) 701 (15) 823 (13) 417 (8) 323 (7)
sköterska, slutenvård
28 (< 1) 109 (2) 258 (4) 70 (1) 124 (2)
sköterska, ej spec
583 (12) 0 18 (< 1) 18 (< 1) 5 (< 1)
Övriga 24 (< 1) 57 (1) 42 (< 1) 27 (< 1) 32 (< 1)

Tabell IX. Antal rapporter från sjuksköterske- rapportering.
År Antal rapporter
2006 1 712
2007 1 209
2008 1 161
2009 1 491
2010 917
2011 638
I Tabell X redovisas rapporter från 2011 om allvarliga bi-verkningar till vacciner inom barnvaccinationsprogrammet. Totalt kom 45 rapporter om sådana biverkningar in. Tabell XI visar vilka vacciner som oftast förekommer i rapporter om allvarliga biverkningar. Summan av antal rapporterade vacciner blir fler än antal rapporter i Tabell X eftersom samma individ ibland har fått mer än ett vaccin.
Tabell X. Ålders- och könsfördelning för allvarliga rapporter.
Ålder (år) Män
% av total Kvinnor
% av total Totalt %
0–4 17 38 14 31 31 69
5–9 5 11 2 4 7 15
10–14 0 0 5 11 5 11
15–19 0 0 2 4 2 4
totalt 22 49 23 51 45 100
Tabell XI. Mest inrapporterade vacciner (allvarliga biverkningar).
Vaccin Antal rapporterade vacciner
synflorix 11
Pentavac 11
bCG-vaccin ssI 6
Prevenar 13 5
Infanrix hexa 5
m-m-rvaxpro 4
Priorix 4
Gardasil 4
Prevenar 3
Infanrix – Polio+HIb 3
bCG-medac 3
I Tabell XII redovisas de vanligaste allvarliga biverkningarna till de vacciner som ingår i barnvaccinationsprogrammet.
Tabell XII. ”Tio i topp” biverkningsdiagnoser (allvarliga rapporter).
Biverkningsdiagnos Antal
feberkramper 3
kramptillstånd 5
bCG-infektion 4
feber 4
kollaps (HHe) 4
exantem 3
temperaturstegring 3
apné 2
Lymfkörtelabscess 2
mjukdelsabscess 2
stor lokal reaktion (> 5 cm i diameter) 2
Urtikaria 2
24 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Tabell XIII. Ålders- och könsfördelning för ej allvarliga rapporter.
Ålder (år) Män % av total Kvinnor % av total Okänt % av total Totalt %
0–4 93 28 78 24 1 < 1 172 52
5–9 72 22 49 15 0 0 121 37
10–14 9 3 24 7 0 0 33 10
15–19 0 0 4 1 0 0 4 1
totalt 174 53 155 47 1 < 1 330 100

Tabell XIII–XV tar upp rapporter om icke allvarliga biverk-ningar till vacciner inom barnvaccinationsprogrammet. To-talt kom 330 sådana in under 2011.
Tabell XIV. Mest inrapporterade vacciner (ej allvarliga rapporter).
Vaccin Antal inrapporterade vacciner
tetravac 69
Infanrix polio 59
Pentavac 51
synflorix 45
Infanrix hexa 45
Infanrix – Polio+HIb 30
m-m-rvaxpro 26
Prevenar 13 24
Prevenar 21
Priorix 21
Gardasil 10
bCG-vaccin ssI 9
Prevenar 2
bCG- medac 2
Summan av antal rapporterade vacciner i Tabell XIV är större än antalet rapporter i Tabell XIII eftersom samma in-divid ibland har fått mer än ett vaccin. Motsvarande gäller för Tabell XV, eftersom en individ kan ha mer än en biverk-ningsdiagnos. Totalsumman av antal biverkningsdiagnoser blir därför större än det totala antalet rapporter.
Tabell XV. ”Tio i topp” biverkningsdiagnoser (ej allvarliga rapporter).
Biverkningsdiagnos Antal
stor lokal reaktion (> 5 cm i diameter) 100
feber 90
Lokal reaktion 42
temperaturstegring 22
kräkningar 21
exantem 19
smärta 19
Urtikaria 18
Huvudvärk 16
subkutant infiltrat 16
kontaktdermatit 13
klåda 11
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 25
observanda
Viktigt att veta för tolkningen av biverkningsrapporter
En rapport avser oftast en individ men kan innehålla beskrivningar av flera reaktioner. En bedömning görs av om det kan finnas ett samband mellan läkemedelsbehandlingen och de aktuella reaktionerna. Hänsyn tas bland annat till patientens grundsjukdom, eventuellt samtidigt intag av andra läkemedel samt tidsförloppet.
Allvarlighetsgraden av det inträffade bedöms också. Allvarliga biverkningar definieras vanligen som sådana som är livshotande, medför sjukhusvård eller förlängd sjukhusvård, leder till invalidisering eller medför missbildning eller leder till döden.
I samband med den förstärkta säkerhetsuppföljningen av Pandemrix har hälso- och sjukvården särskilt uppmanats att rapportera alla eventuella misstankar om biverkningar, med undantag för dem som finns angivna som ”vanliga/mycket vanliga” i produktresumén. I och med att man rapporterar redan vid misstanke innebär det att den som rapporterar inte behöver ta ställning till om det utöver tidssamband med vaccinationen även kan finnas ett orsakssamband.
En bra biverkningsrapportering utgör därför ett viktigt komplement till den kunskap om biverkningar som fanns när läkemedlet godkändes och började användas och har stor betydelse för att klargöra dess riskprofil.
Under 2010 och 2011 fick vi till följd av rapporteringen av narkolepsi efter vaccination med Pandemrix en viktig signal som ledde till att produktresumén fick detta som en känd biverkan. Även neurologiska störningar som neuralgi, encefalo-myelit och neurit infördes i produktresumén för Pandemrix.
För övriga produkter har vi inte haft någon signal som har påverkat nytta-riskbalansen på något avgörande sätt. Enstaka produktresuméer har uppdaterats med biverkningsinformation så att de har blivit klarare. Man har i detta arbete jämfört de svenska biverkningarna med rapporter från andra länder vilket är möjligt i den europeiska biverkningsdatabasen Eudra-Vigilance. Detta arbete bedrivs framför allt på europeisk nivå.

Varje år sammanställer Läkemedelsverket de läkemedels-biverkningar som rapporterats till myndigheten. Under 2011 tog Läkemedelsverket emot 597 biverkningsrappor-ter från allmänheten.
Sedan 2008 har patienter och andra läkemedelskonsumenter möjlighet att rapportera misstänkta läkemedelsbiverkningar till Läkemedelsverket. Rapporteringen sker framför allt via Läkemedelsverkets webbplats och i mindre omfattning brevledes. Rapporteringen från patienter/konsumenter ger en värdefull möjlighet att från allmänhetens synvinkel bidra med information om läkemedelsproblem som kan behöva åtgärdas och kompletterar på ett bra sätt rapporteringen från sjukvården. Inkomna uppgifter lagras i en databas och analyseras. Andelen konsumentrapporter uppgår till cirka en tiondel av den totala biverkningsrapporteringen under året 2011.
Varför behövs biverkningsrapporter?Vid godkännandet av nya läkemedel är endast de vanligaste biverkningarna kända hos utvalda grupper av patienter. Kunskapen om mer ovanliga biverkningar eller till exempel biverkningar i kombination med andra läkemedel är ofta mycket begränsad. En bra biverkningsrapportering och analys av densamma är därför av stor betydelse för att klar-göra riskprofilen hos nya läkemedel när de kommit i normalt bruk av patienter.
Vad ska rapporteras? Vem kan rapportera? Varje misstänkt biverkning av läkemedel eller naturläkeme-del kan rapporteras, oavsett om medlet skrivits ut på recept, köpts receptfritt och oberoende av inköpsställe. I första hand bör rapportering ske av den som själv upplevt biverkan. Om detta av något skäl inte är möjligt kan, i andra hand, närstående rapportera. Rapporteringen omfattas av såväl sekretessregler som offentlighetsprincip vilket i korthet innebär att enbart allmänna uppgifter på gruppnivå om rapporteringen kan lämnas ut till tredje part. Eftersom inga särskilda råd för vad som bör rapporteras finns inom konsu-mentområdet (till skillnad från för rapporteringen från sjukvården) rapporteras en hel del redan välkända biverk-ningar. För misstänkta biverkningar av receptfria läkemedel och naturmedel för egenvård ges med konsumentrapporte-ringen en naturlig direkt rapporteringsmöjlighet – detta alltså utan att behöva gå omvägen kring sjukvården.
Hur kan uppgifterna från rapporteringen tolkas?Uppgifterna från konsumentrapporteringen kan sällan an-vändas för att dra direkta slutsatser om ett visst läkemedel har orsakat en specifik biverkning eller orsakat ett dödsfall.
Den rapporterade reaktionen kan vara orsakad av läkemedlet i fråga men kan även vara orsakad av andra läkemedel som patienten behandlas med. Den kan också vara orsakad av patientens bakomliggande sjukdom eller uppkommit obero-ende av läkemedlet.
Den enskilda biverkningsrapporten ska ses som en pus-selbit där man kan dra slutsatser om ett eventuellt samband med läkemedlet först efter analys av andra liknande rappor-ter från omvärlden och från resultat i vetenskapliga under-sökningar.
Rapporteringsstatistik 2011, konsumentrapporterDe sammanlagt 597 rapporterna fördelar sig enligt nedan avseende allvarlighetsgrad (Tabell I). Hälften av rapporterna har av Läkemedelsverket klassats som allvarliga vilket är en något större andel än för rapporteringen som helhet.
Tabell I. Konsumentrapporter fördelade på allvarlighetsgrad.
Rapporternas allvarlighetsgrad Antal rapporter
allvarliga – ej dödsfall 297
allvarliga – dödsfall 3
ej allvarliga 297
En allvarlig biverkning definieras som en sådan som leder till döden, är livshotande, nödvändiggör sjukhusvård eller förlängd sjukhusvård, leder till invalidisering eller medför missbildning. För dödsfallen har sambandet mellan reaktionen och den fatala utgången bedömts som möjlig eller ej utesluten.
Ålders- och könsfördelning för samtliga konsumentrapporterRapporteringen domineras av kvinnor som står för mer än två tredjedelar av rapporteringen. Yngre vuxna står totalt sett för en stor del av rapporteringen även om denna grupp endast står för en mindre andel av läkemedelsanvändningen. En möjlig förklaring till detta är den större datorvanan och datortillgången i denna åldersgrupp vilket gör rapporte-ring smidig och enkel. Barn och äldre står för en mindre del av rapporteringen (Tabell II). Rapporteringsmönstret skiljer sig alltså något mellan konsument- och sjukvårds-rapporteringen.
26 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Allmänhetens inrapporterade misstänkta läkemedelsbiverkningar 2011

