Embed Size (px)
Citation preview

Physikalische Chemie IV
Biophysikalische Chemie
Stefan Seeger
Skript: Michael Rankl
Physikalisch-Chemisches Institut der Universität Zürich
SS 2002

___________________________________________________________________________ 2
Einführung:
Allgemein beschreibt die biophysikalische Chemie die physikalisch-chemischen Grundlagen
biologischer Prozesse und die Methoden, die in den Biowissenschaften Anwendung finden.
Ziel dabei ist eine umfassende Beschreibung der Lebensvorgänge auf der Grundlage
molekularer und supramolekularer Strukturen und Wechselwirkungen. Unter molekularen
Strukturen mit spezifischen Funktionen und Aufgaben versteht man insbesondere biologische
Makromoleküle, die sogenannten Biopolymere. Supramolekulare Strukturen sind
entscheidend für die Definition eines Organismus, dessen Kompartimentierung und die
mechanischen Eigenschaften. Als Beispiel seien Zellwände, das Cytoskelett und
Biomembranen als strukturbildende Elemente von Zellen erwähnt. Im Rahmen dieser
Vorlesung beschränken wir uns deshalb auf die molekulare Struktur und die Dynamik von
Biopolymeren, Methoden zur Analyse der Struktur und Funktion biologisch relevanter
Moleküle und deren Wechselwirkungen sowie supramolekulare Strukturen wie z.B.
Biomembranen.
1. Biopolymere
1.1 Was sind Biopolymere?
Als Polymere bezeichnet man allgemein chemische Verbindungen, die durch Verknüpfung
von mehreren Einzelmolekülen als Grundbausteinen oder Monomeren entstehen. Häufig
werden Polymere aus weniger als 20 Monomeren als Oligomere bezeichnet. Die Monomer-
Einheiten können entweder alle identisch (Homopolymere) oder aber verschieden
(Copolymere) voneinander sein. Periodisch sich wiederholende Molekülgruppen aus zwei
oder mehreren Grundbausteinen bezeichnet man als Strukturelemente.
Zu den wichtigsten Biopolymeren gehören:
- Polypeptide (Proteine)
- Polysaccharide
- Polynukleotide (Nukleinsäuren)
- Polyisoprene
- Lignin

___________________________________________________________________________ 3
Hiervon stellen die Polyisoprene im allgemeinen Homopolymere dar. Ebenso sind die auch
mengenmässig sehr bedeutenden Polysaccharide Cellulose und Stärke im wesentlichen aus
einem einzigen Grundbaustein, der D-Glucose, aufgebaut. Polypeptide und Proteine können
aus bis zu etwa 20 verschiedenen Grundbausteinen, den L-Aminosäuren, bestehen, während
die Nukleinsäuren aus mehreren Strukturelementen, den Nucleotiden zusammengesetzt sind.
Bei Polymeren, die nur einen Grundbaustein enthalten, werden die Eigenschaften im
wesentlichen durch drei Parameter beeinflusst, und zwar:
- durch die Art der Verknüpfung der Monomere
- durch die Zahl der Grundbausteine pro Molekül, d.h. Polymerisationsgrad bzw.
Molekülmasse
- durch Verzweigung bzw. Vernetzung linearer Ketten
Homopolymere dienen in der Natur im allgemeinen als Gerüst- und Speichersubstanzen, so
dass ihre vergleichsweise geringe Variabilität von Struktur und Eigenschaften kein Nachteil
ist.
Bei Makromolekülen, die aus mehreren Grundbausteinen bestehen, sind die Eigenschaften
zusätzlich noch von der Reihenfolge, der Sequenz, der Monomereinheiten abhängig. Daraus
folgt, dass man bei den Proteinen mit der grössten Zahl verschiedener Grundbausteine (~20)
auch die grösste Variationsmöglichkeit findet.
1.2 Polyisoprene: Kautschuk, Guttapercha, Balata
Der Name Kautschuk entstammt der indianischen Tupi-Sprache und bedeutet soviel wie
„weinender Baum“, da er in dem Milchsaft von Bäumen enthalten ist, so dass im Prinzip unter
Kautschuk das Naturprodukt zu verstehen ist.
Chemisch handelt es sich beim Kautschuk um 1,4 cis-Polyisopren. Die 1,4 trans-Polyisoprene
kommen als Guttapercha und Balata ebenfalls im Milchsaft verschiedener Pflanzen vor.
Kautschuk, 1,4-cis-Polyisopren
1,4-trans-Polyisopren,Guttapercha, Balata

___________________________________________________________________________ 4
1.3 Lignin
Wie der Name andeutet (lat.: lignum = Holz) wird Lignin bei der Verholzung pflanzlicher
Gewebe in Interzellularspalten und interfibrillären Zwischenräumen gebildet. Es wird in allen
höheren Pflanzen einschliesslich der Farne gefunden. Lignin kann man als die druckfeste
Matrix des Holzes betrachten, in die die zugfesten Cellulosefibrillen eingelagert sind. Das
Lignin ist kovalent mit Cellulose und Hemicellulosen verbunden und wird von Pflanzen in
grossen Mengen synthetisiert. Es handelt sich bei Lignin im wesentlichen um dreidimensional
vernetzte Mischpolymerisate von Phenylpropan-Derivaten. Grundkörper ist z.B. beim
Fichtenholzlignin nahezu ausschliesslich der Coniferylalkohol (4-Hydroxy-3-methoxy-
zimtalkohol):
OHH
CH2OH
OMe
1.4 Grösse und Form der Biopolymere
Obwohl viele Gesetze der Physikalischen Chemie für Biopolymere genauso gelten wie für
kleine Moleküle, treten bei den Makromolekülen spezielle Fragestellungen und Probleme auf.
Dazu gehören auch die Bestimmung ihrer Grösse, Form und der mittleren Molekülmassen.
Die Bestimmung der Molekülmasse wird häufig dadurch erschwert, dass die Probe aus
Molekülen mit verschiedenen Massen bestehen kann. Ein reines Protein ist beispielsweise
eine definierte Substanz mit einer einheitlichen Molmasse. Die Massenverteilung nennt man
dann monodispers. Ein synthetisches Polymeres ist dagegen eine Mischung aus Molekülen
verschiedener Kettenlänge und damit verschiedenen Molekülmassen. Man spricht dann von
einem polydispersen System. Je nach Verfahren der Molekülmassenbestimmung kann es
dabei zu verschiedenen Mittelwertbildungen kommen.

___________________________________________________________________________ 5
- Osmometrie
Aus dem osmotischen Druck erhält man die über die Teilchenzahl gemittelte relative
Molekülmasse NM (zahlengewichtete mittlere Molmasse). Man nimmt an es seien Ni
Moleküle mit der relativen Molekülmasse Mr,i vorhanden, und insgesamt seien es N
Moleküle. Dann ergibt sich die über die Teilchenzahl gemittelte relative Molekülmasse durch
Addition aller relativen Molekülmassen, jeweils gewichtet mit dem Faktor NNi , der angibt,
welcher Bruchteil aller Moleküle gerade die entsprechende Molekülmasse hat:
- Viskosimetrie
Aus viskosimetrischen Messungen erhält man die viskositätsgewichtete mittlere Molmasse
VM .
- Lichstreuung
Mit der Methode der Lichtstreuung ermittelte Molmassen nennt man massengewichtete
mittlere Molmassen MM .
- Sedimentation
Sedimentationsexperimente führen zur sogenannten Z-gewichteten Molmasse ZM .
2. Kolligative und hydrodynamische Eigenschaften von Biopolymeren
2.1 Osmose
Die klassischen Methoden zur Bestimmung der relativen Molekülmassen beruhen auf ihren
kolligativen Eigenschaften, d.h. die Eigenschaften der Lösung hängen nur von der Anzahl der
Teilchen des gelösten Stoffes und nicht deren Art ab. Bei Makromolekülen ist die Anzahl der
Moleküle in einer Lösung in der Regel sehr klein, auch wenn gewichtsmässig hohe
Konzentrationen vorliegen. Deshalb ist lediglich die Osmometrie noch genügend empfindlich.
Das Phänomen der Osmose ist das Bestreben eines Lösungsmittels, durch eine semipermeable
Membran in eine Lösung hineinzuwandern. Im Versuch (siehe Abbildung 2-1) bewirkt die
Osmose, dass Wasser in die Lösung hineinfliesst. Man kann die Richtung des Flusses
umkehren, wenn man auf die Lösung Druck ausübt. Der osmotische Druck Π ist der Druck,
der diesen Fluss gerade zum Stillstand bringt. In Abbildung 2-1b erkennt man, wie die

___________________________________________________________________________ 6
Osmose die Lösung ansteigen lässt und so selbst einen Gegendruck erzeugt. Das
Gleichgewicht ist erreicht, wenn der hydrostatische Druck der Lösung gerade gleich dem
osmotischen Druck ist. Das Problem in diesem Versuchsaufbau ist die Verdünnung der
Lösung durch einströmendes Lösungsmittel.
Abbildung 2-1: Osmose
Zur thermodynamischen Betrachtung der Osmose betrachtet man den Aufbau in Abbildung 2-
1. Im Gleichgewicht muss das chemische Potential des Lösungsmittels auf beiden Seiten der
Membran gleich Null sein. Auf der Seite des reinen Lösungsmittels ist sein chemisches
Potential , auf der anderen Seite ist es erniedrigt aufgrund der Anwesenheit der
gelösten Substanz und erhöht aufgrund des höheren Druckes. Im Gleichgewicht lässt sich
deshalb schreiben:
)(* pAµ
),()(* Π+= pxp AAA µµ
Unter Berücksichtigung der gelösten Substanz wird daraus:
)ln()(),( *AAAA xRTppx +Π+=Π+ µµ
und mit der Druckabhängigkeit des chemischen Potentials erhält man:
∫Π+
+=Π+p
pMAA dpVpp )()( ** µµ

___________________________________________________________________________ 7
Aus diesen drei Gleichungen ergibt sich dann:
∫Π+
=−p
pMA dpVxRT ln
Diese Gleichung beschreibt die Abhängigkeit des osmotischen Drucks vom Molenbruch der
gelösten Substanz (xB = 1- xA). Für stark verdünnte Lösungen, d.h. xA → 1, kann man ln xA
durch ln(1-xB) ≈ -xB ersetzen. Setzt man noch voraus, dass der Druck sich im
Integrationsbereich nicht wesentlich verändert, kann man das Molvolumen VM des
Lösungsmittels als konstant ansehen und man bekommt:
MB VRTx ∏=
Der Molenbruch ist definitionsgemäss )( BAB nnn + , was für eine verdünnte Lösung
praktisch dasselbe ist wie AB nn . Das Volumen des Lösungsmittels ist V . AM nV=
Damit vereinfacht sich obige Gleichung zu:
RTnV B=∏
Diese Formel nennt man die van’t Hoffsche Gleichung.
Schreibt man ][BVB =n für die Konzentration der gelösten Substanz, so kann man die van’t
Hoffsche Gleichung einfacher darstellen als:
RTB][=∏
Diese Gleichung gilt nur für verdünnte Lösungen die sich ideal Verhalten. Für
Makromoleküle ist die van’t Hoffsche Gleichung nur das Anfangsglied einer
Virialentwicklung
...][1][ ++=∏ BRTB β
bei der die weiteren Glieder die Abweichungen vom idealen Verhalten beschreiben.

___________________________________________________________________________ 8
Die Konzentration des Makromolekül [B] hängt über [B]=cp/Mm von der
Massenkonzentration cp ab, wobei Mm die Molmasse des Polymers ist.
Die van’t Hoffsche Gleichung wird damit zu:
...)/(1)/( ++=∏ pmmp cMMRTc β
Wenn man pc∏ gegen aufträgt und gegen =0 extrapoliert, erhält man aus dem
Achsenabschnitt die Molmasse M
pc pc
m und der Steigung der Kurve den osmotischen
Virialkoeffizienten β.
Lösungen von Makromolekülen weichen stark von idealem Verhalten ab. Das liegt zum Teil
daran, dass die grossen Moleküle jeweils eine grosse Lösungsmittelmenge verdrängen,
während kleine Moleküle nur einzelne Lösungsmittelmoleküle ersetzen und damit die
Struktur der Flüssigkeit viel weniger stören. Ein gelöstes Makromolekül kann sich aber auch
viel weniger frei bewegen als ein kleines Molekül, weil der Raum, der von anderen ähnlich
grossen Molekülen besetzt ist, nicht mehr zur Verfügung steht. Man spricht deshalb auch vom
besetzten Volumen. Thermodynamisch gesehen spielt daher beim Auflösen von
Makromolekülen die Entropieänderung eine entscheidende Rolle. Daneben tragen aber auch
enthalpische Effekte zur Nicht-Idealität makromolekularer Lösungen bei (anziehende und
abstossende Kräfte zwischen den Molekülen). In den meisten Lösungssystemen gibt es eine
Temperatur, bei der sich diese beiden Effekte gerade aufheben. Diese Temperatur ist ein
Analogon zur Boyle-Temperatur der realen Gase. Sie heisst Flory-Temperatur und ist
definiert als diejenige Temperatur, bei der der osmotische Virialkoeffizient gleich Null ist.
2.2 Dialyse von Polyelektrolyten
Eine Komplikation bei der osmometrischen Molmassenbestimmung ergibt sich aus der
möglichen elektrischen Ladung mancher Makromoleküle. Dabei können sich sowohl
Polyanionen, Polykationen oder Polyampholyte bilden. Bei Polyelektrolyten muss man daher
den Ionisationszustand kennen um die Ergebnisse osmotischer Messungen quantitativ
interpretieren zu können.
Wir nehmen an, eine Lösung eines Natriumsalzes eines Polyelektrolyten NaνP enthalte
zusätzliches NaCl und stehe über eine semipermeable Membran mit einer anderen Salzlösung
in Kontakt. Die Membran sei sowohl für das Lösungsmittel als auch für die Salz-Ionen

___________________________________________________________________________ 9
durchlässig, aber undurchlässig für das Polyanion. Damit lässt sich der Einfluss des Salzes auf
den osmotischen Druck leicht berechnen und der Ionisationsgrad des Polyelektrolyten
bestimmen, da wegen des Elektroneutralitätsprinzips Anionen und Kationen nicht in
beliebiger Menge durch die Membran wandern dürfen, sondern mit jedem Anion auch ein
Kation durch die Membran wandern muss.
Liegt NaνP auf der einen Seite der Membran in der Konzentration [P] vor und auf beiden
Seiten wird NaCl zugefügt, dann befinden sich auf der linken Seite (L) die Ionen Pν-, Na+, Cl-
und auf der rechten Seite (R) gibt es nur Na+ und Cl-. Im Gleichgewicht ist das chemische
Potential von NaCl auf beiden Seiten der Membran gleich. Deshalb müssen Na+ und Cl—
Ionen wandern, bis die Gleichgewichtsbedingung erfüllt ist. )()( RL NaClNaClΘΘ = µµ
Es muss also gelten:
RClNaNaClLClNaNaCl aaRTaaRT )ln()ln( −+−+ +=+ ΘΘ µµ
Wenn man die Aktivitätskoeffizienten vernachlässigt, ist das gleichbedeutend mit:
[ ][ ] [ ][ ] RL ClNaClNa −+−+ =
Die Natrium-Ionen stammen sowohl vom Polyelektrolyten als auch vom zugegebenen
Kochsalz. Damit die Elektroneutralität erhalten bleibt, müssen die Bedingungen:
[Na+]L = [Cl-]L + ν[P] und [Na+]R = [Cl-]R
erfüllt sein. Wenn man diese Beziehungen miteinander kombiniert, erhält man Formeln für
die Konzentrationsunterschiede der Ionen beiderseits der Membran:
][][2]][[
][][]][[][][
PClNaP
NaNaNaPNaNa L
RL
LRL ν
νν+
=+
=− −
+
++
+++
][2]][[
][][]][[][][ −
−
−−
−−− −
=+
−=−
ClClP
ClClClPClCl L
RL
LRL
νν

___________________________________________________________________________ 10
Dabei ist :
RL ClClCl ][][][ 21 −−− +=
Berücksichtigt man nun noch dass der osmotische Druck vom Unterschied in der
Teilchenzahl auf beiden Seiten der Membran abhängt, so folgt aus der van’t Hoffschen
Gleichung:
][1][)][]([)][][]([ PPRTClNaClNaPRT RRLL β+=−++=Π −+−+
mit: ][][4(2 PCl ννβ += −
Ist die zugegebene Salzmenge so gross, dass 1][ <<Pβ gilt, so vereinfacht sich diese
Gleichung zu Π , und das ist unabhängig von ν. Wenn man also den osmotischen
Druck in Gegenwart hoher Salzkonzentrationen misst, kann man die relative Molekülmasse
direkt bestimmen.
][PRT=
2.3 Sedimentation
Im Schwerefeld der Erde setzen sich schwere Teilchen allmählich im unteren Teil einer
Lösung ab. Wie schnell diese Sedimentation erfolgt, hängt von der Stärke des
Gravitationsfeldes, der Masse der Teilchen und deren Form ab. DNA z.B. sedimentiert in der
natürlichen Helix-Form viel schneller als wenn sie zu einem ungeordneten Knäuel denaturiert
ist. Die Sedimentationsgeschwindigkeit kann somit auch zur Untersuchung von
Denaturierungsprozessen eingesetzt werden. Da die Höhenverteilung der Teilchen im
Gleichgewicht einer Boltzmann-Verteilung entspricht und von der Masse der Teilchen
abhängt, eignet sich die Sedimentation ebenfalls zur Bestimmung von Molekülmassen.
Normalerweise verläuft die Sedimentation sehr langsam. Man kann sie aber beschleunigen,
indem man das Gravitationsfeld durch ein Zentrifugalfeld, z.B. das einer Ultrazentrifuge,
ersetzt. Eine Ultrazentrifuge (siehe Abbildung 2-2) besteht im Prinzip aus einem Zylinder, der
mit hoher Geschwindigkeit um seine Achse rotiert.

