Embed Size (px)
Citation preview

TREACHER COLLINS SYNDROME
(MANDIBULOFACIAL DYSOSTOSIS )
Omamah Asif Jiman
Demonstrator
Department of Genetic Medicine
Faculty of Medicine, KAU
09/01/2012 Paediatric Grand Round
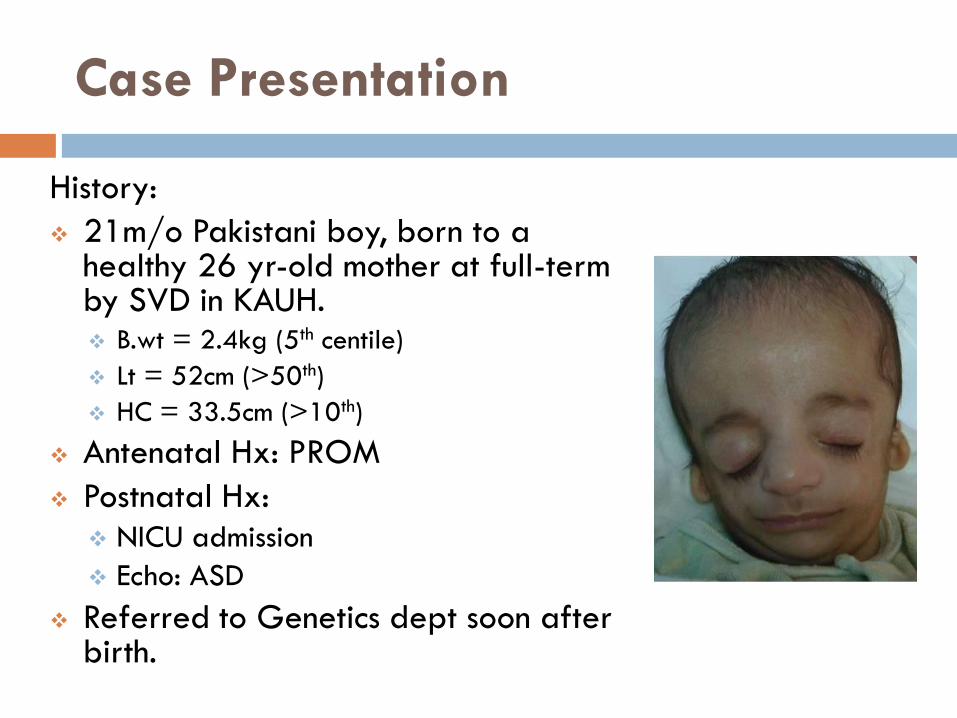
Case Presentation
History:
21m/o Pakistani boy, born to a healthy 26 yr-old mother at full-term by SVD in KAUH. B.wt = 2.4kg (5th centile)
Lt = 52cm (>50th)
HC = 33.5cm (>10th)
Antenatal Hx: PROM
Postnatal Hx: NICU admission
Echo: ASD
Referred to Genetics dept soon after birth.
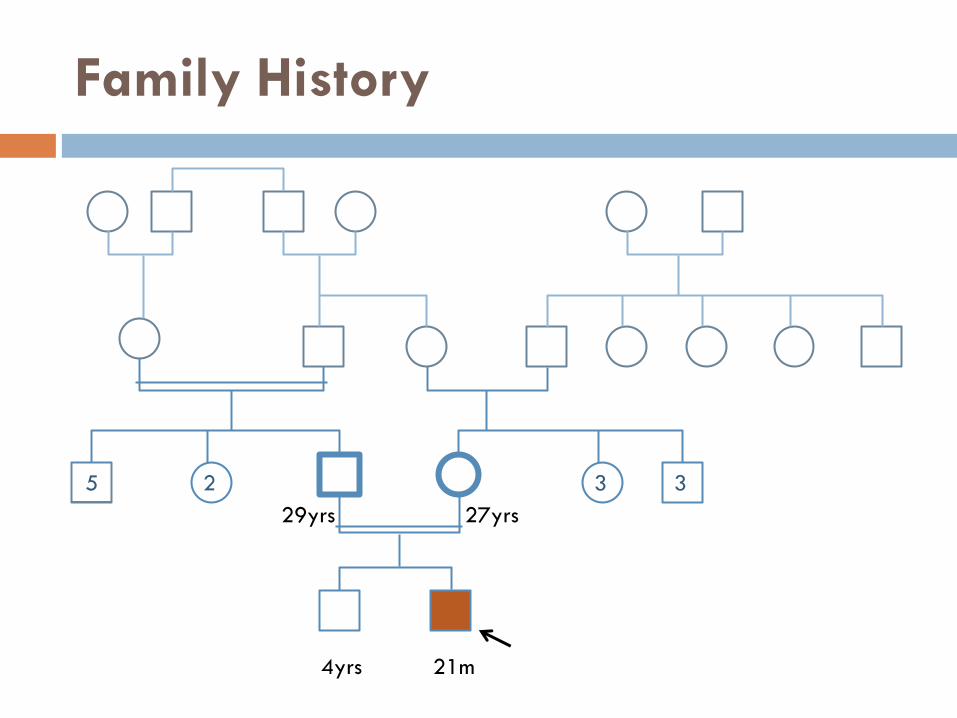
Family History
5 2 3 3
27yrs 29yrs
21m 4yrs