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 27
observanda
Tabell II. Konsumentrapporter fördelade på kön och ålder.
Ålder (år) Män % av total Kvinnor % av total Totalt %
0–20 31 5 54 9 85 14
21–40 37 6 146 25 183 31
41–60 49 8 122 20 171 28
61–80 57 10 86 14 143 24
81–100 9 2 6 1 15 3
totalt 183 31 414 69 597 100
Tabell III. Konsumentrapporter efter 4-ställig ATC-kod.
Läkemedelsgrupp ATC-kod Allvarlig Ej allvarlig Totalt
vacciner mot virusinfektioner J07b 133 18 151
antidepressiva medel n06a 22 21 43
antikonceptionella medel G03a 11 21 32
Icke-steroida antiinflammatoriska/antireumatiska medel, nsaId
m01a 8 20 28
medel som påverkar serumlipidnivåerna C10a 7 16 23
neuroleptika n05a 16 5 21
antibakteriella betalaktamer, penicilliner J01C 4 14 18
Hosthämmande medel, exkl.kombinationer med expektorantia
r05d 1 16 17
medel vid magsår ochgastroesofageal refluxsjukdom
a02b 4 12 16
opioider n02a 7 6 13
Mest rapporterade läkemedelsgrupperDå antalet konsumentrapporter i förhållande till antalet läkemedel är begränsat har varje enskilt läkemedel med få undantag endast rapporterats enstaka gånger. Läkemedlen har i Tabell III, för att ge en bra översikt, grupperats efter s.k. 4-ställig ATC-kod. På detta sätt ses tydligare hur rap-porteringen fördelar sig på grupper av likartade läkemedel.
Mest rapporterade är vacciner mot virusinfektioner som utgör en knapp fjärdedel av rapporteringen och nära hälften av rapporteringen av allvarliga biverkningar. En stor andel av konsumentrapporteringen utgörs också av medel som påver-kar nervsystemet där medel mot psykiska sjukdomar samt centralt verkande medel mot smärta där opioider/morfinbe-släktade preparat ingår. Fördelningen mellan allvarliga och icke allvarliga reaktioner varierar stort beroende på läkeme-delsgrupp där till exempel hostmediciner och penicilliner hör till de mest rapporterade grupperna medan bara få av dessa rapporter har bedömts som allvarliga.
Mest rapporterade läkemedel (oavsett allvarlighetsgrad)Pandemrix fortsätter att vara det i särklass vanligast rappor-terade läkemedlet i konsumentrapporteringssammanhang (Tabell IV). En tredjedel av dessa rapporter handlar om någon form av sömnstörning, varav hälften narkolepsi. Van-ligt rapporterat för Pandemrix är också allmänna symtom såsom orkeslöshet samt led- och muskelbesvär och sjukdomar som kan hänföras till autoimmuna tillstånd. En del av sådana misstänkta biverkningar av Pandemrix är för närvarande fö-remål för större studier för att utreda ett eventuellt orsaks-samband på gruppnivå.

28 • InformatIon fr ån L äkemedeLsverket 4 : 2012
observanda
Tabell IV. De vanligast förekommande preparaten i konsumentrapporter.
Läkemedel (ATC-grupp) Antal rapporter (%)
Pandemrix (J) 128 (21)
nipaxon (r) 9 (2)
Levaxin (H) 9 (2)
roaccutan (d) 8 (1)
doxyferm (J) 8 (1)
simvastatin (C) 8 (1)
enalapril (C) 6 (1)
vaxigrip (J) 5 (0,8)
Cocillana-etyfin (r) 5 (0,8)
singulair (r) 5 (0,8)
atarax (n) 5 (0,8)
Implanon (G) 5 (0,8)
Heracillin (J) 5 (0,8)
mirena (G) 5 (0,8)
seroquel depot (n) 5 (0,8)
noskapin aCo (r) 5 (0,8)
Tre hostmediciner finns med i tabellen (Nipaxon, Cocillana-Etyfin och Noskapin ACO) och de biverkningar som helt dominerar för dessa preparat är övergående bröst/buksmär-tor, en otrevlig men välkänd biverkan för flera hostmediciner vilket kan ha sitt ursprung i gallvägskramper. För nummer tre på listan, Levaxin, syns inget tydligt mönster i rapporte-ringen även om det är tydligt att vissa av de rapporterade biverkningarna är klassiska symtom på under- respektive överskott av sköldkörtelhormon.
Roaccutan, ett preparat mot svår akne, är inte godkänt i Sverige på grund av sin fosterskadande potential men för-skrivs på licens i utvalda fall. De biverkningar som är inrap-porterade är företrädesvis kända sådana. Tilläggas bör att inga fosterskador finns biverkningsrapporterade i Sverige. För simvastatin rapporteras framför allt muskelvärk och olika överkänslighetsrektioner, båda välbeskrivna kända bi-verkningar.
Rapporterna för Vaxigrip (säsongsinfluensavaccin) är svåra att bedöma eftersom man i nästan alla fall beskriver samtidig behandling med annat vaccin, vanligen Pandemrix. För övriga preparat på listan rör de inrapporterade biverk-ningarna till största del redan kända möjliga bieffekter av behandling med preparaten i fråga.
De konsumentrapporterade biverkningarna utgör ett alltmer värdefullt tillskott till säkerhetsuppföljningen av lä-kemedel i Sverige. De bidrar med konsumenters/patienters upplevelse av den typ av biverkningar – inklusive sjukvårds-krävande sådana – som rapporteras från sjukvården. Utöver detta har de dessutom fördelen av att de ger viktig säkerhets-information som aldrig kommer till sjukvårdens kännedom men som likväl kan ha stor betydelse för läkemedelsanvänd-ningens genomförbarhet.
All biverkningsrapportering bidrar till att öka kunskapen om läkemedels säkerhet och kunskapen om hur tillgängliga läkemedel kan användas på mest ändamålsenliga sätt.
Samband mellan narkolepsi och Pandemrix på IrlandI april presenterades en irländsk utredning som stärker sambandet mellan en ökad risk för narkolepsi hos barn och ungdomar och vaccination med Pandemrix.
Läkemedelsverket har fått ta del av en rapport från de irländ-ska hälsovårdsmyndigheterna som visat en 13-faldigt ökad risk av narkolepsi hos barn och ungdomar som vaccinerats med Pandemrix vilket motsvarar cirka fem fall narkolepsi per 100 000 vaccinerade barn och ungdomar.
Undersökningen, som täcker perioden april 2009 till decem-ber 2011, har ännu ej granskats i detalj av läkemedelsmyn-digheterna, men data förefaller styrka tidigare undersök-ningar från Finland och Sverige.
Rapporten finns att läsa via det irländska Department of Health, www.dohc.ie.