___________________________________________________________________________ 11
Abbildung 2-2: Zentifugation
Die zu untersuchende Probe befindet sich in einer Zelle, die im äusseren Teil des Zylinders
angebracht ist. Dabei sind Beschleunigungen bis zum 105-fachen der Erdbeschleunigung
erreichbar. Zu Beginn ist die Probe homogen, und die obere Grenzfläche der Lösung bewegt
sich bei der Sedimentation allmählich nach aussen. Verfolgen lässt sich dies am besten an
Hand der Änderung des Brechungsindex der Lösung, der von der Konzentration abhängt.
Eine Möglichkeit dazu bietet die sogenannte Interferenz-Technik. Dabei werden zwei
Strahlen miteinander zur Interferenz gebracht, der eine, nachdem er durch die Probe gelaufen
ist, der andere, ohne dass er ein Hindernis durchläuft. Eine räumliche Änderung des
Brechungsindexes erzeugt dann ein charakteristisches Muster.
2.4 Sedimentationsgeschwindigkeit
Wegen des Auftriebs hat ein Teilchen der Masse m in einer Lösung nur die effektive Masse
meff =b·m mit b=1-ρνs·ρ ist die Dichte der Lösung, νs das spezifische Volumen der gelösten
Substanz. Wenn die Zentrifuge mit der Winkelgeschwindigkeit ω rotiert und sich die Probe
im Abstand r von der Achse befindet, so wirkt auf die Probe die Zentrifugalkraft meff·r·ω2.
Dieser nach aussen gerichteten Kraft wirkt die Reibung entgegen, die proportional zur
Geschwindigkeit s der Teilchen gegenüber dem Medium ist. Wenn wir mit f den
Reibungskoeffizienten bezeichnen, so ist f·s die Reibungskraft. Wenn die Reibungskraft

___________________________________________________________________________ 12
gerade gleich der Zentrifugalkraft ist, kann man aus der Gleichung meff·r·ω2=f·s die
Wanderungsgeschwindigkeit s berechnen:
fmbr
frm
s eff22 ωω
==
Sie hängt von der Winkelgeschwindigkeit und vom Radius ab. Man benützt deshalb
üblicherweise die Sedimentationskonstante S:
A
m
fNbM
rsS == 2ω
Für kugelförmige Teilchen mit dem Radius a in einem Lösungsmittel mit der Viskosität η
wird der Reibungskoeffizient f durch die Stokes'sche Gleichung gegeben:
f=6·π·a·η
Dann gilt:
A
m
aNbM
Sπη6
=
und man kann aus S entweder Mm oder a berechnen. Wenn die Teilchen nicht kugelförmig
sind, hat man für f andere Werte einzusetzen.
Wenn man aus der Sedimentationsgeschwindigkeit die relative Molekülmasse berechnen will,
braucht man eigentlich den Molekülradius a und auch den Reibungskoeffizienten f. Dieses
Problem löst sich unter Verwendung der Stokes-Einsteinschen-Beziehung:
DkTf =
Der Diffusionskoeffizient D gibt an, wie schnell Moleküle in der Richtung eines
Konzentrationsgradienten wandern.

___________________________________________________________________________ 13
Aus obiger Gleichung für S folgt dann:
bDSRT
bDkTSN
bfSNM AA
m ===
Das Ergebnis ist unabhängig von der Form der gelösten Moleküle und erlaubt es, die
Molmasse zu bestimmen, wenn die Sedimentations- und Diffusionsgeschwindigkeiten
gemessen wurden.
2.5 Das Sedimentationsgleichgewicht
Die Bestimmung der relativen Molmasse über die Sedimentationsgeschwindigkeit ist etwas
problematisch, weil sich die Diffusionskoeffizienten oft nicht genau bestimmen lassen. So
wird z.B. die Beobachtung der Grenzflächen in vielen Fällen durch Konvektionsströme
gestört. Man kann diese Schwierigkeit umgehen, wenn man das System im Gleichgewicht
untersucht, da D dann keine Rolle mehr spielt. Weil die Anzahl der gelösten Moleküle mit der
potentiellen Energie E proportional zu kTE
e−
ist, kann man aus dem Verhältnis der
Konzentrationen in verschiedenen Höhen bzw. in der Zentrifuge in verschiedenen Abständen
von der Achse, ihre Masse berechnen. Im Zentifugalfeld mit der Winkelgeschwindigkeit ω
hat ein Molekül der Masse meff im Abstand r die potentielle Energie 22
21 ωrmeff . Das
Verhältnis der Konzentrationen bei r1 und r2 ist danach gegeben durch:
kTrrbm
kTE
kTE
ee
eNN
cc 2
)(
2
1
2
1
21
22
2
2
1−
−
−
−
===ω
Damit erhält man:
−
=1
222
12
2
ln)(
2cc
brrRTMm ω

___________________________________________________________________________ 14
2.6 Elektrophorese
Viele Makromoleküle tragen elektrische Ladungen und wandern somit im elektrischen Feld.
Diesen Vorgang nennt man Elektrophorese. Die Lösung wird dabei entweder auf Papier
aufgebracht oder wie bei der Gel-Elektrophorese auf ein vernetztes Polyacrylamid-Gel. Die
Beweglichkeit der Makromoleküle hängt von der Masse und von ihrer Form ab. Wenn die auf
die Teilchen wirkende Kraft e·z·E (z ist die Anzahl der Ladungen an einem Molekül, E ist die
Feldstärke) gleich der Reibungskraft f·s ist, beobachtet man eine konstante
Wanderungsgeschwindigkeit fqEs = ( zeq ⋅= ). Die Wanderungsgeschwindigkeit ist somit
proportional der angelegten Spannung. Als charakteristische Grösse für die Mobilität des
Moleküls führt man die elektrophoretische Beweglichkeit ein:
fq
Esu ==
Der Reibungskoeffizient f hängt sowohl von der Masse und der Form des wandernden
Moleküls als auch von der Viskosität des umgebenden Mediums ab.
Das Protein Albumin z.B. hat eine elektrophoretische Beweglichkeit von u = 4·10-9 m2 V-1 s-1
bei pH = 4. Mit einer Spannung U = 2000 V und einer Laufstrecke von d = 20 cm ergibt sich
hieraus eine Wanderungsgeschwindigkeit von s = 4·10-5 m s-1 = 14.4 cm h-1 auf dem Gel.
Empirisch wurde für die Beweglichkeit der Komponente i folgender Zusammenhang
gefunden:
Geliirelirel ckuu −= )lg()lg( 0,,
mit , der elektrophoretischen Beweglichkeit ohne den retardierenden
Matrixeinfluss des Gels. In den sog. Ferguson-Plots wird dieser Zusammenhang graphisch
ersichtlich.
)0(,0
, == Gelirelirel cuu
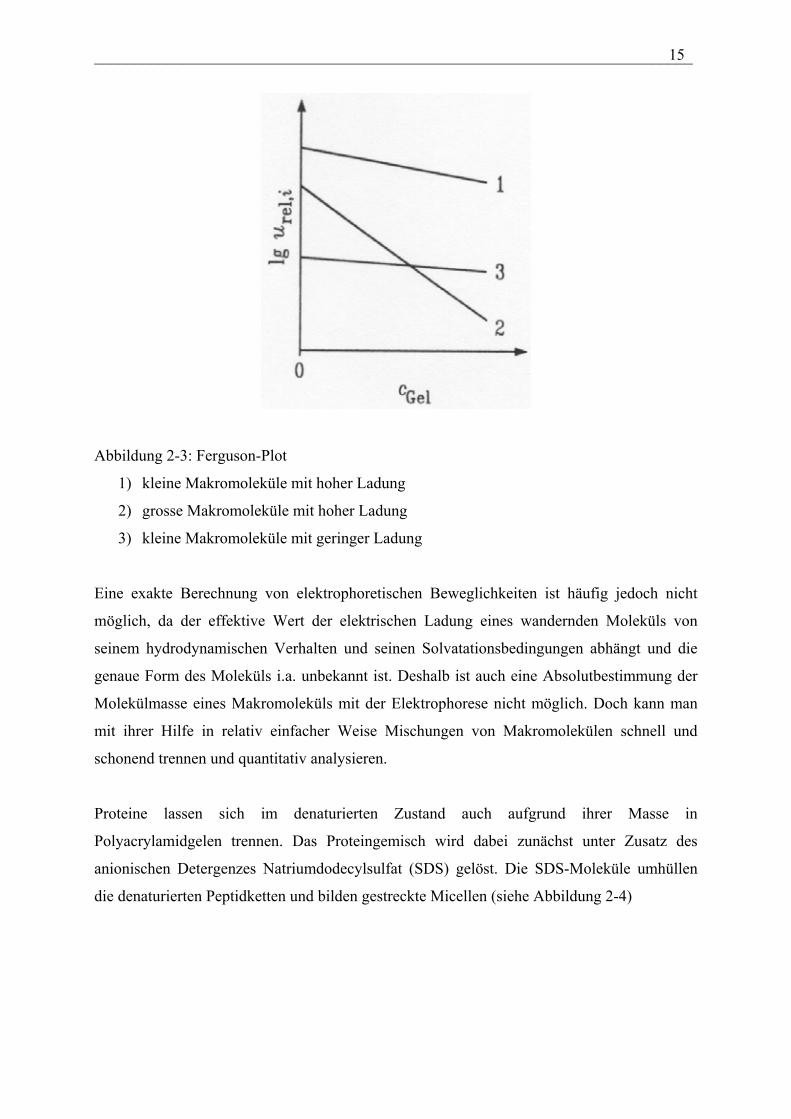
___________________________________________________________________________ 15
Abbildung 2-3: Ferguson-Plot
1) kleine Makromoleküle mit hoher Ladung
2) grosse Makromoleküle mit hoher Ladung
3) kleine Makromoleküle mit geringer Ladung
Eine exakte Berechnung von elektrophoretischen Beweglichkeiten ist häufig jedoch nicht
möglich, da der effektive Wert der elektrischen Ladung eines wandernden Moleküls von
seinem hydrodynamischen Verhalten und seinen Solvatationsbedingungen abhängt und die
genaue Form des Moleküls i.a. unbekannt ist. Deshalb ist auch eine Absolutbestimmung der
Molekülmasse eines Makromoleküls mit der Elektrophorese nicht möglich. Doch kann man
mit ihrer Hilfe in relativ einfacher Weise Mischungen von Makromolekülen schnell und
schonend trennen und quantitativ analysieren.
Proteine lassen sich im denaturierten Zustand auch aufgrund ihrer Masse in
Polyacrylamidgelen trennen. Das Proteingemisch wird dabei zunächst unter Zusatz des
anionischen Detergenzes Natriumdodecylsulfat (SDS) gelöst. Die SDS-Moleküle umhüllen
die denaturierten Peptidketten und bilden gestreckte Micellen (siehe Abbildung 2-4)

___________________________________________________________________________ 16
Abbildung 2-4: Proteinmolekül in SDS-Lösung
Die SDS-Moleküle schirmen die Proteinladungen im Inneren der Micelle ab. Es entsteht dabei
ein Komplex aus SDS und Protein, dessen stark negative Ladung der Masse des Proteins in
etwa proportional ist. Kleine Moleküle wandern dann rasch durch das Gel, während die
grossen Moleküle kleine Wanderungsgeschwindigkeiten haben. Mit Hilfe von
Relativmessungen lässt sich somit die Molekülmasse des unbekannten Proteins ermitteln.
2.7 Isoelektrische Fokussierung
Proteine lassen sich auch aufgrund ihres relativen Gehalts an sauren und basischen Resten
elektrophoretisch trennen. Am isoelektrischen Punkt eines Proteins, also dem pH-Wert, bei
dem seine Nettoladung Null ist, wird die elektrophoretische Beweglichkeit u des Teilchens
gleich Null. Betrachten wir die Elektrophorese eines Proteingemisches nun in einem pH-
Gradienten. Jedes Protein wandert so weit, bis es die Position im Gel erreicht hat, deren pH-
Wert seinem isoelektrischen Punkt entspricht. Diese Methode der Proteintrennung aufgrund
ihrer isoelektrischen Punkte nennt man isoelektrische Fokussierung. Der pH-Gradient im Gel
lässt sich mit Trägerampholyten erzeugen oder durch sogenannte Immobiline die in die
Trägermatrix einpolymerisiert werden (siehe Abbildung 2-5)
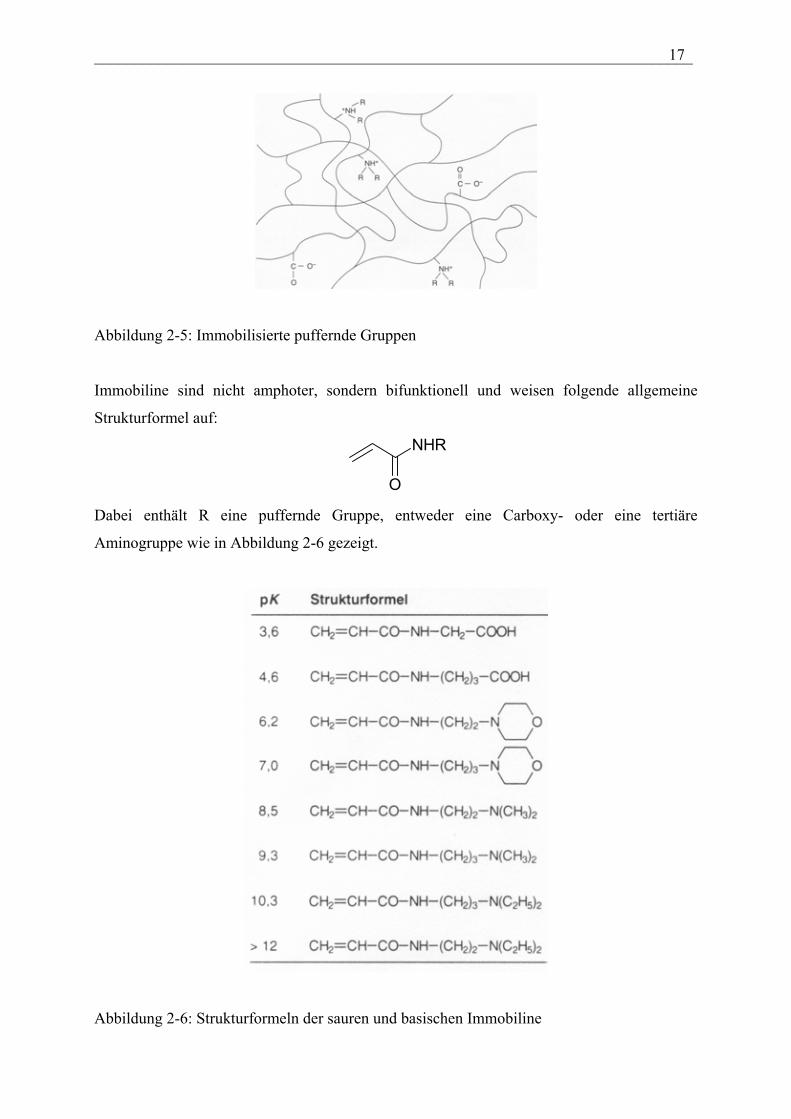
___________________________________________________________________________ 17
Abbildung 2-5: Immobilisierte puffernde Gruppen
Immobiline sind nicht amphoter, sondern bifunktionell und weisen folgende allgemeine
Strukturformel auf:
O
NHR
Dabei enthält R eine puffernde Gruppe, entweder eine Carboxy- oder eine tertiäre
Aminogruppe wie in Abbildung 2-6 gezeigt.
Abbildung 2-6: Strukturformeln der sauren und basischen Immobiline

___________________________________________________________________________ 18
Diese Immobiline sind Acrylamidderivate und zugleich schwache Säuren oder schwache
Basen. Um einen bestimmten pH-Wert puffern zu können, benötigt man eine Mischung von
mindestens zwei verschiedenen Immobilinen, einer Säure und einer Base.
2.8 Das Zeta-Potential
Bei Kenntnis der Partikelgeschwindigkeit kann man auch die Nettoladung bzw. das
Oberflächenpotential der Teilchen relativ zur umgebenden Lösungsmittelphase bestimmen.
An der Oberfläche elektrisch geladener Teilchen mit Oberflächenpotential Ψ0 bildet sich
durch Adsorption von Gegenionen oder durch Adsorption von Dipolen aus der Umgebung
eine sog. elektrische Doppelschicht aus (siehe Abbildung 2-7), die relativ fest mit dem
Teilchen verbunden ist (STERNsche Doppelschicht). Sie wird von einer beweglichen,
diffusen Ionenschicht (Gouy-Chapman-Schicht) umhüllt. An diese diffuse Doppelschicht
schliesst sich das elektrisch neutrale Lösungsmittelmedium an. Das Oberflächenpotential Ψ0
ist hier auf den Wert Null abgesunken.
Abbildung 2-7: Oberflächenpotentiale eines negativen Dispersionsteilchens

___________________________________________________________________________ 19
Bei der Diffusionsbewegung der Teilchen im elektrischen Feld wird aufgrund der
Reibungskräfte ein Teil der diffusen Doppelschicht abgestreift, und es entsteht eine
Abreissebene. Das Potential ζ an dieser Stelle bezeichnet man Zetapotential. Es kann
näherungsweise aus der Wanderungsgeschwindigkeit s der Teilchen im elektrischen Feld
nach Helmholt-Smoluchowski ermittelt werden:
Es
εηζ ≈
Dabei ist ε = εr ε0 die Dielektrizitätskonstante des Lösungsmittels, η der Viskositätskoeffizient
des Lösungsmittels und E die elektrische Feldstärke.
2.9 Viskosität
Jeder konvektive Transport ist mit Reibung verbunden. sie äussert sich durch den widerstand,
den das strömende fluide Medium der Antriebskraft entgegensetzt. durch die Reibung wird
die Strömungsgeschwindigkeit begrenzt und damit die Einstellung eines stationären
Strömungszustandes ermöglicht. Zur Erläuterung des von Newton aufgestellten
Elementargesetzes der inneren Reibung soll zunächst die laminare Strömung einer Flüssigkeit
über einer ebenen Bodenfläche betrachtet werden (siehe Abbildung 2-8).
Abbildung 2-8: Innere Reibung
Die in der Abbildung durch die Länge der parallelen Pfeile anschaulich dargestellte
Strömungsgeschwindigkeit w nimmt in Richtung der y-Achse zu, wobei die Geschwindigkeit
der unmittelbar an der Bodenfläche haftenden Flüssigkeitsschicht gleich Null zu setzen ist.
Durch die Impulsübertragung zwischen den Teilchen der mit verschiedener Geschwindigkeit

___________________________________________________________________________ 20
aneinander vorbeigleitenden Schichten entsteht innere Reibung. Sie bewirkt, dass die
oberhalb einer Fläche A befindliche Schicht auf die darunter liegende Schicht beschleunigend
einwirkt, wobei die Bewegung der oberen Schicht durch die hemmende Wirkung der unteren
Schicht verzögert wird. Nach Newton ist die an der Fläche A angreifende Kraft durch die
Gleichung:
dydwAK η=
gegeben. K ist proportional der Fläche A und der Änderung der Geschwindigkeit in Richtung
der Flächennormalen. Der Proportionalitätsfaktor η wird als Viskositätskoeffizient bezeichnet.
Unter den einfachen Bedingungen der ebenen Strömung bildet sich im Strömungsfeld ein
lineares Geschwindigkeitsgefälle aus, d.h. es gilt:
dw
AK 0η=
wenn man die Geschwindigkeit der obersten Flüssigkeitsschicht mit w0 und ihren Abstand
von der Bodenfläche mit d bezeichnet. Für den Viskositätskoeffizienten gilt damit:
sPabzwmNs
AwKd
⋅
= .2
0
η
Soweit η unabhängig von w0 ist, liegt eine Newtonsche Flüssigkeit vor.
Bei Viskositätsmessungen an Lösungen makromolekularer Stoffe zeigen sich deutliche
Unterschiede zur Viskosität des reinen Lösungsmittels. Als Konzentrationsmass wählt man
bei diesen Messungen in der Regel die in g/cm3 angegebene Massenkonzentration cm der
gelösten makromolekularen Komponente. Es nach Einstein für kugelförmige Teilchen die
Beziehung:
ϕηη
ηηKsp ==
−
0
0
mit η0 als Viskosität des reinen Lösungsmittels.