What is Treacher Collins Syndrome (TCS) ?
It is a genetic condition in which cheek bones and
jawbones are under-developed.
Named after Dr. Edward Treacher Collins, a British
opthalomlogist, who described the condition in an
affected individual in 1900.
Although Thompson reported the first case in 1846.
Franceschetti and Klein made extensive reports on
the condition in 1940s and called it
Mandibulofacial Dysostosis.

Overveiw of Treacher Collins Syndrome
Frequency between 1:10,000-50,000
Male= Female
Most of cases are the result of new mutation
Non-penetrance is rare
Variable expressivity
Mild cases may go undiagnosed
Mutation on the TCOF-1 gene
Affected structures are those derived from
the first and second brachial arches

Why TCS Happens ?
TCS is an Autosomal Dominant disorder.
40% of affected population are familial cases.
60% of cases represent de novo gene mutation
associated with older paternal age.
Teratogenic dose of vitamin A and isotretinoin given
in animal models produced malformations of the
craniofacial skeleton that closely resemble the
syndromic features of TCS.
A possible human phenocopy induced by vitamin A
was reported by Lungarotti et al (1987).
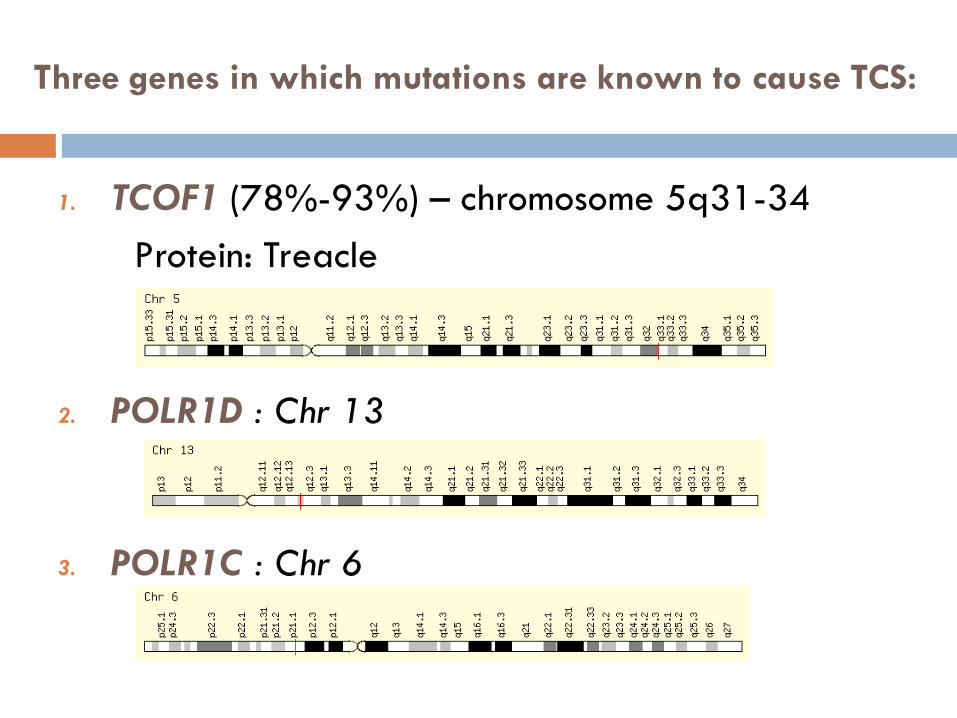
1. TCOF1 (78%-93%) – chromosome 5q31-34
Protein: Treacle
2. POLR1D : Chr 13
3. POLR1C : Chr 6
Three genes in which mutations are known to cause TCS:

