Embed Size (px)
Citation preview

I966 H . H . Inhoffen, R. Jonas, H . Krosche und U. Eder 19
Untersuchungen an hochsubstituierten Athylenen und Glykolen, IV 1)
Darstellung von hochsubstituierten Athylenen mit Hilfe der Staudinger-Pfenninger-Reak tion
von Hans Herloff Inhoffen*), Rochus Jonas, Henning Krosche und Ulrich Eder
Aus dem Institut fur Organische Chemie der Technischen Hochschule Braunschweig
Eingegangen am 20. Oktober 1965
Die Darstellung hochsubstituierter Athylene gelang durch Anwendung der Staudinger- Pfenninger-Reaktion: Ketonhydrazone werden rnit HgO zu Diazoverbindungen dehydriert, diese rnit SO2 in Thiadiazoline (3) iibergefuhrt und letztere durch Pyrolyse unter Abspaltung von SO2 und Nz in tetrasubstituierte Athylene umgewandelt.
Die Darstellung des tetrasubstituierten Athylens 1 d, des 3.4-Bis-4-[oxo-cyclohexyl]- hexens-(3), gelang uns nach zwei Methoden : 1) Aus 3.4-Bis-[4.4-athylendioxy-cyclo- hexyl]-hexadiin-(l.5)-diol-(3.4) durch Kuhn-Winterstein-Reaktion 2) rnit anschliekn-
R' i?
k' la-g
R' CaH5; R
b R' = CzH,; R
+ d: R' C2H5; R =
Id
der Hydrierung und Ketalspaltung; 2) aus 3.4-Bis-[4.4-athylendioxy-cyclohexyl]- hexandioL(3.4) durch Wasserabspaltung rnit Tic13 und nachfolgender partieller Re- duktionl).
*) Vlado Prelog in freundschaftlicher Verbundenheit zum 60. Geburtstag gewidmet. 1) 111. Mitteilung: H. H. Inhofen, K . Radscheit und H . Dettmer, Liebigs Ann. Chem. 692,
2) H. H . Inhoffen, K . Radscheit, U. Stache und V . Koppe, Liebigs Ann. Chem. 684,24 (1965). 66 (1966).
2'

20 H . H . Inhoffen, R . Jonas, H. Kr5.whe und U. Eder Bd. 694
Z u r Darstel lung weiterer te t rasubst i tuier ter Athylene (1 a - g) h a b e n wir e inen zusatzlichen W e g beschritten, narnlich eine Synthese d u r c h sfaudinger-Pfenninger- Reakfiod) .
Die Staudinger-Pfenninger-Reaktion Diese interessante Reakt ion besteht aus 3 Stufen : I ) Dehydr ie rung eines Hydrazons
(2) rnit gelbern HgO z u r Diazoverbindung, 2) Urnsetzung d e r Diazoverb indung rnit SO2 zu einem Thiadiazol in (3) und 3) thermische Zersetzung des Thiadiazol ins unter Abspal tung von SO2 u n d N2 zurn Athylenderivat .
Die Struktur der intermediar gebildeten Thiadiazoline, die in der hydroaromatischen Reihe recht bestandig sind, ist noch nicht eindeutig geklart. Smuditiger und fjenninger-’) formulierten die Zwischenprodukte ohne nahere Begriindung als I . I-Dioxo-A2- I .2.3-thiadiazoline (4), wahrend Hesse und Reichold4a) aufgrund der beim lungsumen Erwlrmen unter Schwefeldioxid- abspaltung erfolgenden Azin-Riickbildung die Entstehung eines I . I -Dioxo-A3-1-3.4-thiadia- zolins (3) vorschlugen. In ubereinstimmung mit dem Strukturvorschlag von Hesse und Reichold4a) mochten wir den Bildungsmechanismus des Thiadiazolins 3 folgendermaflen formulieren : I ) Addition von Schwefeldioxid an die Diazoverbindung unter Eliminierung von Stickstoff und Bildung des Sulfens4b). 2) Umsetzung des Sulfens mit einer weiteren Molekel Diazoverbindung in einer 1.3-dipolaren Additon nach Huisge115). aus der das Thiadiazolin 3 resultiert.
* ) Die Reste R und R’ haben in den Formeln 2 - 5 die gleiche Bedeutung wie bei l a - g . 3) H. Sraudinger und F . ffenninger, Ber. dtsch. chem. Ges. 49, 1941 (1916). 4) 4a) G. Hesse und E . Reirhold, Chem. Ber. 90, 2101 (1957). -- 4b) G. Hesse, E . Reichold und
S . Mujmudur, ebenda 90, 2106 (1957). 5 ) R. Huisgen, Angew. Chem. 75, 604 (1963).

1966 Untersuchungen an hochsubstituierten Athylenen und Glykolen, IV 21
Synthese der hochsubstituierten Athylene la---g Hesse und Reichold4") erzielten bei der Darstellung des 3.4-Bis-cyclohexyl-hexens-(3)
(1 a ) nach Staudinger-Pfenninger nur eine Ausbeute von 8 %. Wir suchten deshalb zunachst nach optimalen Reaktiorisbedingungeti fur die entscheidende Stufe, namlich die Pyrolyse der Thiadiazoline, wobei wir als Testreaktion die Synthese des 3.4-IBis- cyclohexen-(3)-yl-( I)]-hexens-(3) (1 b) aus [Cyclohexen-(2)-yl-( I)]-athyl-keton be- nutzten. Das Hexen 1 b konnte durch katalytische Hydrierung zu 1 a identifiziert werden 4a).
