Embed Size (px)
Citation preview

JMed Genet 1996;33:603-606
Prenatal diagnosis in Treacher Collinssyndrome using combined linkage analysis andultrasound imaging
S J Edwards, A Fowlie, M P Cust, D T Y Liu, I D Young, Michael J Dixon
AbstractTreacher Collins syndrome is an auto-somal dominant disorder of facial de-velopment, the features of which includeconductive hearing loss and cleft palate. Inthe current investigation, linkage analysishas been used to make first trimester diag-nostic predictions in a pregnancy at highrisk of producing an affected child. Theresults of this analysis predicted that thechild would be affected. As predictions ofthe severity of the disease were not pos-sible, the pregnancy was also assessed byultrasound imaging. This confirmed theaffected diagnosis and predicted that thechild would be severely affected.(J7Med Genet 1996;33:603-606)
Key words: Treacher Collins syndrome; human chro-mosome 5; prenatal diagnosis.
School of BiologicalSciences andDepartments of DentalMedicine and Surgery,3.239, StopfordBuilding, University ofManchester,Manchester M13 9PT,UKS J EdwardsM J Dixon
Department ofObstetrics andGynaecology, DerbyCity General Hospital,Derby, UKA FowlieM P Cust
Department ofObstetrics andGynaecology,City Hospital,Nottingham, UKD T Y Liu
Department ofClinical Genetics,City Hospital,Nottingham, UKI D Young
Correspondence to:Dr Dixon.
Received 6 December 1995Revised version acceptedfor publication21 February 1996
Treacher Collins syndrome (TCOF1) affectsapproximately 1 in 50000 live births and isinherited in an autosomal dominant fashion.Forty percent of affected subjects have a pre-vious family history, the remaining 60% of casesarising as a result of a de novo mutation. Theclinical characteristics of the disease include(1) hypoplasia of the mandible and zygomaticcomplex; (2) abnormalities of the pinnae, oftenassociated with atresia of the external auditorycanals and anomalies of the middle ear ossiclesleading to a conductive hearing loss'; (3) down-ward slanting palpebral fissures with colobo-mata of the lower eyelids; (4) cleft palate.While the clinical features are generally bi-laterally symmetrical, expression of the mut-ated gene is highly variable. At one extremethe features can be so mild that it may bedifficult to reach a diagnosis.2 At the otherextreme the facial complex may be so severelyhypoplastic that perinatal death ensues as a
result of compromisation of the airway. Rarely,non-penetrance may occur,2 although in thevast majority of cases where this is suspectedcareful examination of the obligate carrier willshow minor stigmata of the disorder.3As part of the continuing attempts to isolate
the mutated gene, TCOF1 has been mappedto 5q31.3-32"8 and a high resolution geneticmap of short tandem repeat polymorphisms(STRPs) encompassing the disease locus hasbeen produced.9 All the families that have beenanalysed to date (approximately 50) supportlinkage of the disease locus to markers in thesame region of the genome, with none showing
unequivocal evidence of non-linkage.-'0 Thesedata support genetic homogeneity.
Postnatal diagnostic predictions have beenmade in mildly affected, and apparently un-affected, subjects using linked STRPs.2However, prenatal diagnosis has only been per-formed in families with a history of TCOF1using either fetoscopy" or ultrasound imagingin the second trimester.'2'3 The procedure re-lated fetal mortality rate for fetoscopy is low(approximately 2%) and is acceptable for themajority of patients with a high recurrencerisk.'4 While the quality of ultrasound imaginghas improved markedly in recent years, al-lowing non-invasive prenatal diagnosis to bemade, it can still be difficult to make a positivediagnosis where the fetus is mildly affected.Prenatal diagnosis using either fetoscopy orultrasound imaging is not possible until thesecond trimester of pregnancy (approximately18 weeks). At this time termination of preg-nancy is a particularly traumatic procedurepsychologically as it involves the induction oflabour. First trimester prenatal diagnosis wouldtherefore seem to be preferable, particularly ifthe family feel that termination of pregnancyis desirable in the event that the fetus is affected.Nevertheless, chorionic villus sampling conveysthe twin disadvantages of a miscarriage riskand a lack ofinformation about disease severity,which must be discussed with the parents be-fore any invasive testing. Consequently, parentswho opt for chorionic villus sampling, andsubsequently receive an unfavourable result,may then quite understandably defer their de-cision pending the outcome of detailed ultra-sound scanning. Even then the decision can bean extremely difficult one, as illustrated by thisfirst report ofthe prenatal diagnosis ofTreacherCollins syndrome using molecular methods.