How TCS Happens?
TCOF1gene encodes for a low complexity,
serine/alanine-rich nucleolar phosphoprotein,
Treacle.
The clinical phenotype of Treacher Collins syndrome
suggests that the responsible gene plays a
fundamental role in early embryonic development,
particularly in development of the craniofacial
complex.

Mutation analysis of TCOF1 has resulted in the
identification of over 120 mutations that are
spread throughout the gene.
Mutations include splicing mutations, insertions
and non-sense mutations, the majority are
deletions.
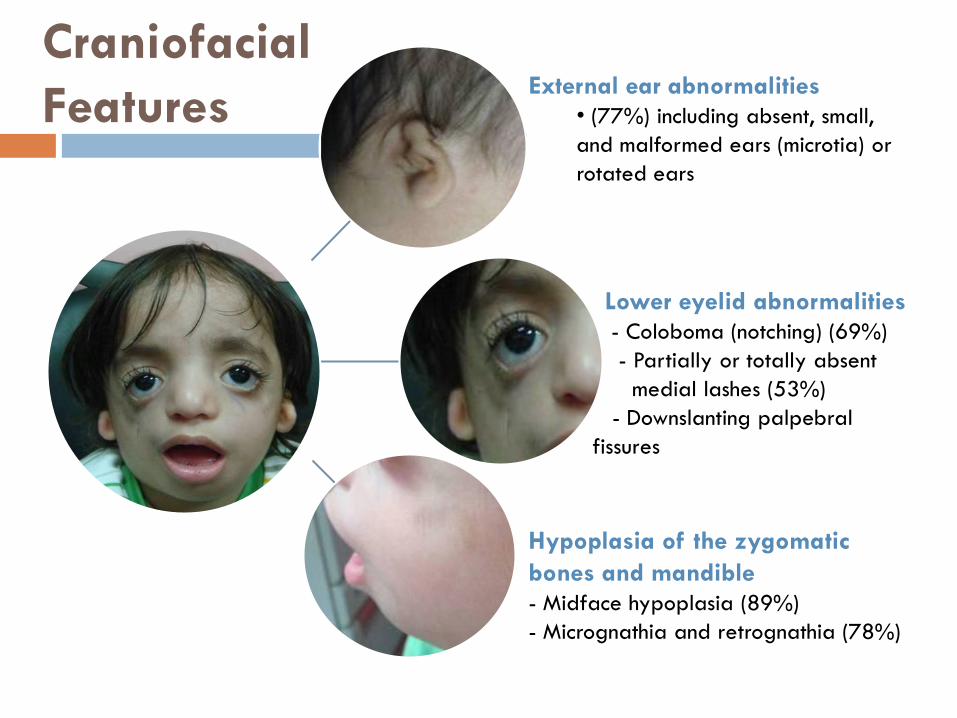
Craniofacial
Features
Lower eyelid abnormalities
- Coloboma (notching) (69%)
- Partially or totally absent
medial lashes (53%)
- Downslanting palpebral
fissures
External ear abnormalities
• (77%) including absent, small,
and malformed ears (microtia) or
rotated ears
Hypoplasia of the zygomatic
bones and mandible
- Midface hypoplasia (89%)
- Micrognathia and retrognathia (78%)