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 29
observanda
Q-Med upphör med Macrolane för behandling av bröst
Tillverkaren Q-Med har beslutat att Macrolane inte ska användas för behandling av bröst. Orsaken är att produkten försvårar och ibland omöjliggör tolkningen av mammografibilder. Läkemedelsverkets rapport visar också att produkten i en del fall orsakat knölar som lett till onödig utredning och oro.
I maj 2010 uppmärksammades Läkemedelsverket på svårig-heterna att tolka mammografibilder från kvinnor med Ma-crolaneinjektioner i brösten. Med anledning av detta startade Läkemedelsverket en utredning och 2011 gavs uppdraget att utvärdera problemets omfattning till en extern expertgrupp. Expertgruppen presenterade sin rapport i mars 2012 och rapporten skickades till Q-Med i form av ett föreläggande. Svarstiden gick ut den 16 april 2012.
Läkemedelsverket framförde i föreläggandet att den medicinska nyttan med produkten bedöms vara låg. Det ska ställas emot att produkten, när den injicerats i bröst, försvårar och i vissa fall omöjliggör tolkningen av mammografibil-derna. Enligt Socialstyrelsens rekommendationer ska alla kvinnor i åldrarna 40–74 år erbjudas regelbunden mammo-grafiscreening. Det ska betonas att undersökning med ultra-
ljud och magnetkamera inte är screeningmetoder utan an-vänds i samband med utredningar. Av rapporten framgår också att Macrolane i en del fall orsakat knölar som lett till onödig utredning och oro.
Sammanfattningsvis framförde Läkemedelsverket i före-läggandet att verkets bedömning är att sådana oönskade ris-ker och sidoeffekter inte är godtagbara i förhållande till produktens nytta och avsedda prestanda. Läkemedelsverket anser att Macrolane inte ska användas för bröstförstoring respektive kroppskonturering av bröst. Q-Med fick möjlighet att bemöta Läkemedelsverkets bedömning.
Q-Med inkom med ett svar till Läkemedelsverket och i samband med sitt svar publicerade de ett pressmeddelande där man anger att Q-Med upphör med Macrolane för be-handling av bröst. I svaret till Läkemedelsverket har Q-Med dessutom uppgett att arbete pågår med att uppdatera pro-duktens märkning och bruksanvisning och att information kommer att skickas till alla berörda. Läkemedelsverket kom-mer nu att följa upp de åtgärder som presenterats av Q-Med.
Äldre Evab-patientsängar kan störa elektronisk utrustning
Läkemedelsverket vill uppmärksamma användare av äldre Evab-patientsängar på att det kan uppstå stör-ningar på elektronisk utrustning vid användning av sängarna. Sängarna tillverkas av Proton Engineering AB/Caretec.
Störningar på elektroniska utrustningar kan uppkomma vid användning av patientsängar av märket Evab. De berörda sängarna är tillverkade före 1999 av tillverkaren Proton Engineering AB/Caretec. Sängarna manövreras med elek-tronik och hydraulik, bland annat för att höja och sänka sängen eller delar av den.
Tillverkaren rekommenderar att sängar som är äldre än tio år inte ska användas i närheten av känsliga mätapparater.
Eventuella störningar kan enligt tillverkaren avhjälpas med en jordsladd som kan erhållas mot kostnad från tillverkaren.
Läkemedelsverket vill uppmärksamma hälso- och sjuk-vården samt övriga användare av Evab-patientsängar på att eventuella förekommande negativa händelser och tillbud ska anmälas både till tillverkaren och till Läkemedelsverket. Rapporteringsrutinerna är likadana för alla medicintekniska produkter och sker enligt SOSFS 2008:1 (www.lakemedels-verket.se under Hälso- & sjukvård och vidare till Medicinteknisk säkerhetsinformation).
Vid eventuella frågor om sängarna ska tillverkaren kontaktas.