Patients and methodsPATIENTSThe relevant part of the family pedigree isshown in fig 1. The pregnancy at risk is in-dicated as II-3. The father (I2) and half sister(II 1) are relatively mildly affected with mod-erate maxillary and mandibular hypoplasia,lower eyelid colobomata, small ears, and nar-row external auditory meati. The mother's pre-vious pregnancy resulted in the prematuredelivery of a severely affected male infant (II-2),who died at the age of 4 weeks. This child hadsevere micrognathia with a small mouth andcleft palate, these being factors which con-tributed to severe respiratory problems to which
603
on 26 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.33.7.603 on 1 July 1996. D
ownloaded from
Edwards, Fowlie, Cust, Liu, Young, Dixon
I -2
1 23 14 33 22 32 32 2
/ 2
3
11422113411211
11
1
113144143132412~121
3
1 23 1414 2131112142 j12 1
Order ofprobes
(1) D5S372(2) CSF1R(3) RPS14(4) D5S519(5) ANX6(6) SPARC(7) D5S378
Figure 1 The family pedigree. The STRP haplotype linked to the disease is indicated on
the left and is boxed.
he eventually succumbed. Venous bloodsamples were taken from the family memberswith informed consent.
DNA ANALYSISGenomic DNA was extracted from peripheralblood leucocytes or chorionic villus samplesusing standard procedures and PCR amplified
--1 in 5 p1l reaction volumes containing 4 ng gen-
; omic DNA; 10 pmol of each primer, 200 ,umol/1 each of dCTP, dGTP, dTTP, and 25 imol/lof dATP (Pharmacia); 2 jiCi 35S-dATP at 500
D5S372 Ci/mmol (NEN); 10 mmol/l Tris-HCl pH 8 3,50mmol/I KC1, 1 mmol/l MgCl2, and 0-01%gelatin. The samples were overlaid with mineral
12 11 11 11 oil, heated to 96°C for 10 minutes, and cooledto 55°C. After addition of 0-15 U Taq DNA
CSF 1 R polymerase, the samples were processed
13 24 34 34 through 35 amplification cycles of 92°C for 30
seconds, primer annealing temperature (table)I;-+ for 30 seconds, 72°C for 30 seconds using a
Hybaid thermal cycler. The final extension stepRPS14 was lengthened to 10 minutes. Negative con-
trols were established for all reactions. Theamplified products were extracted once with
34 12 44 24 chloroform, 2 tl was mixed with an equal vol-ume of formamide loading buffer, heated to
80°C, and the alleles resolved on a 6% de-D5S519 .^ naturing polyacrylamide gel. The gels were
23 13 33 13
SPARC
23 12 12 12
Figure 2 Results offive ofthe seven STRPs typed inthe family. The genotypesare indicated below eachSTRP
Short tandem repeat polymorphisms used in the currentstudyProbe Locus Heterozygosity Reference
N5.61 D5S372 0-75 9CSF1R CSF1R 0-86 21RPS14 RPS14 0-66 9IG90-3 D5S519 0-82 6p68 ANX6 0-72 22SPARC SPARC 0-80 62G10 D5S378 0-75 23
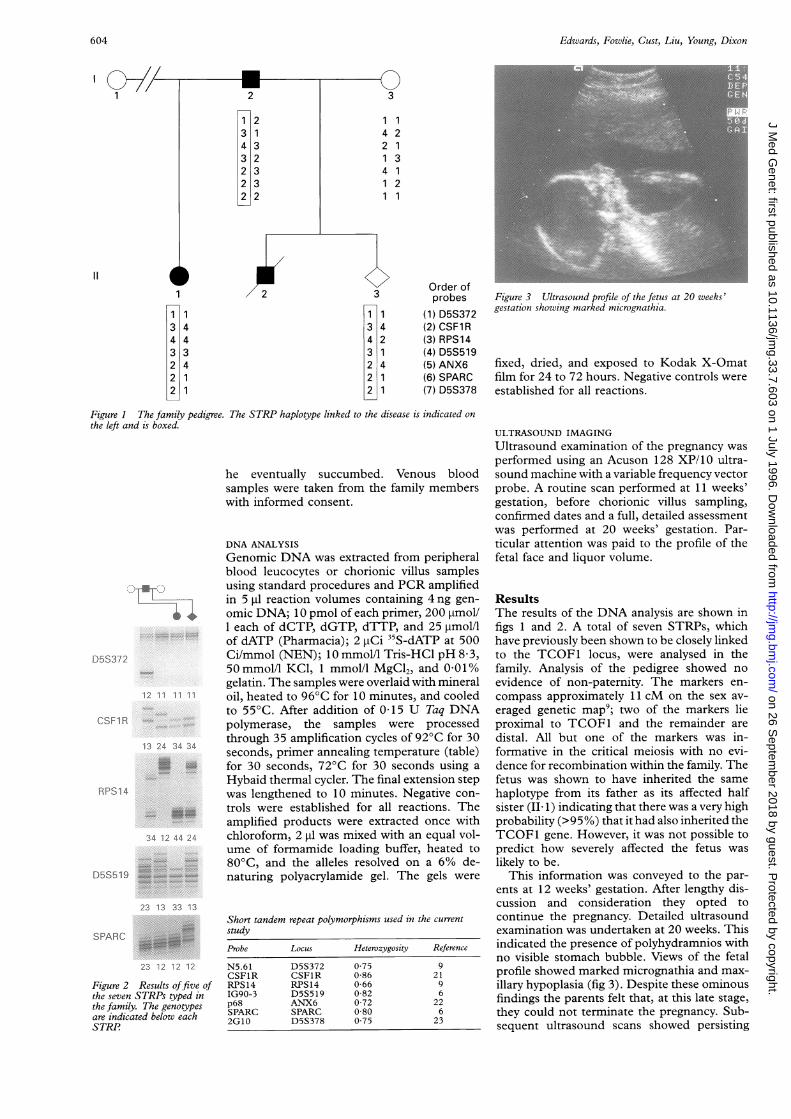
Figure 3 Ultrasound profile of the fetus at 20 weeks'gestation showing marked micrognathia.