Minor Clinical features
Ear abnormalities including atresia or stenosis of the
external auditory canals (36%)
Conductive hearing loss (40%-50%) attributed
mostly to hypoplasia, or absence of the ossicles and
hypoplasia of the middle ear cavities.
Ophthalmologic defects
Vision loss (37%), amblyopia (33%), refractive errors (58%),
anisometropia (17%), strabismus (37%)

Minor Clinical features(contd.)
Cleft palate with or without cleft lip (28%)
Preauricular hair displacement (26%), in which hair growth extends in front of the ear to the lateral cheekbones
Airway abnormalities including the following:
Tracheostoma
Uni- or bilateral choanal stenosis or atresia
Congenital Heart defects
Cryptorchidism
Blind fistulas and skin tags between auricle and angle of mouth
Absence of parotid gland and dacrostenosis

Diagnosis
The diagnosis of Treacher Collins syndrome (TCS) is
based upon a characteristic pattern of
malformation and radiographic findings.
TCS is distinguished from the other genetically
determined disorders with mandibulofacial
dysostoses by the absence of limb anomalies.

Testing Strategy
To confirm/establish the diagnosis in a proband.
Sequence analysis of TCOF1 is performed first for:
Those with a family history of TCS consistent with autosomal dominant inheritance; and
Those who represent simplex cases (i.e., a single occurrence in a family).
If a mutation in TCOF1 is not identified, POLR1D sequence analysis should be considered next.
POLR1C sequence analysis should be considered:
In families with multiple affected sibs and/or consanguinity; or
In those who represent simplex cases who do not have a TCOF1 or POLR1D mutation.

Prenatal Diagnosis
Ultrasound has allowed successful prenatal
diagnosis and should help identify those infants with
severe craniofacial involvement in the second
trimester of pregnancy (18wks).
Prenatal diagnosis and preimplantation genetic
diagnosis (PGD) for at-risk pregnancies are
available but require prior identification of the
disease-causing mutation in the family.