___________________________________________________________________________ 21
Den Quotienten 0
0
ηηη − nennt man die spezifische Viskosität. Der Faktor φ ist der
Volumenbruchteil des gelösten Stoffes, d.h. es gilt:
vNvp=ϕ
wobei N die Teilchenzahl, v das Volumen und vp das Eigenvolumen der gelösten Teilchen
darstellt.
Mit:
Mvc
NN mL=
erhält man:
pmL
sp vMcN
K=η
Diese Beziehung für kugelförmige Teilchen lässt sich auch auf ein zylinderförmiges
Fadenmolekül anwenden, indem man das Eigenvolumen vp durch das Volumen π(d/2)2h
ersetzt, mit d als Durchmesser und h als der Höhe des Fadenmoleküls. Man erhält:
hdMcN
K mLsp
2)2/(πη =
Dabei ist h dem Molekulargewicht M proportional. Bei konstantem Durchmesser d lassen sich
alle konstanten Faktoren auf der rechten Seite der Gleichung zu einer neuen Konstanten
zusammenfassen und man kann die Gleichung mit der auf das Grundmol bezogenen
Konzentration cgm in der einfachen Form
gmsp cK '=η
schreiben. Nach dieser Gleichung sollte die spezifische Viskosität einer Lösung von
Fadenmolekülen nur von der Konzentration, nicht aber der Kettenlänge der Moleküle
abhängig sein. Die Praxis zeigt jedoch, dass dies nicht der Fall ist. Vielmehr ist die
spezifische Viskosität bei gleicher Grundmolarität dem Molekulargewicht des Gelösten direkt
proportional, wenn die gelösten Moleküle in Form lockerer, gut solvatisierter Knäuel
vorliegen. Dann ist die Annahme zulässig, dass das mittlere Knäuelvolumen mit dem Quadrat

___________________________________________________________________________ 22
der Moleküllänge wächst. In diesem Fall lässt sich vp durch das Volumen einer Scheibe mit
der Grundfläche π(h/2)2 und der Dicke d beschreiben Man erhält dann für die spezifische
Viskosität:
dhMcN
K mLsp )4/( 2πη =
Da h bei Fadenmolekülen proportional M ist und alle anderen Faktoren auf der rechten Seite
der Gleichung mit Ausnahme von cm zu einer Konstanten Km zusammengefasst werden
können, ergibt sich die einfache Gleichung:
McK mmsp =η
Km wird als Viskositätmolekulargewichtskonstante bezeichnet. Da bei endlichen
Konzentrationen zwischenmolekulare Wechselwirkungen zwischen den Knäueln auftreten, ist
der Quotient msp c/η konzentrationsabhängig. Für den Grenzfall der ideal verdünnten Lösung
resultiert daraus die Beziehung:
MKc mmspcm==
→][)/(lim
0ηη
D.h. es besteht eine Proportionalität zwischen dem Molekulargewicht und der
Grenzviskositätszahl [η]. Diese Gleichung wurde zuerst empirisch von Staudinger gefunden.
Eine genaue Betrachtung des Knäuel-Zustandes gelöster Makromoleküle erfordert allerdings
die statistische Berücksichtigung der durch die Segmentbeweglichkeit der Kettenelemente
vorgegebenen Vielfalt von Anordnungsmöglichkeiten. In einem Fadenmolekül sind der
Abstand zwischen zwei Kettenatomen und der Valenzwinkel im Bereich von drei
aufeinanderfolgenden Kettenatomen festgelegt. In einem einfachen Segmentmodell, das die
behinderte Rotation benachbarter Gruppen und das Eigenvolumen der Kettenelemente
vernachlässigt, muss jedes Segment so viele Strukturelemente enthalten, dass es die
Bedingung der freien Orientierung seines Endpunktes im Raum erfüllt. Dies bedeutet, dass
die Länge l eines Kettensegments von der Kettensteifigkeit des jeweiligen Polymeren
abhängt.
−

___________________________________________________________________________ 23
Für das mittlere Anstandsquadrat der Fadenenden eines aus n frei orientierbaren
Segmenten bestehenden Fadenmoleküls gilt die Beziehung:
−2s
−−
= 22 lns
Die maximale Länge h der gestreckt gedachten Fadenmoleküle ist gegeben durch: −
= lnh
Daraus folgt:
2/12/12 2/1
)( hls−−
=
Die maximale Fadenlänge h ist dem Molekulargewicht M proportional. Für eine vorgegebene
konstante Segmentlänge ergibt sich damit das Kuhn’sche Wurzelgesetz: −
l
Mconsts ⋅=−2
Nach diesem Gesetz sind der wahrscheinlichste Fadenabstand und damit auch der
Trägheitsradius r proportional der Wurzel aus dem Molekulargewicht. Demnach hat man in
der Beziehung für die spezifische Viskosität für das Eigenvolumen vp eines statistisch
geknäuelten Fadenmoleküls einen zu M3/2 proportionalen Faktor einzusetzen. Man gelangt
damit zu einer Beziehung der Form:
2/1][ MKθθη =
Diese Gleichung heisst auch das Kuhn’sche Viskositätsgesetz für Lösungen idealer
statistischer Knäuel. Es gilt für ein gegebenes Polymeres in bestimmten Lösungsmitteln nur
bei einer definierten Temperatur Θ (Flory-Temperatur). Mit zunehmender Temperatur steigt
der Wert des Exponenten von M an, das Knäuel erfährt eine Aufweitung.
Mark und Houwink haben den Einfluss des Lösungsmittels und der Temperatur auf die
Knäuel-Konformation durch die Einführung eines allgemeinen Exponenten a in die
Beziehung:
a
mMK=][η
berücksichtigt.
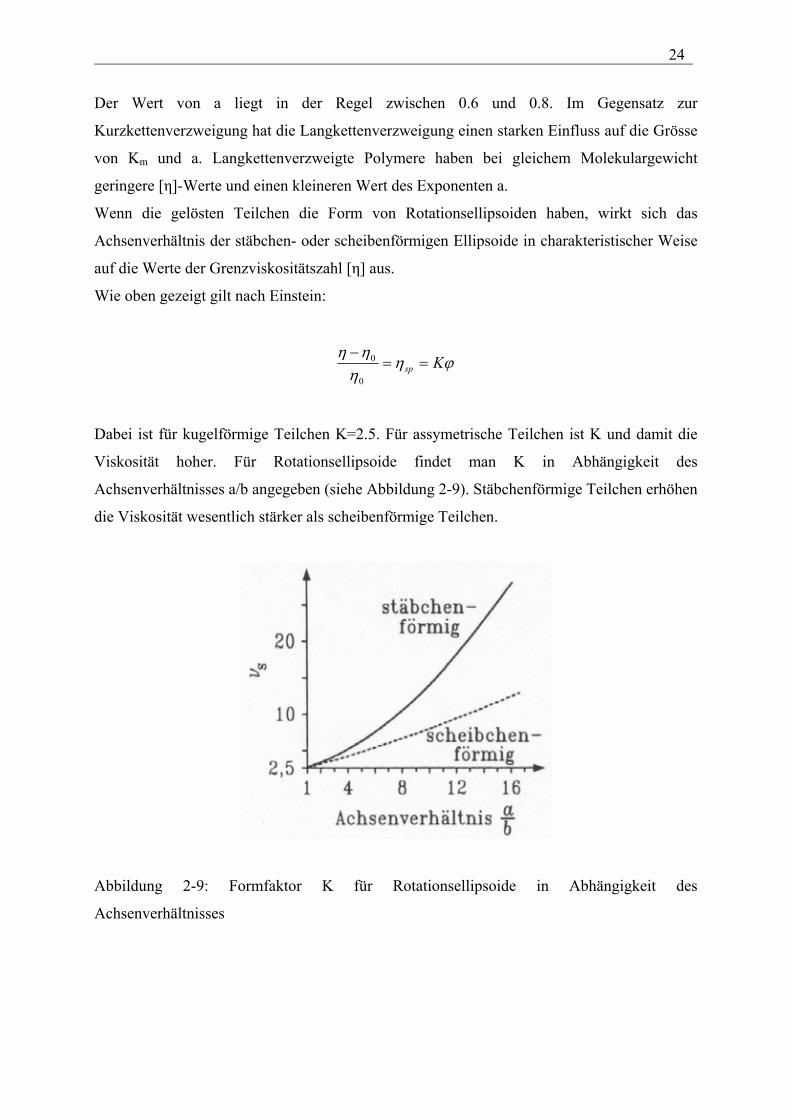
___________________________________________________________________________ 24
Der Wert von a liegt in der Regel zwischen 0.6 und 0.8. Im Gegensatz zur
Kurzkettenverzweigung hat die Langkettenverzweigung einen starken Einfluss auf die Grösse
von Km und a. Langkettenverzweigte Polymere haben bei gleichem Molekulargewicht
geringere [η]-Werte und einen kleineren Wert des Exponenten a.
Wenn die gelösten Teilchen die Form von Rotationsellipsoiden haben, wirkt sich das
Achsenverhältnis der stäbchen- oder scheibenförmigen Ellipsoide in charakteristischer Weise
auf die Werte der Grenzviskositätszahl [η] aus.
Wie oben gezeigt gilt nach Einstein:
ϕηη
ηηKsp ==
−
0
0
Dabei ist für kugelförmige Teilchen K=2.5. Für assymetrische Teilchen ist K und damit die
Viskosität hoher. Für Rotationsellipsoide findet man K in Abhängigkeit des
Achsenverhältnisses a/b angegeben (siehe Abbildung 2-9). Stäbchenförmige Teilchen erhöhen
die Viskosität wesentlich stärker als scheibenförmige Teilchen.
Abbildung 2-9: Formfaktor K für Rotationsellipsoide in Abhängigkeit des
Achsenverhältnisses

___________________________________________________________________________ 25
3 Konformationsumwandlungen von Biopolymeren
Die meisten Biopolymere besitzen im nativen Zustand eine wohl definierte Konformation.
Alle die Wechselwirkungen, die die Sekundär- und Tertärstrukturen der Biomoleküle und
damit die Konformation der Moleküle stabilisieren, hängen vom Lösungsmittel, dem pH-
Wert, der Ionenstärke, der Temperatur und dem Druck ab. Die native Konformation ist
entscheidend für die biochemische Funktion des Biomoleküls. So ist z.B. mit dem Verlust der
nativen Konformation bei der Proteindenaturierung ein Verlust der Enzymaktivität
verbunden. Untersuchungen der Konformationsumwandlungen biochemischer Systeme
liefern wertvolle Informationen über die Stabilität der Systeme und über molekulare
Mechanismen biochemischer Prozesse.
Die Konformationsumwandlungen biologischer Makromoleküle lassen sich mit einer Reihe
experimenteller Methoden, wie z.B. spektroskopisch oder kalorimetrisch, verfolgen.
Als Beispiel für eine Konformationsumwandlung betrachten wir die durch eine
Temperaturänderung hervorgerufene Knäuel-Helix Umwandlung eines Homoplypeptids wir
in Abbildung 3-1 dargestellt.
Abbildung 3-1: Knäuel-Helix-Umwandlung eines Polypeptids
Dazu nimmt man an, dass die Umwandlung eines Segments vom ungeordneten Knäuel-
Zustand A in die α-Helix-Konformation B als ein Zwei-Zustands-Gleichgewicht
A B
angesehen werden kann. A steht dabei für das Segment im ungeordneten Knäuel und B für
das Segment im α-Helix-Zustand.

___________________________________________________________________________ 26
Wendet man das Massenwirkungsgesetz auf dieses Gleichgewicht an, dann ist die
Gleichgewichtskonstante dieses Gleichgewichts gegeben durch:
A
B
nnK =
Führt man noch einen Helixbildungsgrad φ ein, der beschreibt wie gross der Anteil der
Segmente im Helixzustand im Vergleich zur Gesamtzahl an Segmenten ist, dann ergibt sich
für die Gleichgewichtskonstante:
ϕϕ−
=1
K
Die Temperaturabhängigkeit der Gleichgewichtskonstanten ist durch die van’t Hoff’sche
Gleichung gegeben:
2
0lnRTH
TK mU∆
=∂
∂
Sie ist mit der molaren Umwandlungsenthalpie im Standardzustand verknüpft. Man
bezeichnet die dem Helixbildungsgrad φ = 0.5 (hier ist K=1) zugeordnete Temperatur T
0mUH∆
U als
Umwandlungstempeartur. Bei dieser Temperatur liegt die Hälfte aller Segmente in der
Helixkonformation vor.
Ein möglicher Verlauf von φ als Funktion der Temperatur ist in Abbildung 3-2 dargestellt.
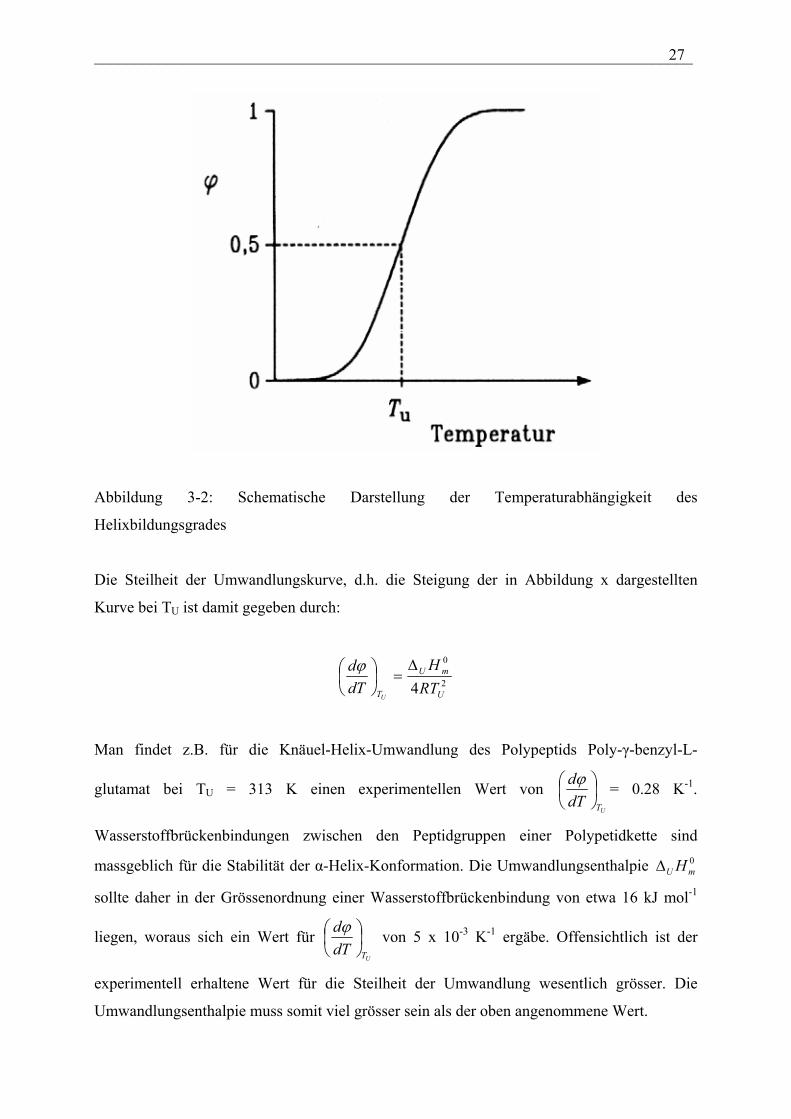
___________________________________________________________________________ 27
Abbildung 3-2: Schematische Darstellung der Temperaturabhängigkeit des
Helixbildungsgrades
Die Steilheit der Umwandlungskurve, d.h. die Steigung der in Abbildung x dargestellten
Kurve bei TU ist damit gegeben durch:
2
0
4 U
mU
T RTH
dTd
U
∆=
ϕ
Man findet z.B. für die Knäuel-Helix-Umwandlung des Polypeptids Poly-γ-benzyl-L-
glutamat bei TU = 313 K einen experimentellen Wert von UT
dTd
ϕ = 0.28 K-1.
Wasserstoffbrückenbindungen zwischen den Peptidgruppen einer Polypetidkette sind
massgeblich für die Stabilität der α-Helix-Konformation. Die Umwandlungsenthalpie
sollte daher in der Grössenordnung einer Wasserstoffbrückenbindung von etwa 16 kJ mol
0mUH∆
-1
liegen, woraus sich ein Wert für UT
dTd
ϕ von 5 x 10-3 K-1 ergäbe. Offensichtlich ist der
experimentell erhaltene Wert für die Steilheit der Umwandlung wesentlich grösser. Die
Umwandlungsenthalpie muss somit viel grösser sein als der oben angenommene Wert.