fixed, dried, and exposed to Kodak X-Omatfilm for 24 to 72 hours. Negative controls wereestablished for all reactions.
ULTRASOUND IMAGINGUltrasound examination of the pregnancy wasperformed using an Acuson 128 XP/10 ultra-sound machine with a variable frequency vectorprobe. A routine scan performed at 11 weeks'gestation, before chorionic villus sampling,confirmed dates and a full, detailed assessmentwas performed at 20 weeks' gestation. Par-ticular attention was paid to the profile of thefetal face and liquor volume.
ResultsThe results of the DNA analysis are shown infigs 1 and 2. A total of seven STRPs, whichhave previously been shown to be closely linkedto the TCOF1 locus, were analysed in thefamily. Analysis of the pedigree showed noevidence of non-paternity. The markers en-compass approximately 11 cM on the sex av-eraged genetic map9; two of the markers lieproximal to TCOF1 and the remainder aredistal. All but one of the markers was in-formative in the critical meiosis with no evi-dence for recombination within the family. Thefetus was shown to have inherited the samehaplotype from its father as its affected halfsister (II- 1) indicating that there was a very highprobability (>95%) that it had also inherited theTCOF1 gene. However, it was not possible topredict how severely affected the fetus waslikely to be.
This information was conveyed to the par-ents at 12 weeks' gestation. After lengthy dis-cussion and consideration they opted tocontinue the pregnancy. Detailed ultrasoundexamination was undertaken at 20 weeks. Thisindicated the presence of polyhydramnios withno visible stomach bubble. Views of the fetalprofile showed marked micrognathia and max-illary hypoplasia (fig 3). Despite these ominousfindings the parents felt that, at this late stage,they could not terminate the pregnancy. Sub-sequent ultrasound scans showed persisting
604
on 26 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.33.7.603 on 1 July 1996. D
ownloaded from

Prenatal diagnosis in Treacher Collins syndrome
Figure 4 The baby in the immediately postnatal period.She shows severe features of Treacher Collins syndromeincluding mandibular and zygomatic hypoplasia,downward slanting palpebral fissures, and severeanomalies of the external ears. (Photograph published withparental consent.)
polyhydramnios, with a stomach bubble notappearing until 30 weeks' gestation.Spontaneous onset of labour occurred at 36
weeks' gestation when the baby, a female giventhe name Emily, was delivered by caesareansection with birth weight 2510g. The infantwas in good condition at birth but showedobvious severe features of Treacher Collinssyndrome (fig 4). Despite the presence of ex-perienced paediatric and anaesthetic staff, thebaby could not be intubated and could notmaintain her own airway. Death was confirmedat the age of 30 minutes.