Tabell I.
Grad Klinisk beskrivning av intensitetsgradering
0 Ingen Ingen ak-lesion finns, varken synlig eller palpabel.
1 Lindrig Platta, rosa fläckar utan tecken på hyperkeratos och erytem, svag palpabilitet, där ak är lättare att känna än att se.
2 måttlig rosa till rödaktiga papler och erytematösa plack med hyperkeratotisk yta, måttligt tjocka ak som lätt kan ses och kännas.
3 svår mycket tjocka och/eller tydliga ak.
30 • InformatIon fr ån L äkemedeLsverket 4 : 2012
monoGr afIer
Läkemedelsmonografier
Ameluz (5-aminolevulinsyra)ATC-kod: L01XD04Gel, 78 mg/gBiofrontera Bioscience GmbH
Godkännandedatum: 2011-12-14. Central procedur.
IndikationBehandling av aktinisk keratos av mild till måttlig grad i ansiktet och hårbotten (Olsen grad 1 till 2).
DoseringAmeluz används vid fotodynamisk behandling av enstaka eller flera lesioner vid ett behandlingstillfälle. Gelen ska täcka lesionerna och ungefär 5 mm av det omgivande områ-det i ett lager som är omkring 1 mm tjockt. Hela behand-lingsområdet belyses med en röd ljuskälla, antingen med ett smalt spektrum på omkring 630 nm och en ljusdos på ungefär 37 J/cm2 eller ett bredare och kontinuerligt spektrum i inter-vallet 570 till 670 nm med en ljusdos på 75 till 200 J/cm2. Det är viktigt att säkerställa att korrekt ljusdos ges för att uppnå optimal klinisk effekt.
Lesionerna ska utvärderas tre månader efter behandling och de som inte eller bara delvis svarat på behandling ska behandlas på nytt.
BakgrundAmeluz är en gelformulering av 5-aminolevulinat (5-ALA) i en nanoemulsion vilken ska ge en kemisk stabilitet av den ak-tiva komponenten, samt öka penetrationen genom dermis.
Efter applicering av 5-ALA metaboliseras substansen in-tracellulärt till protoporfyrin IX, en fotoaktiv förening som ackumuleras i lesionen. Protoporfyrin IX aktiveras vid belys-ning med rött ljus av lämplig våglängd och energi, och i närvaro av syre bildas reaktiva syreföreningar som skadar cellkomponenterna och förstör cellerna i lesionen.
De aktiniska keratoslesionernas intensitet graderades en-ligt den skala som beskrivits av Olsen, et al. 1991.
Sammanfattning av kliniska studierStudiedataDen kliniska effekten av Ameluz utvärderades i två randomi-serade Fas III-studier. I den första studien med 571 patienter jämfördes Ameluz med behandling med Metvix (metylami-nolevulinat; godkänd som fotodynamisk terapi vid behand-ling av bland annat aktiniska keratoser sedan 2001) samt även mot placebo. Alla patienter hade fyra till åtta lindrigt till måttligt svåra aktiniska keratoslesioner i ansiktet och/eller hårbotten. De röda ljuskällor som användes var antin-gen en smalspektrumlampa (Aktilite CL 128 eller Omnilux PDT) eller en lampa med bredare och kontinuerligt spek-trum (Waldmann PDT 1200 L, eller Hydrosun Photodyn 505 eller 750). Om lesionerna inte hade läkt ut helt tolv veckor efter den första behandlingen, behandlades de en andra gång med identisk behandlingsregim. Det primära effektmåttet var fullständig utläkning av patienternas lesio-ner tolv veckor efter den sista fotodynamiska behandlingen.
Patienter behandlade med Ameluz uppvisade utläkta le-sioner i 78 % av fallen, jämfört med 64 % med Metvix och 17 % med placebo. En skillnad i procentuell fullständig ut-läkning sågs alltså i denna studie till Amulez fördel. Studien visade också att både utläkningsfrekvens och tolerabilitet var beroende av den använda belysningskällan. Tabellen II visar utläkning i procent och vanliga lokala biverkningar (erytem och tillfällig smärta) på appliceringsstället under behand-lingen med olika ljuskällor. Patienter som fick placebo upp-visade utläkning i 13 % och 22 % när de behandlades med ljuskälla med smalt respektive brett spektrum.
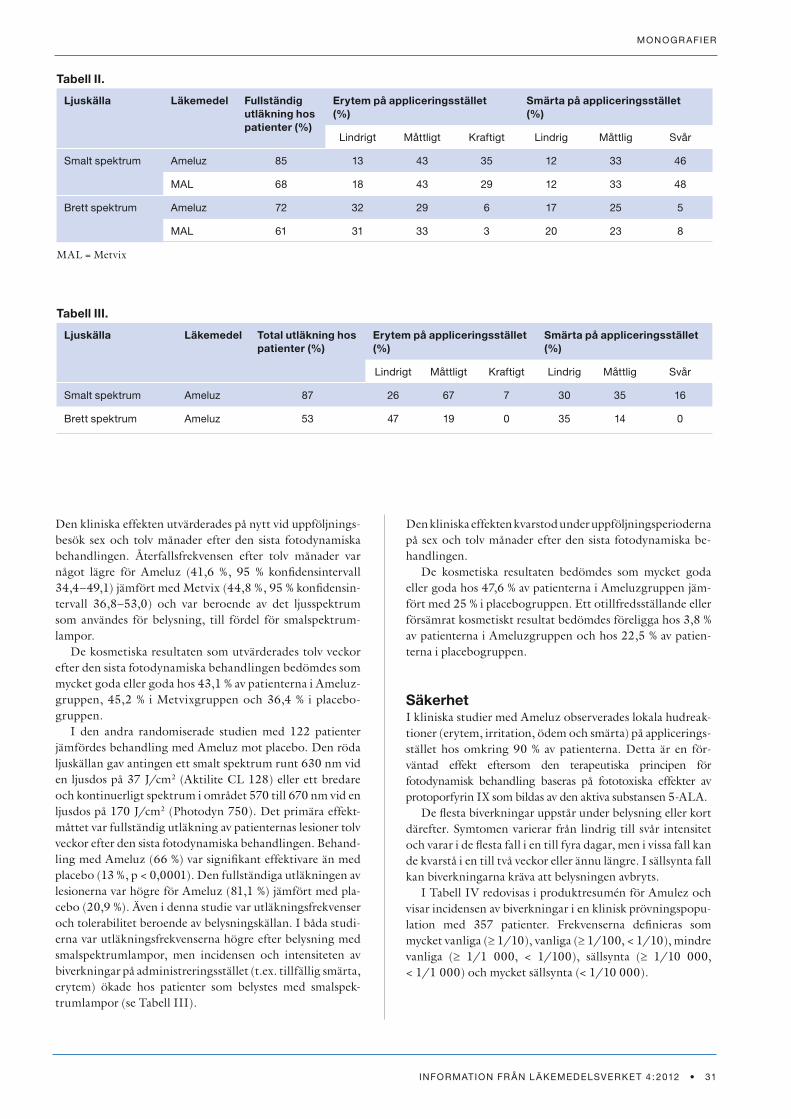
Tabell II.
Ljuskälla Läkemedel Fullständig utläkning hos patienter (%)
Erytem på appliceringsstället(%)
Smärta på appliceringsstället(%)
Lindrigt måttligt kraftigt Lindrig måttlig svår
smalt spektrum ameluz 85 13 43 35 12 33 46
maL 68 18 43 29 12 33 48
brett spektrum ameluz 72 32 29 6 17 25 5
maL 61 31 33 3 20 23 8
MAL = Metvix
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 31
monoGr afIer
Den kliniska effekten utvärderades på nytt vid uppföljnings-besök sex och tolv månader efter den sista fotodynamiska behandlingen. Återfallsfrekvensen efter tolv månader var något lägre för Ameluz (41,6 %, 95 % konfidensintervall 34,4–49,1) jämfört med Metvix (44,8 %, 95 % konfidensin-tervall 36,8–53,0) och var beroende av det ljusspektrum som användes för belysning, till fördel för smalspektrum-lampor.
De kosmetiska resultaten som utvärderades tolv veckor efter den sista fotodynamiska behandlingen bedömdes som mycket goda eller goda hos 43,1 % av patienterna i Ameluz-gruppen, 45,2 % i Metvixgruppen och 36,4 % i placebo-gruppen.
I den andra randomiserade studien med 122 patienter jämfördes behandling med Ameluz mot placebo. Den röda ljuskällan gav antingen ett smalt spektrum runt 630 nm vid en ljusdos på 37 J/cm2 (Aktilite CL 128) eller ett bredare och kontinuerligt spektrum i området 570 till 670 nm vid en ljusdos på 170 J/cm2 (Photodyn 750). Det primära effekt-måttet var fullständig utläkning av patienternas lesioner tolv veckor efter den sista fotodynamiska behandlingen. Behand-ling med Ameluz (66 %) var signifikant effektivare än med placebo (13 %, p < 0,0001). Den fullständiga utläkningen av lesionerna var högre för Ameluz (81,1 %) jämfört med pla-cebo (20,9 %). Även i denna studie var utläkningsfrekvenser och tolerabilitet beroende av belysningskällan. I båda studi-erna var utläkningsfrekvenserna högre efter belysning med smalspektrumlampor, men incidensen och intensiteten av biverkningar på administreringsstället (t.ex. tillfällig smärta, erytem) ökade hos patienter som belystes med smalspek-trumlampor (se Tabell III).
Den kliniska effekten kvarstod under uppföljningsperioderna på sex och tolv månader efter den sista fotodynamiska be-handlingen.
De kosmetiska resultaten bedömdes som mycket goda eller goda hos 47,6 % av patienterna i Ameluzgruppen jäm-fört med 25 % i placebogruppen. Ett otillfredsställande eller försämrat kosmetiskt resultat bedömdes föreligga hos 3,8 % av patienterna i Ameluzgruppen och hos 22,5 % av patien-terna i placebogruppen.
SäkerhetI kliniska studier med Ameluz observerades lokala hudreak-tioner (erytem, irritation, ödem och smärta) på applicerings-stället hos omkring 90 % av patienterna. Detta är en för-väntad effekt eftersom den terapeutiska principen för fotodynamisk behandling baseras på fototoxiska effekter av protoporfyrin IX som bildas av den aktiva substansen 5-ALA.
De flesta biverkningar uppstår under belysning eller kort därefter. Symtomen varierar från lindrig till svår intensitet och varar i de flesta fall i en till fyra dagar, men i vissa fall kan de kvarstå i en till två veckor eller ännu längre. I sällsynta fall kan biverkningarna kräva att belysningen avbryts.
I Tabell IV redovisas i produktresumén för Amulez och visar incidensen av biverkningar i en klinisk prövningspopu-lation med 357 patienter. Frekvenserna definieras som mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), sällsynta (≥ 1/10 000, < 1/1 000) och mycket sällsynta (< 1/10 000).
Tabell III.
Ljuskälla Läkemedel Total utläkning hos patienter (%)
Erytem på appliceringsstället(%)
Smärta på appliceringsstället(%)
Lindrigt måttligt kraftigt Lindrig måttlig svår
smalt spektrum ameluz 87 26 67 7 30 35 16
brett spektrum ameluz 53 47 19 0 35 14 0
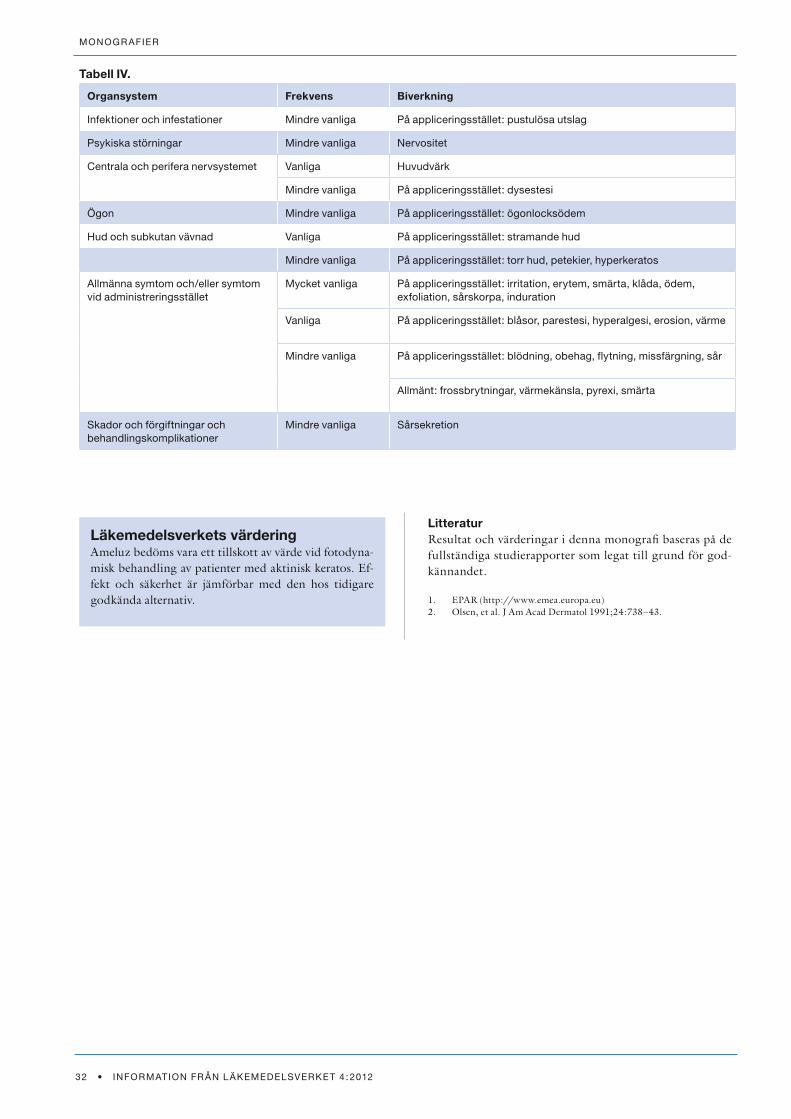
32 • InformatIon fr ån L äkemedeLsverket 4 : 2012
monoGr afIer
Tabell IV.
Organsystem Frekvens Biverkning
Infektioner och infestationer mindre vanliga På appliceringsstället: pustulösa utslag
Psykiska störningar mindre vanliga nervositet
Centrala och perifera nervsystemet vanliga Huvudvärk
mindre vanliga På appliceringsstället: dysestesi
Ögon mindre vanliga På appliceringsstället: ögonlocksödem
Hud och subkutan vävnad vanliga På appliceringsstället: stramande hud
mindre vanliga På appliceringsstället: torr hud, petekier, hyperkeratos
allmänna symtom och/eller symtom vid administreringsstället
mycket vanliga På appliceringsstället: irritation, erytem, smärta, klåda, ödem, exfoliation, sårskorpa, induration
vanliga På appliceringsstället: blåsor, parestesi, hyperalgesi, erosion, värme
mindre vanliga På appliceringsstället: blödning, obehag, flytning, missfärgning, sår
allmänt: frossbrytningar, värmekänsla, pyrexi, smärta
skador och förgiftningar och behandlingskomplikationer
mindre vanliga sårsekretion
LitteraturResultat och värderingar i denna monografi baseras på de fullständiga studierapporter som legat till grund för god-kännandet.
EPAR (http://www.emea.europa.eu) 1. Olsen, et al. J Am Acad Dermatol 1991;24:738–43.2.
Läkemedelsverkets värderingAmeluz bedöms vara ett tillskott av värde vid fotodyna-misk behandling av patienter med aktinisk keratos. Ef-fekt och säkerhet är jämförbar med den hos tidigare godkända alternativ.