___________________________________________________________________________ 28
Das bedeutet, dass der oben angenommene Mechanismus einer Einzelsegmentumwandlung
nicht richtig ist. Die Wechselwirkung mit den Nachbarsegmenten der Polypeptidkette bei der
Umwandlung ist offensichtlich nicht zu vernachlässigen. dies bedeutet, dass benachbarte
Segmente der Kette gemeinsam (kooperativ) aus dem Knäuel- in den Helix-Zustand
übergehen.
Unter Beibehaltung des bisherigen Formalismus führt man zur Beschreibung der
experimentellen Daten die sogenannte mittlere kooperative Wellenlänge N0 ein. sie entspricht
der mittleren Anzahl der bei TU kooperativ vom Knäuel- in den Helix-Zustand übergehenden
Segmente und kann damit als stöchiometrische Einheit für die kooperative
Konformationsumwandlung bezeichnet werden. Die in der van’t Hoffschen Gleichung auf das
einzelne Segment bezogene molare Umwandlungsenthalpie muss durch das Produkt
, die sogenannte ‚scheinbare’ Umwandlungsenthalpie (pro Mol kooperative
Einheiten), ersetzt werden. Die van’t Hoffsche Umwandlungsenthalpie ist somit
und es gilt:
0mUH∆
00 mU HN ∆⋅
00vH NH =∆ 0
mU H∆⋅
2
0lnRTH
TK vH∆
=∂
∂
Die wahre, auf ein Mol Einzelsegmente bezogene Umwandlungsenthalpie ∆ erhält man
aus kalorimetrischen Messungen. Da durch die Enthalpieänderung dH der sich pro
Mol umwandelnden Segmente A gegeben ist:
0mUH
0mUH∆
AmU dn
dHH =∆ 0
und da sich mit der Gesamtzahl n = nA + nB an Segmenten sowie ϕ
ϕ−
=1
K und A
B
nnK =
folgender Zusammenhang ergibt:
dTdn
ndTd B1
=ϕ
erhält man für den auf ein Mol bezogenen Umwandlungsteil der Wärmekapazität C:
___________________________________________________________________________ 29
dTdH
dTdH
nC muum
ϕ0,
1∆==
Die Steilheit des Übergangs bei TU ist gegeben durch:
2
00
4 U
mU
T RTHN
dTd
U
∆=
ϕ
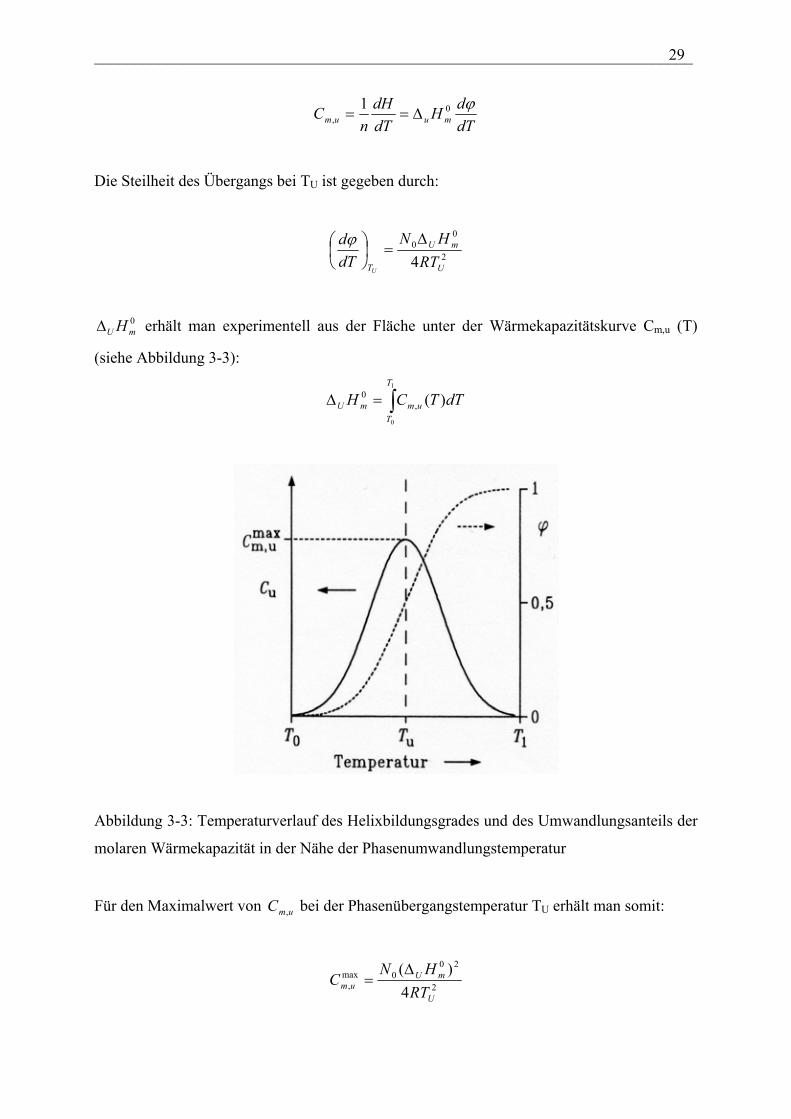
0mUH∆ erhält man experimentell aus der Fläche unter der Wärmekapazitätskurve Cm,u (T)
(siehe Abbildung 3-3):
∫=∆1
0
)(,0
T
TummU dTTCH
Abbildung 3-3: Temperaturverlauf des Helixbildungsgrades und des Umwandlungsanteils der
molaren Wärmekapazität in der Nähe der Phasenumwandlungstemperatur
Für den Maximalwert von C bei der Phasenübergangstemperatur Tum, U erhält man somit:
2
200max
, 4)(
U
mUum RT
HNC
∆=

___________________________________________________________________________ 30
Aus den kalorimetrisch bestimmbaren Grössen TU, und lässt sich somit die
kooperative Einheit N
max,umC
0mUH∆
0 bestimmen.
Der Prozess der Proteindenaturierung ist ein kooperativer Prozess und man findet Werte für
N0 bis zu 104. Die Kooperativität und damit die Steilheit des Übergangs ist umso grösser, je
länger die Polypeptidkette ist. Die thermisch induzierte Helix-Knäuel-Denaturierung wird
häufig mit einem Schmelzvorgang verglichen, obwohl es sich hierbei streng genommen nicht
um einen Phasenübergang erster Ordnung handelt. Aus den experimentell bestimmbaren
Denaturierungskurven lassen sich Aussagen über die relative Stabilität von Proteinstrukturen
ableiten. Die meisten Proteine haben TU-Werte weit unter 373 K. Ausnahmen bilden Proteine
thermophiler Bakterien, die heisse Quellen mit Temperaturen von annähernd der
Siedetemperatur des Wassers gewohnt sind.
Ein weiteres Beispiel für eine kooperative Phasenumwandlung von Biomolekülen ist das
durch pH-Änderung oder Temperaturerhöhung induzierte Aufschmelzen (Aufwinden) der
DNA-Doppelhelix (DS) in zwei Einzelstränge (ES):
DS 2 ES
Dabei werden die Wasserstoffbrückenbindungen zwischen den Stickstoffbasen der beiden
DNA-Stränge aufgebrochen. Der Übergang lässt sich mit Hilfe spektroskopischer Messungen
leicht verfolgen, da Doppel- und Einzelstrang bei einer Wellenlänge von 260 nm
unterschiedlich stark adsorbieren.
4 Konformation und Konfiguration von Biopolymeren
Unter der Primär-Struktur eines Makromoleküls versteht man die Folge der chemischen
Bausteine, aus denen die Kette aufgebaut ist. die Sekundärstruktur beschreibt die räumliche
Anordnung der Struktureinheiten eines Moleküls und die Tertärstruktur beschreibt den
gesamten dreidimensionalen Aufbau des Moleküls. Die Quartärstruktur gibt an, in welcher
Weise manche Moleküle durch Zusammenlagerung anderer Moleküle entstehen.

___________________________________________________________________________ 31
4.1 Ungeordnete Knäuel
Bei der Besprechung der verschiedenen Strukturmerkmale betrachten wir die
wahrscheinlichste Struktur, die eine Kette aus identischen Bausteinen annimmt, die weder
Wasserstoffbrücken noch irgendwelche anderen speziellen Bindungen ausbilden können. Als
Modell legen wir eine bewegliche Kette zugrunde, bei der nur die Abstände der Bausteine
vorgegeben sind, während die Bindungswinkel völlig beliebig sind (siehe Abbildung 4-1).
Abbildung 4-1: bewegliche Molekülkette
Das ist natürlich eine starke Vereinfachung, denn in der Regel sind die Bindungswinkel fest
vorgegeben, und die Beweglichkeit der Kette kommt durch die Drehbarkeit der Molekülteile
um die Bindungen zustande.
Die Wahrscheinlichkeit, dass die Enden der Kette einen Abstand voneinander haben, der
zwischen R und R+dR liegt ist gleich f dR mit:
22232/1 )/(4 RaeRaf −= ππ und )2/(3 22 Nla =
Bei der ungeordneten Bewegung ist das die Wahrscheinlichkeit, dass das wandernde Teilchen
in einer beliebigen Richtung eine Strecke zwischen R und R+dR zurückgelegt hat. N ist die
Anzahl der Bindungen und l die Länge eines Bausteins.
Nach dieser Gleichung gibt es Knäuel, deren Enden weit voneinander entfernt sind (wie viele
das sind, gibt der Wert von f für grosses R an), aber auch Knäuel, deren Enden nahe
beieinander liegen. Danach geht ein Knäuel ständig von einer Konformation in die andere
über, und f dR gibt die Wahrscheinlichkeit an, dass in einem gegebenen Augenblick der
Abstand der Enden gerade zwischen R und R+dR liegt.

___________________________________________________________________________ 32
Der quadratisch gemittelte Abstand Rrms (rms =root mean square) ist ein Mass für den
durchschnittlichen Abstand der Enden eines ungeordneten Knäuels.
Rrms ist die Quadratwurzel aus dem Mittelwert von R2, der berechnet wird, in dem jeder
mögliche Wert von R2 mit der Wahrscheinlichkeit f, dass R auftritt, gewichtet wird:
lNfdRRRrms2/1
0
2 == ∫∞
Dieses Modell des ungeordneten Knäuels ist nur eine grobe Näherung, da die Annahme
beliebiger Bindungswinkel und die Annahme dass die Atome keine Ausdehnung haben noch
im Modell enthalten sind und bei realen Systemen zu einem zu kleinen Wert für Rrms führt.
4.2 Helix- und Faltblattstruktur
Für die Sekundärstrukturen von Proteinen gelten die von Linus Pauling und Robert Corey
formulierten Regeln, die im wesentlichen darauf beruhen, dass die Strukturen durch
Wasserstoffbrückenbindungen stabilisiert werden, an denen die Peptid-Bindungen beteiligt
sind. Die Peptidbindungen können als H-Donoren oder als H-Akzeptoren wirken.
Die Regeln lauten:
(1) Die Atome einer Peptid-Bindung liegen in einer Ebene
(2) Die Atome N, H und O einer Wasserstoffbrücke liegen auf einer Geraden, wobei
Auslenkungen von H bis zu 30° von der N-O-Linie toleriert werden
(3) Alle NH- und CO-Gruppen sind an den Bindungen beteiligt
Wenn die Wasserstoffbrücken zwischen den Peptid-Bindungen derselben Kette gebildet
werden, sprechen wir von einer α-Helix. Wenn die Wasserstoffbrücken zwischen den Peptid-
Bindungen verschiedener Ketten bestehen, sprechen wir von einem ß-Faltblatt.
Die Geometrie einer Kette wird durch zwei Winkel beschrieben, den Torsionswinkel Φ der N-
C-Bindung und den Torsionswinkel Ψ der C-C-Bindung. Abbildung 4-2 zeigt die Winkel an
einem α-L-Polypeptid.

___________________________________________________________________________ 33
Abbildung 4-2: Torsionswinkel Φ und Ψ in einer Kette
In dieser Abbildung ist die all-trans-Form des Polypeptids wiedergegeben, in der Φ = Ψ =
180° ist. Die Vorzeichenkonvention lautet, dass bei einem positiven Winkel das vordere Atom
im Uhrzeigersinn zu bewegen ist, damit die Bindungen am vorderen Atom mit denjenigen am
hinteren Atom zur Deckung kommen. Wenn alle Ψ und alle Φ gleich sind, erhält man eine
Helix. Für eine Rechts- α-Helix ist Φ=-57° und Ψ = -47°, für eine Links- α-Helix sind beide
Winkel positiv.
5 Thermisch-kalorische Messverfahren
Phasenumwandlungen von Biopolymeren sind im allgemeinen thermisch induzierbar. Zur
Untersuchung dieser Umwandlungen liegt es daher nahe, kalorimetrische Messeverfahren
einzusetzen. Dazu gehören die Differenz-Thermoanalyse (DTA) und die Difference Scanning
Calorimetry (DSC).
5.1 Differenz-Thermoanalyse (DTA)
Messgrösse bei der DTA ist die Temperaturdifferenz ∆T zwischen der Messprobe und einer
Vergleichsprobe. Abbildung 5-1 zeigt schematisch den Aufbau eines DTA-Geräts.

___________________________________________________________________________ 34
Abbildung 5-1: Schematische Darstellung eines DTA-Geräts
Referenz und Probe werden in Reaktionsgefässe gefüllt, die in einen beheizbaren Ofenblock
eingepasst sind. Zur Messungen von Proben- und Referenztemperatur befinden sich
Thermoelemente an den Reaktionsgefässen. Über eine programmierbare Regelungseinheit
wird die Heizung des Ofenblocks derart angesteuert, dass seine Temperatur langsam linear
ansteigt oder abnimmt. Dazu wird die Temperatur des Blocks über ein weiteres
Thermoelement gemessen und mit dem momentanen Sollwert verglichen. Die Heizleistung
wird entsprechend der Differenz der beiden Werte geregelt. Zu Beginn einer Messung
besitzen Probe und Referenz dieselbe Temperatur. Wenn beim Aufheizen in der Probe z.B.
ein endothermer Phasenübergang erfolgt, hinkt die Probentemperatur solange der
Referenztemperatur hinterher, bis die Phasenumwandlung abgeschlossen ist. Eine von Null
verschiedene Temperaturdifferenz ∆T tritt auf, die elektronisch verstärkt und von einem
Computer aufgezeichnet wird. Die Darstellung der Temperaturdifferenz erfolgt in der Regel
als Funktion der Probentemperatur oder der Zeit. Durch das Auftreten der
Temperaturdifferenz ändern sich auch die Wärmeströme im Ofenblock. Bis ∆T wieder Null
ist, wird der Probe zusätzlich die Umwandlungswärme zugeführt. Daher ist die Fläche unter
dem Messsignal ein Mass für die zusätzlich zugeführte Wärme, allerdings nur qualitativ, da
bei den meisten DTA-Geräten die Wärmeübergänge in der Messanordnung nicht definiert
erfolgen.
Abbildung 5-2 zeigt als Beispiel die DTA-Messkurve einer Phosphatidylcholin-Dispersion.
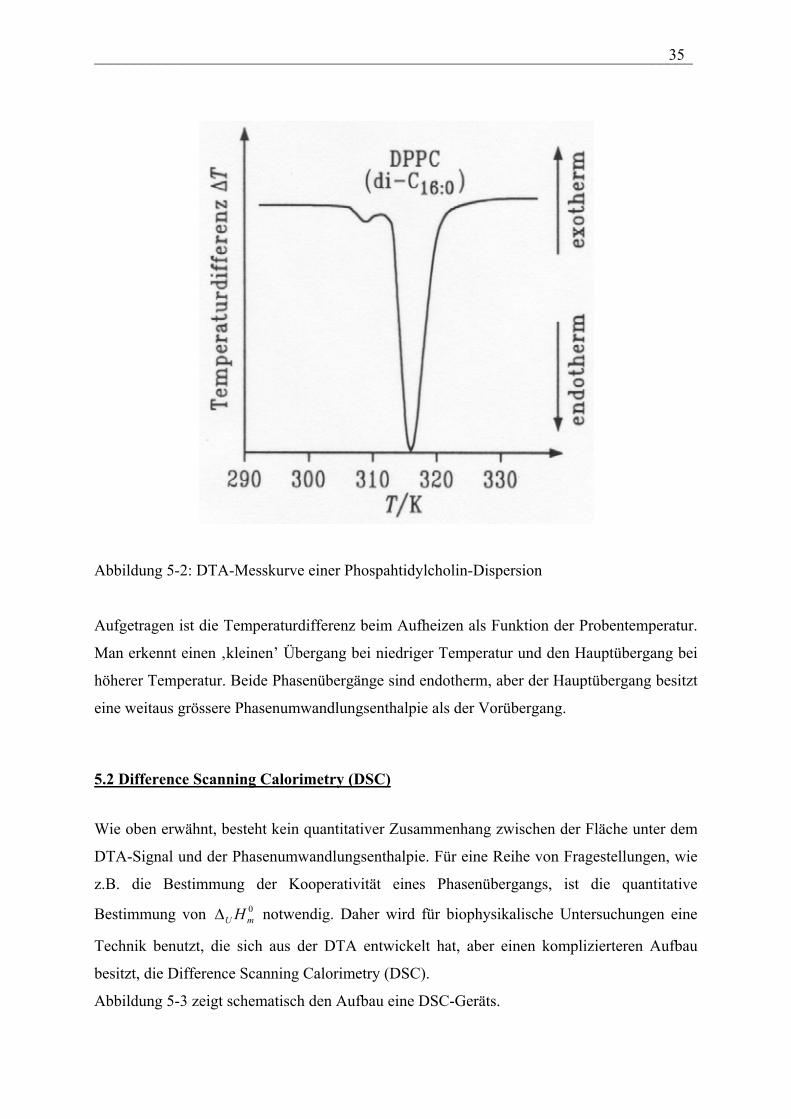
___________________________________________________________________________ 35
Abbildung 5-2: DTA-Messkurve einer Phospahtidylcholin-Dispersion
Aufgetragen ist die Temperaturdifferenz beim Aufheizen als Funktion der Probentemperatur.
Man erkennt einen ‚kleinen’ Übergang bei niedriger Temperatur und den Hauptübergang bei
höherer Temperatur. Beide Phasenübergänge sind endotherm, aber der Hauptübergang besitzt
eine weitaus grössere Phasenumwandlungsenthalpie als der Vorübergang.
5.2 Difference Scanning Calorimetry (DSC)
Wie oben erwähnt, besteht kein quantitativer Zusammenhang zwischen der Fläche unter dem
DTA-Signal und der Phasenumwandlungsenthalpie. Für eine Reihe von Fragestellungen, wie
z.B. die Bestimmung der Kooperativität eines Phasenübergangs, ist die quantitative
Bestimmung von notwendig. Daher wird für biophysikalische Untersuchungen eine
Technik benutzt, die sich aus der DTA entwickelt hat, aber einen komplizierteren Aufbau
besitzt, die Difference Scanning Calorimetry (DSC).
0mUH∆
Abbildung 5-3 zeigt schematisch den Aufbau eine DSC-Geräts.