Zunachst wurde das Ketonhydrazon 2b nach Hesse und Reichold4') rnit Queck- silber(l1)-oxid zur Diazoverbindung dehydriert. Andere Oxydationsmittel [MnOz, Pb(OAc)4, PbOz] oder HgO in flussigern Schwefeldioxid eigneten sich nicht. Eine Dirnerisierung des durch therrnische Zersetzung der Diazoverbindung gebildeten Carbens, wie sie beirn Diazoessigester beschrieben wurdes), konnte nicht nachgewiesen werden. Durch Aufblasen oder vorsichtiges Einleiten von SO2-Gas unter starkem Ruhren bei -50" wurde I-[Cyclohexen-(3)-yl-( 1)J-1 -diazo-propan in das entsprechende Thiadiazolin 3 b ubergefuhrt, von dern zwei isornere Formen rnit identischen IR- und UV-Spektren isoliert wurden. Moglicherweise liegen cisltrans-Isornere vor, wie es schon bei dern analogen Sulfon 3a angenomrnen wurde4". Die therrnische Zer- setzung der Isorneren ergab jeweils den gleichen Kohlenwasserstoff 1 b. Die Aus- beute an Thiadiazolin betrug ca. 26%, jedoch nur bei genauer Einhaltung der Re- aktionsbedingungen.
Die Pyrolyse des Thiadiazolins, von Hrsse und Reichold4") durch Eintauchen in ein 250" heiBes Metallbad ausgefuhrt, konnten wir wesentlich verbessern. Hierzu spruhten wir die benzolische Losung in ein stufenlos beheizbares Metallrohr ein, das mit Kupfer oder Kupfer-Quarz als Katalysator gefullt war. Die Ausbeute an 1 b erhohte sich rnit steigender Ternperatur und abnehrnender Konzentration und erreichte bei 500-550" und 0.5-proz. Losung ein Maximum von 26 04. Durch Strornungspyrolyse IPBt sich also eine nennenswerte Steigerung der Ausbeute erreichen, wodurch die Staudinger- Pfenninger-Reaktion fur die Synthese syrnmetrischer hochsubstituierter Athylene interessant wird.
Das fur den Aufbau von 1 d benotigte [4.4-Athylendioxy-cyclohexyl]-athyl-keton (5c) lieB sich durch Glykolspaltung von 3.4-Bis-[4.4-athyIendioxy-cyclohexyl]- hexandioL(3.4) in 95-proz. Ausbeute darstellen. Die Urnsetzung von 5 c rnit Hydrazin- hydrat lieferte das kristalline Ketazin 6c, das mit absol. Hydrazin im Bornbenrohr das Hydrazon 2c in 85-proz. Ausbeute ergab.
5a- g 6b.c.t 6 ) A . Loose, J . prakt. Chem. ['I 79, 505 (1909).
22 H. H. Inhofen, R. Jonas, H. Krosche und U. Eder Bd. 694
Nach Dehydrierung mit HgO in siedendem k h e r wurde auf die Losung der Diazo- verbindung bei -50" ein kraftiger Schwefeldioxid-Strom geblasen: Unter heftiger Stickstoff-Entwicklung trat Entfarbung ein. Das aus dem Rohprodukt isolierte Thia- diazolin 3c vom Schmp. 185 - 186" zeigte in seinem IR-Spektrum die fur die Sulfon- Gruppierung charakteristischen Banden bei 1300 und 11 50 cm-1. Die Ketalspaltung mit athanolischer Schwefelsaure ergab das freie Keton der Forrnel 3d. Eine Isomeren- Trennung war wegen der Zersetzlichkeit der Substanz nicht moglich. Die Stromungs- pyrolyse lieferte neben Ketazin das Athylen I d in 27-proz. Ausbeute. Der Struktur- beweis wurde durch Mischprobe und durch Ozonabbau zum [4-Oxo-cyclohexyl]- athyl-keton (5d) erbracht. Das cis-konfigurierte Athylen I d 1) wurde nicht aufge- funden.
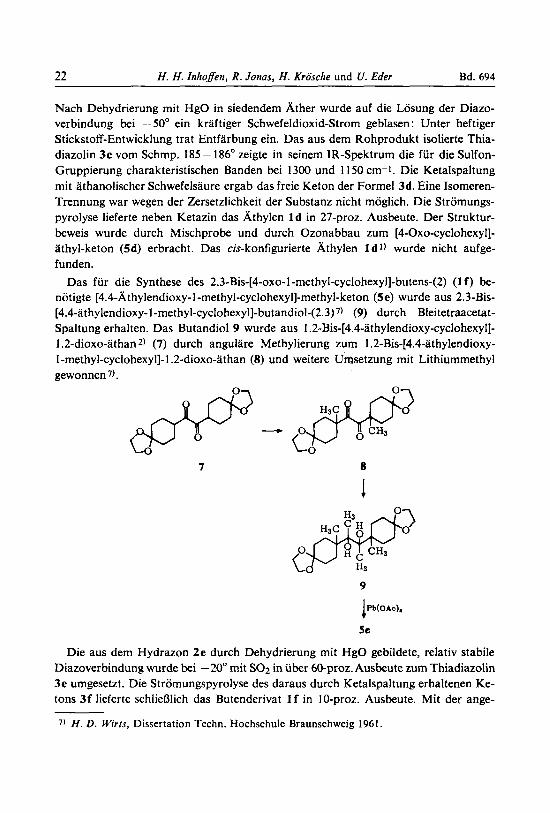
Das fur die Synthese des 2.3-Bis-[4-oxo-l -methyl-~yclohexyl]-butens-(2) (1 f ) be- notigte [4.4-~thylendioxy-1-methyl-cyclohexyl]-methyl-keton (5e) wurde aus 2.3-Bis- [4.4-athylendioxy-l-rnethyl-cyclohexyl]-butandiol-(2.3~7~ (9) durch Bleitetraacetat- Spaltung erhalten. Das Butandiol 9 wurde aus 1.2-Bis-[4.4-athylendioxy-cyclohexyl]- 1.2-dioxo-athan 2) (7) durch angulare Methylierung zum 1.2-Bis-[4.4-athylendioxy- 1 -methyl-cyclohexyl]-1.2-dioxo-athan (8) und weitere Umsetzung mit Lithiummethyl gewonnen 7).