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 33
monoGr afIer
Dexdor (dexmedetomidin)ATC-kod: N05CM18Koncentrat till infusionsvätska, lösning 100 mikrog/mLOrion Pharma
Godkännandedatum: 2011-09-16. Central procedur.
Godkänd indikationFör sedering av vuxna intensivvårdspatienter vilka behöver en sederingsnivå som inte är djupare än att de kan väckas av verbal stimulans (motsvarande Richmond Agitation-Seda-tion Scale (RASS) 0 till -3.
DoseringDexdor ska enbart administreras av sjukvårdspersonal med erfarenhet av att vårda intensivvårdspatienter. Patienter som redan är intuberade och sederade kan byta till Dexdor med en initial infusionshastighet på 0,7 mikrogram/kg/timme. Infusionshastigheten kan sedan justeras stegvis inom dosin-tervallet 0,2–1,4 mikrogram/kg/timme för att uppnå öns-kad grad av sedering. En lägre initial infusionshastighet bör övervägas för patienter med nedsatt allmäntillstånd. Ladd-ningsdos av dexmedetomidin rekommenderas inte och är associerat med ökade biverkningar. Vid behov ges propofol eller midazolam tills den kliniska effekten av dexdor är upp-nådd.
Tillräckliga data för behandling med dexmedetomidin i åldersgruppen under 18 år saknas.
Sammanfattning av kliniska studierEffektDexmedetomidins sederande effekt har utvärderats i fem placebokontrollerade studier (två Fas II, tre Fas III) där re-spiratorbehandlade postoperativa patienter (n = 911) hölls sövda i upp till 72 timmar. Behovet av ”rescue”-medicinering (morfin – midazolam – propofol) var lägre hos de patienter som behandlades med dexmedetomidin. För att utvärdera dexmedetomidins effekter jämfört med midazolam och propofol i ett mer representativt urval av respiratorbehand-lade intensivvårdspatienter och under längre tid (från 24 timmar upp till 14 dagar) har tre Fas III-studier inklude-rande aktiv kontroll genomförts. Patientpopulationen ut-gjordes av typiska medicinska och kirurgiska IVA-behandla-de patienter. Huvudsakliga exklusionskriterier var akut allvarlig intrakraniell patologi, obehandlad hypotension (MAP < 55 mmHg trots vätske- och vasokonstriktorbe-handling), bradykardi (hjärtfrekvens < 50 hjärtslag per minut), AV-block II–III om patienten inte hade pacemaker och svår leversvikt med bilirubin > 101 μmol/L.
Det primära målet för studierna var att visa att effekten av dexmedetomidin är jämförbar (non-inferiority) med stan-
dardbehandling när det gäller att hålla patienten inom målet för sederingsdjup (RASS 0 till -3) utan behov av extradoser av sedativa. I de tre studierna fick totalt 538 patienter huvud-sedering med dexmedetomidin och 539 antingen med mida-zolam eller propofol. Behandlingsgrupperna var likvärdiga avseende kön, ålder, vikt och orsak till respiratorbehandling (kirurgi, medicin och trauma). Totalt fanns en viss övervikt av medicinska patienter.
I dessa studier uppfyllde dexmedetomidin kravet på non-inferiority. Kvoten för tiden för välfungerande sedering med dexmedetomidin/midazolam var 1,07 (95 % konfidensinter-vall; 0,971–1,176) och för dexmedetomidin/propofol 1,00 (95 % konfidensintervall; 0,922–1,075) vilket talar för stor likhet avseende de testade sederande behandlingarna. Sam-manvägda data visade att dexmedetomidin förkortade tiden i respirator jämfört med standardsedering, likaså tiden till extubering efter att sederingsinfusionerna avslutats men ej någon skillnad avseende vårdtid på IVA. Omvårdnads-aspekter som möjlighet att kunna få kontakt med patienten, patientens förmåga att meddela smärta och patientens sam-arbetsförmåga vid omvårdnadsåtgärder rapporterades som positiva till dexmedetomidins fördel.
FarmakodynamikDexmedetomidin är en selektiv alfa-2-receptoragonist med breda farmakologiska egenskaper. Substansen har en sympa-tolytisk effekt genom en minskad noradrenalinfrisättning i sympatiska nervändar.
Den sedativa effekten sker via minskad aktivering av locus coeruleus, den dominerande noradrenerga kärnan i hjärn-stammen.
De kardiovaskulära effekterna är dosberoende. Vid lägre infusionshastigheter dominerar de centralt medierade effek-terna vilket leder till minskad puls och lägre blodtryck. Vid högre doser, dominerar de perifera kärlsammandragande effekterna vilket leder till en ökning av systemvaskulär resis-tans och blodtryck, samtidigt som bradykardieffekten ytter-ligare förstärks.
Dexmedetomidin har relativt liten påverkan på andningen.Konventionella sedativa preparat som används för inten-
sivvårdssedering (exempelvis midazolam och propofol) är GABA-receptoragonister. Sederingskvaliteten medierad av dexmedetomidin rapporteras skilja sig från sedering fram-kallad av GABA-receptoragonister.

34 • InformatIon fr ån L äkemedeLsverket 4 : 2012
monoGr afIer
SäkerhetSäkerhetsdatabasen för dexmedetomidin innehåller 106 kliniska studier. I dessa har 4 765 individer exponerats för substansen. Av dessa var 2 103 IVA-patienter varav 778 i studier med aktiv kontroll. Jämfört med konventionell sede-ring hade de dexmedetomidinbehandlade patienterna en överrepresentation av kardiovaskulära händelser (hypoten-sion, bradykardi och hypertension). Noteras bör att patienter med kända kardiovaskulära riskfaktorer ändå var exklude-rade i dessa studier. Dessa händelser kunde i de flesta fall hanteras genom dossänkning De vanligaste anledningarna till att avbryta behandling i dexmedetomidingruppen var bradykardi, agitation och hypotension. Säkerhetsdata avse-ende längre tids sedering (> 14 dygn) och högre doser (> 1,4 mikrogram/kg/timme) är fortfarande begränsade.
I länder där dexmedetomidin är godkänt (bland annat USA) beräknas antalet behandlingsdygn med preparatet för perioden från december 1999 till april 2010 ligga på drygt 1,5 miljoner. Antalet biverkningsrapporter som redovisats till tillverkaren för denna period var 978, representerande totalt 1 497 enskilda biverkningar. De vanligaste rapporte-rade biverkningarna var hypotension och bradykardi.
LitteraturResultat och värdering i denna monografi baseras på de fullständiga studierapporter som legat till grund för god-kännandet. Nedanstående publicerade referenser har inte granskats av Läkemedelsverket.
EPAR (Central procedur) (http://www.ema.europa.eu).1. Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs mida-2. zolam for sedation of critically ill patients: a randomized trial. JAMA 2009;301:489–99. Ruokonen E, Parviainen I, Jakob SM, et al. Dexmedetomidine versus 3. propofol/midazolam for long-term sedation during mechanical venti-lation. Intensive Care Med 2009;35:282–90.
Läkemedelsverkets värderingDexdor (dexmedetomidin) är inom humanmedicinen det första godkända sederingsmedlet som utövar sin effekt via alfa-2 receptoragonism. Med bibehållen god tolerans för respiratorbehandling finns möjlighet till viss kommunikation med patienten. Biverkningarna är framför allt kopplade till preparatets sympatolytiska ef-fekter och Dexdor är kontraindicerat vid AV-block II och III (om patienten inte har pacemaker) och vid obe-handlad hypotension. Inom intensivvården blir prepara-tet ett värdefullt alternativ till kardiovaskulärt stabila patienter när djup sedation i samband med respiratorbe-handling inte är ett krav.
Du vet väl att samtliga läkemedelsmonografier finns påwww.lakemedelsverket.se

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 35
monoGr afIer
Plenadren (hydrokortison)ATC-kod: H02AB09Tablett med modifierad frisättning, 5 mg och 20 mgDuoCort Pharma AB
Godkännandedatum: 2011-11-03. Central procedur.
IndikationBehandling av binjurebarksvikt hos vuxna.
Sammanfattning av kliniska studierPlenadren är en ny beredningsform av hydrokortison, som kombinerar omedelbar och fördröjd frisättning för behand-ling av binjurebarksvikt hos vuxna. Tabletten har ett yttre hölje som ger omedelbar frisättning och snabb absorption av läkemedlet samt en kärna som ger en förlängd plasmaprofil för kortisol.
Detta möjliggör dosering en gång per dag till skillnad från behandling med konventionella hydrokortisonpreparat som måste doseras två till tre gånger dagligen.
StudiedataGodkännandet baseras på en randomiserad, sex månader lång, öppen multicenterstudie (2). Studien var upplagd i två behandlingsperioder om tre månader och patienterna bytte behandling efter tre månader (crossover-design). Studien inkluderade 64 patienter med primär binjurebarksvikt, varav elva även hade diabetes mellitus och elva hade hypertoni. Huvudsyftet med studien var att jämföra Plenadren givet en gång dagligen med konventionella tabletter tre gånger dagli-gen med avseende på farmakokinetiska parametrar. Patien-
terna bibehöll sin ordinarie dygnsdos av hydrokortison (20–40 mg) under hela studien. Efter den initiala randomi-serade perioden, erbjöds alla patienter att fortsätta i en sex månader lång uppföljningsstudie där alla behandlades med Plenadren. Av de ursprungligen 64 patienterna valde 59 pa-tienter att delta i uppföljningsstudien och av dessa fullföljde 57 patienter ett års behandling.
Jämfört med konventionella tabletter som togs tre gånger dagligen resulterade behandling med Plenadren i ökad kor-tisolexponering under de första fyra timmarna efter admi-nistreringen på morgonen men minskad exponering under sen eftermiddag/kväll och över hela dygnet (Figur 1). Bio-tillgängligheten är cirka 20 % lägre med Plenadren jämfört med konventionella hydrokortisontabletter.
Behandlingarna var likvärdiga med avseende på tolerabi-litet utvärderat genom frågeformulär. Efter tolv veckors be-handling uppgav 95,1 % som fick Plenadren och 96,7 % av dem som fick konventionell behandling att behandlingen fungerade acceptabelt, bra, eller mycket bra. På frågan om vilken behandling man föredrog svarade 61,1 % av patien-terna att man föredrog Plenadren medan 8,3 % föredrog konventionell behandling. Studien visade ingen skillnad i livskvalitet mellan behandlingarna.
Figur 1. Observerad medelkoncentration av kortisol i serum jämfört med klockslag efter en respektive flera doser hos patienter med primär binjurebarksvikt (n = 62) efter oral administrering av Plenadren en gång dagligen respektive hydrokortison tre gånger dagligen.
Plenadren
Hydrokortisontablett med omedelbar frisättning
800
600
400
200
008.00 12.00 16.00 20.00 klockslag
kortisolkoncentration (nm)