___________________________________________________________________________ 36
Abbildung 5-3: schematische Darstellung einer DSC-Apparatur (1 Probe, 2 Referenz, 3
Heizplatten, 4 thermisch isolierte Gefässe, 5 isolierter Mantel, 6 Temperaturmessstellen)
Proben- und Referenz-Gefäss befinden sich jeweils auf einer Heizplatte. sie enthalten eine
Lösung der zu untersuchenden Substanz bzw. das reine Lösungsmittel. Beide Gefässe
befinden sich in einem thermisch isolierten Gefäss und ein thermisch isolierter Mantel sorgt
dafür, dass keine Wärme das System nach aussen verlassen oder von aussen aufgenommen
werden kann. Beide Gefässe werden getrennt beheizt und ihre Temperaturen von je einem
Temperaturfühler erfasst. Das Aufheizen von Probe und Referenz erfolgt mit einer vor
Messbeginn eingestellten Heizrate β = ∆T/ ∆t. Die Temperatur des Systems ist damit gegeben
als:
T = T0 + β t mit T0 Temperatur zum Zeitpunkt t = 0
Das Messprinzip der DSC-Methode fordert, dass die Temperaturen von Probe (TP) und
Referenz (TR) während der Messung gleich bleiben, d.h.:
T = TP = TR

___________________________________________________________________________ 37
Findet in der Probe z.B. eine endotherme Phasenumwandlung statt, muss diese im Vergleich
zur Referenz stärker aufgeheizt werden, damit die Temperaturen gleich gehalten werden
können. Die Heizleistung für die Probe (PP) wird also grösser als die Referenz (PR). die daraus
resultierende Differenz ∆P der Heizleistung ist die Messgrösse:
∆P = PP - PR
Diese Differenz ist dem Unterschied ∆C(T) der Wärmekapazitäten proportional:
∆C(T) = CP – CR = β
)(TP∆
In DSC-Themogrammen wird ∆P gegen T aufgetragen. Bei bekannter Heizrate lässt sich aus
den Thermogrammen die Wärmekapazitätsdifferenz ∆C(T) zwischen Probe und Referenz
bestimmen.
5.3 Beispiele für kalorimetrische Messungen
Typische Beispiele für die Anwendung kalorimetrischer Messverfahren sind z.B. die
Untersuchungen von Phasenumwandlungen von Lipid-Doppelschichten die als
Modellmembran-Systeme dienen. Auch die Denaturierung von Proteinen bei Temperaturen
oberhalb von 330 K lässt sich mit diesen Verfahren gut messen. Aus Messungen der
Denaturierungsenthalpie lassen sich wichtige Hinweise über die Stabilität von
Proteinstrukturen und die Natur der Helix-Knäuel-Phasenumwandlung gewinnen. Auch bei
Poly-Nucleotiden erhält man wichtige Informationen über deren Primär- und
Sekundärstruktur. So liess sich z.B. für DNA-Moleküle ein systematischer Zusammenhang
zwischen der Doppelhelix-Einzelstrang-Umwandlungsenthalpie und dem Gehalt an den
Nucleinsäuren Guanin und Cytosin feststellen. Ausserdem können kalorimetrisch erhaltene
Enthalpiewerte als analytische Methode zur Bestimmung der relativen
Basenzusammensetzung von Polynucleotiden herangezogen werden.

___________________________________________________________________________ 38
6 Rastertunnel- und Rasterkraftmikroskopie
Einen grossen Fortschritt beim Vordringen in die direkte Abbildung atomarer Dimensionen
brachte die Entwicklung des Rastertunnelmikroskops (Scanning Tunneling Microscope,
STM) durch G. Binning und H. Rohrer im Jahr 1981. Mit diesem Instrument gelang es den
atomaren Aufbau der Materie direkt abzubilden. In der Folgezeit wurde eine ganze Reihe von
Varianten entwickelt, von denen insbesondere das Rasterkraftmikroskop (Atomic Force
Microscope, AFM) für Untersuchungen biologischer Proben eine weite Anwendung gefunden
hat.
Die Kernstücke von STM und AFM sind sehr ähnlich. Zunächst ist dies eine sehr feine Spitze,
die in einem geringen Abstand d der Probenoberfläche gegenübersteht. Abbildung 6-1 zeigt
den schematischen Aufbau eines Rastertunnelmikroskops.
Abbildung 6-1: Schematische Darstellung des Rastertunnelmikroskops
Der Abstand der Spitze von der Probe kann durch einen piezoelektrischen Antrieb sehr genau
eingestellt werden. Zwei weitere piezokeramische Stellelemente erlauben eine sehr genaue
Positionierung der Probe in x-y-Richtung und damit ein rasterförmiges Abtasten.
Beim STM ist der Abstand der metallischen Spitze und der ebenfalls elektrisch leitenden
Probe so klein, dass die Elektronenwolken der Atome von Spitze und Probe sich sehr nahe
kommen. Wird zwischen beiden eine kleine Spannung angelegt, so fliesst trotz dieses
Abstands ein geringer Strom. Ursache hierfür ist der quantenmechanische Tunneleffekt. Es
fliesst ein Tunnelstrom It der exponentiell vom Abstand d abhängt. Wird die Probe relativ zur
Spitze bewegt, dann variiert dieser Tunnelstrom entsprechend dem Oberflächenprofil und auf

___________________________________________________________________________ 39
diese Weise wird ein Bild der Höhenkontur der Oberfläche erzeugt. die Anwendung des
Tunnelmikroskops ist jedoch auf leitende und halbleitende Materialien beschränkt.
Um das Messprinzip auch zur Untersuchung von Isolatoroberflächen anzuwenden wurde das
STM zum Rasterkraftmikroskop modifiziert. Diese Modifikation bestand darin, dass zwischen
Tunnelspitze und Probe eine mir Federkraft auf die Probe gedrückte Spitze gebracht wird.
Diese Spitze ist in unmittelbarem Kontakt mit der Oberfläche der Probe. Beim Abrastern der
Oberfläche folgt die Spitze dem Oberflächenprofil und bewegt sich auf und ab. Diese
Bewegung wurde bei den ersten AFM mit Hilfe einer aufgesetzten Tunnelspitze gemessen.
Bei den heute kommerziell erhältlichen Geräten hat sich als Nachweismethode der Bewegung
der Spitze eine optische Methode durchgesetzt (siehe Abbildung 6-2).
Abbildung 6-2: Ablenkung des Kraftsensors eines Rasterkraftmikroskops
Der Lichtstrahl eines Lasers wird von der Blattfeder reflektiert und von einer meist
viergeteilten Photodiode detektiert. Die Bewegung der Spitze und damit auch der Feder führt
zu einer Veränderung des Reflexionswinkels. Folglich liefern die einzelnen Segmente der
Photodiode ein Differenzsignal, dass in eine Höheninformation umgerechnet werden kann.
Mit diesem Kontakt-AFM ist eine nahezu atomare Auflösung der Oberfläche erreichbar. Der
grosse Vorteil der AFM-Methode ist, dass die nichtleitenden biologischen Proben nicht mit

___________________________________________________________________________ 40
leitfähigen Materialien bedampft werden müssen, wodurch eine direkte und unverfälschte
Beobachtung möglich ist.
Eine andere Variante des AFM (siehe Abbildung 6-3) arbeitet nahezu kontaktfrei.
Abbildung 6-3: Aufbau eines kontaktfreien Rasterkraftmikroskops
Diese Gerät besitzt ebenfalls eine federn aufgehängte Spitze, jedoch in einem Abstand zur
Oberfläche in dem zwischen Probe und Spitze anziehende van-der-Waals-Kräfte wirken. Die
Abstandsabhängigkeit des van-der-Waals-Potentials wird in analoger Weise wie die
Abstandsabhängigkeit des Tunnelstroms beim STM verwendet. Dabei geht man so vor, dass
man nicht das Potential oder die daraus resultierende Anziehungskraft direkt misst, sondern
man verwendet eine dynamische Methode, bei der es auf den Kraftgradienten des van-der-
Waals-Potentials, also dessen zweite Ableitung nach dem abstand r, ankommt. Diesen kann
man mit einer schwingenden Spitze messen. Bringt man die Spitze nahe oder bei der
Eigenfrequenz in einem Abstand von der Oberfläche zum Schwingen, der ausserhalb des
Wirkungsbereichs des Potentials liegt, so ist die Amplitude der Schwingung durch die
Federkonstante und die Anregungsenergie der Biegefeder gegeben. Nähert man nun die

___________________________________________________________________________ 41
Spitze der Oberfläche, so überlagert sich diesem Spitzenpotential der Eigenfrequenz das van-
der-Waals-Potential. Dies führt zu einer Erniedrigung der Resonanzfrequenz und die
Amplitude der Schwingung bei der Eigenfrequenz nimmt ebenfalls ab. Die
Schwingungsamplitude ist damit ein mass für den Abstand der Spitze von der Oberfläche.
Eine andere Weiterentwicklung der Rastersondemikroskopie stellt auch das optische
Rasternahfeldmikroskop (Scanning Near Field Optical Microscope, SNOM) dar. Hier besteht
die Messsonde aus einer nach vorn zulaufenden Glasfaser, deren Wand mit einer
Aluminiumschicht bedampft ist. Das vorderste Ende wird dabei nicht mit Al beschichtet, so
dass eine Öffnung mit einem Durchmesser von ca. 50 nm bleibt, durch die in die Glasfaser
eingekoppeltes Laserlicht wieder austreten kann. Bewegt man diese Sonde in einem Abstand
von einigen nm über eine Probe, kann man Mikroskopie mit einer im Vergleich zum gängigen
Lichtmikroskop weitaus höheren Auflösung betreiben. Begrenzt wird die Auflösung durch
den Durchmesser der Sondenöffnung. Mit dieser Methode ist dann sogar Spektroskopie an
einzelnen Molekülen möglich.
7 Sequenzanalyse von Biopolymeren
Die Primärstruktur von Polysacchariden, Proteinen und Nukleinsäuren entsteht wie bereits
erwähnt aus kovalenten Bindungen von Monomereinheiten und lässt sich leicht durch
geeignete Reagenzien rückgängig machen. Deshalb gehen die Verfahren zur Aufklärung der
Primärstruktur von Biopolymeren in den meisten Fällen von einer hydrolytischen Spaltung
der Bindungen aus, da die physikalisch-chemischen Eigenschaften eines Biopolymers
weitgehend von der Sequenz unabhängig sind und eine Sequenzanalyse nur so möglich ist.
Dabei wird immer mit Markierungsmethoden gearbeitet, um die einzelnen Positionen exakt
lokalisieren und zuordnen zu können. Gängige Markierungssubstanzen sind
Fluoreszenzfarbstoffe oder Radionukleotide.
7.1 Sequenzanalyse von Proteinen
Zur schrittweisen Sequenzbestimmung von Proteinen benutzt man heute meist den Edman-
Abbau. Wie in Abbildung 7-1 dargestellt wird dabei ein Polypeptid mit Phenylisothiocyanat
umgesetzt.
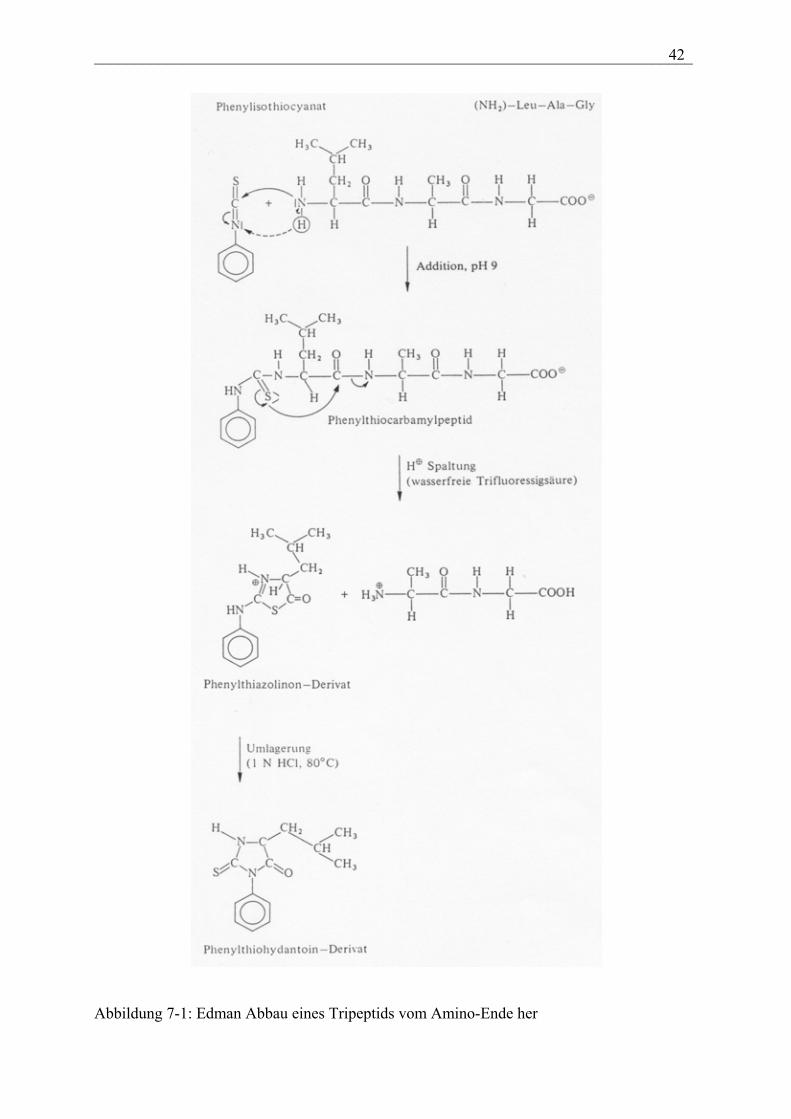
___________________________________________________________________________ 42
Abbildung 7-1: Edman Abbau eines Tripeptids vom Amino-Ende her

___________________________________________________________________________ 43
Die Reaktionsfolge kann mehrmals wiederholt werden, so dass im Prinzip schrittweise vom
Amino-Ende her eine Aminosäure nach der anderen als Phenylthiohydantonin-Dervat
abgespalten wird. Man hat die Methode technisch noch verfeinert, indem man die
Carboxylgruppe der C–terminalen Aminosäure mit einem makromolekularen Träger
verknüpft, wodurch die Abtrennung de verbleibenden Peptids aus den einzelnen
Reaktionsansätzen sehr erleichtert wird. Das Verfahren kann weitgehend automatisch in
sogenannten Sequenzern durchgeführt werden.
Da bei der Sequenzbestimmung längerer Peptide fast immer Schwierigkeiten infolge
unerwünschter Nebenreaktionen auftreten, wird man ein zu sequenzierendes Peptid durch
mindestens zwei unterschiedliche Partialhydrolysen möglichst spezifisch in kleinere Peptide
zerlegen, deren Sequenz man dann bestimmt.
7.2 Sequenzanalyse von DNA
Bei der Sequenzanalyse von DNA haben sich in den letzten Jahren besonders zwei Methoden
herauskristallisiert: die basenspezifische chemische Spaltung nach Maxam und Gilbert und
die mit Primern gestartete enzymatische Synthese nach Sanger.
7.2.1 Sequenzanalyse von DNA nach Maxam und Gilbert
Ausgangsmaterial für die chemische Spaltmethode von Maxam und Gilbert sind
einzelsträngige DNA-Fragmente, die an einem Ende radioaktiv markiert werden. In vier
getrennten Ansätzen modifiziert man jeweils Purin- oder Pyrimidinbasen oder nur Guanin
bzw. Thymin durch spezifische chemische Reaktionen. Anschliessend wird an diesen Stellen
die n-glykosidische Bindung gespalten, wodurch es hier sekundär zu einem Strangbruch
kommt, also an der Position der ursprünglichen A+G-, G-, C+T- oder C-Nukleotide. Wie in
Abbildung 7-2 gezeigt, erfolgt die Trennung der Fragmente in parallelen Lauflinien
entsprechend den vier Ansätzen. Nach Autoradiographie kann die DNS-Sequenz anhand des
Elektrophoresegels direkt in aufsteigender Reihenfolge ermittelt werden. Bei DNA-
Fragmenten liegt die Genauigkeit der Trennung noch unter ± 1 Nukleotid.