Differential Dagnosis
1. Nager syndrome:
Preaxial acrofacial dysostosis
Similar craniofacial
malformations as TCS
Radial limb reduction defects
varying from mild thenar
hypoplasia to absent radius with
a radial club hand
AD and AR inheritance reported
Differential Dx (contd.)
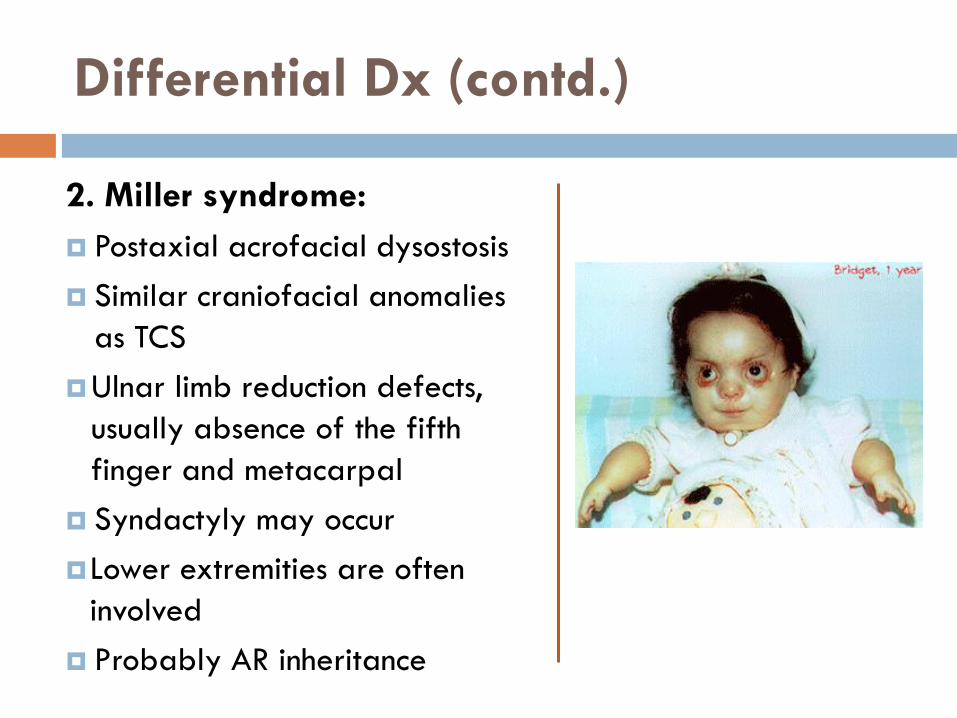
2. Miller syndrome:
Postaxial acrofacial dysostosis
Similar craniofacial anomalies
as TCS
Ulnar limb reduction defects,
usually absence of the fifth
finger and metacarpal
Syndactyly may occur
Lower extremities are often
involved
Probably AR inheritance
Differential Dx (contd.)
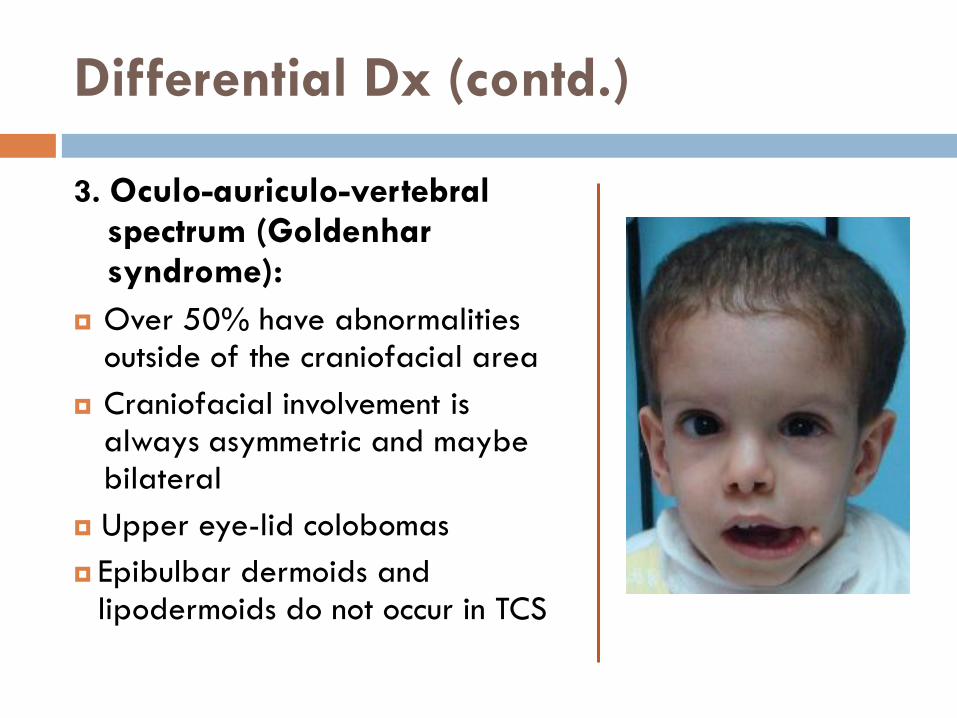
3. Oculo-auriculo-vertebral spectrum (Goldenhar syndrome):
Over 50% have abnormalities outside of the craniofacial area
Craniofacial involvement is always asymmetric and maybe bilateral
Upper eye-lid colobomas
Epibulbar dermoids and lipodermoids do not occur in TCS