7 8
1
9
1Pb(O*o),
5e
Die aus dem Hydrazon 2e durch Dehydrierung mit HgO gebildete, relativ stabile Diazoverbindung wurde bei -20" rnit SO2 in uber 60-proz. Ausbeute zum Thiadiazolin 3e umgesetzt. Die Stromungspyrolyse des daraus durch Ketalspaltung erhaltenen Ke- tons 3f lieferte schliefllich das Butenderivat 1 f in 10-proz. Ausbeute. Mit der ange-
7) H . D. Wir/s, Dissertation Techn. Hochschule Braunschweig 1961.

1966 Untersuchungen an hochsubstituierten Athylenen und Glykolen, IV 23
gebenen Struktur sind das IR- und vor allem das NMR-Spektrum in Einklang, eine Mischprobe mit einer auf unabhangigem Wege synthetisierten Probe ergab keine Depression. Fur die Konfiguration an der zentralen Doppelbindung in I f ist in Analogie zu dem Ergebnis der thermischen Zersetzung des Thiadiazolins 3d die trans- Anordnung sehr wahrscheinlich.
Uber die raumliche Anordnung der Atome in dem hhylen 1 f wird die Rontgenstruktur- analyse Auskunft geben.
Aus [I -MethyI-~yclohexen-(2)-yl-( I)]-methyl-keton, das sich aus I-Acetyl-cyclohexen durch angulare Methylierung 8) gewinnen lieB, konnte 2.3-Bis-[ 1 -methyl-cyclohexen- (2)-yl-(I)]-buten-(2) (1 g) nach dem gleichen Verfahren in nur 2-proz. Ausbeute er- halten werden. Seine Abtrennung aus dem oligen Rohprodukt bereitete g r o k Schwie- rigkeiten. Seine Struktur wurde durch Massenspektrum und NMR-Spektrum belegt. Das NMR-Spektrum bestatigt die Lage der Ringdoppelbindungen in l g durch ein AB-Spektrum der Vinyl-Protonen bei T = 4.4 (J = 10.5 Hz) und eine Aufspaltung des bei hoherem Feld liegenden Dubletts zu je einem Triplett (J = 2.2 Hz). Die Protonen der an der mittleren Doppelbindung stehenden Methylgruppen bei T = 8.33 und die Protonen der angularen Methylgruppen bei 7 = 8.87 erscheinen als Singulett.
Im Rahmen ihrer Untersuchung iiber sterisch behinderten, bivalenten Kohlenstoff beob- achteten Zimmerman und Puskovichg) bei der Bestrahlung von Dimesityl-diazomethan die quantitative Dimerisierung des Dimesityl-carbens zum Tetramesityl-athylen. Dies veranlabte uns, 1-[4.4-Athylendioxy-1-methyl-cyclohexyl]-diazoathan unter den gleichen Bedingungen zu bestrahlen. Allerdings war bei der Ubertragung auf das hydroaromatische System von vornherein mit einer intramolekularen Absattigung des Carbens zu rechnen. Tatsachlich fiihrte die Bestrahlung in n-Hexan nur in sehr geringer Ausbeute (0.5 %) zum Athylen le , das diinnschichtchromatographisch nachgewiesen wurde. Das Hauptprodukt konnte als 6e identifiziert werden. - Auch bei der Bestrahlung des Thiadiazolins 3 e konnte das Athylen l e (< 0.5 %) nur diinnschichtchromatographisch neben dem Ketazin 6e als Haupt- produkt nachgewiesen werden.
AbschlieBend sol1 noch kurz eine Methode zur Einfuhrung von Doppelbindungen in die Cyclohexanon-Ringe erwahnt werden. Zur Darstellung des Trien-diketons der Formel 12 aus I f wurde letzteres in das Bis-enolacetat 10 iibergefuhrt, dieses bromiert und aus dern Bis-[a-bromketon] 11 mit LiBr + Li2CO3 in Dimethylformamid in iiber 70-proz. Ausbeiite HBr abgespalten. Das Bromketon 11 konnte durch Bro- mierung des Bis-enolacetats 10 mit Brom bei -20" gewonnen werden. Die quantitative Umsetzung des k h y l e n s 1 f zum Bis-enolacetat I0 erforderte langere Reaktionszeiten (3 Tage). Bei der chromatographischen Abtrennung trat ein teilweiser Zerfall ein, so daB sich nur eine maximale Ausbeute an 12 von 28 % erzielen lieB.
8) Nach einer Vorschrift von J. A . Burlrrop und A . C . Duy, Tetrahedron [London] 14, 310
9 ) H. E. Zimmerman und D. H. Paskovich, J. h e r . chem. SOC. 86, 2149 (1964). (1961).

24 H . H. Inhofen, R . Jonas, H . Krosche und U. Eder Bd. 694
B r A -0Ac --
10 Br
11
12
Der Stt'ftung Volkswugenwerk und der Deutschen Forscliungsgemeinschaft sowie den Farb- werken Hoechst und der Badischen Anilin- & Soda-Fabrik sei fur die stete Forderung unserer Arbeiten ergebenst gedankt.
Beschreibung der Versuche
Die Schmelzpunkte sind unkorrigiert und wurden auf dem Kofler-Mikroheiztisch RCH der Fa. Reichert, die Mischproben im Paraffinbad bestimmt. - Die IR-Spekrren wurden teils in CHCI,, teils in KBr rnit dem Gerat Beckman IR 5, die UV-Spektren rnit einem Beckman- Spektralphotometer, Modell DU, gemessen. - Die NMR-Spekrretr wurden mit den Spektro- photometern Varian A 60 oder H A 100 in CDCI3 (Tetramethylsilan als interner Standard) aufgenommen. - Molekulnrgew,ichte wurden rnit dem Gerat MS 9 der Fa. AEI bestimmt. Fur die Aufnahme und Deutung der N M R - und Massen-Spektren danken wir den Herren Professor H . H. Perkampus, Dr. G. Foster und Dr. L. Pohl sowie Dr. J . Buchler und cand. chem. v. d. Haar. - Die Analysen wurden durch A . Bernhardr, Mulheim/Ruhr, ausgefuhrt. - Zur Saulenchromatographie wurde neutrales Aluminiumoxid der Fa. Merck verwendet. - Ubliche Aufurbeirung heiBt : Ausschutteln mil einem organischen Losungsmittel, Neutral- waschen der organischen Phase rnit NaHCOj-Losung und rnit Wasser bei sauren, nur rnit Wasser bei alkalischen Reaktionsmischungen.