___________________________________________________________________________ 44
Abbildung 7-2: Sequenzierung nach Maxam und Gilbert
In Abbildung 7-2 wird schematisch das Prinzip der Sequenzierung des DNA-Fragments
AACAGGTC mit Hilfe der Gelelektrophorese gezeigt. Die Laufrichtung ist durch die Pfeile
markiert. Im oberen Teil der Abbildung sind diejenigen Nukleotide angegeben, an denen die
chemischen Spaltungen stattfanden. Rechts ist die Sequenz aller produzierbaren radioaktiv
markierten DNA-Fragmente angegeben, mit angenommener Markierung durch 32P am 5’-
Ende. Die Linien symbolisieren die Lage der tatsächlich gefundenen Fraktionen. Da die
Wanderungslänge streng der Kettenlänge proportional ist, gibt die jeweilige Laufstrecke die
Grösse der Fragmente an. Das 5’-terminale Nukleotid (hier A) muss gesondert identifiziert
werden.
7.2.2 Sequenzanalyse von DNA nach Sanger
Im Kettenabbruchverfahren nach Sanger wird das zu sequenzierende einzelsträngige DNA-
Stück zusammen mit einem kurzen Primer, den vier Deoxyribonucleosidtriphosphatasen von
denen je eines in vier Ansätzen radioaktiv markiert ist und der DNA-Polymerase inkubiert.
Jedes Inkubationsgemisch enthält zusätzlich ein 2’,3’-Dideoxyanalogon eines der
Nucleosidtriphosphate. Wird das Analogon eingebaut, kommt es zum Kettenabbruch. Jedes
Gemisch enthält nun verschieden lange radioaktiv markierte Oligonucleotide, die ein
gemeinsames 5’-Ende haben, sich aber in der Länge unterscheiden. Die entstandenen

___________________________________________________________________________ 45
komplementären Fragmente werden ebenfalls elektrophoretisch nach ihrer Grösse
aufgetrennt, ihre Sequenz ist dem Autoradiogramm zu entnehmen.
Abbildung 7-3: Sequenzierung nach Sanger
Im oberen Teil der Abbildung sind diejenigen Nukleotide angegeben, deren 2’, 3’-
Didesoxyanaloga den Abbruch der Kettenverlängerung bewirkt haben. Im Gegensatz zur
Methode von Maxam und Gilbert wird hier die Sequenz des komplementären Stranges der zu
untersuchenden DNA bestimmt. Ein Primer der mindestens aus zwei Nukleotiden bestehen
muss und gleichfalls komplementär zur Matrizen-DNA ist, muss bereits vorhanden sein.
Hieran werden die weiteren im Bild bezeichneten Nukleotide nacheinander gebunden. Bei der
Positionierung der einzelnen DNA-Fragmente wurde angenommen, dass in den vier Ansätzen
jeweils nur das am oberen Ende angegebene 2’, 3’-Dideoxyanlogon radioaktiv markiert war.
7.3 Einzelmolekülsequenzierungskonzepte
Die zeitaufwendige Sequenzbestimmung der herkömmlichen Methoden und Fortschritte in
der ultrasensitiven Laserfluoreszenzspektroskopie veranlassten Keller 1998 dazu, ein neues
DNA-Sequenzierungsverfahren basierend auf der Einzelmoleküldetektion zu entwickeln.
Dabei wird ein einzelner DNA-Strang bestehend aus mit Farbstoff markierten Nukleotiden in
einer Strömungsapparatur auf einem Träger befestigt. Dazu gibt man eine Exonuclease,

___________________________________________________________________________ 46
welche die DNA in Einzelnukleotide zerlegt. Die markierten Nukleotide strömen dann in
Abbaureihenfolge durch ein Messvolumen, in dem sie mit Laserlicht bestrahlt und zur
Fluoreszenz angeregt werden. Die Fluoreszenzsignale werden durch einen Detektor
aufgefangen und die Strömungsreihenfolge der Nukleotide bestimmt die Sequenz der
abgebauten DNA.
Abbildung 7-4: Schema der DNA-Sequenzierung nach der Abbaumethode
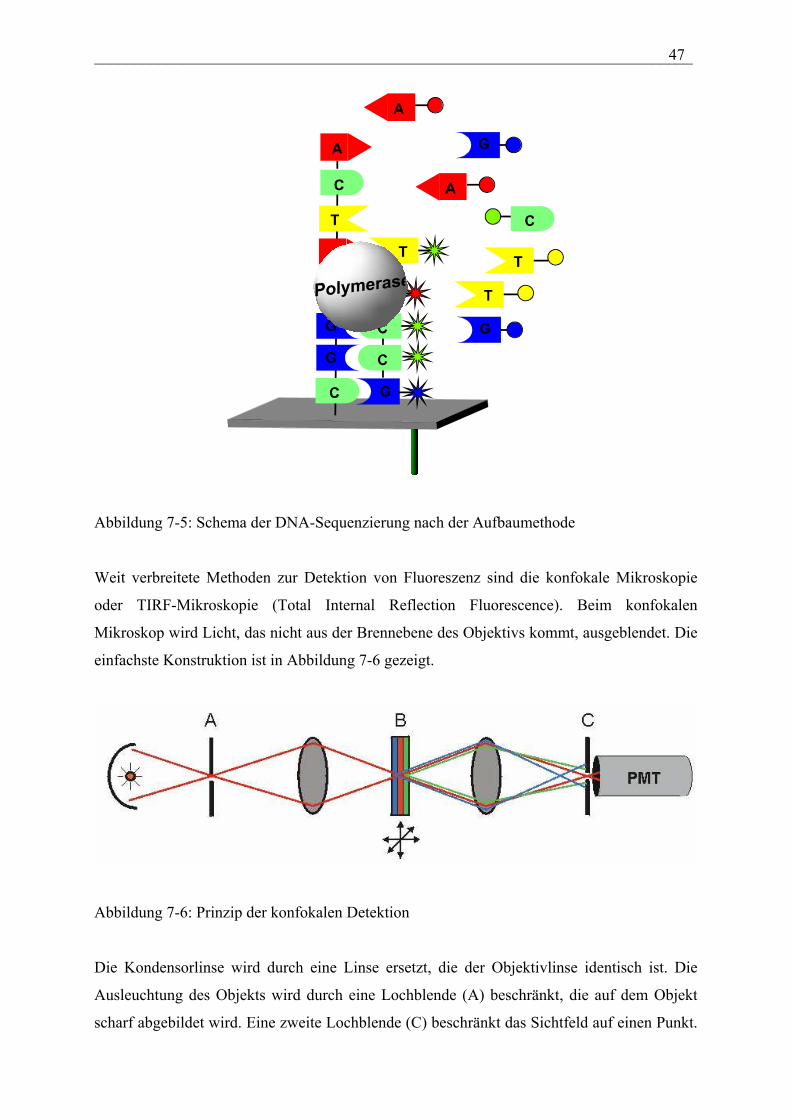
Ein weiterer Ansatz zur Einzelmolekülsequenzierung beruht auf dem Aufbau eines
komplementären DNA-Stranges mit Hilfe von fluoreszenzgelabelten dNTPs. Die zu
sequenzierende DNA wird auf der Oberfläche als Einzelstrang fixiert und der Einbau der
fluoreszenzmarkierten dNTPs durch die Polymerase detektiert. Die Reihenfolge der
Fluoreszenzsignale ergibt die Sequenz der DNA.
___________________________________________________________________________ 47
Abbildung 7-5: Schema der DNA-Sequenzierung nach der Aufbaumethode
Weit verbreitete Methoden zur Detektion von Fluoreszenz sind die konfokale Mikroskopie
oder TIRF-Mikroskopie (Total Internal Reflection Fluorescence). Beim konfokalen
Mikroskop wird Licht, das nicht aus der Brennebene des Objektivs kommt, ausgeblendet. Die
einfachste Konstruktion ist in Abbildung 7-6 gezeigt.
Abbildung 7-6: Prinzip der konfokalen Detektion
Die Kondensorlinse wird durch eine Linse ersetzt, die der Objektivlinse identisch ist. Die
Ausleuchtung des Objekts wird durch eine Lochblende (A) beschränkt, die auf dem Objekt
scharf abgebildet wird. Eine zweite Lochblende (C) beschränkt das Sichtfeld auf einen Punkt.

___________________________________________________________________________ 48
Durch den symmetrischen Aufbau dieses Systems sind beide Blenden und ein Punkt des
Objekts in der Brennebene der Linsen konfokal. Der Durchmesser der Blenden wird so klein
gewählt, daß Licht aus Bereichen des Objekts, die nicht in der Brennebene liegen, nicht in die
Apertur der Blende C fallen und damit ausgeblendet werden (hier: grüne und blaue Strahlen).
In den Photomultiplier (PMT) gelangt deshalb nur Licht aus der Brennebene des Objekts.
Im Unterschied zum konventionellen Mikroskop erzeugt das konfokale Mikroskop also
zunächst nur einen Bildpunkt, der allerdings genau einen Punkt aus der Brennebene des
Objektivs darstellt.
8 Massenspektroskopische Methoden in der Bioanalytik
Für die Peptid- und Proteinanalytik wird inzwischen verstärkt die Massenspektroskopie (MS)
eingesetzt. Die MS ist eine Analysemethode zur Ermittlung der Molekülmasse freier Ionen im
Hochvakuum. Massenspektrometer bestehen aus einer Ionenquelle, in der aus der Probe
gasförmige Ionen erzeugt werden. Im Massenanalysator werden die Ionen nach ihrem
Masse/Ladungs- Quotienten (m/z) aufgetrennt. Ein Detektor liefert ein Massenspektrum, aus
dem abgelesen werden kann, welche Ionen in welchen relativen Mengen erzeugt werden. Die
zur Erzeugung der Ionen verwendeten Energien waren lange für die Peptidanalytik
unbrauchbar, weil sie die biologischen Proben zerstörten. Mit der Entwicklung schonenderer
Techniken, bspw. MALDI (matrix assisted laser desorption ionisation), wurde die MS zu
einer wichtigen Protein- Analysemethode. Bei bekannter Aminosäuresequenz kann bspw. aus
dem Vergleich zwischen der theoretischen Masse und der Masse des untersuchten "nativen"
Proteins auf posttranslationale Modifikationen geschlossen werden.
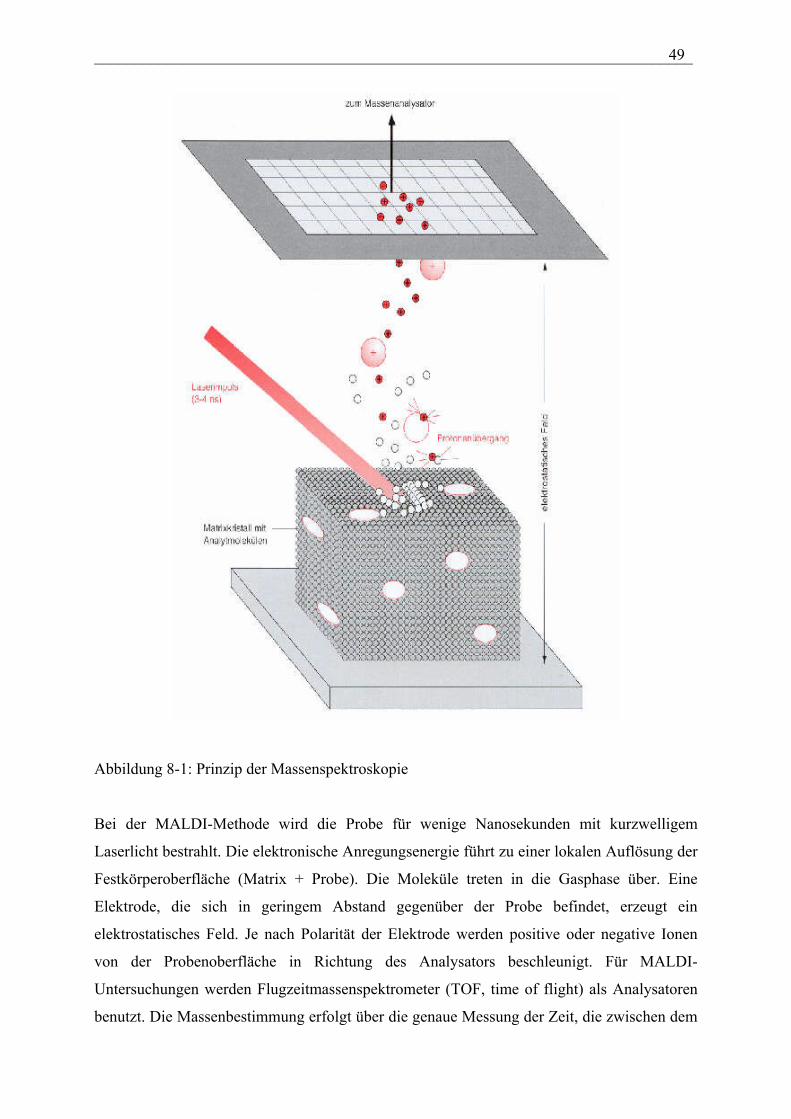
___________________________________________________________________________ 49
Abbildung 8-1: Prinzip der Massenspektroskopie
Bei der MALDI-Methode wird die Probe für wenige Nanosekunden mit kurzwelligem
Laserlicht bestrahlt. Die elektronische Anregungsenergie führt zu einer lokalen Auflösung der
Festkörperoberfläche (Matrix + Probe). Die Moleküle treten in die Gasphase über. Eine
Elektrode, die sich in geringem Abstand gegenüber der Probe befindet, erzeugt ein
elektrostatisches Feld. Je nach Polarität der Elektrode werden positive oder negative Ionen
von der Probenoberfläche in Richtung des Analysators beschleunigt. Für MALDI-
Untersuchungen werden Flugzeitmassenspektrometer (TOF, time of flight) als Analysatoren
benutzt. Die Massenbestimmung erfolgt über die genaue Messung der Zeit, die zwischen dem
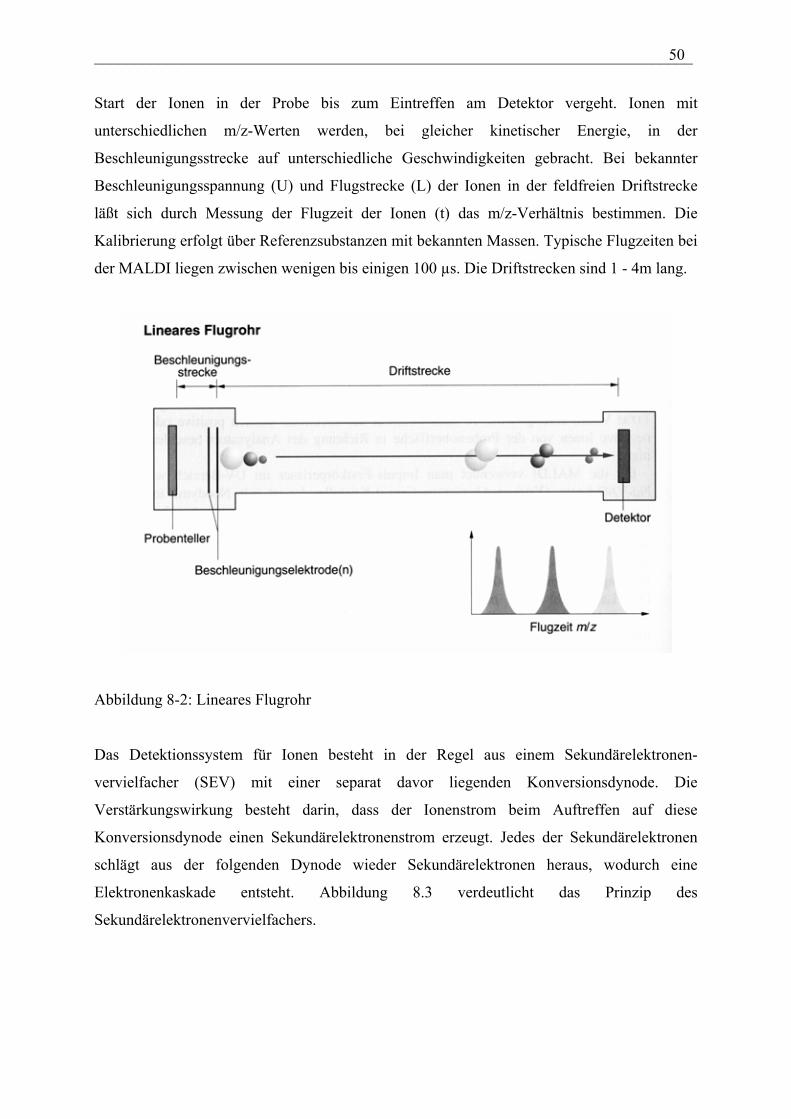
___________________________________________________________________________ 50
Start der Ionen in der Probe bis zum Eintreffen am Detektor vergeht. Ionen mit
unterschiedlichen m/z-Werten werden, bei gleicher kinetischer Energie, in der
Beschleunigungsstrecke auf unterschiedliche Geschwindigkeiten gebracht. Bei bekannter
Beschleunigungsspannung (U) und Flugstrecke (L) der Ionen in der feldfreien Driftstrecke
läßt sich durch Messung der Flugzeit der Ionen (t) das m/z-Verhältnis bestimmen. Die
Kalibrierung erfolgt über Referenzsubstanzen mit bekannten Massen. Typische Flugzeiten bei
der MALDI liegen zwischen wenigen bis einigen 100 µs. Die Driftstrecken sind 1 - 4m lang.
Abbildung 8-2: Lineares Flugrohr
Das Detektionssystem für Ionen besteht in der Regel aus einem Sekundärelektronen-
vervielfacher (SEV) mit einer separat davor liegenden Konversionsdynode. Die
Verstärkungswirkung besteht darin, dass der Ionenstrom beim Auftreffen auf diese
Konversionsdynode einen Sekundärelektronenstrom erzeugt. Jedes der Sekundärelektronen
schlägt aus der folgenden Dynode wieder Sekundärelektronen heraus, wodurch eine
Elektronenkaskade entsteht. Abbildung 8.3 verdeutlicht das Prinzip des
Sekundärelektronenvervielfachers.