SäkerhetI studien var frekvensen och typen av biverkningar generellt likartade för Plenadren och konventionella hydrokortisonta-bletter. En initial ökning av frekvensen av biverkningar ob-serverades hos ungefär en av fem patienter i upp till åtta veckor efter det första bytet från konventionella hydrokorti-sontabletter till Plenadren. De aktuella biverkningarna (buksmärta, diarré, illamående, trötthet och muskuloskele-tala symtom) var dock lindriga eller måttliga, övergående och kortvariga, och har tolkats som en upplevd hypokortiso-lism i samband med övergång från konventionell behandling till Plenadren. Dosen kan behöva justeras i samband med övergång till Plenadren, även om de sparsamma data som finns rörande dostitrering tyder på att majoriteten av patien-terna bibehåller den dos de tidigare haft.
Behovet av extradoser av kortison i samband med fysisk och psykisk stress (inkluderande infektioner) studerades specifikt under studien. Det var ingen skillnad i antalet ex-tradoser eller mängd extra hydrokortison. Även om något fler patienter drabbades av annan sjukdom under behandling med Plenadren än under konventionell behandling så finns det inget som tyder på att Plenadren är sämre än konventio-nell behandling vid annan tillfällig sjukdom eller psykisk stress. Under fortsatt behandling med Plenadren sågs en lägre frekvens av annan sjukdom, motsvarande den som rapporterades för konventionell behandling i den inledande studien.
Plenadren rekommenderas inte för patienter med ökad motilitet i magsäcken, t.ex. kronisk diarré. På grund av ris-ken för nedsatt kortisolexponering ska dessa patienter be-handlas med andra läkemedelsformer av hydrokortison. Inga data finns tillgängliga för patienter med bekräftade sjukdomar/tillstånd som ger långsam tömning av magsäcken eller minskad motilitet i magsäcken. Då upptaget av Plena-dren kan vara påverkat hos dessa patienter ska det kliniska svaret övervakas vid dessa sjukdomar/tillstånd för att upp-täcka behov av dosjustering eller byte till konventionell be-handling.
DoseringPlenadren är avsett för underhållsbehandling av binjure-barksvikt och dosen måste anpassas individuellt efter det kliniska svaret. En vanlig underhållsdos är 20–30 mg Plena-dren per dag, en gång dagligen på morgonen. 40 mg är den högsta underhållsdos av Plenadren som studerats. Den lägsta möjliga underhållsdosen ska användas. I situationer där kroppen är utsatt för stor fysisk och/eller psykisk stress kan patienter behöva ytterligare ersättningsbehandling med hy-drokortisontabletter med omedelbar frisättning, särskilt på eftermiddagen/kvällen.
Byte från konventionell oral glukokortikoid- behandling till PlenadrenNär patienter ska byta från konventionell oral ersättningsbe-handling med hydrokortison tre gånger dagligen till Plen-adren, kan en identiskt total dygnsdos ges. Då Plenadren har en lägre biotillgänglighet än konventionella hydrokortison-tabletter, måste det kliniska svaret övervakas och ytterligare individuell anpassning av dosen kan behövas. Vid byte till Plenadren från behandling med hydrokortisontabletter som ges två gånger dagligen, eller från andra kortisonpreparat rekommenderas en hydrokortisonekvivalent dygnsdos av Plenadren; ytterligare individuell anpassning av dosen kan behövas.
Användning vid tillfällig annan sjukdomI mindre svåra fall, när parenteral administrering av hydro-kortison inte krävs, t.ex. vid lindriga infektioner, feber oav-sett orsak och i stressande situationer som mindre kirurgiska ingrepp ska den normala orala dagliga ersättningsdosen ökas tillfälligt. Den totala dagliga Plenadrendosen ska ökas genom att underhållsdosen ges två eller tre gånger dagligen med 8 ± 2 timmars intervall (ökat antal administreringar, inte en ökning av morgondosen). Man kan även välja att ordinera hydrokortisontabletter med omedelbar frisättning i stället för Plenadren eller som tillägg till Plenadren. När episoden av tillfällig annan sjukdom är över kan patienten återgå till den normala underhållsdosen av Plenadren.
För fullständig doseringstext hänvisas till produktinfor-mationen.
LitteraturResultat och värderingar i denna monografi baseras på de fullständiga studierapporter som legat till grund för god-kännandet. Den citerade artikeln har ej granskats av Läke-medelsverket.
EPAR (http://www.emea.europa.eu).1. Johannsson G, et al. J Clin Endocrin Metab 2012;97(2):473–81.2.
36 • InformatIon fr ån L äkemedeLsverket 4 : 2012
monoGr afIer
Läkemedelsverkets värderingVid binjurebarksvikt måste doseringen av hydrokorti-son anpassas efter hur individen svarar på behandlingen. Plenadren är ett värdefullt tillskott i behandlingen av patienter med binjurebarksvikt då beredningsformen med sin annorlunda profil innebär möjlighet att ytterli-gare individualisera behandlingen. Effekt och säkerhet är jämförbar med den hos tidigare godkända alternativ men den enklare doseringen kan innebära en fördel för patienten.

IndikationBehandling av svår, aktiv ulcerös kolit hos barn 6–17 år gamla, som svarat otillräckligt på konventionell behandling inklusive kortikosteroider och immunsuppressiva läkemedel (6-merkaptopurin, azatioprin) eller har intolerans mot eller kontraindikationer mot sådana behandlingar.
Dosering Infliximab, 5 mg/kg ges som en intravenös infusion under en tvåtimmarsperiod som åtföljs av ytterligare infusioner med doser på 5 mg/kg, två och sex veckor efter den första infusionen. Därefter ges infusioner var åttonde vecka.
Sammanfattning av kliniska studierRemicade är sedan tidigare godkänt för behandling av vuxna och barn med Crohns sjukdom, för vuxna med ulcerös kolit, reumatoid artrit, ankyloserande spondylit, psoriasisartrit och psoriasis.
Effekt och säkerhet av infliximabbehandling av barn med ulcerös kolit har utvärderats i en öppen randomiserad multi-center studie. Sextio barn mellan 6 och 17 år med måttlig till svår ulcerös kolit (Mayo-score 6–12, endoskopi subscore ≥ 2) inkluderades. Patienterna hade aktiv sjukdom trots adekvat behandling med konventionell terapi (perorala kor-tikosteroider, aminosalicylater och/eller immunomodule-rare [6-MP, AZA]). Samtidig behandling med stabila doser perorala aminosalicylater, kortikosteroider och/eller im-munomodulerare var tillåten. Cirka 62 % av patienterna stod på steroider vid inklusion, 53 % hade immunmodulerare och 53 % 5-ASA-preparat. Nedtrappning av steroider och im-munomodulerare var tillåten efter vecka 0.
Samtliga patienter gavs infliximab 5 mg/kg vecka 0, 2 och 6. De patienter som inte svarat på behandlingen vecka 8 fick ingen ytterligare behandling (n = 15). Resterande pa-tienter (n = 45) randomiserades vecka 8 och gavs underhålls-behandling med infliximab 5 mg/kg var 8:e eller var 12:e vecka under 42 alternativt 46 veckor. Primärt effektmått var respons vecka 8. Respons karakteriserades som en minskning av Mayo-score med minst 30 % (minst 3 poäng). Remission definierades som Mayo-score på högst 2 och ingen eller obetydlig blödning från tarmen och som Pediatric Ulcerative Colitis Activity Index (PUCAI) score < 10.
Effekt Behandlingen ledde till klinisk respons hos 73 % av patien-terna vecka 8 och till remission för 40 % (Mayo-score).
Av de 45 patienter som randomiserades vecka 8, fanns endoskopi subscore tillgängligt för endast nio av patienterna vecka 54. Totalt fanns det 43 patienter med PUCAI score vecka 54 och 28 % av dem var i remission vid denna tidpunkt. En större andel av patienterna i gruppen med doseringsinter-vall var 8:e vecka var i remission vecka 30 och 54 i jämförelse med patienter som doserades var 12:e vecka. Vecka 30 var 40 % av barnen med doseringsintervall var 8:e vecka i remis-sion jämfört med 19 % av barn med dosering var 12:e vecka. Motsvarande siffror vecka 54 var 38 % och 18 %.
Det genomsnittliga intaget av kortikosteroider minskade fram till vecka 8 i båda behandlingsgrupperna. Endast hos barn med doseringsintervall var 8:e vecka var den dagliga dosen fortfarande lägre vecka 54 jämfört med vid studiestarten. Det fanns inga skillnader i kliniskt svar vecka 8 mellan patienter med eller utan immunomodulatorer vid studiestarten.
Fem av de nio yngre barnen med kliniskt svar vecka 8, randomiserades till behandling var 8:e vecka. I den äldre gruppen var motsvarande siffra 17 av 35 individer.
Effekt av behandling i olika åldersgrupper ses i Tabell I.
Tabell I. Effekt av infliximab i olika åldersgrupper.
Infliximab 5 mg/kg 6–11 år 12–17 år
antal patienter 15 45
kliniskt svar vecka 8 9 (60 %) 35 (78 %)
klinisk remission vecka 8 (mayo-score)
7 (47 %) 17 (38 %)
slemhinneläkning vecka 8 8 (53 %) 33 (73 %)
Patienter med PUCaI score v 8 12 39
klinisk remission v 8 (PUCaI) 4 (33 %) 13 (33 %)
Patienter med PUCaI score vecka 54 9 34
klinisk remission v 54 (PUCaI) 3 (33 %) 9 (26 %)
Förutom resultaten från den beskrivna barnstudien ger studier på vuxna med ulcerös kolit stöd till induktions- och under-hållsbehandling av barn. Den rekommenderade underhålls-dosen för barn är, som för vuxna, 5 mg/kg var 8:e vecka.
InformatIon fr ån L äkemedeLsverket 4 : 2012 • 37
monoGr afIer
Remicade (infliximab) – ny indikationATC-kod: L04AA12100 mg pulver till koncentrat till infusionsvätska, lösningJanssen Biologics B.V.
Godkännandedatum: 2012-02-21. Central procedur.