Hydrazon 2b. - 50 g [ C y c l o h e x e ~ t - ( 3 / - y l - ( I j ! - a t l i y l - k ~ ~ 0 1 1 ~ ~ ~ (5b) und 12.2 g 100-proz. Hydrazitthydrat in 35 ccm absol. Athanol wurden 20 Stdn. unter RiickfluB gekocht. Hierauf wurde i. Vak. abgedampft und der Ruckstand i . Hochvak. destilliert. I . Fraktion: 12 g (22%) Hydrazon 2 b vom Sdp.o.5 88-90", v(NH2) = 3370-3375 cm-1; 2. Fraktion: 35.2 g (72%) Kerazin 6 b vom Sdp.o.2 I17 - 120". A,,, == 235 mp (E = - 3500). Die erneute Umsetzung von 6 b mit Hydrnzinhydrar ergab in 78-proz. Ausbeute 2b.
Thindiazolin 3b. - 20 g Hydrnzon Zb in 300 ccm Petrolather (40- 80") wurden unter Nz bei -5" unter Ruhren rnit 2 ccm I n methanol. Kalilauge und 7 0 g gelbem Queck-
10) J . Doucet und P . Rumpf, Bull. SOC. chim. France 1954, 610.

I966 Untersuchungen an hochsubstituierten Athylenen und Glykolen, IV 25
silber(II)-oxid portionsweise versetzt; nach 2 -3 Stdn. war die Dehydrierung beendet. Die orangerote Losung der Diazoivrbindung filtrierte man iiber Na2S04 in einen auf -50" ge- kiihlten Kolben. Auf die Oberflache der Losung wurde ein kraftiger SOz-Strom geblasen; unter Nz-Entwicklung trat Entfarbung ein. Das Losungsmittel wurde bei Raumtemperatur i . Vak. abgesaugt und der ohge Riickstand aus Methanol/Benzol (3 : I ) durch llngeres Stehen bei - 20" zur Kristallisation gebracht. Die fraktionierte Kristallisation ergab 2 lsomere: I ) 4.9 g vom Schmp. 177- 178" und 2) 0.6 g vom Schmp. 144- 145". - IR-Spektrrrm: 1295 bis 1300, 1136- 1156 cm--l (Sulfon). - UV-Spektrrtm (in CHzC12): A,,, = 369 m p ( E = 165). Die 1R- und UV-Spektren beider lsomeren waren identisch.
ClsHz8Nz02.5 (336.4) Ber. C 64.26 H 8.39 N 8.33 S 9.51 Gef. 64.82 8.70 8.12 9.01
3.4-Bis-[cyclohexen-(3;-yl-(l) j-hexen-(3) (1 b). - a) Durch Pyrolyse nach Hesse find Reichold4a): 2 g Thiadiazoliri 3 b wurden in vier Portionen von 500 mg mit je 100 mg Kirpferpulwr gut vermischt und in einem 100 ccm Rundkolben mit aufgesetztem Steigrohr durch Eintauchen in ein Metallbad von 250" zersetzt. Es entstand eine griinliche Schmelze, nach einigen Sekunden setzte heftige Gasentwicklung ein und ein farbloses 01 destillierte hoch. Nach dem Abkiihlen wurde mehrmals mit Petrolather ausgekocht, die vereinigten Petrolather- ausziige wurden auf 3 ccm eingeengt und a n Aluminiumoxid chromatographiert. Beim Eluieren mit Petrolather erhielt man folgende Fraktionen: 1 ) 303 mg farbloses 01, 2) 30 mg farbloses 0 1 und 3) 9 m g farbloses 0 1 . Mit Ather wurden noch 412 mg braunes 01 eluiert. Die erste Fraktion kristallisierte fast vollig durch: Aus Methanol 21 I mg (14.5 7 ; ) 1 b, farblose Prismen vom Schmp. 84". - Das zweite Isomere 3 b ergab bei der thermischen Zersetzung ebenfalls 1 b.
C I ~ H ? ~ (244.4) Ber. C 88.45 H 11.55 Gef. C 88.48 H 11.34
Die katalytische Hydrierung von 1 b mit PdiKohle in Athanol lieferte in 96-proz. Ausbeute 3.4-Bis-cyclohexyl-hexen-(3)4a) vom Schmp. 76" (Mischprobe) 1 1 ) .
b) Durch Strumurigspyrolyse: Die Pyrolyse-Apparatur bestand aus einem stufenlos elektrisch heizbaren Messingrohr (C :- 3 cm, Llnge: 40cm), das mit dem Katalysator (ca. 200 g Kupfer oder Kupfer/Quarz) gefiillt war. Bei 50 - 100 Torr wurde das geloste Thiadiazolin durch eine Diise in das mit Nz gespiilte Metallrohr eingesaugt. Die Reaktionsprodukte wurden unterhalb des senkrecht stehenden Heizrohres in einer auf -~ 20" gekiihlten Vorlage kondensiert. Die Temperatur im Kontaktrohr wurde mit einem Thermoelement gemessen. - Zur Durchfiihrung der Zersetzung wurde zunachst Benzol mit der fur die Zersetzung vorge- sehenen Geschwindigkeit solange durch die Apparatur geleitet, bis sich die Temperatur nicht mehr anderte. Dann wurde eine henzol. Lusung des Thiridicrzolins durch das Rohr gespriiht und anschlieBend mit Benzol nachgespiilt. Die in der Kiihlfalle aufgefangene Losung wurde i. Vak. eingeengt, der Ruckstand in Petrolather an Aluminiumoxid chromatographiert (s. 0.) und aus Methanol umkristallisiert. Die eingesetzten Mengen a n Thiadiazolin betrugen 600 mg (I-proz. Losung) und 300 mg (0.5- bzw. 0.25-proz. Losung). Einen Uberblick iiber Reaktions- bedingungen und die Ausbeuten gibt Tabelle 1.