___________________________________________________________________________ 51
Abbildung 8.3: Sekundärelektronenvervielfacher
Eine wichtige Eigenschaft eines Massenanalysators ist das Auflösungsvermögen, d.h. Ionen
mit geringen Massendifferenzen noch getrennt voneinander registrieren zu können.
Unter dem Begriff Auflösungsvermögen R wird im allgemeinen das Verhältnis von einer
Ionenmasse m zur Massendifferenz m verstanden:
R = m/∆m
∆m gibt den kleinsten Massenabstand, bei dem Massenlinien gleicher Intensität noch getrennt
erscheinen. Um verschiedene Auflösungen miteinander vergleichen zu können, muss die
Trennung zunächst definiert werden. In der hochauflösenden Massenspektrometrie wird
normalerweise eine Auflösungsdefinition verwendet, bei der das Tal zwischen zwei gleich
großen Signalen nicht mehr als 10 % der Signalhöhe ausmachen darf. In der
niederauflösenden Massenspektrometrie wird dagegen oft der Begriff der Peakhalbwertsbreite
(auch FWHM = full width at half maximum) für m (also bei 50 % Peakhöhe) verwendet.
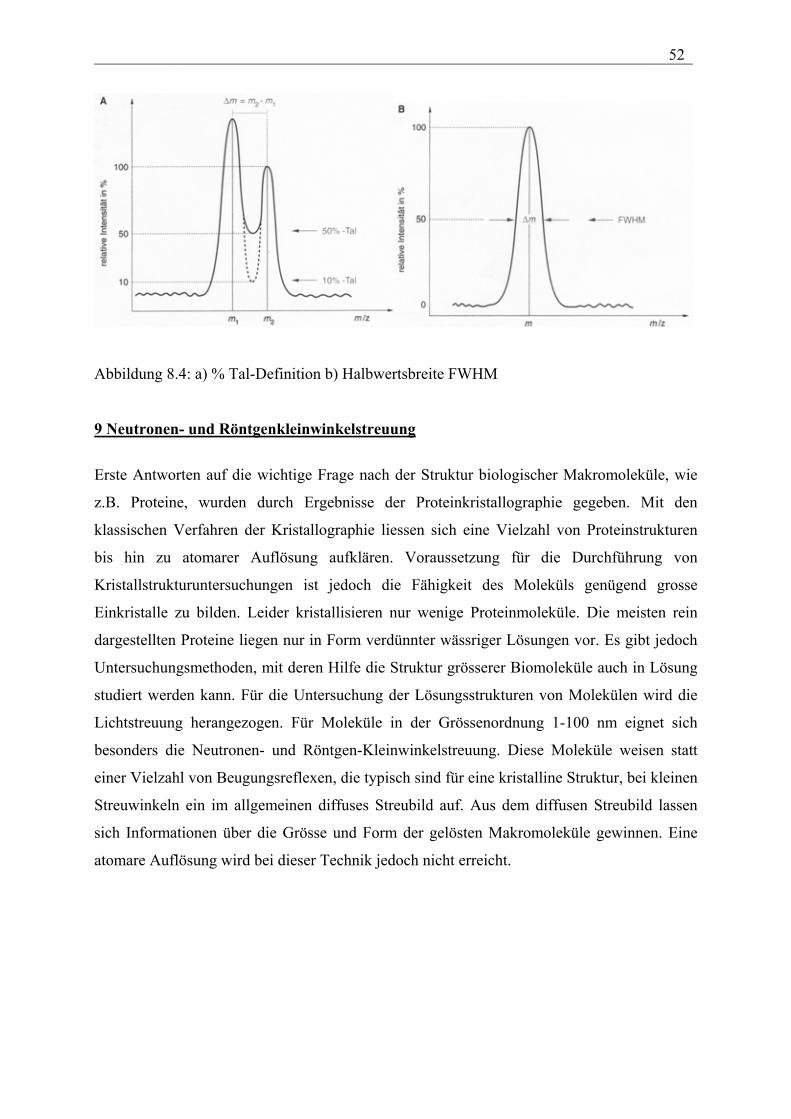
___________________________________________________________________________ 52
Abbildung 8.4: a) % Tal-Definition b) Halbwertsbreite FWHM
9 Neutronen- und Röntgenkleinwinkelstreuung
Erste Antworten auf die wichtige Frage nach der Struktur biologischer Makromoleküle, wie
z.B. Proteine, wurden durch Ergebnisse der Proteinkristallographie gegeben. Mit den
klassischen Verfahren der Kristallographie liessen sich eine Vielzahl von Proteinstrukturen
bis hin zu atomarer Auflösung aufklären. Voraussetzung für die Durchführung von
Kristallstrukturuntersuchungen ist jedoch die Fähigkeit des Moleküls genügend grosse
Einkristalle zu bilden. Leider kristallisieren nur wenige Proteinmoleküle. Die meisten rein
dargestellten Proteine liegen nur in Form verdünnter wässriger Lösungen vor. Es gibt jedoch
Untersuchungsmethoden, mit deren Hilfe die Struktur grösserer Biomoleküle auch in Lösung
studiert werden kann. Für die Untersuchung der Lösungsstrukturen von Molekülen wird die
Lichtstreuung herangezogen. Für Moleküle in der Grössenordnung 1-100 nm eignet sich
besonders die Neutronen- und Röntgen-Kleinwinkelstreuung. Diese Moleküle weisen statt
einer Vielzahl von Beugungsreflexen, die typisch sind für eine kristalline Struktur, bei kleinen
Streuwinkeln ein im allgemeinen diffuses Streubild auf. Aus dem diffusen Streubild lassen
sich Informationen über die Grösse und Form der gelösten Makromoleküle gewinnen. Eine
atomare Auflösung wird bei dieser Technik jedoch nicht erreicht.

___________________________________________________________________________ 53
9.1 Prinzip des Streuexperiments
Das Prinzip eines Streuexperiments ist in Abbildung 9-1 dargestellt.
Abbildung 9-1: Prinzip eines Streuexperiments
Der einfallende Strahl mit Wellenvektor (0
→
k λπ2=
→
k ), Wellenlänge λ und Energie E0 trifft
auf die zu untersuchende Probe. Gemessen wird die Intensität der gestreuten Strahlung mit
Wellenvektor und Energie E1
→
k 1 unter dem Streuwinkel 2θ im Raumwinkelelement dΩ. aus
der gemessenen Streuintensität I(Q,ħω) in Abhängigkeit vom Impulsübertrag h und
Energieübertrag ħω mit:
→
Q
)( 01
→→→
−= kkQ hh
E∆=−= )( 01 ωωω hh
erhält man Informationen über die strukturellen und dynamischen Eigenschaften der Probe.
Für den Fall der elastischen Streuung (∆E=0, →→
= 01 kk ) gilt:
Θ=Θ=→
sin4sin2 0 λπkQ
___________________________________________________________________________ 54
Strukturuntersuchungen erfolgen stets aufgrund elastischer Streuung. Im allgemeinen wird der
differentielle Streuquerschnitt dσ/dΩ im elastischen Streuexperiment ermittelt. Er ist definiert
als das Verhältnis aus der Anzahl der in das Raumwinkelelement dΩ pro Zeiteinheit
gestreuten Teilchen zum Produkt aus dem einfallenden Teilchenstrom und dΩ.
In vielen Fällen besteht ein einfacher Zusammenhang zwischen der Messgrösse dσ/dΩ und
den strukturellen Eigenschaften der Probe. Der differentielle Streuquerschnitt kann allgemein
beschrieben werden als:
ktorStrukturfafNdd
s ⋅Θ∝Ω
2)2(σ
Der sogenannte Strukturfaktor charakterisiert die Struktur der Probe. der atomare Streufaktor
f(2Θ) ist abhängig von der Streusonde (Neutronen- bzw. Röntgenstrahlen) und ist ein Mass
für die Streukraft der einzelnen Streuzentren (Ns Anzahl der Streuzentren).
Abbildung 9-2 zeigt die Abhängigkeit des Streufaktors für Neutronen und Röntgenstrahlen.
Abbildung 9-2: Abhängigkeit des atomaren Streufaktors

___________________________________________________________________________ 55
Abbildung 9-3 gibt einen Überblick über die Unterschiede zwischen Neutronen und
Röntgenstrahlen.
größere Eindringtiefe, größere Probenµ ~ 0.1cm-1µ ~ 102-103cm-1Absorption
1Å: ~4000m/scGeschwindigkeit
Energieübertrag meßbar
Energieübertrag vernachlässigbar
Inelastische Streuung
Spektroskopie mit Neutronen (Phononen, Schwingung, Rotation, Diffusion)
0.0816eV12400eVEnergie bei 1Å
Leichtatome sichtbar, Elementunterscheidung (Al⇔Si, Fe⇔Co⇔Ni), Rückstreuung
„zufällig“ verteilt, θ-unabhängig
~ Z, θ-abhängig (Atomformfaktor)
Atomare Streukraft
Mit Neutronen sind Isotope unterscheidbarVariation des Streukontrasts möglich,
insbesondere H⇔D
1) „Starke Wechselwirkung“ 2) Magnet. Dipol-wechselwirkung
Elektromagnet. Feld der Strahlung wechselwirkt
mit Elektronen
Wechselwirkung
µ = -1.91 µN-Magnet. Moment
Ideale Sonde für Spin- und magnetische Strukturen
1/21Spin
Teilchen (λ = h/p)ElektromagnetischStrahlungsart
NeutronenRöntgen
Abbildung 9-3: Vergleich Röntgen-Neutronen
10 Fluoreszenzspektroskopische Methoden in der Bioanalytik
In den letzten 15 Jahren hat die Fluoreszenzspektroskopie eine weite Verbreitung bei
biologischen und biochemischen Anwendungen erfahren. Moderne Anwendungsgebiete der
Fluoreszenzspektroskopie sind z.B.:
- DNA-Sequenzing
- FISH (Fluorescence In Situ Hybridization)
- Antigen-Antikörper-Reaktionen (Immunoassays)
- Klinisch chemische Nachweisverfahren
- Zellbiologie (z.B Cytometrie)
Die Fluoreszenzspektroskopie profitiert gegenüber vergleichbaren Techniken wie
Radioassays oder ELISA-Tests (Enzyme Linked Immunosorbent Assay) vor allem durch
hohe Sensitivität, geringere Kosten und im Vergleich zu den Radioassays vor allem von der
Entsorgungsproblematik die mit radioaktiven Markern verbunden ist.

___________________________________________________________________________ 56
10.1 Definition der Fluoreszenz
Unter Fluoreszenz versteht man die spontane Emission von Licht bei der Rückkehr eines
Moleküls aus einem elektronisch angeregten Singulettzustandzustand, in welchen es durch
Lichtabsorption angeregt wurde, in den elektronischen Grundzustand. Dabei bleibt der
Elektronenspin erhalten. Die Lebensdauer τ des angeregten Zustandes beträgt um die 10 ns,
was einer Emissionsrate von 108 s-1 entspricht. Daraus folgt, daß die Lichterscheinung nach
Abstellen der Anregungsquelle sofort erlischt.
Ein Fluoreszenz(emissions)spektrum ist die Darstellung von Fluoreszenzintensität gegen die
Wellenlänge des Fluoreszenzlichtes bei Anregung mit einer konstanten Wellenlänge.
10.2 Fluorophore
Voraussetzung für Fluoreszenz ist, daß das eingestrahlte Licht von Molekülen absorbiert
werden kann und dadurch Elektronenübergänge induziert werden. Derartige Moleküle
besitzen im allgemeinen ein delokalisiertes π-Elektronensystem. Viele Biomoleküle enthalten
natürliche Fluorophore, manche sind jedoch fluoreszenzinaktiv und müssen mit
Fluoreszenzfarbstoffen markiert werden, um detektiert werden zu können. Abbildung 10-1
zeigt einige typische Moleküle mit Fluoreszenzeigenschaften.
Abbildung 10-1: Typische fluoreszierende Moleküle

___________________________________________________________________________ 57
10.3 Elektronenübergänge
Fluoreszenz fällt unter den Oberbegriff der Lumineszenz, welche allgemein die Emission von
Photonen aus elektronisch angeregten Zuständen charakterisiert. Ein weiterer Typ von
Lumineszenz ist die Phosphoreszenz. Phosphoreszenz ist die Lichtemission, die aus
Übergängen zwischen Zuständen verschiedener Spinmultiplizität resultiert. Der angeregte
Zustand ist in der Regel ein Triplettzustand und eine Rückkehr in den elektronischen
Grundzustand muß unter Spinumkehr erfolgen, damit die Spins gepaart sind. Ein solcher
Übergang ist im Grunde verboten und deswegen zeitlich verzögert. Typische Lebensdauern
betragen Millisekunden bis zu Sekunden und werden eher durch mögliche
Deaktivierungsprozesse begrenzt als durch die eigentliche Emission.
Unter der Lebensdauer τ versteht man die durchschnittliche Zeit, die ein Fluorophor (ein
Molekül, das Fluoreszenz zeigt) im angeregten Zustand verbleibt. Da die Fluoreszenz ein
zufälliges Ereignis ist, wird es wenige Moleküle geben, die tatsächlich zum Zeitpunkt t = τ
ihre Photonen emittieren. Für einen einfachen exponentiellen Abfall der Fluoreszenzintensität
gilt beispielsweise, daß 63% der Moleküle vor t = τ emittieren und 37% später als die
Lebensdauer es angibt.
Die elektronischen Prozesse, die der Fluoreszenz und Phosphoreszenz zugrunde liegen,
werden im Jablonski-Diagramm (Abbildung 10-2) dargestellt.
Abbildung 10-2: Jablonski-Diagramm

___________________________________________________________________________ 58
Die ersten elektronischen Singulettzustände sind mit S0, S1, S2 bezeichnet, entsprechend
kennzeichnet T1 den ersten angeregten Triplettzustand. Ein Fluorophor in einem bestimmten
elektronischen Zustand kann eine Vielzahl von Schwingungszuständen einnehmen. Unter
internal conversion (innere Umwandlung) versteht man einen strahlungslosen Übergang, der
zwischen zwei Zuständen gleichen Spins (S2 → S1) vom niedrigsten Schwingungszustand
eines höheren elektronische Zustands in einen energiegleichen höheren Schwingungszustand
des nächst niederen Elektronenzustand stattfindet. Intersystem crossing ist der strahlungslose
Übergang von einem angeregten Singulettzustand in einen energiegleichen Triplettzustand.
10.4 Franck-Condon Prinzip
Ein wesentliches Charakteristikum von elektronischen Übergängen, ist, daß sie vertikal sind,
d.h. ohne Änderung der Kernpositionen. Da die Atomkerne sehr viel schwerer sind als die
Elektronen (mp = 1,672 10-27 kg, me = 9,109 10-31 kg), sind elektronische Übergänge wie
Anregung durch Lichtabsorption (10-15 s), internal conversion (10-12 s) und
Fluoreszenzemission (10-8 s) sehr viel schneller als die Kerne sich an die neue elektronische
Umgebung anpassen können. Dieses Merkmal der Elektronenübergänge entspricht dem
Franck-Condon Prinzip.
Bei Raumtemperatur sind die meisten Moleküle in ihrem Schwingungsgrundzustand, d.h. die
Absorption von Licht geeigneter Wellenlänge wird zu einer vertikalen Anregung aus diesem
Schwingungsniveau führen. Es sind jedoch (vertikale) Übergänge in verschiedene
Schwingungszustände des angeregten elektronischen Zustands möglich, was eine Feinstruktur
im Absorptionsspektrum bewirken kann.
Die Wahrscheinlichkeit der jeweiligen Übergänge und damit die Intensität des
Absorptionssignales werden durch die Schwingungswellenfunktionen des angeregten
elektronischen Zustands bestimmt. Der Übergang erfolgt von der Kernlage der maximalen
Amplitude der Schwingungsgrundzustandswellenfunktion in ein Schwingungsniveau des
angeregten elektronischen Zustandes, wo eine maximale Amplitude der Wellenfunktion bei
derselben Kernlage auftritt. Derjenige Übergang, bei dem im angeregten Zustand die
Amplitude der Schwingungswellenfunktion maximal ist, hat die größte Intensität. In welches
Schwingungsniveau angeregt wird hängt damit von der relativen Verschiebung der
betreffenden Potentialkurven für den Grund- und angeregten Zustand ab. Da in anderen
Schwingungsniveaus aber auch signifikante Aufenthaltswahrscheinlichkeiten bestehen