38 • InformatIon fr ån L äkemedeLsverket 4 : 2012
monoGr afIer
Säkerhet Biverkningarna som rapporterades i studien vid pediatrisk ulcerös kolit och studierna vid vuxen ulcerös kolit var jäm-förbara. Vanliga biverkningar i barnstudien var övre luft-vägsinfektion, faryngit, buksmärta, feber och huvudvärk. Den vanligaste biverkningen var försämring av ulcerös kolit, incidensen av denna var högre hos patienter med dosering var 12:e vecka jämfört med var 8:e vecka.
Infektioner rapporterades hos 31 (52 %) av 60 behandlade patienter och 22 (37 %) av dessa behövde oral eller parenteral antimikrobiell behandling. Allvarliga infektioner rapporte-rades hos 12 % (7/60) av alla behandlade patienter.
En större andel patienter fick allvarliga biverkningar och avbröt på grund av biverkningar i den yngre åldersgruppen än i den äldre åldersgruppen. Andelen patienter med infek-tioner var också större i den yngre åldersgruppen medan andelen allvarliga infektioner var jämförbar i de två ålders-grupperna. Sammantaget var antalet biverkningar och infu-sionsreaktioner jämförbara mellan åldersgrupperna 6–11 och 12–17 år.
Patienter som utvecklar antikroppar mot infliximab är mer benägna (ungefär två- till trefaldigt) att utveckla infu-sionsrelaterade reaktioner, dock oftast milda. Antikroppar mot infliximab påvisades hos fyra (7,7 %) av patienterna till och med vecka 54. Inga infusionsreaktioner som var så all-varliga att behandlingen måste avslutas rapporterades.
Vid klinisk användning av infliximab har det rapporterats om en ökad risk för en sällsynt, mycket svårbehandlad, vari-ant av lymfom (hepatosplenärt T-cellslymfom) hos ungdo-mar och yngre vuxna patienter med Crohns sjukdom och ulcerös kolit som har behandlats med infliximab. Dessa pa-tienter har också samtidigt haft behandling med immun-suppression (azatioprin eller merkaptopurin). Information har tidigare gått ut till berörda förskrivare. Uppföljnings-program, både för barn och vuxna pågår för att följa lång-tidssäkerhet. Med hänsyn till de beskrivna riskerna bör pa-tienter uppmärksamt kontrolleras under behandlingen.
Litteratur Resultat och värdering i denna monografi baseras på de fullständiga studierapporter som legat till grund för god-kännandet.
EPAR (Central procedur) (http://www.ema.europa.eu) alternativt 1. PAR (Ömsesidig procedur) relevant internetadress: (www.lakeme-delsverket.se) eller (http://www.hma.eu/mri.html).
Dokumentation ej granskad av Läkemedelsverket
Hyams J, Damaraju L, Blank M, et al. Induction and Maintenance 1. Therapy with Infliximab for Children with Moderate to Severe Ulce-rative Colitis. Clin. Gastroenterol. Hepatol. Clin Gastroenterol He-patol 2012;10(4):391–9.
Läkemedelsverkets värderingRemicade är ett värdefullt tillskott vid behandling av en grupp barnpatienter med svårare skov av ulcerös kolit som inte svarar på behandling med 5-ASA, kortikoste-roider och annan immunsuppression. Då anti-TNFα-behandlingens säkerhetsprofil kan vara allvarlig, med ökad risk för infektioner och malignitet, bör behand-lingen begränsas till ovanstående patientkategorier.

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 39
tLv
För dessa sidor ansvarar TLV, Tandvårds- och läkemedelsförmånsverketHelena Roslund, Box 22520, 104 22 StockholmKontakt vid frågor: [email protected]
Oförändrad ersättning till apoteken enligt TLVs uppföljning av apoteksmarknadenVi har genomfört vår första översyn av den svenska apoteks-marknaden. Vi har fått över 300 nya apotek, längre öppet-tider och en mångfald aktörer. Samtidigt har inte försälj-ningen av receptbelagda läkemedel ökat – det är fler apotek som delar på den ersättning staten betalar ut. Trots detta uppvisar de flesta apotek ett positivt resultat. Därför har vi beslutat att inte ändra nivån på apotekens handelsmarginal.
Läs mer: http: //www.tlv.se/apotek/apotekets-marginaler/oversyn-handelsmarginal/
Insulinpumpar utesluts ur läkemedels- förmånerna TLV tycker att insulinpumpar är bra och viktiga produkter men på grund av att de har för lång hållbarhet kan de inte betraktas som förbrukningsartiklar. Landstingen kommer att fortsätta att ha ansvaret för att tillhandahålla insulin-pumparna, som utesluts ur förmånerna från och med den 1 december 2013.
Faktor VIII-preparat kvarstår i högkostnads-skyddet efter prissänkningarEfter prissänkningar från företagen har TLV beslutat att av-sluta omprövningen av de läkemedel som används vid hemo-fili typ A. Samtliga preparat kvarstår i högkostnadsskyddet. TLV anser, i likhet med SBU, att ett nationellt kvalitets- register behöver införas inom området.
De företag som marknadsför rekombinant framställda faktor VIII-preparat har ansökt om nya lägre priser. Prissänk-ningarna ligger mellan 3 och 12 procent. De plasmaderiverade preparaten kostar fortfarande mindre än rekombinant fram-ställda preparat. TLV kommer att följa kostnadsutvecklingen för faktor VIII-preparaten och kan starta en ytterligare om-prövning beroende på hur användningen och kostnaderna utvecklas över tid.
Mot denna bakgrund kvarstår samtliga faktor VIII-preparat i högkostnadsskyddet.
Cymbalta fortfarande subventionerat som tredjehandsalternativ vid depression och ångest Vid behandling av depression och generaliserat ångestsyn-drom är Cymbalta som tidigare subventionerat tidigast som tredjehandsalternativ eller senare. Efter TLV:s nya ompröv-ning av Cymbalta har priset för läkemedlet sänkts något. Cymbalta är dock ett tio gånger dyrare alternativ. Det är därför viktigt att begränsningen följs. Cymbalta ingår återigen i högkostnadsskyddet vid behandling av diabetesneuropati.
Beslutet träder i kraft den 1 augusti 2012.
Ny form av Viramune ingår i högkostnads-skyddetViramune (nevirapin) depottablett används i kombinations-behandling av HIV-1-infektion hos vuxna, ungdomar och barn från tre år som kan svälja tabletter. Läkemedlet ingår sedan tidigare i högkostnadsskyddet i beredningsformerna tablett samt oral suspension. TLV bedömer att den nya be-redningsformen, depottablett, har likvärdig medicinsk effekt och behandlingskostnad som den redan subventionerade beredningsformen tablett.
Mot denna bakgrund beslutar TLV att Viramune depot tablett ska vara subventionerad och ingå i högkostnadsskyddet.
Beslutet gäller från och med den 23 mars 2012.
Dexametason Rosemont ingår i högkostnads-skyddet med begränsningDexametason är en potent kortikosteroid med en mycket vid indikation. Dexametason Rosemont är dyrare än betameta-son, som är den mest jämförbara kortikosteroiden. Företaget har inte kommit in med något underlag som visar att dexa-metason skulle vara kostnadseffektivt jämfört med andra kortikosteroider generellt sett.
Det kan dock finnas situationer då det saknas alternativ till dexametasonbehandling, exempelvis vid screening för Cushings syndrom samt vid behandling av återfall i multipelt myelom med lenalidimid (Revlimid).
TLV beslutar mot denna bakgrund att Dexametason Rose-mont ska vara subventionerat och ingå i högkostnadsskyddet i de fall när andra kortikosteroider inte kan användas.
Beslutet gäller från och med den 23 mars 2012.
Tandvårds- och läkemedelsförmånsverketTLV, Tandvårds- och läkemedelsförmånsverket, är den myndighet som beslutar vilka läkemedel och förbrukningsartiklar som ska subventioneras av samhället. Besluten fattas av en nämnd som finns inom myndigheten. Nämnden består av en ordförande och tio ledamöter. Ledamöterna har tillsammans en bred medicinsk, såväl praktisk som vetenskaplig, och hälsoekonomisk kompetens. Två av ledamöterna har erfarenhet från brukargrupper.