1 1 ) Herrn Professor G . Hesse danken wir fur das Vergleichsprlparat.

26 H. H. Inhoffen, R . Jonas, H . Krosche und U. Eder Bd. 694
Tabelle 1 Reaktionsbedingungen und Ausbeuten an 1 b bei der Stromungspyrolyse ~~ -
Konzentration Reaktionsdauer Temperatur Ausbeute an 1 b
1 % 10 Min 350"/450" 10.0/18.0~~ ___. ~ ~ - - - -- --
0.5 5 .5 420"/490"/590" 21.2/25.4/24.3 1 1.5/5/10 400" 4.711 3.7118.0 0.5 519 450" 16.5121.9
I /0.5/0.25 5 400" 13.7/21.2/22 110.5 5 500" 18.0125.8
[4.4-Arhylendioxy-cyclohexylJ-afhyl-keton (5c). - Zu 10 g 3.4-Bis-[4.4-arhylendioxy-cyclo- hexylJ-hexandiol-(3.4) 2 ) in 250 ccm absol. Benzol gab man unter Schiitteln portionsweise 1 I g Bleirerruacetat. Nach 2 Stdn. Schiitteln wurde abfiltriert, die benzol. Losung rnit NaHCO3- Losung neutralisiert, getrocknet und eingedampft. Das erhaltene 61 wurde i. Hochvak. destilliert: 9.1 g (91 %) vom Sdp.o.os 95-98". - IR-Banden bei 1709 und 1100 cm-1.
[4-Oxo-cyclohexyl]-arhyl-keron (5d). - 1 g Keron 5 c in I5 ccrn Athanol, 20 ccm Wasser und 4 Tropfen konz. Schwefelsaure wurden kurz unter RiickfluB gekocht. Nach dem Auf- arbeiten rnit Ather verblieben 716 mg farbloses 61. - IR-Bande bei 1705--1710cm-1. 2.4-Dinirro-phenylhydrazon: Schmp. 160- 162".
CzlH22N808 (514.5) Ber. C49.04 H4.31 N 21.79 Gef. C49.44 H4.69 N 20.73
Keruzin 6c. - 5 g Keron 5 c in 5 ccm Athanol und I .8 ccm 100-proz. Hydrazinhydraf ergaben nach 8stdg. Kochen neben sehr wenig Hydrazon 2c 4.2 g (84 %) Ketazin 6 c vom Schmp. 89". - IR-Bande bei 1630cm-I. - UV-Spektrum (CHIOH): A,,, = 234my (E = 4000).
C2zHlsN204 (392.5) Ber. C67.31 H9.24 N 7.14 Gef. C 67.45 H9.07 N 7.38
Hydrazon 2c. - 5 g Kerazin 6 c wurden rnit 1.3 ccm absol. Hydrazin 5 Stdn. im Bombenrohr auf 130" erwarmt. Nach Abziehen des iiberschiissigen Hydrazins wurde i. Hochvak. destilliert. 4.2 g ( 8 5 % ) vom Sdp.1 130- 135" (teilweise Zers.). - IR-Bande bei 3370-3380 cm-1.
Thiadiazolin 3c. - 5 g Hydrazon 2c in 125 ccm absol. Ather wurden nach Zugabe von 1 ccrn 0.5-proz. methanol. Kaliluuge und 0.2 ccm Triufhylamin rnit 20 g gelbem Queck- silber(II)-oxid bei 35" dehydriert (Dauer : 45 Min.). Nach Umsetzung der tiefroten Losung rnit SO2 (S. 25) wurde das erhaltene gelbliche 61 nach Zusatz von 100 ccm Aceton bei -20" stehengelassen: 0.94 g (19.5%) 3 c vom Schmp. 185-186". - IR-Banden bei 1295 bis 1300, 1136-1156 (Sulfon) und l l00cm-I.
CzzH36N206S (456.3) Ber. C 57.88 H 7.95 N 6.14 S 7.01 Gef. 57.70 7.54 6.47 7.41
Thiadiazolin 3d. - 4.5 g oliges Kerul 3 c in 50ccm Methanol wurden nach Zusatz von 50 ccm 2-proz. Schwefelsuure 5 Min. gekocht. Nach iiblicher Aufarbeitung erhielt man 3.8 g gelbes 61. Nach Zusatz von 100 ccm Ather kristallisierten 1.2 g 3d vom Schmp. 217-219" (Zers.) aus. - IR-Banden bei 1709, 1295-1300, 1136--1156cm-~.
C18H28Nr04S (368.5) Ber. C 58.67 H 7.66 N 7.60 S 8.70 Gef. 58.60 7.94 7.52 8.48

I966 Untersuchungen an hochsubstituierten Athylenen und Glykolen, IV 27
3.4-Bis-[4-oxo-cyclohexyll-hexen-(3) (1 a). - Die Stromungspyrolyse (S. 25) von I .02 g 3d (0.5-proz. Losung, 550°, 40 ccm/Min.) lieferte nach Chromatographie in Benzol an Alu- miniumoxid 201 mg Id vom Schmp. 199-200"2) (aus Ather).