Differential Dx (contd.)
4. Autosomal dominant mandibulofacial dysostosis
(Bauru type): similar but no eyelid coloboma or
zygomatic arch hypolasia
5. Autosomal dominant mandibulofacial dysostosis
(Hedera type): Similar features but linkage to TCS
locus was excluded

What problems can be expected?
The most common difficulties involve:
Breathing
Hearing
Vision

Causes of breathing problems
Micrognathia
Cleft palate
Choanal atresia/stenosis
Glossoptosis
Pharyngeal hypoplasia

Airway (evaluation and treatment)
Intubation maybe difficult due to abnormal
anatomy of the airway… may need long-term
tracheostomy.
Observe all neonates in a supine sleep position to
assess for lesser degrees of obstruction.
Monitor O2 saturation if signs or symptoms of
hypoxia develop.
A formal sleep study may be necessary.

Ophthalmologic
Evaluate the adequacy of lid closure in the newborn
period (large lower lid colobomas and zygomatic
deficency corneal exposure)
Referral to ophthalmologist
Rarely , an affected child will need surgery early in
life to provide adequate coverage of the eye
closure

ENT & Hearing
All neonates with TCS need formal ENT and hearing assessment
CT scan of the petrous temporal bone to evaluate middle ear and adjacent structures if hearing loss is bilateral
Early amplification is essential
Bone-anchored hearing aides have been used successfully
Early intervention programs are critical
External ear reconstruction

Craniofacial
Craniofacial malformations in TCS are stable over
time and non-progressive.
Cleft palate repair, typically at less than a year of
age.
Distraction osteogenesis to lengthen the mandible.
Zygomatic and orbital
reconstruction.

Growth and feeding
Growth potential is normal
Feeding difficulty secondary to craniofacial malformation
Gastroesophageal reflux occurs in TCS
Management:
Soft squeeze devices for isolated clefts, gavage-assisted
feeding and gastrostomy tube can be considered
Feeding specialist or occupational therapist should be
involved in the care of a child with feeding difficulty

Development and behaviour
Majority are cognitively normal
With proper attention to the hearing loss, language
development should be normal.
Evaluation: Longitudinal monitoring of speech is
critical.
Treatment: Augmentative systems for communication
such as sign and picture exchange should be
implemented early in those with long term
tracheostomies.

Anaesthesia
The craniofacial structural alterations in TCS present
a variety of challenges for which the
anaesthesiologists need to be prepared.
Laryngeal mask airways and fiberoptic devices
have been advocated.
Postoperative airway obstruction should be
anticipated.

Patients with auricular and facial abnormalities
need to be carefully evaluated for skeletal and
visceral anomalies.
Early intervention and recognition of hearing
has paramount importance
Family support and counselling
Learning points

References
Management of Genetic Syndromes, Second Edition, edited by Suzanne B. Cassidy and Judith E. Allanson (2005)
Gorlin’s Syndromes of the heaqd and neck, 5th edition, edited by Raoul Hennekam, Ian Krantz and Judith Allanson (2010)
GeneReviews, Pagon RA, Bird TD, Dolan CR, et al., editors. : http://www.ncbi.nlm.nih.gov/books/NBK1532/
OMIM-Online Mendelian Inheritance in Man: http://www.ncbi.nlm.nih.gov/omim
Smith’s Recognizable Patterns of Human Malformation, Jones, 6th edition, 2010
Dixon J., Trainor P. and Dixon M. (2007), Treacher Collins Syndrome. Orthodontics & Craniofacial Research, 10:88-95.
Thompson A. Notic of several cases of malformation of the external ear, together with experiments on the state of hearing in such persons. Monthly J Med Sci 1846 ; 7:420
Treacher Collins E. Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones. Trans Ophthalmol Soc UK 1900; 20:190-2
Franceschetti A, KleinD. Mandibulo-facial dysostosis: new hereditary syndrome. Acta Ophthalmol 1949; 27:143-224

THANK YOU !
Special Thanks to the parents of our patient for their cooperation





