Firmagon ingår inte i högkostnadsskyddetFirmagon (degarelix) för behandling av prostatacancer kommer inte att ingå i högkostnadsskyddet. Företaget har inte visat att Firmagon har en bättre effekt än GNRH-ana-logen leuprorelin med initialt flareskydd. Det begärda priset, och även administrationskostnaderna, är högre än för det jämförda alternativet.
Mot denna bakgrund beslutar TLV att Firmagon inte ska subventioneras och inte ingå i högkostnadsskyddet.
Beslutet gäller från och med den 27 mars 2012.
Votubia ingår i högkostnadsskyddetVotubia (everolimus) är avsedd för patienter med växande symptomatisk SEGA, subependymalt jättecellsastrocytom, där det bedöms nödvändigt med en behandlingsåtgärd men för vilka operation inte är lämplig.
När SEGA är svåropererbart blir operationskostnaden mycket hög. Den totala kostnaden kan bli lägre med Votubia än med operation.
Det finns en betydande osäkerhet kring hur det faktiska utfallet kommer att bli i klinisk praxis. Läkemedlet har ett högt pris och om kostnadsbesparingar inte kommer till stånd kan frågan om läkemedlets kostnadseffektivitet komma i ett annat läge. TLV:s beslut förenas därför med ett villkor för företaget att inkomma med en hälsoekonomisk analys som inkluderar data från studierna som ska visa Votu-bias långvariga effekt och från användningen av Votubia i klinisk praxis.
Mot denna bakgrund beslutar TLV att Votubia ska vara subventionerat och ingå i högkostnadsskyddet.
Beslutet gäller från och med den 28 april 2012.
Premalex ingår i högkostnadsskyddetPremalex (escitalopram) för behandling av PMDS, premen-struellt dysforiskt syndrom, ingår i högkostnadsskyddet. Läkemedlet tillhör läkemedelsgruppen selektiva serotonin-återupptagshämmare, SSRI. Läkemedlet är det första i denna grupp som fått indikationen PMDS. Premalex har visats vara kostnadseffektiv i jämförelse med ingen behand-ling alls.
Mot denna bakgrund beslutar TLV att Premalex ska vara subventionerat och ingå i högkostnadsskyddet.
Beslutet gäller från och med den 4 maj 2012.
Snabbguide till TLVs beslutBeviljas generell subventionViramune i beredningsformen depottablett för kombina-tionsbehandling av HIV-1-infektion ingår i högkostnads-skyddet sedan den 23 mars 2012.
Votubia för behandling av växande symptomatisk SEGA ingår i högkostnadsskyddet sedan den 28 april 2012.
Premalex för behandling av PMDS ingår i högkostnads-skyddet sedan den 4 maj 2012.
Beslut om begränsad subventionKortikosteroiden Dexametason Rosemont ingår i högkost-nadsskyddet i de fall när andra kortikosteroider inte kan användas. Beslutet gäller sedan den 23 mars 2012.
Avslag och uteslutningarFirmagon för behandling av prostatacancer ingår inte i högkostnadsskyddet. Beslutet fattades den 27 mars 2012.
Utträde ur förmånerna den 1 juni 2012Grifols Nordic ABXepol, infusionsvätska, lösning, 50 mg/mL, glasflaska 100 mL.
Xepol, infusionsvätska, lösning, 50 mg/mL, glasflaska 200 mL.
Leo Pharma ABEtalpha, injektionsvätska, lösning, 2 mikrog/mL, ampull 10 × 1 mL.
Etalpha, injektionsvätska, lösning, 2 mikrog/mL, ampull 10 × 0,5 mL.
Novartis Sverige ABDiovan, filmdragerad tablett, 160 mg, blister, 98 tabletter (kalenderförp).
Diovan, filmdragerad tablett, 160 mg, burk, 250 tabletter (dosdisp).
Diovan, filmdragerad tablett, 160 mg, blister, 28 tabletter (kalenderförp).
Diovan, filmdragerad tablett, 160 mg, blister, 98 × 1 tabletter (endos).
Diovan, filmdragerad tablett, 160 mg, burk, 100 tabletter (dosdisp).
Diovan, filmdragerad tablett, 320 mg, blister, 98 × 1 tabletter (endos).
Diovan, filmdragerad tablett, 320 mg, blister, 28 tabletter (kalenderförp).
Diovan, filmdragerad tablett, 320 mg, blister, 98 tabletter (kalenderförp).
Diovan, filmdragerad tablett, 40 mg, blister, 28 tabletter.Diovan, filmdragerad tablett, 40 mg, blister, 14 tabletter.Diovan, filmdragerad tablett, 80 mg, blister, 98 tabletter
(kalenderförp).Diovan, filmdragerad tablett, 80 mg, blister, 28 tabletter
(kalenderförp).Diovan Comp, filmdragerad tablett, 160 mg/12,5 mg,
blister, 98 tabletter (kalenderförp).Diovan Comp, filmdragerad tablett, 160 mg/12,5 mg,
blister, 28 tabletter (kalenderförp).Diovan Comp, filmdragerad tablett, 160 mg/25 mg,
blister, 98 tabletter (kalenderförp).
40 • InformatIon fr ån L äkemedeLsverket 4 : 2012
tLv
För dessa sidor ansvarar TLV, Tandvårds- och läkemedelsförmånsverketHelena Roslund, Box 22520, 104 22 StockholmKontakt vid frågor: [email protected]

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 41
tLv
För dessa sidor ansvarar TLV, Tandvårds- och läkemedelsförmånsverketHelena Roslund, Box 22520, 104 22 StockholmKontakt vid frågor: [email protected]
De nya läkemedelsförmånerna – ett produktinriktat system med två subventionsmöjligheter.
• Generell subvention innebär att ett läkemedel är subventionerat för hela det godkända användningsområdet.
• begränsad subvention innebär att ett läkemedel bara är subventionerat för ett visst användningsområde.
Prenumerera på nyheter om tandvård och läkemedel via e-post och RSS
du kan prenumerera på nyheter om tandvård och läkemedel från tLv. Gå in på www.tlv.se/nyhetsmejl. I en meny till vänster kan du välja att få beslut eller nyhetsbrev till din e-postadress, eller att prenumerera på nyheter via rss. det kostar dig ingenting och du kan självklart avsluta prenumerationen när du vill.
Diovan Comp, filmdragerad tablett, 160 mg/25 mg, blister, 28 tabletter (kalenderförp).
Diovan Comp, filmdragerad tablett, 320 mg/12,5 mg, blister, 98 tabletter (kalenderförp).
Diovan Comp, filmdragerad tablett, 320 mg/25 mg, blister, 98 tabletter (kalenderförp).
Diovan Comp, filmdragerad tablett, 80 mg/12,5 mg, blister, 98 tabletter (kalenderförp).
Diovan Comp, filmdragerad tablett, 80 mg/12,5 mg, blister, 28 tabletter (kalenderförp).



44 • InformatIon fr ån L äkemedeLsverket 4 : 2012

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 45

46 • InformatIon fr ån L äkemedeLsverket 4 : 2012

InformatIon fr ån L äkemedeLsverket 4 : 2012 • 47
blanketten skickas till:
[email protected]äkemedelsverketmedicinteknikbox 26751 03 Uppsalawww.lakemedelsverket.se

B
approvals • authorisation • clinical trials • communication • competence • cosmetics • dialogue • directives • efficacy • environment • evaluation • guidelines • harmonisation • health economics • herbals • homeopathics • information • inspection laboratory analysis • market surveillance • medicinal products • medical devices • narcotics • public health • quality • registration • regulations • reliability • risk/benefit • safety • scientific • standar-disation • transparency • vigilance • approvals • authorisation • clinical trials • communication • competence • cosmetics • dialogue • directives • efficacy • environment • evaluation • guidelines • harmonisation • health econo-mics • herbals • homeopathics • information • inspection • laboratory analysis • market surveillance • medicinal
Postadress/Postal address: P.o. box 26, se-751 03 Uppsala, sWeden. besöksadress/visiting address: dag Hammarskjölds väg 42, Uppsala
telefon/Phone: +46 (0)18 17 46 00 fax +46 (0)18 54 85 66 Internet: www.lakemedelsverket.se e-mail: [email protected]
Tidigare utgivna nummer
Läkemedelsrekommendation:Ingen rekommendation i detta nummer.
Monografier:Edurant (rilpivirin)Sativex (nabiximols)Procox (emodepsid och toltrazuril)
Nummer 1: 2012
Information från Läkemedelsverket 2012(23)1
Läkemedelsrekommendation:Läkemedelsbehandling av psoriasis.
Monografier:Azyter (azitromycin)Halaven (eribulin)Pradaxa (dabigatranetexilat) – kompletterande monografiTrobalt (retigabin)Firmagon (degarelix)Plenaxis (abarelix)
Nummer 4: 2011
Information från Läkemedelsverket 2011(22)4
Läkemedelsrekommendation:Behandling av och profylax mot influensa med antivirala medel.
Monografier:Bydureon (exenatid)Lucentis (ranibizumab) – ny indikationYellox (bromfenak)Zoely/IOA (nomegestrolacetat, östradiol)Xeplion (paliperidonpalmitat)
Nummer 5: 2011
Information från Läkemedelsverket 2011(22)5
Läkemedelsrekommendation:Läkemedelsbehandling av hepatit C-virusinfektion hos vuxna och barn.
Monografier:Detremin (kolekalciferol)Ozurdex (dexametason) – ny indikationXgeva (denosumab)
Nummer 6: 2011
Information från Läkemedelsverket 2011(22)6
Läkemedelsrekommendation:Läkemedelsbehandling vid inflammatorisk tarmsjukdom (IBD).
Monografier:Buccolam (midazolam)Jetvana (cabazitaxcel)Prevenar 13
Nummer 2: 2012
Information från Läkemedelsverket 2012(23)2
Läkemedelsrekommendation:Ingen rekommendation i detta nummer.
Monografier:Dificlir (fidaxomicin)Herceptin (trastuzumab) – ny indikationLaif (extrakt av Hypericum perforatum L, herba [johannesört])
Nummer 3: 2012
Information från Läkemedelsverket 2012(23)3





