1.2-Bis-[4.4-afhylendioxy-l-merhyl-cyclohexyl]-l,2-dioxo-afl1an~~ (8). - 200 g a-Diketon 72) in 2 labsol. tert.-Butanol wurden mit 2.4 I 1 m Kalium-ferf.-bufylat versetzt (Temperatur 10 bis 20"). Unter kraftigem Riihren tropfte man 240 ccm Methybodid im Laufe 1 Stde. ein. Nach 2stdg. Riihren wurde das gleiche Volumen Benzol hinzugefiigt und i. Vak. (Bad: 50") abge- dampft. Den Riickstand zersetzte man mit gesatt. NhCl-Losung, nahm mit 2 I Benzol auf, trennte die organische Phase ab und extrahierte den walk. Anteil mehrmals rnit Benzol. Die vereinigten Benzollosungen wurden rnit einer 5-proz. Na&O3-Losung geschiittelt, neutral gewaschen, iiber NazS04 getrocknet und eingedampft. Der gelbe Riickstand lieferte aus Methanol 155 g (69 %) Kristalle vom Schmp. 105 - 106". - IR-Banden bei 1695, I 1 0 0 cm-1. - UV-Spekfrum: 368-372 (19.7), 287 (53.8) m p (E).
CzoH3o06 (366.4) Ber. C 65.55 H 8.25 Gef. C 65.45 H 8.25
2.3-Bis-[4.4-afhylendioxy-l-mefhyl-cyclohexyl]-bufandiol-(2.3) 7) (9). - In die Suspension von 60 g Lithium-Granular in 2.7 I absol. Ather wurde unter Wasserkiihlung und Riihren bis zur vollstandigen Umsetzung des Lithiums Merhylbromid eingeleitet. Bei Raumtemperatur tropfte man 60 g Dikefon 8 in 1 I Benzol innerhalb von 20 Min. zu. Es wurde 2 Stdn. geriihrt und nach 12 Stdn. vorsichtig bei -20" mit gesatt. NH4CI-LOsurig zersetzt. Nach Zusatz von 2 I Essigester wurde aufgearbeitet und aus Methanol umkristallisiert: 61.8 g (95 %) vom Schmp. 152- 153". - IR-Banden bei 3400, 1100 cm-1.
Ber. C 66.30 H 9.61 CzzH3806 (398.5) Gef. C 66.70 H 9.72
[4.4-Afhylendioxy-1-mefhyl-cyclohexyl]-mefhyl-kefon (5e). - 60 g Diol 9 in 1.8 I Benzol wurden unter Riihren rnit 70 g Bleiferraacefaf versetzt und 2 Stdn. kraftig geriihrt. Das nach dem Aufarbeiten erhaltene farblose 81 wurde i. Hochvak. destilfiert: 57 g (95 %) vom Sdp.o.3 100". - IR-Banden bei 1701, IlOOcm-*.
Kefalspalfung: Diese ergab in 89-proz. Ausbeute [4-0xo-1-rnethyl-cyclohexyl]-methyl-keton (5f) vom Sdp.3 100". - IR-Bande bei 1702 cm-1. - 2.4-Dinifro-phenylhydrazon: Schmp. 21 1-212".
Hydrazon Ze. - 50 g Keton 5e in 110 ccm absol. Athanol wurden rnit 40 g 100-proz. Hydrazinhydraf 3 Stdn. gekocht. Man destillierte das Losungsmittel i. Vak. ab und nahm in 1.2 I Methylenchlorid auf. Nach Stehenlassen iiber N a ~ S 0 4 wurde abdestilliert. Das resul- tierende bl lieferte aus Ather 52 g (97%) Kristalle vom Schmp. 78-79". - IR-Banden bei 3378, 3257 cm-1 (NHz). - NMR-Spekfrum: i = 5.1 (2 H, Multiplett, NHz), 6.9 (4 H, Singu- lett, Ketal), 8.29 (3 H, Singulett), 8.95 ppm (3 H, Singulett, angulares CH3).
CllHzoNrOz (212.3) Ber. C62.63 H9.50 N 13.20 Gef. C62.45 H 9.47 N 12.37
Thiadiazolin 3e. - 5 g Hydrazon Ze, gelost in l2Occm Petrolather (40-60"), wurden mit 30 g Quecksilber(II)-oxid in der Siedehitze dehydriert (S. 25) (Dauer: 10 Min.). Auf die filtrierte, tiefrote Losung wurde bei -20" unter Riihren ein sehr langsamer SOz-Sfrom geleitet. Ohne merkliche Stickstoff-Entwicklung trat innerhalb von 20 Min. Entfarbung ein. Danach wurde bei Raumtemperatur i. Vak. abgedampft. Aus Methylenchlorid/Ather kristallisierten

28 H. H. Inhoffen, R . Jonas, H. Krosche und U. Eder Bd. 694
3 4 g (63 ":) 3e mit einem Schmelzbereich von 21 5 235". Die Umkristallisation ergab keine Verbesserung des Schmelzpunkts. -- IR-Burrden bei 1285, 1130 (Sulfon), I100cm-1.
C Z ~ H ~ ~ N ~ O ~ S (456.6) Ber. C 57.87 H 7.95 N 6.14 S 7.02 Gef. 57.48 7.95 6.00 6.58
Der Ruckstand der Mutterlauge lieferte aus Aceton/Ather in wechselnder Ausbeute I -[4.4-Athylendioxy- I -methyl-cyclohexyl]-athansulfons~uremethylester vom Schmp. 68 - 69". IR-Spektrum: 1351, 1 I70 (Sulfonsaureester), 1 100 c m 1. - NMR-Spektrum: 5 - 6.08 (4 H, Singulett, Ketal), 6.15 (3 H , Singulett, OCH,), 6.75 ( I H , Quartett, -CHCH,), 8.58 (3 H, Dublett, CH,CH), 8.82 ppm ( 3 H, Singulett, CH,C). - - Mossenspektrirm: Molmasse 278, intensive Fragmente: 248/249/155/99.