DiscussionIn the current investigation, seven STRPs havebeen used to make first trimester prenatal diag-nostic predictions in a family with a history ofTreacher Collins syndrome. The highly in-formative nature of STRPs"5 makes them ex-cellent markers for use in prenatal diagnosis,where it is important to maximise the amountof linkage information that can be extractedfrom the family under investigation, par-ticularly where the pedigree structure is notideal. This was the case in the current studywhere all but one of the markers was in-formative in the critical meiosis, despite thefact that the father had remarried and his firstwife was not available for investigation. Thefact that STRPs are formatted for use with thePCR is also important given that only a limitedamount of CVS may be available for analysis.Moreover, application of the PCR to type poly-morphic markers is faster than standard blot-
ting and hybridisation. In addition, the abilityto use this technique to amplify several differentmarkers simultaneously means that it is possibleto type a large number of markers in a relativelyshort time. Nevertheless, STRPs have beenshown to exhibit a relatively high mutation rate,particularly in DNA samples extracted fromtransformed lymphoblastoid cell lines.'6 How-ever, this is unlikely to present a problem wheremultiple markers are used to analyse DNAfreshly extracted from either blood or a CVS.
Ideally, diagnostic predictions of the typeundertaken in the present study should onlybe performed in families showing significantevidence of linkage to markers in the region of5q31.3-32 or when the possibility of hetero-geneity has been further minimised by the studyof additional families. In this regard TCOF1has been associated with a number of differentchromosomal anomalies: two apparently bal-anced translocations, t(6;16)(p21 .31;p13. 1 1)3and t(5;13)(q1l;pl1),1' and two interstitialdeletions, del(4)(p15.32pl4)18 and del(3)(p23p24.12)19 which raise the possibility thatthe disorder may be heterogeneous. However,in each of these cases, linkage analysis with aseries of familial cases of well documentedTCOF1 families failed to show cosegregationwith markers for the relevant region. Moreover,the chromosome 6 translocation did not ul-timately completely cosegregate with the dis-ease phenotype,3 while in the remaining casesthe facial gestalt of the patients did not entirelyconform to the TCOF1 clinical criteria. Fur-thermore, while genetic heterogeneity inTCOF1 can not be excluded, all of the 50families that have been analysed to date supportlinkage of the disease locus to the region en-compassed by the markers used in the currentstudy with none showing unequivocal evidenceof non-linkage.`7-0These parents specifically requested prenatal
diagnosis because of their previous experienceof losing a severely affected child in the peri-natal period. They had been fully informedabout the risks and limitations of a first tri-mester prenatal diagnosis and were aware thatthis would not indicate the extent to which ababy with the "high risk" haplotype would beaffected. When informed of the CVS results,they decided, quite understandably, to defer adecision in the hope that subsequent detailedultrasound scanning would show a relativelynormal facial profile. Although this proved notto be so, the parents made the agonising de-cision to continue the pregnancy in what provedto be a forlorn hope that the baby's respiratoryproblems would not be overwhelming.
This unhappy experience illustrates the di-lemma faced by all parents who embark uponprenatal diagnosis in a much wanted pregnancy.It is not surprising that attitudes to terminationcan alter as the pregnancy proceeds, despiteaccumulating evidence for a potentially un-happy outcome. To insist on a predeterminedcourse of action given an adverse result wouldbe totally contradictory to the principle ofpatient autonomy. It would be ideal if non-invasive ultrasound scanning could provide anearly prediction of disease severity, but despite
605
on 26 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.33.7.603 on 1 July 1996. D
ownloaded from

Edwards, Fowlie, Cust, Liu, Young, Dixon
continuing improvement in ultrasound defin-ition, this is unlikely within the near future. Therecent identification of the Treacher Collinssyndrome gene" may lead to a better under-standing of the relationship between genotypeand phenotype, but it is likely to be a con-
siderable time before molecular analysis shedslight on the elusive mechanism underlying thewell documented intrafamilial variation in se-
verity which is seen in many families with a
history of Treacher Collins syndrome.
We should like to thank the family concerned for their permissionto publish the results of this study, and the photograph of Emily,and all ofthe Treacher Collins syndrome families without whosehelp the study would not have been possible. The financialsupport of the Wellcome Trust (grant numbers 036797/Z/92/Zand 044684/Z/95/Z), the Hearing Research Trust (grant num-bers 073:MAN:MD and 150:MAN:MD), and the IndependentOrder of Odd Fellows is gratefully acknowledged.
1 Phelps PD, Poswillo D, Lloyd GAS. The ear deformitiesin mandibulofacial dysostosis. Clin Otolaryngol 1981;6:15-28.
2 Dixon MJ, Marres HAM, Edwards SJ, Dixon J, CremersCWRJ. Treacher Collins syndrome: correlation betweenclinical and genetic linkage studies. Clin Dysmorphol 1994;3:96-103.