___________________________________________________________________________ 59
können, finden auch diese Übergänge statt, wenn auch mit geringerer Wahrscheinlichkeit und
damit Intensität.
Der Lichtabsorption können nun viele Prozesse folgen. Zum einen tritt eine schnelle
strahlungslose Relaxation (innerhalb von 10-12 s) des Elektrons aus einem angeregten Zustand
in den Schwingungsgrundzustand des angeregten elektronischen Zustandes ein (internal
conversion). Aus diesem Zustand erfolgt dann die Lichtemission. Die bei der strahlungslosen
Relaxation freiwerdende Energie wird in Form von Wärme an das System abgegeben.
Anstelle einer sofortigen Lichtemission kann auch ein intersystem crossing in einen
Triplettzustand gleicher elektronischer Energie stattfinden, aus dem dann Phosphoreszenz
resultieren kann. Tritt Fluoreszenz ein, so erfolgt ein Elektronenübergang aus dem
Schwingungsgrundzustand des elektronisch angeregten Zustands in ein Schwingungsniveau
des elektronischen Grundzustands.
10.5 Spiegelbildregel
Aus diesen Betrachtungen für die Absorption und Emission eines Fluorophoren folgt, daß das
Absorptionsspektrum des Moleküls die Schwingungsniveaus des elektronisch angeregten
Zustands widerspiegelt, das Emissionsspektrum die des elektronischen Grundzustands.
Hieraus ergibt sich, daß ein Fluoreszenzemissionsspektrum oftmals ein Spiegelbild des
entsprechenden Absorptionsspektrums (S0 → S1) zu sein scheint. Dies liegt darin begründet,
daß die Schwingungsniveaus eines elektronisch angeregten Zustands sich nicht sehr von
denen des Grundzustands unterscheiden, so daß der wahrscheinlichste Absorptionsprozeß
auch gleich der wahrscheinlichste Emissionsübergang ist. Ist der Absorptionsübergang im
Spektrum relativ höherenergetisch, so ist der Emissionsübergang im Emissionsspektrum
relativ niederenergetisch. Ein Beispiel ist z.B. das Absorptions- und Emissionsspektrum von
Perylen (Abbildung 10-3).
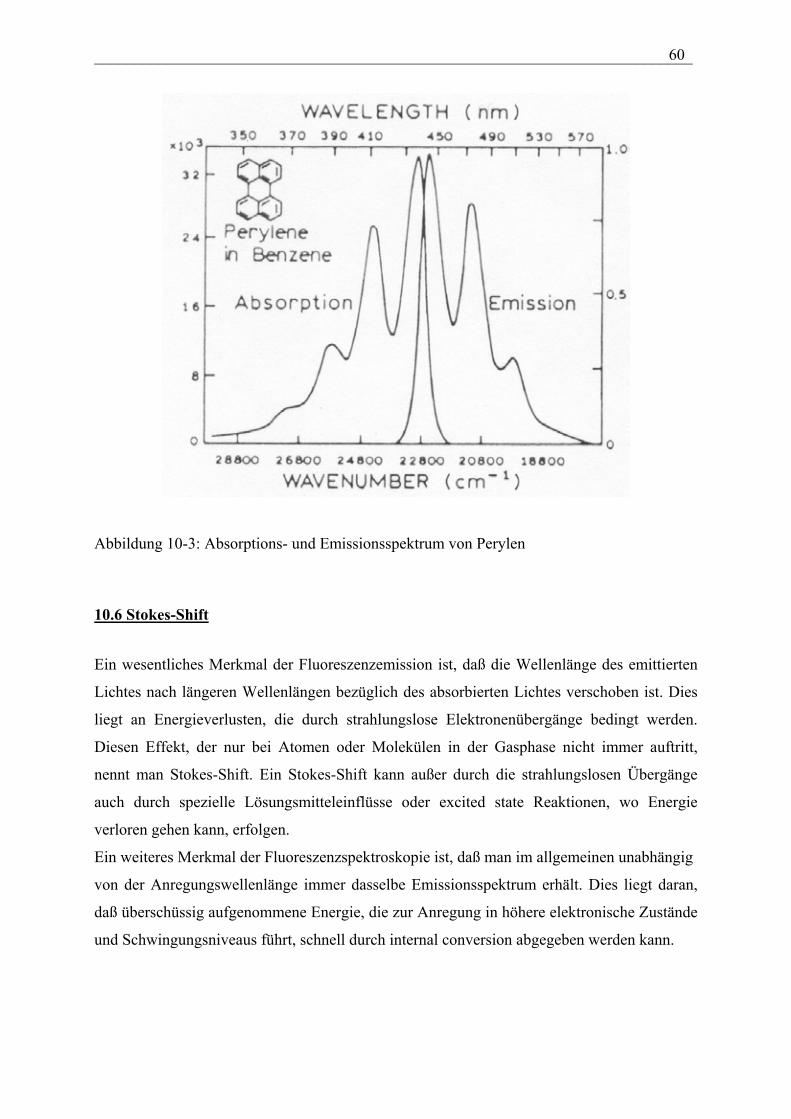
___________________________________________________________________________ 60
Abbildung 10-3: Absorptions- und Emissionsspektrum von Perylen
10.6 Stokes-Shift
Ein wesentliches Merkmal der Fluoreszenzemission ist, daß die Wellenlänge des emittierten
Lichtes nach längeren Wellenlängen bezüglich des absorbierten Lichtes verschoben ist. Dies
liegt an Energieverlusten, die durch strahlungslose Elektronenübergänge bedingt werden.
Diesen Effekt, der nur bei Atomen oder Molekülen in der Gasphase nicht immer auftritt,
nennt man Stokes-Shift. Ein Stokes-Shift kann außer durch die strahlungslosen Übergänge
auch durch spezielle Lösungsmitteleinflüsse oder excited state Reaktionen, wo Energie
verloren gehen kann, erfolgen.
Ein weiteres Merkmal der Fluoreszenzspektroskopie ist, daß man im allgemeinen unabhängig
von der Anregungswellenlänge immer dasselbe Emissionsspektrum erhält. Dies liegt daran,
daß überschüssig aufgenommene Energie, die zur Anregung in höhere elektronische Zustände
und Schwingungsniveaus führt, schnell durch internal conversion abgegeben werden kann.

___________________________________________________________________________ 61
10.7 Quantenausbeute und Fluoreszenzlebensdauer
Die Fluoreszenz Quantenausbeute Q ist das Verhältnis der Anzahl emittierter Photonen zur
Anzahl der absorbierten Photonen. Es gilt:
ICF
F
kkkQ+
=
wobei kF die Photonenemissionsrate des Fluorophors ist und kIC die Rate der strahlungslosen
Übergänge von S1 nach S0.
Mit den Raten kF und kIC ergibt sich für die Lebensdauer des Fluorophors (die mittlere Zeit,
die ein Molekül im angeregten Zustand bleibt):
)(1
ICF kk +=τ
10.8 Fluoreszenzlöschung (Quenching) Unter Fluoreszenzlöschung versteht man die strahlungslose Desaktivierung des angeregten
Zustands des Fluorophors. Quenching setzt also die Fluoreszenz-Quantenausbeute und die
Fluoreszenzintensität herab und kann durch verschiedene Prozesse erfolgen, z.B. durch
Energietransfer, Elektronen- bzw. Protonentransfer, und chemische Reaktionen im
Anregungszustand. Man unterscheidet zwei Arten von Löschprozessen: die dynamische
Fluoreszenz-Löschung, die sich durch Kollisionsprozesse des Fluorophors mit den
Quenchermolekülen ergibt, und die statische Fluoreszenz-Löschung, die auf Komplexbildung
des Fluorophors mit den Quenchern beruht. Die Effektivität beider Prozesse ist von der
Konzentration der Löschermoleküle abhängig. In beiden Prozessen ist der direkte molekulare
Kontakt zwischen Fluorophor und Quencher erforderlich. Eine Vielzahl von Verbindungen
eignen sich als Fluoreszenzquencher. Typische, häufig verwendete Quencher sind Iodid, O2,
Acrylamid und Amine.
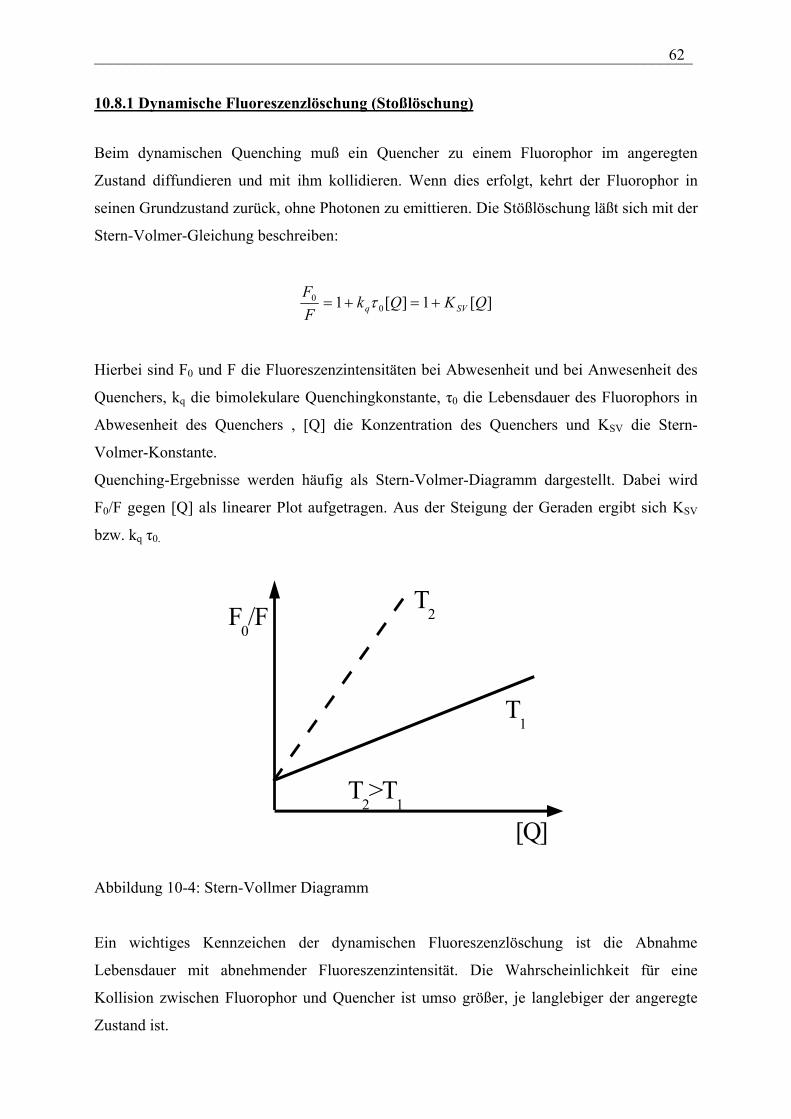
___________________________________________________________________________ 62
10.8.1 Dynamische Fluoreszenzlöschung (Stoßlöschung)
Beim dynamischen Quenching muß ein Quencher zu einem Fluorophor im angeregten
Zustand diffundieren und mit ihm kollidieren. Wenn dies erfolgt, kehrt der Fluorophor in
seinen Grundzustand zurück, ohne Photonen zu emittieren. Die Stößlöschung läßt sich mit der
Stern-Volmer-Gleichung beschreiben:
][1][1 00 QKQkFF
SVq +=+= τ
Hierbei sind F0 und F die Fluoreszenzintensitäten bei Abwesenheit und bei Anwesenheit des
Quenchers, kq die bimolekulare Quenchingkonstante, τ0 die Lebensdauer des Fluorophors in
Abwesenheit des Quenchers , [Q] die Konzentration des Quenchers und KSV die Stern-
Volmer-Konstante.
Quenching-Ergebnisse werden häufig als Stern-Volmer-Diagramm dargestellt. Dabei wird
F0/F gegen [Q] als linearer Plot aufgetragen. Aus der Steigung der Geraden ergibt sich KSV
bzw. kq τ0.
T1
T2
[Q]
F0/F
T2>T
1
Abbildung 10-4: Stern-Vollmer Diagramm
Ein wichtiges Kennzeichen der dynamischen Fluoreszenzlöschung ist die Abnahme
Lebensdauer mit abnehmender Fluoreszenzintensität. Die Wahrscheinlichkeit für eine
Kollision zwischen Fluorophor und Quencher ist umso größer, je langlebiger der angeregte
Zustand ist.

___________________________________________________________________________ 63
ττ 0
0
=FF
Die bimolekulare Quenching-Konstante kann durch die Smoluchowski-Gleichung berechnet
werden.
))((10004
QFQFA
q DDRRNk ++=π
γ
Dabei sind R die Stoßradien von Fluorophor F und Quencher Q, und D die Summe der
Diffusionskoeffizienten von F und Q, γ ist die Quench-Effizienz. Die Diffusionskoeffizienten
wiederum sind von der Temperatur und der Viskosität η der Lösung abhängig und durch die
Einstein-Stokes-Gleichung beschreibbar.
RkTDπη6
=
Insgesamt ist die Stern-Volmer-Konstante also temperatur- und viskositätsabhängig. Bei
höherer Temperatur steigt auch KSV.
10.8.2 Statisches Quenching
Im statischen Quenching-Prozeß bildet sich zwischen Fluorophor F und Quencher Q ein
nichtfluoreszierender Grundzustandskomplex FQ. Wenn dieser Komplex Licht absorbiert,
kehrt er sofort wieder in den Grundzustand zurück ohne Photonen zu emittieren. Für die
Komplexbildung kann die Komplexbildungskonstante formuliert werden:
]][[][QF
FQKS =
Auch das statisches Quenching läßt sich durch die Stern-Volmer-Gleichung beschreiben, in
diesem Fall ist KS aber die Komplexbildungskonstante und nicht die diffusionskontrollierte
Stern-Volmer-Konstante KSV.
Im Unterschied zum dynamischen Quenching wird KS meistens bei Temperaturerhöhung
kleiner, da dadurch die Stabilität des Komplexes sinkt. Außerdem bleibt die Fluoreszenz-

___________________________________________________________________________ 64
Lebensdauer konstant (τ0/τ = 1), weil die Komplexbildung lediglich die Konzentration an
freiem Fluorophor reduziert und die Lebensdauer der angeregten Moleküle nicht beeinflußt.
In vielen Fällen kann der Fluorophor durch Kombination von dynamischer und statischer
Fluoreszenzlöschung gequencht werden. Dann ergibt sich kein linearer, sondern ein
kurvenartiger Verlauf im Stern-Volmer-Diagramm, da die kombinierte Stern-Volmer-
Gleichung zweiter Ordnung bezüglich der Konzentration des Quenchers ist.

___________________________________________________________________________ 65
Inhaltsverzeichnis
Einführung:................................................................................................................................. 2 1. Biopolymere ........................................................................................................................... 2
1.1 Was sind Biopolymere? ................................................................................................... 2 1.2 Polyisoprene: Kautschuk, Guttapercha, Balata ................................................................ 3 1.3 Lignin ............................................................................................................................... 4 1.4 Grösse und Form der Biopolymere .................................................................................. 4
2. Kolligative und hydrodynamische Eigenschaften von Biopolymeren................................... 5 2.1 Osmose ............................................................................................................................. 5 2.2 Dialyse von Polyelektrolyten ........................................................................................... 8 2.3 Sedimentation................................................................................................................. 10 2.4 Sedimentationsgeschwindigkeit ..................................................................................... 11 2.5 Das Sedimentationsgleichgewicht.................................................................................. 13 2.6 Elektrophorese................................................................................................................ 14 2.7 Isoelektrische Fokussierung ........................................................................................... 16 2.8 Das Zeta-Potential .......................................................................................................... 18 2.9 Viskosität........................................................................................................................ 19
3 Konformationsumwandlungen von Biopolymeren ............................................................... 25 4 Konformation und Konfiguration von Biopolymeren........................................................... 30
4.1 Ungeordnete Knäuel....................................................................................................... 31 4.2 Helix- und Faltblattstruktur ............................................................................................ 32
5 Thermisch-kalorische Messverfahren ................................................................................... 33 5.1 Differenz-Thermoanalyse (DTA)................................................................................... 33 5.2 Difference Scanning Calorimetry (DSC) ....................................................................... 35 5.3 Beispiele für kalorimetrische Messungen ...................................................................... 37
6 Rastertunnel- und Rasterkraftmikroskopie............................................................................ 38 7 Sequenzanalyse von Biopolymeren ...................................................................................... 41
7.1 Sequenzanalyse von Proteinen ....................................................................................... 41 7.2 Sequenzanalyse von DNA.............................................................................................. 43
7.2.1 Sequenzanalyse von DNA nach Maxam und Gilbert.............................................. 43 7.2.2 Sequenzanalyse von DNA nach Sanger .................................................................. 44
7.3 Einzelmolekülsequenzierungskonzepte ......................................................................... 45 8 Massenspektroskopische Methoden in der Bioanalytik ........................................................ 48 9 Neutronen- und Röntgenkleinwinkelstreuung ...................................................................... 52
9.1 Prinzip des Streuexperiments ......................................................................................... 53 10 Fluoreszenzspektroskopische Methoden in der Bioanalytik............................................... 55
10.1 Definition der Fluoreszenz ........................................................................................... 56 10.2 Fluorophore .................................................................................................................. 56 10.3 Elektronenübergänge.................................................................................................... 57 10.4 Franck-Condon Prinzip ................................................................................................ 58 10.5 Spiegelbildregel............................................................................................................ 59 10.6 Stokes-Shift .................................................................................................................. 60 10.7 Quantenausbeute und Fluoreszenzlebensdauer............................................................ 61 10.8 Fluoreszenzlöschung (Quenching) ............................................................................... 61
10.8.1 Dynamische Fluoreszenzlöschung (Stoßlöschung)............................................... 62 10.8.2 Statisches Quenching ............................................................................................ 63





