ClzH220SS (278.1) Ber. C 51.60 H 7.96 Gef. C 51.13 H 7.57
Thiodiazolin 3f. - - Zu 7 g 3e, gelost in 200ccm Methylenchlorid -r I I Aceton, gab man in der Siedehitze 50ccm Wasser und 20ccm 9n H z S 0 4 . Die klare Losung wurde I5 Min. gekocht. Nach dem Aufarbeiten erhielt man ein 01, das aus Aceton kristallisierte: 5.08 g (80%) vom Schmp. 195 -197". - IR-Botrden bei 1718, 1287, I140cm 1. - - U V - Spektrrrm (CHzC12): - = 370 (170). 285 (48) m p (E).
C I ~ H ~ ~ N ~ O ~ S (368.5) Ber. C 58.69 H 7.65 N 7.60 s 8.69 Gef. 58.71 7.76 7.51 8.87
2.3-Bis-~4-oxo-I-merl1yl-cyclol1exyli-br1tet1-~2) ( 1 f ) . - 25 g 3f wurden durch Stromungs- pyrolyse (vgl. S. 25) zersetzt (0.5-proz. Losung, 520--550, 100 ccm/3 Min.). Nach Abdestillieren des Benzols i . Vak. erhielt man 6.7 g braunes 0 1 , das von leichtfliichtigen Bestandteilen bei lOO'/O. 1 Torr befreit wurde. Der Ruckstand (5.82 g ) wurde an 600 g Alu- miniumoxid chromatographiert. Nach mehreren Vorfraktionen (insgesamt I .7 g ) eluierte man mit Benzol/Essigester ( 1 9 : l ) 1.99 g (10.7'4) I f vom Schmp. 138-140 (aus Ather, Misch- probe12)). - IR-Bun& bei 1712 cm-1. - NMR-Spekrrrrm: 5 - 7.3-7.9 (12 H , Multiplett), 8.12 (6 H , Singulett), 8.0-8.5 ( 4 H, Multiplett), 8.76 ppm (6 H, Singulett).
C18H2802 (276.4) Ber. C 78.2 H 10.21 Gef. C 77.96 H 10.07
-'.3-Bis-~4-crceroxy-I-ttrerhyl-cycluhexetr-(3)-yl-(I) ;-briten-/2) (10). -- Zu I g 1 f in 70 ccm I.sopropenylacetrrt gab man eine Spatelspitze p-Tolvolsrrljottsiirrre und erhitzte 6 Tage zum Sieden (Wasserabscheider). Man erhielt nach dem Aufarbeiten I .6 g dunkelbraunes 01, das a n 75 g Kieselgel chromatographiert wurde. Nach einigen Vorfraktionen (mit Benzol/Petrol- ither-Gemischen) wurde das Bis-enolacetat 10 (mit Benzol) eluiert. Die Ausbeute an diinn- schichtchromatographisch einheitlichem Produkt betrug 330 mg (28 7;). Aus Ather bei - 80' weiBe Kristalle voni Schrnp. 60 ~ 62". -- IR-Bunden bei 1748, 1225, 1684 c m I .
C ~ ~ H Q O J (360.5) Ber. C 73.30 H 8.95 Gef. C 72.71 H 8.95
Bromierirrrg: 305 mg 10 in 40 ccm Methylenchlorid versetzte man bei - 30" unter Ruhren mit 8.1 ccm einer Losung von 0.5 ccm Brom in 50 ccm Methylenchlorid (augenblickliche Ent- farbung). Nach Neutralwaschen mit Wasser wurde getrocknet, filtriert und abgedampft: 375 mg helles 0 1 (11) mit einer IR-Bande bei 1727cm 1.
12) Unabhlngige Synthese der Vergleichsprobe s. Lit.]).

I966 Untersuchungen an hochsubstituierten Athylenen und Glykolen, 1V 29
2.3-Bis-iI-uxo-I-niethyl-rycluhe.ren-i2)-.~1-( I)i-hirtrn-(2) (12). .- 375 mg I1 in 50 ccm Dimethylformamid wurden mit 300 mg LiBr -1 270 mg LizC03 versetzt, 1 Stde. leicht er- warmt und I Stde. zum Sieden erhitzt. Nach 40 Stdn. bei Raumtemperatur wurde in Me- thylenchlorid aufgenommen, mit Wasser mehrmals gewaschen, getrocknet und filtriert. Nach vorsichtigem Abdampfen i. Hochvak. blieben 240 mg eines teilweise kristallisierenden 0 1 s zuriick (A,,, = 230 mp, E = 15400). Es wurde mit Benzol/Methylenchlorid iiber 1 5 g Alu- miniumoxid (Aktiv.-Stufe I I ) filtriert: 165 mg (il, das aus Ather kristallisierte; Schmp. I17 bis 122". - IR-Brrndetr hei 1669, 1605 cm-1. -- UV-Spektruni (Methanol): A,,, = 232 mp, E = 23500. - NMR-Spekrrunrt 7 = 2.97 ( 2 H, Dublett), 4.16 (2 H, Dublett), 7.4-8.1 (8 H, Multiplett), 8.22 (3 H, Singulett), 8.28 (3 H , Singulett), 8.65 ppm (6 H, Singulett). - M~issenspekrrirni;Molmasse 272 (Ber. 272.2). Intensive Fragmente hei 257, 163 und 109.
C 1 8 H 2 4 0 2 (272.2) Ber. C 79.37 H 8.88 Gef. C 78.15 H 8.71
Hvdrazon 2g. - 8 g 5g in 20 ccm absol. Athanol und 12 ccm Hydrazinhydrat wurden 2 Stdn. gekocht. Die Aufarbeitung ergab 8.4 g (95 :<) farbloses 0 1 , das i. Vak. nur unter Zersetzung destilliert werden konnte. Sdp.1 78 --82". - IR-Btrnden bei 3170, 3320 cm-1.