3 Dixon MJ, Haan E, Baker E, et al. Association of TreacherCollins syndrome and translocation 6p2l.31/16pl3.11:exclusion of the locus from these candidate regions. AmJf Hum Genet 1991;48:274-80.
4 Dixon MJ, Read AP, Donnai D, Colley A, Dixon J, Wil-liamson R. The gene for Treacher Collins syndrome mapsto the long arm of chromosome 5. Am Hum Genet 199 1;49:17-22.
5 Dixon MJ, Dixon J, Raskova D, et al. Genetic and physicalmapping of the Treacher Collins syndrome locus. Re-finement of the localization to chromosome 5q32-33.2.Hum Mol Genet 1992;1:249-53.
6 Dixon MJ, Dixon J, Houseal T, et al. Narrowing the positionof the Treacher Collins syndrome locus to a small intervalbetween three new microsatellite markers at 5q32-33. 1.Am _7 Hum Genet 1993;52:907-14.
7 Jabs EW, Li X, Coss CA, Taylor EW, Meyers DA, WeberJL. Mapping the Treacher Collins syndrome locus to5q31.3-q33.3. Genomics 1991;11:193-8.
8 Jabs EW, Li X, Lovett M, et al. Genetic and physicalmapping of the Treacher Collins syndrome locus withrespect to loci in the chromosome 5q3 region. Genomics1993;18:7-13.
9 Loftus SK, Edwards SJ, Scherpbier-Heddema T, BuetowKH, Wasmuth JJ, Dixon MJ. A combined genetic andradiation hybrid map surrounding the Treacher Collinssyndrome locus on chromosome 5q. Hum Mol Genet 1993;2:1785-92.
10 Edery P, Manach Y, Le Merrer M, et al. Apparent genetichomogeneity of the Treacher Collins-Franceshetti syn-drome. Am Jt Med Genet 1994;52:174-7.
11 Nicolaides KH, Johansson D, Donnai D, Rodeck CH.Prenatal diagnosis of mandibulofacial dysostosis. PrenatDiagn 1984;4:201-5.
12 Meizner I, Carmi R, Katz M. Prenatal ultrasonic diagnosisof mandibulofacial dysostosis (Treacher Collins syn-drome). J Clin Ultrasound 199 1;19:124-7.
13 Milligan DA, Harlass FE, DuffP, Kopelman JN. Recurrenceof Treacher Collins syndrome with sonographic findings.Mil Med 1994;159:250-2.
14 Rodeck CH, Nicolaides KH. Fetoscopy and fetal tissuesampling. Br Med Bull 1983;39:332-7.
15 Weber JL. Informativeness of human (dC-dA)n.(dG.dT)npolymorphisms. Genomics 1990;7:524-30.
16 Weber JL, Wong C. Mutation of human short tandemrepeats. Hum Mol Genet 1993;2:1123-8.
17 Balestrazzi P, Baeteman MA, Mattei MG, Mattei JF. France-shetti syndrome in a child with a de novo balanced trans-location (5;13)(ql l;pl 1) and significant decrease ofhexosaminidase B. Hum Genet 1983;64:305-8.
18 Jabs EW, Coss CA, Hayflick SJ, et al. Chromosomal deletion4pl5.32-p14 in a Treacher Collins syndrome patient:exclusion of the disease locus from and mapping of an-onymous DNA sequences to this region. Genomics 1991;11:188-92.
19 Am PH, Mankinen C, Jabs EW. Mild mandibulofacialdysostosis in a child with a deletion of 3p. Am J7 MedGenet 1993;46:534-6.
20 The Treacher Collins Syndrome Collaborative Group. Po-sitional cloning of a gene involved in the pathogenesis ofTreacher Collins syndrome. Nature Genet 1996;12:130-6.
21 Polymeropoulos MH, Xiao H, Rath DS, Merril CR. Di-nucleotide repeat polymorphism at the human c-fmsprotooncogene for the CFS-1 receptor (CFS1R). NucleicAcids Res 1991;19:1160.
22 Loftus SK, Dixon J, Koprivnikar K, Dixon MJ, WasmuthJJ. Transcriptional map of the Treacher Collins candidategene region. Genome Res 1996;6:26-34.
23 Ryan SG, Dixon MJ, Nigro MA, et al. Genetic and radiationhybrid mapping of the hyperekplexia region on chro-mosome 5q. Am J Hum Genet 1992;51:1334-43.
606
on 26 Septem
ber 2018 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.33.7.603 on 1 July 1996. D
ownloaded from