Thiadiazolin 3g. - 5 g Hydrcrzon 2g, in 80ccm Petrolither (40-60") gelost, wurden mit 30 g gelbem Qitacksilher~ll)-oxid bei Siedetemperatur dehydriert (Dauer: 10 Min.). Nach Abfiltrieren, Abkiihlen und Uberleiten von SO*-Gus bei -20" wurde aufgearbeitet (S. 25). Das erhaltene 0 1 wurde durch Zugabe von SO ccm Methanol zur Kristallisation gebracht. Die fraktionierte Kristallisation ergab 2 lsomere: I ) I .4 g vom Schmp. 128.- 130"; IR-Bnnden bei I 150, I300 cm 1 ; U V-Spektrum (CHzC12) : ).mar = 364 m p (E = 163).
Cl8H28NzOzS (336.1) Ber. c 64.35 H 8.34 N 8.34 s 9.51 Gef. 65.01 8.42 8.1 I 9.28
2) 0.154 g vom Schmp. 103- 105". 1R-spektroskoprsch identisch mit der I . Fraktion. Gef. 64.35 8.39 8.27 9.73
2.3-Bis-~I-nrcrhyl-cyclohexeri-~~)-yl-( I),!-hurert-i2/ ( 1 g). -. 3.9 g Sulfun 3g wurden der Stromungspyrolyse (S. 25) unterworfen (0.5-proz. Losung, 510 -530", 100 ccm/3.Min.). Es wurde i. Vak. zuerst bei ca. 20", dann i. Hochvak. bei 45" abgedampft. Das restliche 01 wurde in 10 ccm Petrollther aufgenommen und an I50 g Aluminiumoxid chromatographiert. Die I . Fraktion ergab 410 mg hellgelbes 0 1 , das in der Hitze in 30 ccm Methanol gelost wurde. Nach llngerem Stehen bei -20" erhielt man 59 mg (2.1 y,;) l g vom Schmp. 33-34". - -
IR-Bundebei 1640cm-I. - NMR-Spekrrum:~ = 4 . 4 ( 4 H, =CH-- ) ,8 .03 (4H,C=C-CH?) , 8.3 ( 14 H), 8.87 ppm (6 H, angullre CH3-Gruppen). - Massenspekrrrini: Molmasse 244 (CI 8H28).
I-[4.4-Athylendioxy- I-methyl-cyclohexy1,'-diazoarho,i. - Eine Losung der aus dem Hydra- zun 2 e mit HgO erhaltenen Diazoverbindung zeigte IR-Banden bei 2041, 1085, 1033 cm 1 und ein A,,, = 491 m y (n-Hexan). Beim llngeren Stehenlassen in Losung lieferte die Diazo- verbindung das stabilere Ketazin 6e, das durch Umkristallisieren aus Ather/Petrollther rein isoliert werden konnte; Schmp. 75-77', - IR-Bunden bei 1621, 1085, 1033 cm--l . -- U V - Spekrrum (Methanol): A,,, = 230 mp, E = 3050.
CzrH36Nz04 (392.5) Ber. C 67.40 H 9.18 N 7.30 Gef. C 67.43 H 9.10 N 7.30

30 H . H. Inhofen, R. Jonas, H . Krosche und U. Eder Bd. 694
Besrrahlung: Die Appararur bestand aus einer Quecksilber-Hochdrucklampe Q 8 1 der Quarz- lampengesellschaft Hanau, die in einen mit Luft durchspiilten Quarzmantel tauchte, der iiber einen 55er Schliff in ein 300 ccm fassendes BestrahlungsgefaB eingefiihrt wurde. Durch eine Glasfritte am Boden des GefaBes wurde Stickstoff eingeblasen, der gleichzeitig eine gute Durchmischung der Losung bewirkte. Zwei seitlich angebrachte 14.5er Schliffe ermoglichten den Anbau eines Dimrothkiihlers und eines Thermometers. - Ausfiihrung: Die durch De- hydrierung von I2 g Hydrazon 2e in l5Occm n-Hexan hergestellte tiefrote Losung der Diazoverbindung wurde in das BestrahlungsgefaB eingefiillt und bei - 30" unter Stickstoff bis zur vollstandigen Entfarbung bestrahlt ( 3 0 Stdn.). Das Losungsmittel wurde i. Vak. bei Raumtemperatur abdestilliert. Das erhaltene bl (9.7 g) wurde i. Hochvak. bei 90"/0.1 Torr destilliert : 7.05 g farbloses 01 gingen iiber und I .42 g gelbes bl blieben zuriick. 1.4g des oligen Riickstandes wurden an 40 g Aluminiumoxid (Aktiv.-St. 11) in Benzol chrornatographiert. Nach einem Vorlauf von 0.35 g wurden mit Benzol/Essigester (9 : I ) 650 mg Kerazin 6 e vom Schmp. 75 --77" und A,,, = 230 m p (E = 3000) isoliert. Mit Chloroform konnten noch 2 3 0 mg sehr polare Substanzen von unbekannter Struktur eluiert werden. l e konnte diinn- schichtchromatographisch nur in Spuren nachgewiesen werden.
Bestrahlung des Thiadiazolins 3 e mir UV-Lichr: Eine Losung von 800 mg 3 e in l5Occm absol. Tetrahydrofuran wurde bei Raumtemperatur solange bestrahlt (siehe voranstehende Vorschrift), bis irn IR-Spektrum keine Sulfon-Banden mehr vorhanden waren (2 Stdn.). Es trat ein starker Geruch nach H2S auf. Nach dem Abdestillieren des Losungsmittels erhielt man 600 mg helles 01, das an 20 g Aluminiumoxid (Aktiv.-St. 11) chromatographiert wurde. Die mittleren Fraktionen ergaben ein teilweise kristallisierendes 61, das nach dem UV- Spektrum zu 70% aus dem Kerazin 6 e und nach dem Diinnschichtchromatogramm aus Ketazin 6 e und sehr wenig k'rhylen l e bestand. [ 1871651





























